

Cellular senescence limits proliferation of potentially detrimental cells, preventing tumorigenesis and restricting tissue damage. However, the function of senescence in nonpathological conditions is unknown. Here, Krizhanovsky and colleagues discover a new pathway to activate senescence cell fusion. The authors find that fusion-induced senescence occurs during embryonic development in the placenta. A counterpart of this process is also observed after infection by the measles virus. The results suggest that fusion-induced senescence is essential during development, and reuse of this program later in life protects agains viral infections.
Keywords: cellular senescence, cell fusion, placenta, ERVWE1, p53, pRb
Abstract
Cellular senescence limits proliferation of potentially detrimental cells, preventing tumorigenesis and restricting tissue damage. However, the function of senescence in nonpathological conditions is unknown. We found that the human placental syncytiotrophoblast exhibited the phenotype and expressed molecular markers of cellular senescence. During embryonic development, ERVWE1-mediated cell fusion results in formation of the syncytiotrophoblast, which serves as the maternal/fetal interface at the placenta. Expression of ERVWE1 caused cell fusion in normal and cancer cells, leading to formation of hyperploid syncytia exhibiting features of cellular senescence. Infection by the measles virus, which leads to cell fusion, also induced cellular senescence in normal and cancer cells. The fused cells activated the main molecular pathways of senescence, the p53- and p16–pRb-dependent pathways; the senescence-associated secretory phenotype; and immune surveillance-related proteins. Thus, fusion-induced senescence might be needed for proper syncytiotrophoblast function during embryonic development, and reuse of this senescence program later in life protects against pathological expression of endogenous fusogens and fusogenic viral infections.
Cellular senescence is a robust mechanism that permanently arrests cell cycle progression of potentially harmful cells (Kuilman et al. 2010; Campisi 2011). It thus prevents tumorigenesis and limits short-term tissue damage but also contributes to aging of the tissues (Serrano et al. 1997; Krizhanovsky et al. 2008; Baker et al. 2011). Senescence can be induced by a variety of triggers, including telomere shortening, DNA damage, expression of activated oncogenes, and other forms of cellular stress (Allsopp and Harley 1995; Serrano et al. 1997; Knudsen et al. 2000; te Poele et al. 2002). These triggers as well as others yet unidentified lead to senescent cell accumulation in aged tissues, premalignant lesions, tumors after therapy, and tissue damage (Dimri et al. 1995; Schmitt et al. 2002; Krizhanovsky et al. 2008; Collado and Serrano 2010; Jun and Lau 2010; Kuilman et al. 2010).
Phenotypically, senescent cells are identified by their flattened and enlarged morphology, stability in culture, and increased senescence-associated β-galactosidase (SA-β-gal) activity (Campisi 2011). On the molecular level, cellular senescence is regulated by two major tumor suppressor pathways controlled by p53 and p16. The p53 protein can be induced by various forms of stress, such as DNA damage or activation of ARF or LATS2 tumor suppressors by oncogenes and cytoskeletal damage (Aylon et al. 2006). Upon activation, p53 mediates cell cycle arrest mainly through induction of its transcriptional target, p21. Both p21 and p16 are cyclin-dependent kinase (CDK) inhibitors that maintain the tumor suppressor protein pRB in its inactive hypophosphorylated state, thereby preventing the transcription factor E2F from transcribing genes that promote cell cycle progression (Narita et al. 2003, 2006). Thus, the senescence program is regulated coordinately by the p53 and p16–pRB pathways (Campisi and d'Adda di Fagagna 2007; Krizhanovsky et al. 2008; Krizhanovsky and Lowe 2009).
In addition to the molecular events aimed at cell cycle arrest, a prominent feature of senescent cells is their altered interaction with the environment. Senescent cells secrete a variety of senescence-associated secretory phenotype (SASP) proteins, including proinflammatory cytokines, growth factors, and proteases (Acosta et al. 2008; Coppe et al. 2008; Kuilman et al. 2008; Wajapeyee et al. 2008). SASP components were shown to either inhibit or promote cancer progression in a cell-nonautonomous manner in different settings (Canino et al. 2012; Lujambio et al. 2013). They also play a role in the interaction of senescent cells with the immune system and reinforce senescence-associated arrest of proliferation in an autocrine fashion (Acosta et al. 2008; Krizhanovsky et al. 2008; Kuilman et al. 2008). Besides secreting proteins, senescent cells also up-regulate membrane molecules that mediate the interaction of these cells with the immune system and promote their recognition and clearance, thereby facilitating the resolution of tissue damage and of tumors bearing senescent cells (Xue et al. 2007; Krizhanovsky et al. 2008; Soriani et al. 2009; Sagiv et al. 2013).
Cell cycle arrest, the main feature of cellular senescence, can be induced by cell fusion (Duelli and Lazebnik 2007). Seminal experiments >40 years ago demonstrated that fusion between a normal cell and a cancer cell leads to a reduction in tumorigenic potential (Harris et al. 1969). Moreover, intratumoral delivery of fusogenic agents or drugs that induce polyploidy has been suggested as a means of cancer treatment (Lin et al. 2010; Tovar et al. 2010). Cell fusion is an essential event in many developmental and physiological processes (Ogle et al. 2005; Duelli and Lazebnik 2007; Sapir et al. 2008). In mammals, cell fusion is vital for the proper functioning of muscles, osteoclasts, and the placenta. The mechanism of physiological cell fusion relies on the activity of fusogenic proteins (fusogens) (Sapir et al. 2008). Aberrant expression of fusogens in normal cells leads to unprogrammed illicit cell fusion (Duelli and Lazebnik 2007; Larsson et al. 2008).
One such endogenous fusogen, termed ERVWE1, mediates cell fusion required for formation of the syncytiotrophoblast, the outer layer of the placenta, as part of normal embryonic development (Blond et al. 2000; Mi et al. 2000). ERVWE1 is an endogenous retroviral locus that became incorporated into the human genome during the course of evolution and encodes a highly fusogenic glycoprotein (Blond et al. 2000). Like the endogenous viruses, several pathogenic viruses use cell fusion as a mechanism for increasing the spread of the virus in the infected organism. One such example is the measles virus (MV), which infects >40 million people annually despite the existence of an effective vaccine. Infection of human cells with MV leads to cell fusion, consequently leading to cell cycle arrest or apoptosis of the fused cells (Delpeut et al. 2012). However, the nature of the cell cycle arrest following cell fusion triggered by MV and the mechanisms governing the arrest as well as an effect of ERVWE1-mediated fusion on cell proliferation are not completely understood.
In this study, we examined the hypothesis that cell fusion induces cellular senescence, a process that serves two distinct purposes in mammals: one during embryonic development and the other later in life. First, we found that the placental syncytiotrophoblast exhibits multiple markers of cellular senescence and, second, that expression of ERVWE1 or infection by the fusogenic MV in normal or cancer cells leads to illicit cell fusion and induction of a p53- or pRB-dependent cell senescence program. These findings led us to conclude that, during embryonic development, cell fusion-induced senescence provides a functional interface between the mother and the developing fetus and that the senescent phenotype of the syncytiotrophoblast might contribute to physiological placental function. Later in life, illicit cell fusion caused by the expression of endogenous fusogens or by fusogenic viral infection induces a program of cellular senescence that protects the organism against short-term tissue damage and tumorigenesis.
Results
ERVWE1 expression in normal fibroblasts causes cell fusion
One of the endogenous fusogens that drives formation of the syncytiotrophoblast and is aberrantly expressed in human cancers is ERVWE1 (Mi et al. 2000; Strick et al. 2007). To investigate the cellular consequences of ERVWE1-induced cell fusion, we expressed ERVWE1 in normal and cancer cells. The expression of ERVWE1 in normal human diploid fibroblasts (IMR-90 cells) led to formation of multinuclear cells (Fig. 1A). To determine whether the multinuclear cells observed following ERVWE1 expression result from cell fusion, we cocultured GFP-expressing and mCherry-expressing IMR-90 cells and transduced the mixed population with ERVWE1. Expression of ERVWE1 in these IMR-90 cells led to the emergence of fused multinuclear cells with overlapping expression of both fluorescent proteins (Fig. 1A). Therefore, expression of ERVWE1 in normal fibroblasts causes cell fusion and formation of multinuclear cells.
Figure 1.

Expression of ERVWE1 in normal cells leads to cell fusion, proliferative arrest, and increased SA-β-gal activity. (A) Cocultured IMR-90 cells expressing GFP or mCherry, transduced with ERVWE1, and stained with DAPI. Arrowheads indicate a multinuclear fused cell. Bar, 50 μm. (B,C) BrdU incorporation was assessed in IMR-90 cells expressing ERVWE1 in syncytium (Syn) and mononuclear cells (Mono). (D,E) IMR-90 cells expressing ERVWE1, growing or triggered to undergo DNA damage-induced senescence (DIS), were assessed for SA-β-gal activity. Percentage of SA-β-gal-positive cells is shown. Arrowheads indicate syncytia cells. Bar, 100 μm. Values are means + SEM; (*) P < 0.05; (**) P < 0.01.
ERVWE1-mediated cell fusion of normal cells leads to cellular senescence
To investigate the effect of the ERVWE1-induced cell fusion in normal cells, we compared the proliferative capacities of fused and nonfused IMR-90 cells. Transfection of the cells with ERVWE1 resulted in emergence of a mixed population of mononuclear and fused multinuclear cells. We found that most of the multinuclear cells were BrdU-negative and that the overall syncytia cell population showed a reduction in BrdU incorporation of at least fivefold compared with the mononuclear cell population (P < 0.05) (Fig. 1B,C). In some of the multinuclear cells, only one of the several nuclei in the cell was BrdU-positive. These cells were considered positive in our analysis but were not proliferative and did not divide, indicating that illicit cell fusion induced by ERVWE1 significantly inhibited proliferation of normal cells.
The marked reduction in proliferative capacity observed in the fused IMR-90 cell population led us to investigate whether illicit cell fusion leads to cellular senescence. Compared with the mononuclear cell population, the ERVWE1-transduced multinuclear IMR-90 cells exhibited characteristic features of senescent cells; namely, flattened, enlarged morphology and a marked increase in SA-β-gal activity (P < 0.01; 91% vs. 6% of SA-β-gal-positive cells for syncytia and mononuclear cells, respectively) (Fig. 1D,E). Similar results were obtained when cell fusion was induced by ERVWE1 expression in immortalized human epithelial MCF-10A cells (Supplemental Fig. S1). Cellular senescence is a condition of stable cell cycle arrest, and senescent cells can remain viable in culture for long periods (Campisi and d'Adda di Fagagna 2007). Multinuclear cells were present in ERVWE1-expressing IMR-90 cells after 30 d in culture, suggesting that the illicitly fused cells were exceptionally stable and could be maintained in a nonproliferative state throughout long-term culture.
The molecular machinery of cellular senescence is regulated by p53 and p16–pRB tumor suppressor pathways (Campisi and d'Adda di Fagagna 2007; Krizhanovsky and Lowe 2009). We therefore evaluated the activation of these pathways in our ERVWE1-expressing fused cells. Expression of the effectors of these pathways, the CDK inhibitors p21 and p16, were found to be elevated at both the protein and mRNA levels (Fig. 2A,B). Moreover, p53 in the ERVWE1-expressing IMR-90 cells was up-regulated, while pRB was maintained in the hypophosphorylated state (Fig. 2A). pRB in this form is known to bind to the E2F transcription factor and thus prevents transcribing cell cycle-promoting genes (Narita et al. 2003). Consequently, expression levels of the cell cycle-promoting E2F target genes MCM2, Cyclin A, and CDCA8 were down-regulated in the ERVWE1-expressing IMR-90 cells (Fig. 2C). To evaluate the contribution of these molecular events to the cell cycle arrest of the fused cells, we assayed BrdU incorporation in fused and mononuclear cells following pRB knockdown, which was confirmed by immunoblot (Supplemental Fig. S2). We found a twofold increase in BrdU incorporation in the pRB-deficient fused cells (P < 0.05) and only a marginal increase in the mononuclear cell population (Fig. 2D). Similarly to pRB knockdown in oncogene-induced senescent cells (Narita et al. 2003), this increase in BrdU incorporation did not lead to cell proliferation (Supplemental Fig. S3). These results indicated that ERVWE1-induced cell fusion leads to activation of the molecular machinery responsible for the cell cycle arrest of senescent cells. Moreover, it showed that pRB, a principal component of these pathways, is needed in order to maintain the nonproliferative nature of the fused cells.
Figure 2.

ERVWE1-mediated cell fusion activates molecular pathways of cellular senescence. (A) Protein content of ERVWE1, p53, p21, p16, and pRB, was assessed by immunoblotting in IMR-90 cells transduced with ERVWE1 or control vector. Expression of CDK inhibitors p21 and p16 (B) and E2F target genes MCM-2, Cyclin A, and CDCA8 (C) in IMR-90 cells transduced with ERVWE1 or control vector (Ctrl) and assayed by quantitative RT–PCR. (D) BrdU incorporation by ERVWE1-transduced mononuclear (Mono) and syncytial (Syn) cells transfected with siRNA targeting pRB (siRb) or control siRNA (siCon). Expression of LATS2 and ARF (E); IL-6, IL-8, CXCL-1, and CCL5 (F); and MICA and ULBP2 (G) in IMR-90 cells transduced with ERVWE1 or control vector (Ctrl) and assayed by quantitative RT–PCR. (H,I) IMR-90 cells expressing ERVWE1 were immunostained with anti-γH2AX antibody, costained with DAPI, and examined for the presence of γH2AX foci in syncytia (Syn) and mononuclear (Mono) cells. Bar, 20 μm. (J) Expression of p53 phosphorylated at Ser15 in IMR-90 cells transduced with ERVWE1 or control vector and evaluated by immunoblotting. Values are means + SEM; (**) P < 0.01; (*) P < 0.05.
The molecular machinery of senescence, especially the p53 pathway, is known to be activated by a variety of stimuli, including direct DNA damage and expression of oncogenes. We were interested in finding out how this pathway is activated following illicit cell fusion. Two p53 activators, ARF and LATS2, have been implicated in the induction of cell cycle arrest in polyploid cells (Khan et al. 2000; Aylon et al. 2006). We found that the expression levels of ARF and LATS2 in ERVWE1-expressing IMR-90 cells were up-regulated by at least threefold compared with cells transduced with the empty vector control (Fig. 2E). These findings point to LATS2 and ARF as possible upstream regulators of senescence regulatory pathways in ERVWE1-expressing fused cells.
In addition to the molecular changes associated with cell cycle arrest, senescent cells up-regulate components of the SASP and molecules that mediate recognition and clearance of senescent cells by the immune system (Xue et al. 2007; Coppe et al. 2008; Krizhanovsky et al. 2008; Soriani et al. 2009; Sagiv et al. 2013). To investigate the presence of SASP in multinuclear senescent cells, we examined the expression levels of SASP components in ERVWE1-transduced IMR-90 cells. Cytokine array analysis of secreted SASP components revealed increased secretion of cytokines associated with SASP, such as IL-6, IL-8, CXCL1, and CCL-5 (Supplemental Fig. S4). Expression of these genes was also significantly up-regulated at the mRNA level (Fig. 2F). These secreted cytokines have been shown to attract immune cells, including natural killer (NK) cells, to the vicinity of the senescent cells (Hanna et al. 2006). We found that senescent syncytia, like other senescent cells, up-regulated MICA and ULBP2, ligands of the activating NK cell receptor NKG2D (Fig. 2G). The increase in expression of SASP factors and immune ligands might lead to attraction of NK cells to sites harboring fused senescent cells and result in direct interaction between these cells. The in vitro cytotoxicity assay indeed showed an increase in cytotoxicity of NK cells toward fused ERVWE1-transduced IMR-90 cells (Supplemental Fig. S5).
Persistent activation of a DNA damage response (DDR) is important for the establishment and maintenance of cellular senescence and can contribute to an increase in p53 levels (Bartkova et al. 2006; Di Micco et al. 2006; Moiseeva et al. 2006). To examine DDR activation in illicitly fused cells, we immunostained ERVWE1-expressing IMR-90 cells with anti-γH2AX antibody and compared the numbers of γH2AX foci-containing cells in syncytia and mononuclear cell populations. The number of these foci-containing cells was at least threefold higher in the syncytia cell population than in the mononuclear cells (P < 0.01) (Fig. 2H,I). Another marker of DDR activation is the stabilizing phosphorylation of the p53 protein at Ser15, mediated mainly by ATM and ATR kinases (Rodier et al. 2009). Expression of ERVWE1 in IMR-90 cells induced a substantial increase in p53-Ser15 phosphorylation (Fig. 2J). These results suggested that ERVWE1-induced cell fusion induces DDR activation, which plays a significant role in the initiation and maintenance of senescence. One of the causes of persistent DDR in senescent cells is an increase in reactive oxygen species (ROS). In ERVWE1-transduced IMR-90 cells, the ROS levels were increased in syncytia compared with the mononuclear cell population (P < 0.01) (Supplemental Fig. S6). Overall, our results showed that cells fused by ERVWE1 expression demonstrate phenotypical and molecular characteristics of senescent cells.
MV-mediated fusion of normal cells leads to cellular senescence
Endogenous fusogens are not the only possible cause of cell fusion in mammalian organisms. Pathogenic viruses such as MV can infect human cells and cause cell fusion (Oldstone et al. 1999; Sapir et al. 2008; Delpeut et al. 2012). We therefore investigated whether MV-induced fused cells show features of cellular senescence. MV infection of normal human IMR-90 fibroblasts resulted in fusion of infected cells, and the fused cells expressed MV proteins (Fig. 3A). Like the fused cells induced by ERVWE1, the MV-fused cells incorporated fivefold less BrdU than noninfected nonfused cells subjected to the same infection procedure (Fig. 3B,C). The multinuclear MV-infected cells also exhibited increased SA-β-gal activity (2.5% vs. 79% in mononuclear vs. fused cells) (Fig. 3D). Similar results were obtained when fusion of IMR-90 cells was induced by overexpression of another viral fusogen, fusion-associated small trans-membrane (FAST) protein (Supplemental Fig. S7). These results thus suggested that cell fusion induced by viral fusogens leads to induction of the senescence phenotype.
Figure 3.

MV induces cell fusion and senescence in normal human fibroblasts. (A) The MV-infected IMR-90 cells were stained with anti-SSPE antibody that recognizes MV proteins (red) and counterstained with DAPI. BrdU incorporation (B,C) and SA-β-gal activity (D) were assessed in IMR-90 cells infected with MV in syncytium (Syn) and mononuclear cells (Mono). (E) Protein content of p53 and p21 was assessed by immunoblotting in MV-infected or mock-infected (Ctrl) IMR-90 cells. Expression of MCM2, MCM3, CDCA2, and CDCA8 (F); IL-6, IL-8, CXCL-1, and CCL5 (G); and MICA and ULBP2 (H) in MV-infected or mock-infected (Ctrl) IMR-90 cells assayed by quantitative RT–PCR. Values are means + SEM; (*) P < 0.05.
In addition to their phenotypical changes, senescent cells undergo significant changes in their gene expression profiles. We compared the expression of senescence-related genes in MV-infected IMR-90 fibroblasts with that in mock-infected cells. Immunoblot analysis revealed a significant increase in expression of p53 and CDK inhibitor p21 in MV-infected cells (Fig. 3E), indicating activation of the p53 pathway in these cells. To evaluate the effect of the pRB pathway on cell cycle regulators in MV-fused cells, we evaluated the expression of E2F target cell cycle-promoting genes in MV-infected and mock-infected cells by quantitative RT–PCR. Expression levels of MCM2, MCM3, CDCA2, and CDCA8 were significantly down-regulated in the MV-infected IMR-90 cells (Fig. 3F). Therefore, senescence pathways are activated in MV-fused cells and halt cell cycle progression in these cells.
Immune cells respond to MV infection by increased production of inflammatory cytokines, and SASP is an essential part of the senescence phenotype in fibroblasts and epithelial cells (Campisi 2011; Delpeut et al. 2012). We therefore assayed expression of SASP components up-regulated in ERVWE1-fused cells in MV-fused IMR-90 fibroblast cells compared with mock-infected cells. Surprisingly, the expression of these cytokines in MV-infected cells was strongly up-regulated compared with mock-infected cells (Fig. 3G). In fact, the expression of IL-8 and of CCL-5 was up-regulated 107 and 825 times, respectively (Fig. 3G). In addition to the changes in the secretory phenotype, we found that MV-infected cells up-regulated MICA and ULBP2, ligands of the activating NK cell receptor NKG2D (Fig. 3H). Therefore, MV-infected fused cells up-regulate genes that might lead to attraction of immune cells and elimination of the infected cells by the immune system.
Fusion of cancer cells leads to cellular senescence
One of the pathological conditions in which cell fusion occurs is cancer (Duelli and Lazebnik 2007). ERVWE1 is one of the proteins that is expressed in cancer cells of multiple origins and mediates fusion of cells in human tumors (Larsson et al. 2007; Strick et al. 2007; Larsen et al. 2009). Interestingly, both ERVWE1 and MV have been proposed as candidates for the local treatment of cancer via induction of cell fusion (Msaouel et al. 2009; Lin et al. 2010). To examine the effects of ERVWE1-induced and MV-induced fusion of cancer cells, we used the A549 human alveolar adenocarcinoma cell line. These cells harbor wild-type p53 and are able to undergo senescence in response to DNA damage (McKenna et al. 2012). We found that expression of ERVWE1 in these cells resulted in cell fusion, syncytia formation, and an increase in SA-β-gal activity (Fig. 4A,B). ERVWE1 transduction and cell fusion were accompanied by up-regulation of the p53 protein in this cell type (Fig. 4C). For our analysis of the effect of p53 on ERVWE1-induced cell fusion, A549 cancer cells stably expressing shRNA-targeting p53 (sh-p53) or (as a control) sh-LacZ were transduced with ERVWE1. Expression of sh-p53 caused significant knockdown of p53 in these cells (Fig. 4C). SA-β-gal activity in the syncytial population of the cancer cells expressing sh-LacZ was 13-fold higher than in the mononuclear population, while its corresponding increase in cells lacking p53 was only fivefold (Fig. 4B). Thus, p53 knockdown led to a significant reduction in SA-β-gal-positive cells following ERVWE1-induced cell fusion (P < 0.05). These findings suggested that cell fusion might induce cellular senescence in cancer cells and that such induction depends at least in part on p53.
Figure 4.

ERVWE1-induced and MV-induced cell fusion induces the p53-dependent senescence phenotype in cancer cells. (A,B) SA-β-gal activity was examined in a syncytial (Syn) and a mononuclear (Mono) population of A549 cells expressing sh-LacZ or sh-p53 and transduced with ERVWE1 or GFP as a control. Bar, 100 μm. Arrowheads indicate syncytia. (C) Expression of p53 protein in A549 cells expressing sh-LacZ or sh-p53 and transduced with ERVWE1 (ERV) or control vector (Ctrl). (D,E) BrdU incorporation was assayed in a syncytial and a mononuclear population of A549 cells expressing sh-LacZ or sh-p53 and transduced with ERVWE1. Bar, 50 μm. Arrowheads indicate syncytia. (F) Expression of CCL5 and IL-8 in A549 cells expressing sh-LacZ or sh-p53 and transduced with ERVWE1 (ERV) or control vector (Ctrl) was measured by quantitative RT–PCR. (G) Soft agar colony formation assay of A549 cells expressing sh-LacZ or sh-p53 and transduced with ERVWE1 or control vector. SA-β-gal activity (H) and BrdU incorporation (I) were examined in a syncytial (Syn) and a mononuclear (Mono) population of A549 cells expressing sh-LacZ or sh-p53 and infected with MV. Values are means + SEM; (*) P < 0.05; (**) P < 0.01.
If cancer cells do indeed undergo senescence in response to cell fusion, this should be reflected in inhibition of the cell cycle machinery in the syncytia. We therefore compared the BrdU incorporation in ERVWE1-induced syncytia of cancer cells expressing sh-LacZ or sh-p53 and in a mononuclear population of the same cells. BrdU incorporation into the syncytial population of sh-LacZ-expressing cells was at least 20-fold lower than in the mononuclear population, whereas the decrease in sh-p53-expressing syncytia was only minor (Fig. 4D,E). Thus, inhibition of the cell cycle in A549 cells induced by ERVWE1-mediated fusion is p53-dependent.
The SASP is an integral part of the senescent phenotype in both normal and cancer cells (Campisi 2011). We found that the SASP components IL-8 and CCL5 were up-regulated in normal cells after ERVWE1-induced fusion (Fig. 2F). Similarly, in cancer cells transduced with ERVWE1, expression of these genes was significantly higher than in control vector-transduced cells (P < 0.05) (Fig. 4F). Taken together, the above findings suggested that ERVWE1-induced syncytia of cancer cells undergo p53-dependent cellular senescence that limits their proliferative capacity.
Next, we examined the impact of illicit cell fusion on tumorigenicity of A549 cells. For this purpose, we performed a soft agar cell transformation assay of A549 cells expressing sh-LacZ or sh-p53 and transiently transduced with ERVWE1. Surprisingly, cell fusion resulted in elevated colony formation in both cell types compared with the control (Fig. 4G). In addition, growth in the soft agar after ERVWE1 transduction in p53-deficient A549 cells was twofold higher than in p53-expressing A549 cells (P < 0.05) (Fig. 4G). Following cell fusion, the p53-expressing cells showed cell cycle arrest (Fig. 4D,E) but also exhibited an increase in tumorigenicity, which was significantly increased in the absence of p53. The increase in the tumorigenicity despite the nonproliferative nature of senescent cells might be explained by the secretion of cytokines from these cells.
As MV-induced cell fusion induces senescence in normal cells, we examined the effect of MV-induced fusion on A549 cancer cells in the presence and absence of p53. In line with our results in normal cells, SA-β-gal activity in the MV-induced syncytial population of the cancer cells was higher than in the mononuclear population (P < 0.01) (Fig. 4H). Following MV-induced fusion, knockdown of p53 resulted in a significant reduction in SA-β-gal-positive cells (P < 0.05) (Fig. 4H). In addition to this reduction, the MV-induced syncytial cells expressing sh-LacZ showed a decline in BrdU incorporation of at least 15-fold compared with the mononuclear population (P < 0.01), whereas the p53-depleted (sh-p53) syncytia exhibited only a slight decrease (Fig. 4I; Supplemental Fig. S8). Thus, induction of fusion of cancer cells resulted in p53-dependent senescence.
The placental syncytiotrophoblast exhibits characteristics of senescent cells
In addition to its pathological expression in cancer, ERVWE1 is physiologically expressed in the placental syncytiotrophoblast of the developing embryo (Blond et al. 2000; Mi et al. 2000). Given the physiological function of ERVWE1 in mediating the formation of the syncytiotrophoblast from the cytotrophoblast cells by inducing cell fusion, we wanted to find out whether the syncytiotrophoblast exhibits characteristics of cellular senescence. We therefore examined sections of a third trimester post-partum human placenta, which contains syncytiotrophoblast cells, for the presence of markers of cellular senescence. SA-β-gal activity was identified in the multinuclear syncytiotrophoblast cells but was not detectable in the underlying mononuclear cells, blood vessel cells, connective tissue, or other cell layers of the placenta (Fig. 5). To gain an insight into the relationship between the syncytiotrophoblast cells and the molecular pathways of cellular senescence, we analyzed the trophoblast immunohistologically for molecular markers of senescence. We first characterized the syncytiotrophoblast cells as multinuclear cells that express ERVWE1 but not Ki67, a marker of proliferating cells (Fig. 5). The syncytiotrophoblast cells exhibited specific staining for the CDK inhibitors p16 and p21, indicating that both of these central pathways of senescence are activated in these cells. Nuclear staining for p53 strongly supported this view. The nuclear staining of γH2AX (Fig. 5) disclosed activation of DDR, a prominent marker of senescence, in these cells. Yet another senescence marker, DCR2, protects senescent cells from apoptosis induced by the death receptor ligands TRAIL and FASL (Sagiv et al. 2013). We examined the possibility that DCR2 is expressed in the syncytiotrophoblast and might therefore contribute to its resistance to apoptosis. We found that the human syncytiotrophoblast exhibits higher expression of DCR2 than other cell layers of the placenta, including cytotrophoblasts and adjacent connective tissue (Fig. 5). Expression of DCR2 in syncytiotrophoblast cells further supported the activation of a senescence program in these cells. Altogether, our results showed that human syncytiotrophoblast cells exhibit characteristics and activation of the molecular pathways of cellular senescence.
Figure 5.
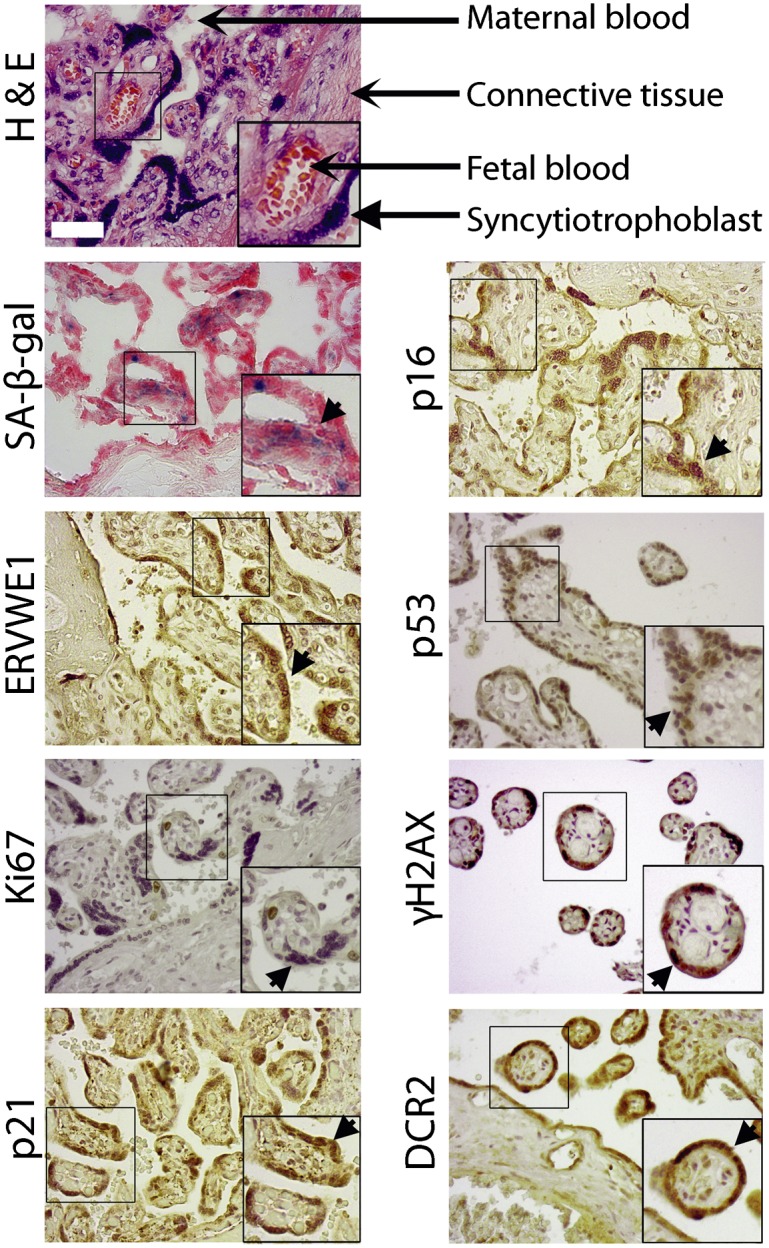
Human placental syncytiotrophoblast cells express markers of cellular senescence. H&E staining demonstrated general structure of the placenta. Sections of human post-partum third trimester placenta were evaluated for SA-β-gal activity (SA-β-gal), expression of ERVWE1, proliferation marker Ki67, and senescence markers p53, DCR2, p16, p21, and γH2AX. Arrowheads indicate syncytiotrophoblast cells.
Discussion
Many viruses induce fusion of infected cells to support their reproduction and dissemination (Sapir et al. 2008). The mammalian organism has found a way to use the fusogenic property of an endogenous virus that was incorporated into the genome during the course of evolution in order to function in placenta formation (Blond et al. 2000; Mi et al. 2000; Dupressoir et al. 2011). In this study, we identified cellular senescence as a mechanism that is activated in cells that have fused due to their expression of endogenous or exogenous viral proteins. Senescence-associated cytokines and growth factors secreted from fused cells are needed for normal placental function. Interaction of the fused cells with the immune system might promote normal maternal–fetal interface and, later in life, might also help to eliminate abnormal cells at sites of pathology or infection.
It remains to be determined why induction of cellular senescence is necessary during syncytiotrophoblast formation and placental development. One possibility is that mechanisms induced in senescent cells help to maintain viability of the syncytium during pregnancy. Senescent cells are known to be resistant to apoptosis, and this resistance might support syncytiotrophoblast viability during pregnancy despite activation of caspase-8 in the cytotrophoblast in the initial stages of syncytiotrophoblast formation (De Falco et al. 2004). Activated caspase-8 can lead to apoptosis in other tissues (Varfolomeev et al. 1998) but becomes inactivated in the syncytiotrophoblast (De Falco et al. 2004). A senescent phenotype of the syncytiotrophoblast, including high DCR2 expression, might contribute to this inactivation and thus support the viability of these cells. Of note, knockout of genes that can regulate senescence (pRb or E2F7) leads to disorganization of the trophoblast in mice and even causes embryonic lethality (Narita et al. 2003; Wu et al. 2003; Aksoy et al. 2012; Ouseph et al. 2012). Placenta-specific ablation of pRb, for example, causes embryonic lethality, and placenta malfunction is the defect responsible for the embryonic lethality of pRb knockout mice (Wu et al. 2003). Therefore, intact molecular machinery of senescence is necessary to support the integrity of the syncytiotrophoblast in order to sustain the function of the placenta. Another possibility is that induction of the flat morphology characteristic of senescent cells is needed to increase the surface area of the syncytiotrophoblast, thus facilitating the transfer of resources from mother to fetus.
In addition to being both flat and resistant to apoptosis, senescent cells produce a variety of cytokines as part of the SASP (Coppe et al. 2008). One of the main SASP components is IL-8, which is strongly up-regulated in fused senescent cells and is essential for normal placental function (Dame and Juul 2000; Gupta et al. 2005). Other SASP cytokines secreted from the fused cells, such as CCL5, attract specific noncytotoxic maternal NK cells that play an important role in maintenance of the placenta (Hanna et al. 2006). Up-regulation of ligands of the NK-activating receptor NKG2D on fused senescent cells further increases cytokine production from the NK cells without increasing cytotoxicity (Hanna et al. 2006). It therefore seems that cellular senescence of the syncytiotrophoblast cells not only prevents DNA replication in these cells but can also support their viability, attract NK cells, and contribute to functionality of the maternal–fetal interface.
Under pathological conditions, cellular senescence might function to prevent the proliferation of polyploid cells resulting from expression of endogenous viral fusogens or viral infection. Proliferation of these cells can be deleterious, as it may produce genetically unstable, aneuploid progenies, thereby promoting cancer development (Ganem and Pellman 2007). Notably, cells with polyploid content undergo p53-dependent senescence (Rieder and Maiato 2004). The mechanism of this process is not fully understood, and it was suggested that it is part of the “tetraploidy checkpoint” whereby cells can “count” their centrosomes or chromosomes. However, not every cell fusion event results in induction of cellular senescence (Duelli and Lazebnik 2007). Therefore, fusion itself is a necessary but not a sufficient condition for induction of the senescence program. Activation of additional mechanisms, which might rely on changes in cell shape rather than the number of the nuclei or the DNA content, might lead to activation of this program.
Polyploid cells undergo major changes in their cytoskeleton. These changes can activate the p53 and pRB pathways, which are the main pathways regulating cellular senescence (Campisi and d'Adda di Fagagna 2007; Ganem and Pellman 2007). One of the proteins that might activate p53 in response to cytoskeletal changes is LATS2. This normally functions as a centrosomal protein, but upon mitotic spindle damage or oncogene activation, it translocates to the nucleus and activates p53 to induce cell death or senescence of polyploid cells (Aylon et al. 2006). Changes in the cytoskeletal organization of senescent cells can lead to pRB hypophosphorylation and LATS2 up-regulation, which were detected in fused senescent cells. Moreover, cell cycle arrest in fused senescent cells is dependent on the p53 and pRB pathways. Therefore, changes in the cytoskeleton might contribute to the cell fusion-induced senescence.
Cell fusion leads to extensive genetic and epigenetic changes and may therefore promote cancer (Ogle et al. 2005; Duelli and Lazebnik 2007). Many tumor cells are fusogenic, and the fusion rate in experimental tumors is estimated at 1% (Duelli and Lazebnik 2003). Moreover, fused cells have been observed in cancer specimens (Duelli and Lazebnik 2007; Strick et al. 2007; Larsen et al. 2009). Despite the oncogenic potential of cell fusion, however, most cases of cell fusion do not lead to cell transformation (Harris et al. 1969; Ogle et al. 2005; Duelli and Lazebnik 2007). Our data suggest that illicitly fused cells might stimulate tumorigenesis, probably in a non-cell-autonomous manner. However, the possibility that senescent syncytia cells affect tumorigenesis by additional mechanisms cannot be excluded.
Cellular senescence of fused cells might be a general phenomenon that occurs as a consequence of misexpression of endogenous fusogens and viral infections. Due to preferential recognition of fused cells by NK cells, it seems likely that the presence of fusion-induced senescent cells is tightly regulated by the immune system in a manner similar to the immune surveillance of senescent cells in other settings (Xue et al. 2007; Krizhanovsky et al. 2008; Kang et al. 2011; Sagiv et al. 2013). A variety of viruses that infect human populations, including HIV, MV, and the more widely spread respiratory syncytia virus and influenza, can induce cell fusion and formation of multinuclear cells following infection (Duelli and Lazebnik 2007). In addition, endogenous fusogens and even putative anti-cancer treatments can cause formation of syncytia (Sapir et al. 2008; Tovar et al. 2010). Our present findings suggest that cellular senescence functions as a protective mechanism that limits proliferation of the syncytia cells and might promote their immune surveillance. If these mechanisms fail, illicitly fused cells persisting in tissues might be able to induce tissue damage and tumorigenesis in a non-cell-autonomous manner.
In contrast to this illicit cell fusion, in the normal environment of the placenta, cellular senescence triggered by physiological cell fusion supports the viability of the syncytiotrophoblast cells, contributes to production of cytokines that are needed for normal placental function, and attracts NK cells that contribute to the functional maternal/fetal interface. We conclude that the cellular senescence mechanism necessary for embryonic development might be adopted later in life by the organism to protect itself from the abnormal and destructive cell fusion induced by misexpression of endogenous fusogens or viral infection.
Materials and methods
Tissue culture and gene transfer
Normal human diploid fibroblasts (IMR-90 cells) and A549 cells (both types from American Type Culture Collection) were cultured in DMEM with 10% fetal bovine serum (FBS) at 7.5% CO2. The medium of the A549 cells also contained 2 ug/mL puromycin. Induction of senescence by DNA damage and SA-β-gal activity assay were performed as described (Krizhanovsky et al. 2008). To induce transient expression, IMR-90 or A549 cells were transfected with the phCMV-HERV-W vector encoding ERVWE1 or, as a control, expressing GFP using Transit-293 transfection reagent (Mirus Bio LLC). To induce stable gene expression, IMR-90 cells were transduced with retroviral pLPC vector with HERV-W or GFP or mCherry (Narita et al. 2003).
For the knockdown experiments, ON-TARGETplus SMARTpool siRNA targeting pRB and the nontargeting (control) pool were transfected into IMR-90 cells with Dharmafect 1 reagent (all from Dharmacon).
For the soft agar transformation assays, 8000 A549 cells per well of 96-well plates were seeded in 0.35% bacto-agar on top of 0.5% bacto-agar (1:1 with DMEM, 20% FCS, 2% penicillin–streptomycin) 7 d post-transfection. Seven days thereafter, Presto Blue reagent (BD Bioscience) was added to each well in a 1:10 volume ratio. After 1 h of incubation with the reagent at 37°C, fluorescence was determined at 600 nm.
For BrdU Incorporation assays, cells were seeded on coverslips and incubated with 0.1 mg/mL 5′-bromo-2-deoxyuridine and 0.01 mg/mL 5′-fluoro-2-deoxyuridine. The incubation periods were 6 h for IMR-90 cells and 3 h for A549 cells. The cells were washed and fixed with 4%PFA followed by immunofluorescence staining.
MV infection and detection procedures
MV stock was prepared from a thrice-purified plaque of Edmonston strain (Gorecki and Rozenblatt 1980). IMR-90 cells were infected at a multiplicity of infection (MOI) of 1:10 as titrated against Vero cells. Medium was changed the next day, and the cells were cultured for 48 h for detection of MV or 5 d for gene expression analysis. For detection of MV, cells were fixed with acetone:methanol 1:1 for 2 min at room temperature and incubated with SSPE serum (which detects MV) (Rozenblatt et al. 1979) and diluted 1:100 in PBS (+3% FBS). Detection was performed with goat anti-human IgG Cy-3 (C2571, Sigma) at a dilution of 1:500.
Detection of gene expression
Protein expression was tested by immunoblotting as described (Krizhanovsky et al. 2008). Detection was performed using α-p53 mAb (a gift from V. Rotter, Weizmann Institute of Science); α-p21 and α-Rb (BD Biosciences); α-p16, α-syncytin, and α-β-tubulin (Santa Cruz Biotechnology); α-p-p53 (Ser-15) (Cell Signaling); and α-β-actin mAb (Sigma) primary antibodies. Immunofluorescence analysis was performed with α-γH2AX mAb (Cell Signaling) or α-BrdU mAb (BD Biosciences). Quantitative RT–PCR was performed as described previously (Sagiv et al. 2013). Primer sequences are available on request.
Tissue collection and analysis
The study was approved by the Institutional Review Board of Meir Medical Center, and Helsinki Committee Approval was received by T.B.-S. Biopsies were obtained from the placentas of 20 uncomplicated pregnancies. All pregnancies were well dated, had normal detailed sonographic assays, and had obtained either normal biochemical screening results for aneuploidy or normal chromosome profiles on amniocentesis. In addition, all neonates had undergone examination and evaluation immediately after birth, with normal findings. Placental tissue samples (∼1 cm3 each) were obtained immediately after delivery under sterile conditions. Biopsies were taken from the intermediate trophoblast from the area midway between cord insertion and the edge of the placenta. The biopsied material was either stored at −80°C or fixed in formalin and paraffin and embedded for later evaluation.
Paraffin-embedded sections were obtained from placental biopsy specimens. Sections were deparaffinized and rehydrated in an ethanol series. Antigen retrieval was performed in a steamer, and sections were blocked for nonspecific binding with 4% horse serum and 1% BSA. Primary antibodies recognizing Ki67 (Abcam); DCR2 (Enzo Life Sciences); p21 (BD Biosciences); and p53, p16, and ERVWE1 (all from Santa Cruz Biotechnology) were applied overnight at 4°C. Staining was developed using DAB (Vector Laboratories) followed by hematoxylin counterstaining. Sections were visualized using an Olympus microscope, and images were analyzed using CellP software (Diagnostic Instruments).
Statistical analysis
All experiments were performed in at least three independent repeats. For the statistical analysis, P-values were calculated using Student's t-test, and data are presented as means + SEM.
Acknowledgments
We are grateful to M. Oren, E. Winocour, and the late D. Givol for comments; D. Burton for helpful comments; and all other members of the Krizhanovsky laboratory for helpful discussions. This work was supported by grants to V.K. from the European Research Council under the European Union's FP7 and from the Israel Science Foundation. V.K. is the incumbent of the Karl and Frances Korn Career Development Chair in Life Sciences.
Footnotes
Supplemental material is available for this article.
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.227512.113.
References
- Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, et al. 2008. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 133: 1006–1018 [DOI] [PubMed] [Google Scholar]
- Aksoy O, Chicas A, Zeng T, Zhao Z, McCurrach M, Wang X, Lowe SW 2012. The atypical E2F family member E2F7 couples the p53 and RB pathways during cellular senescence. Genes Dev 26: 1546–1557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allsopp RC, Harley CB 1995. Evidence for a critical telomere length in senescent human fibroblasts. Exp Cell Res 219: 130–136 [DOI] [PubMed] [Google Scholar]
- Aylon Y, Michael D, Shmueli A, Yabuta N, Nojima H, Oren M 2006. A positive feedback loop between the p53 and Lats2 tumor suppressors prevents tetraploidization. Genes Dev 20: 2687–2700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM 2011. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479: 232–236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, et al. 2006. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444: 633–637 [DOI] [PubMed] [Google Scholar]
- Blond JL, Lavillette D, Cheynet V, Bouton O, Oriol G, Chapel-Fernandes S, Mandrand B, Mallet F, Cosset FL 2000. An envelope glycoprotein of the human endogenous retrovirus HERV-W is expressed in the human placenta and fuses cells expressing the type D mammalian retrovirus receptor. J Virol 74: 3321–3329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J 2011. Cellular senescence: Putting the paradoxes in perspective. Curr Opin Genet Dev 21: 107–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J, d'Adda di Fagagna F 2007. Cellular senescence: When bad things happen to good cells. Nat Rev Mol Cell Biol 8: 729–740 [DOI] [PubMed] [Google Scholar]
- Canino C, Mori F, Cambria A, Diamantini A, Germoni S, Alessandrini G, Borsellino G, Galati R, Battistini L, Blandino R, et al. 2012. SASP mediates chemoresistance and tumor-initiating-activity of mesothelioma cells. Oncogene 31: 3148–3163 [DOI] [PubMed] [Google Scholar]
- Collado M, Serrano M 2010. Senescence in tumours: Evidence from mice and humans. Nat Rev Cancer 10: 51–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J, Nelson PS, Desprez PY, Campisi J 2008. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6: 2853–2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dame JB, Juul SE 2000. The distribution of receptors for the pro-inflammatory cytokines interleukin (IL)-6 and IL-8 in the developing human fetus. Early Hum Dev 58: 25–39 [DOI] [PubMed] [Google Scholar]
- De Falco M, Fedele V, Cobellis L, Mastrogiacomo A, Leone S, Giraldi D, De Luca B, Laforgia V, De Luca A 2004. Immunohistochemical distribution of proteins belonging to the receptor-mediated and the mitochondrial apoptotic pathways in human placenta during gestation. Cell Tissue Res 318: 599–608 [DOI] [PubMed] [Google Scholar]
- Delpeut S, Noyce RS, Siu RW, Richardson CD 2012. Host factors and measles virus replication. Current opinion in virology 2: 773–783 [DOI] [PubMed] [Google Scholar]
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A, et al. 2006. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444: 638–642 [DOI] [PubMed] [Google Scholar]
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, et al. 1995. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci 92: 9363–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duelli D, Lazebnik Y 2003. Cell fusion: A hidden enemy? Cancer Cell 3: 445–448 [DOI] [PubMed] [Google Scholar]
- Duelli D, Lazebnik Y 2007. Cell-to-cell fusion as a link between viruses and cancer. Nat Rev Cancer 7: 968–976 [DOI] [PubMed] [Google Scholar]
- Dupressoir A, Vernochet C, Harper F, Guegan J, Dessen P, Pierron G, Heidmann T 2011. A pair of co-opted retroviral envelope syncytin genes is required for formation of the two-layered murine placental syncytiotrophoblast. Proc Natl Acad Sci 108: E1164–E1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem NJ, Pellman D 2007. Limiting the proliferation of polyploid cells. Cell 131: 437–440 [DOI] [PubMed] [Google Scholar]
- Gorecki M, Rozenblatt S 1980. Cloning of DNA complementary to the measles virus mRNA encoding nucleocapsid protein. Proc Natl Acad Sci 77: 3686–3690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta AK, Hasler P, Holzgreve W, Gebhardt S, Hahn S 2005. Induction of neutrophil extracellular DNA lattices by placental microparticles and IL-8 and their presence in preeclampsia. Hum Immunol 66: 1146–1154 [DOI] [PubMed] [Google Scholar]
- Hanna J, Goldman-Wohl D, Hamani Y, Avraham I, Greenfield C, Natanson-Yaron S, Prus D, Cohen-Daniel L, Arnon TI, Manaster I, et al. 2006. Decidual NK cells regulate key developmental processes at the human fetal-maternal interface. Nat Med 12: 1065–1074 [DOI] [PubMed] [Google Scholar]
- Harris H, Miller OJ, Klein G, Worst P, Tachibana T 1969. Suppression of malignancy by cell fusion. Nature 223: 363–368 [DOI] [PubMed] [Google Scholar]
- Jun JI, Lau LF 2010. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat Cell Biol 12: 676–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, et al. 2011. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 479: 547–551 [DOI] [PubMed] [Google Scholar]
- Khan SH, Moritsugu J, Wahl GM 2000. Differential requirement for p19ARF in the p53-dependent arrest induced by DNA damage, microtubule disruption, and ribonucleotide depletion. Proc Natl Acad Sci 97: 3266–3271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knudsen KE, Booth D, Naderi S, Sever-Chroneos Z, Fribourg AF, Hunton IC, Feramisco JR, Wang JY, Knudsen ES 2000. RB-dependent S-phase response to DNA damage. Mol Cell Biol 20: 7751–7763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krizhanovsky V, Lowe SW 2009. Stem cells: The promises and perils of p53. Nature 460: 1085–1086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krizhanovsky V, Yon M, Dickins RA, Hearn S, Simon J, Miething C, Yee H, Zender L, Lowe SW 2008. Senescence of activated stellate cells limits liver fibrosis. Cell 134: 657–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Vredeveld LC, Douma S, van Doorn R, Desmet CJ, Aarden LA, Mooi WJ, Peeper DS 2008. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 133: 1019–1031 [DOI] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Mooi WJ, Peeper DS 2010. The essence of senescence. Genes Dev 24: 2463–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen JM, Christensen IJ, Nielsen HJ, Hansen U, Bjerregaard B, Talts JF, Larsson LI 2009. Syncytin immunoreactivity in colorectal cancer: Potential prognostic impact. Cancer Lett 280: 44–49 [DOI] [PubMed] [Google Scholar]
- Larsson LI, Holck S, Christensen IJ 2007. Prognostic role of syncytin expression in breast cancer. Hum Pathol 38: 726–731 [DOI] [PubMed] [Google Scholar]
- Larsson LI, Bjerregaard B, Talts JF 2008. Cell fusions in mammals. Histochem Cell Biol 129: 551–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin EH, Salon C, Brambilla E, Lavillette D, Szecsi J, Cosset FL, Coll JL 2010. Fusogenic membrane glycoproteins induce syncytia formation and death in vitro and in vivo: A potential therapy agent for lung cancer. Cancer Gene Ther 17: 256–265 [DOI] [PubMed] [Google Scholar]
- Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE, Zhao Z, Thapar V, Joyce JA, Krizhanovsky V, et al. 2013. Non-cell-autonomous tumor suppression by p53. Cell 153: 449–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna E, Traganos F, Zhao H, Darzynkiewicz Z 2012. Persistent DNA damage caused by low levels of mitomycin C induces irreversible cell senescence. Cell Cycle 11: 3132–3140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi S, Lee X, Li X, Veldman GM, Finnerty H, Racie L, LaVallie E, Tang XY, Edouard P, Howes S, et al. 2000. Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 403: 785–789 [DOI] [PubMed] [Google Scholar]
- Moiseeva O, Mallette FA, Mukhopadhyay UK, Moores A, Ferbeyre G 2006. DNA damage signaling and p53-dependent senescence after prolonged β-interferon stimulation. Mol Biol Cell 17: 1583–1592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Msaouel P, Dispenzieri A, Galanis E 2009. Clinical testing of engineered oncolytic measles virus strains in the treatment of cancer: An overview. Curr Opin Mol Ther 11: 43–53 [PMC free article] [PubMed] [Google Scholar]
- Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW 2003. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113: 703–716 [DOI] [PubMed] [Google Scholar]
- Narita M, Narita M, Krizhanovsky V, Nunez S, Chicas A, Hearn SA, Myers MP, Lowe SW 2006. A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell 126: 503–514 [DOI] [PubMed] [Google Scholar]
- Ogle BM, Cascalho M, Platt JL 2005. Biological implications of cell fusion. Nat Rev Mol Cell Biol 6: 567–575 [DOI] [PubMed] [Google Scholar]
- Oldstone MB, Lewicki H, Thomas D, Tishon A, Dales S, Patterson J, Manchester M, Homann D, Naniche D, Holz A 1999. Measles virus infection in a transgenic model: Virus-induced immunosuppression and central nervous system disease. Cell 98: 629–640 [DOI] [PubMed] [Google Scholar]
- Ouseph MM, Li J, Chen HZ, Pecot T, Wenzel P, Thompson JC, Comstock G, Chokshi V, Byrne M, Forde B, et al. 2012. Atypical E2F repressors and activators coordinate placental development. Dev Cell 22: 849–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder CL, Maiato H 2004. Stuck in division or passing through: What happens when cells cannot satisfy the spindle assembly checkpoint. Dev Cell 7: 637–651 [DOI] [PubMed] [Google Scholar]
- Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR, Freund A, Campeau E, Davalos AR, Campisi J 2009. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol 11: 973–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenblatt S, Gorecki M, Shure H, Prives CL 1979. Characterization of measles virus-specific proteins synthesized in vivo and in vitro from acutely and persistently infected cells. J Virol 29: 1099–1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagiv A, Biran A, Yon M, Simon J, Lowe SW, Krizhanovsky V 2013. Granule exocytosis mediates immune surveillance of senescent cells. Oncogene 32: 1971–1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sapir A, Avinoam O, Podbilewicz B, Chernomordik LV 2008. Viral and developmental cell fusion mechanisms: Conservation and divergence. Dev Cell 14: 11–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, Lowe SW 2002. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 109: 335–346 [DOI] [PubMed] [Google Scholar]
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW 1997. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 88: 593–602 [DOI] [PubMed] [Google Scholar]
- Soriani A, Zingoni A, Cerboni C, Iannitto ML, Ricciardi MR, Di Gialleonardo V, Cippitelli M, Fionda C, Petrucci MT, Guarini A, et al. 2009. ATM–ATR-dependent up-regulation of DNAM-1 and NKG2D ligands on multiple myeloma cells by therapeutic agents results in enhanced NK-cell susceptibility and is associated with a senescent phenotype. Blood 113: 3503–3511 [DOI] [PubMed] [Google Scholar]
- Strick R, Ackermann S, Langbein M, Swiatek J, Schubert SW, Hashemolhosseini S, Koscheck T, Fasching PA, Schild RL, Beckmann MW, et al. 2007. Proliferation and cell–cell fusion of endometrial carcinoma are induced by the human endogenous retroviral Syncytin-1 and regulated by TGF-β. J Mol Med 85: 23–38 [DOI] [PubMed] [Google Scholar]
- te Poele RH, Okorokov AL, Jardine L, Cummings J, Joel SP 2002. DNA damage is able to induce senescence in tumor cells in vitro and in vivo. Cancer Res 62: 1876–1883 [PubMed] [Google Scholar]
- Tovar C, Higgins B, Deo D, Kolinsky K, Liu JJ, Heimbrook DC, Vassilev LT 2010. Small-molecule inducer of cancer cell polyploidy promotes apoptosis or senescence: Implications for therapy. Cell Cycle 9: 3364–3375 [DOI] [PubMed] [Google Scholar]
- Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, Rebrikov D, Brodianski VM, Kemper OC, Kollet O, et al. 1998. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 9: 267–276 [DOI] [PubMed] [Google Scholar]
- Wajapeyee N, Serra RW, Zhu X, Mahalingam M, Green MR 2008. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell 132: 363–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L, de Bruin A, Saavedra HI, Starovic M, Trimboli A, Yang Y, Opavska J, Wilson P, Thompson JC, Ostrowski MC, et al. 2003. Extra-embryonic function of Rb is essential for embryonic development and viability. Nature 421: 942–947 [DOI] [PubMed] [Google Scholar]
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW 2007. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 445: 656–660 [DOI] [PMC free article] [PubMed] [Google Scholar]