

Abstract
The mazEFSa toxin-antitoxin (TA) system is ubiquitous in clinical isolates of Staphylococcus aureus, yet its physiological role is unclear. MazFSa is a sequence-specific endoribonuclease that inhibits the growth of S. aureus and Escherichia coli upon ectopic overexpression. MazFSa preferentially cleaves RNA at UACAU sites, which are overrepresented in genes encoding pathogenicity factors. The exploitation of the inherent toxicity of MazFSa by artificial toxin activation has been proposed as an antibacterial strategy; however, enzymatic activity of endogenous MazFSa has never been detected, and tools for such analyses are lacking. Herein we detail methods for detection of the ribonuclease activity of MazFSa, including a continuous fluorometric assay and a gel-based cleavage assay. Importantly, these methods allowed for the first detection of endogenous MazFSa enzymatic activity in S. aureus lysate. These robust and sensitive assays provide a toolkit for the identification, analysis, and validation of stressors that induce MazF enzymatic activity, and should assist in the discovery of artificial activators of the MazEFSa TA system.
Keywords: MazFSa, toxin-antitoxin, ribonuclease, enzyme assay
Toxin-antitoxin (TA) systems were first discovered on plasmids, where they serve as plasmid maintenance systems via a post-segregational killing (PSK) mechanism [1–3]. In the canonical Type II TA system, plasmid maintenance ensures continued production of the coexpressed inactive complex of antitoxin and toxin; however, if the plasmid is not inherited by a daughter cell, the labile antitoxin succumbs to degradation by the Lon or Clp proteases, releasing the toxin to kill the cell [4–7]. TA systems are also encoded on the chromosomes of nearly all free-living bacteria [8], although here their precise role remains a topic of much debate [9, 10]. As observed for multiple different TA systems, rapid bacterial cell death is induced by the free toxin; these results have led to speculation that artificial activation of toxin proteins from TA systems could be a powerful antibacterial strategy [11–16].
In one study the mazEFSa TA genes were detected in 100% (78 of 78) clinical isolates of MRSA, and the mazEFSa transcript was also detected in these isolates [17]. Ectopic overexpression of MazFSa in S. aureus decreased cell viability by 2-log CFU/mL after 60 min of induction [18]; however, there was only ~27% difference in cell death at the 60 minute time point, suggesting that MazFSa induces stasis and not cell death [18]. MazFSa is a sequence-specific endoribonuclease that cleaves at U↓ACAU [19]. This sequence is highly abundant in certain transcripts, including those that code for virulence factors such as the serine-rich pathogen adhesion factor SraP [19]. Overexpression of MazFSa in S. aureus results in time-dependent cleavage of other virulence transcripts, including hla and spa, whereas the essential housekeeping transcripts recA and gyrB were not cleaved [20]. Thus, the activation of MazFSa could serve as an anti-virulence strategy by enhancing its ability to cleave virulence-factor encoding transcripts. However, the cellular role of MazFSa remains unclear, and the larger question about the cellular role of chromosomally-encoded TA systems has not been answered. In fact, although a number of studies have examined the mazEFSa gene cluster (via PCR) [17], the inducibility of the mazEFSa transcript with various stressors (via Northern blots and RT-PCR) [18, 21], and the presence of the proteins (via Western blot) [22], there have been no studies that assess the enzymatic activity of MazFSa in its cellular context, and tools for such evaluations are lacking. Ultimately, the ribonuclease activity of MazFSa will dictate its ability to restrict growth or kill the cell, and non-enzymatic assessments (e.g., DNA, RNA, or protein levels) are only surrogates that do not directly report on actual enzyme activity. Further complicating matters is the fact that the transcripts for MazESa and MazFSa are typically co-produced [18, 21]; thus, elevation at the mRNA or protein level does not necessitate heightened MazFSa enzymatic activity, as levels of the MazESa antitoxin are also raised.
As described herein, using a fluorogenic substrate we have determined the kinetic parameters for MazFSa. Additionally, an mRNA transcript was engineered to contain one optimal MazFSa cleavage sequence, and this substrate provides a means for clear and rapid assessment of MazFSa activity in vitro. A radiolabeled version of this substrate was used to detect the activity of endogenous MazFSa in S. aureus lysate. This is the first time enzymatic activity of endogenous MazFSa has been evaluated in the cellular milieu, and the tools described herein should facilitate the evaluation of various stressors and assist in the discovery of artificial activators of MazFSa.
Experimental Procedures
Materials
Primers and oligonucleotides were synthesized by IDT. Ni2+-NTA resin was obtained from Qiagen. Pepstatin A, leupeptin, aprotinin, phenylmethanelsulfonyl fluoride (PMSF) and lysozyme were purchased from Sigma. Restriction enzymes, BSA, Low-Range ssRNA Ladder, RNase Inhibitor—Human Placenta, and E. coli strains DH5α and NiCo21(DE3) [23] were purchased from New England Biolabs (NEB). Subcloning Efficiency DH5α Chemically Competent E. coli was purchased from Invitrogen. IPTG and kanamycin were purchased from GoldBio. Lysing Matrix B was obtained from MP Biomedicals. [α-32P]UTP was purchased from Perkin-Elmer. Sypro RED protein stain was purchased from VWR.
Cloning
E. coli DH5α and NiCo21(DE3) were used for cloning and protein expression, respectively. The mazEFSa gene cassette was amplified by PCR using primers mazEFSa -NcoI-F (5′ACACCCATGGATATGTTATCTTTAGTCAAAATAG-3′) and mazEFSa-XhoI-R (5′ CACACTCGAGATTTTTCTGGTGAGCTAC-3′) from the total DNA of the MRSA strain C2 [17]. The amplicon was inserted into the corresponding restriction sites of pET-28a to create pET-28a-mazEFSa, which codes for MazEFSa containing a C-terminal histidine-6 tag on MazFSa. The mazESa antitoxin gene was amplified from the same strain using primers mazESa-NdeI-F (5′ACACCATATGTTATCTTTTATCAAATAGAAG-3′) and mazESa-XhoI-stop-R (5′-ACACCTCGAGTCATTCATTCGTTGAATTAGAA-3′) and cloned into pET-28a, resulting in pET-28a-mazESa, which encodes an N-terminal histidine-6 tagged MazESa. The sequence of all clones was determined by DNA sequencing. E. coli carrying the recombinant plasmids were cultured in LB containing 50 μg/mL kanamycin.
Protein expression and purification
To express MazEFSa(His)6, an overnight culture inoculated with a single colony of E. coli NiCo21(DE3) freshly transformed with pET-28a-mazEFSa was diluted 100-fold into 2 L of LB+Kan. The culture was grown at 37°C until the OD600 reached 0.6–0.8, at which point expression was induced with 1 mM IPTG (final concentration) for 4 hours at 37°C. Expression of (His)6MazESa was performed the same, except IPTG was added when the OD600 reached 0.4–0.6. The 2 L cultures were harvested by centrifugation at 4°C (8,000×g for 500 mL bottles and 10,000×g for 50 mL conical tubes) and stored at −20°C.
MazFSa(His)6 was purified from the MazEFSa(His)6 complex under denaturing conditions. A pellet corresponding to 2 L culture was thawed in a room temperature water bath for 10 minutes. The pellet was resuspended in 20 mL Binding Buffer (10 mM Tris, 500 mM NaCl, 10 mM imidazole, pH 7.9) containing 8 M urea and lysed by 30 min of incubation at room temperature with inversion. Cell debris was pelleted by centrifugation at 40,000×g at room temperature for 15 minutes. The clarified lysate was mixed with 1 mL 1:1 Ni2+-NTA resin slurry and batch loaded for 30 min at room temperature with inversion. The slurry was applied to a gravimetric flow column and the resin was washed with 50 mL binding buffer containing 8 M urea to fully disrupt the MazESa-MazFSa(His)6 complex. On-column refolding of MazFSa(His)6 was achieved with seven washes of 10 mL urea/binding buffer decreasing the concentration of urea by 1 M with each wash. Wash steps containing greater than 4 M urea were performed at room temperature; all subsequent wash steps were performed at 4°C. Refolding was followed with 10 mL washes of binding buffer (containing no urea) and binding buffer containing 60 mM imidazole. MazFSa(His)6 was eluted with 5 mL binding buffer containing 250 mM imidazole. To remove proteins with high intrinsic affinity for Ni2+-NTA resin [23], the eluate was diluted with 5 mL binding buffer and applied to a 5 mL bed volume of chitin resin (NEB) followed by washing with 8 mL binding buffer. The flow-through and wash fractions were combined and concentrated to ~1 mL using an Amicon Ultra-15 3 kDa molecular-weight cutoff spin concentrator (Millipore) at 4°C. After overnight dialysis in PBS (pH 6.5) the purity and concentration MazFSa(His)6 were assessed by SDS-PAGE using 4–20% TGX Mini-Protean gels (Bio-Rad). Concentration was determined by densitometry of protein bands in the gel, and by BCA Assay (Pierce), using lysozyme (molecular mass 14.3 kDa) as the standard for both quantification assays.
(His)6MazESa was purified under native conditions and all steps were performed on ice or at 4°C. A pellet corresponding to 2 L culture was thawed for 5–10 min and resuspended in cold binding buffer containing protease inhibitors (2 μg/mL pepstatin A, 1 μg/mL leupeptin, 1 μg/mL aprotinin and 1 mM PMSF). The cells were lysed by 5 min sonication with 1 s pulse at 50% amplitude. The lysate was cleared by 15 min centrifugation at 40,000×g at 4°C and the supernatant was batch loaded with 0.5 mL 1:1 Ni2+-NTA resin slurry for 30 min at 4°C with inversion. Protease inhibitors were included in the wash and elution buffers. The resin was washed with 20 mL binding buffer, followed with 25 mL binding buffer containing 60 mM imidazole. (His)6MazESa was eluted with 5 mL binding buffer containing 250 mM imidazole, concentrated to ~0.5 mL and dialyzed against 50 mM sodium phosphate, pH 7.0, 150 mM NaCl, 1 mM DTT. Purity and concentration were assessed using the same method described for MazFSa(His)6.
HPLC analysis of oligonucleotide cleavage products
A 10 μL solution of 16 μM MazFSa(His)6 and 32 μM 5-AAGTCrUrACATCAG-3′ (“r” denotes RNA base) was incubated overnight at 37°C in 10 mM Tris, 500 mM imidazole, 20% glycerol (pH 7.9). The reaction was diluted with 20 μL of 0.1 M triethylammonium acetate (TEAA) (pH 7.0) and 10 μL was analyzed by high performance liquid chromatography using an Alliance HPLC System (e2965 Separations Moedule, Waters) with detection at 260 nm (2489 UV/Visible Detector, Waters). The full length oligonucleotide was separated from the cleavage products on a YMCbasic S5 column (4.6 × 150 mm, 5 μm, Waters) with a linear gradient of 0.1 M TEAA to 0.05 M TEAA/50% acetonitrile over 25 min. Fractions were analyzed by MALDI mass spectrometry.
Fluorometric oligonucleotide cleavage assay
The fluorogenic substrate 5′-6FAM-AGTCrUrACATCAG-BHQ-3′ (6-FAM, 6-carboxyfluorescein, BHQ, black hole quencher; “r” denotes RNA base) was diluted in McIlvaine’s phosphate-citrate (PC) buffer (71 mM phosphate, 14.5 mM citrate, pH 6.5). Wells of a 384-well sterile black tissue-culture plate (ThermoFisher) were filled with 15 μL of each dilution (0.025–4 μM, final concentration). After the substrate equilibrated for 30 min at room temperature, 15 μL of MazFSa(His)6 was added to the wells containing intact substrate. The fluorescence of the wells was measured every 30 s for 45 min using a Criterion Analyst AD (Molecular Devices) with 485 ± 15 nm excitation and 530 ± 15 nm emission filters and a 505 nm cutoff dichroic mirror. The fluorophore was excited with a 1000 W continuous lamp with 10 reads per well. The Z-height was set to 1 nm.
A calibration curve of the independently-synthesized substrate halves corresponding to the cleavage products, 5′-6-FAM-AGTCG and ACATCAG-BHQ-3′ [24], was constructed to quantify the amount of cleavage product formed based on the RFU value. Wells containing 0.0625–2 μM of the cleavage fragments were prepared alongside the intact oligonucleotide following the same protocol, except PC buffer was added instead of MazFSa(His)6. The calibration curve was constructed by plotting the average RFU measured over the 45 min experiment against amount of oligonucleotide in the well.
In vitro transcription of an RNA substrate
Quickchange site-directed mutagenesis was performed to modify the MazFSa cleavage sites in the pET-200-mazEF recombinant plasmid, referred to as pKm6EF [24]. MazFSa optimal cleavage site 1 was removed by changing TACAT to GGCAT using the following primers Site1-QC-F (5′-GTTTAACTTTAAGAAGGAGATAGGCATATGCGGGGTTCTCATCATC and Site1-QC-R GATGATGAGAACCCCGCATATGCCTATCTCCTTCTTAAAGTTAAAC-3′, suboptimal cleavage site 2 was removed by changing TACGT to CACAT using Site2-QC-F (5′-GGATCCCGGCCACGTTAATGCAGGCGCTCAATC-3′) and Site2-QC-R (5′-GATTGAGCGCCTGCATTAACGTGGCCGGGATCC-3′), and the suboptimal cleavage site 3 was optimized by changing from TACGT to TACAT using Site3-QC-F (5′-GGTAATGGTAAGCCGATACATACCCGATATGG and Site3-QC-R (5′-CCATATCGGGTATGTATCGGCTTACCATTACC-3′). The quickchange PCR was performed following the guidelines in the QuikChange manual (Stratagene) with annealing temperatures of 58°C for the Site 1 and Site 2 PCRs and 61°C for the Site 3 PCR. The PCR product was purified and digested with DpnI and transformed into Sub-cloning Efficiency CaCl2-treated E. coli following the manufacturer’s instructions except the cells were heat shocked for 30 s. The transformation was plated on LB containing 50 μg/mL kanamycin. Each mutation was confirmed by sequencing of both strands. The plasmid containing the optimized substrate is referred to as pET-200-1Δ2Δ3opt.
To prepare the RNA substrate, the pET-200-1Δ2Δ3opt plasmid was digested with HindIII for 3 hours at 37°C. The digested plasmid was purified using the QiaPrep Spin Column (Qiagen) and eluted with 30 μL DEPC-treated water. The digested plasmid (0.5–1 μg) served as the template for in vitro transcription using the T7 High Yield RNA Synthesis Kit (NEB) according to the manufacturer’s instructions. Each transcription reaction was divided into two equal portions and purified according to the RNeasy Mini handbook (Qiagen) RNA Cleanup protocol using reagents from the Total RNA Kit I (Omega Bio-Tek). RNA was eluted twice with 30 μL 0.1 mM EDTA prepared in DEPC-treated water, and all elution fractions from both columns were pooled. The RNA was quantified by A260 and checked for integrity by gel electrophoresis on a 5% acrylamide 8 M urea 1X TBE gel (denaturing gel) run in 1X TBE (pH 8.3) and post-stained for 15 min with 0.05 μg/mL ethidium bromide.
Synthesis of radiolabeled RNA transcript
Synthesis and purification of the radiolabeled RNA substrate followed the same protocol as described for the unlabeled transcript, except that 3.87 μCi of [α-32P]UTP was added in addition to the standard amounts of unlabeled NTPs.
Gel-based RNA cleavage assay
To assess the cleavage of the 3-site and 1-site substrates, 50 nM MazFSa(His)6 was incubated with 50 nM RNA in Buffer A (71 mM sodium phosphate, 14.5 mM citrate, 137 mM NaCl, 2 mM KCl, pH 6.5) at room temperature. To stop the reaction, a 10 μL aliquot was added to 10 μL SDS-formamide loading dye (87% formamide, 2.75% SDS, 16 mM EDTA, 0.025% bromophenol blue). Samples were heated to 95°C for at least 5 min prior to analysis on a 3.5% acrylamide denaturing gel and visualized with ethidium bromide.
For the time course assay, the same conditions were followed, except 1 nM MazFSa(His)6 and 25 nM RNA were prepared in assay buffer containing 0.01% BSA and the products were separated on 5% acrylamide denaturing gels
Activity against the radiolabeled substrate was assessed following the same conditions, except 1, 5 or 10 nM MazFSa(His)6 was mixed with 100 nM radiolabeled RNA substrate in Buffer A containing 0.05% BSA. The products were separated on 5% acrylamide denaturing gels, which were exposed to an Imaging Screen-K (BioRad) for 10 min prior to visualization.
Detection of MazFSa activity in S. aureus cell lysate
A single colony of MRSA strain NRS26 [17] grown on tryptic-soy agar was inoculated into 10 mL tryptic-soy broth (TSB) and grown overnight at 37°C with aeration. A fresh 22 mL culture was seeded with a 100-fold dilution of the overnight culture and grown to an OD600 of ~1.3, at which point 10 mL portions were harvested by centrifugation at 4°C, flash frozen in liquid nitrogen and stored at −80°C. The pellet was resuspended in 0.3 mL assay buffer and lysed by vortexing for 4 min with 0.15 g Lysing Matrix B silica beads (MP Biomedical). The lysate was cleared by 30 s full speed centrifugation at room temperature and mixed with 1X RNase inhibitor. After 7, 15, 35, and 65 min post-lysis, the radiolabeled RNA substrate was added to 100 nM (final concentration) and the samples were heated at 95°C for 1 min. After 10 min reaction at room temperature with RNA, 10 μL aliquots were quenched with SDS-formamide loading dye, electrophoresed, and imaged as described above. To inhibit endogenous MazFSa activity in the most active lysate, 200 nM purified (His)6MazESa was added to lysate 65 min post-lysis and incubated for 10 min prior to the addition of RNA for a 10 min reaction. For negative and positive controls, 100 nM radiolabeled RNA was incubated without or with 10 nM MazFSa(His)6 in assay buffer with 0.01% BSA at 95°C for 1 min followed by room temperature for 1 min.
Results
Design of a fluorogenic RNA-DNA chimeric substrate for kinetic assessment of MazFSa
MazFSa optimally cleaves at the sequence U↓ACAU in single-stranded RNA and was also shown to cleave the synthetic RNA substrate 5′-AAGUCUACAUCAG-3′ [19]. Based on this substrate, a chimeric DNA-RNA substrate consisting of DNA bases at all positions except for the U and the A surrounding the cleavage site was designed (Fig. 1A). The DNA bases afford enhanced stability and protection from general ribonucleases. The synthetic chimeric substrate 5′-AAGTCrUrACATCAG-3′ was subjected to cleavage by MazFSa(His)6 and analyzed by HPLC. The HPLC trace in Fig. 1B shows the cleavage of approximately 83% of the input chimeric substrate by MazFSa. MALDI mass spectrometry of fractions confirmed the expected mass of the intact substrate and cleavage products (Fig. 1B).
Fig. 1.

Cleavage of an oligomeric substrate by MazFSa(His)6. (A) Sequence of the non-fluorogenic substrate, “r” denotes RNA; MazFSa recognition sequence is underlined. MazFSa cleaves this substrate to the products shown. (B) Products from a 16 hour incubation of the oligonucleotide substrate from Fig. 1A (32 μM) in the absence (black) or presence (blue) of MazFSa(His)6 (16 μM) were separated by HPLC and analyzed by MALDI mass spectrometry. Approxiately 83% of the substrate was cleaved by MazFSa over the course of the incubation. The peak at the longest retention time corresponds to intact oligonucleotide (expected molecular weight [MW] = 3960.6). The peaks at shorter retention times correspond to 5′ and 3′ fragments generated from MazFSa(His)6 cleavage (expected MWs = 1854.3 and 2105.4), respectively). (C) Representative set of progress curves showing cleavage of the fluorogenic substrate derivative (5′-6-FAM-AAGTCrUrACATCAG-BHQ-3′) by MazFSa(His)6. Enzyme (0.8 μM) was added to the indicated concentration of substrate and the fluorescence was monitored over time in 384-well plates. Approximately 2–14% of the substrate was cleaved over 30 min time course. (D) Slopes from the progress curves were plotted against oligonucleotide substrate concentration. The Michaelis-Menten equation was fit to the data and apparent KM and Vmax values were determined using GraphPad. Error bars represent standard deviation, n=3.
A continuous fluorometric assay developed for MazFEc [24] was modified for kinetic evaluation of MazFSa. The chimeric substrate from Fig. 1A was synthesized with a 6-FAM fluorescein on the 5′ end and a quencher (Black Hole Quencher, BHQ) on the 3′ end. Cleavage of this fluorogenic substrate by MazFSa resulted in increased fluorescence over time, allowing progress curves to be generated at various substrate concentrations (Fig. 1C). Over the course of 30 min in the kinetic asays, MazFSa cleaved approximately 2–14% of the fluorogenic substrate. The continuous nature of the fluorometric assay enabled the characterization of MazFSa(His)6 enzyme kinetics (Vmax = 0.12 pmol/min, KM = 0.56 μM, kcat/KM = 0.0079 M−1s−1) (Fig. 1D). As has been noted previously, the absolute rate of processing of oligonucleotide substrates by recombinantly expressed and purified toxins is low and thus requires higher concentrations of enzyme than normally used in standard kinetic assays [24, 25]. Therefore, although the Michaelis-Menten equation was fit to the data to determine an apparent KM and Vmax, the enzymatic parameters observed for MazFSa(His)6 do not follow standard Michaelis-Menten kinetics.
Construction of an RNA substrate for gel-based endoribonuclease activity
Although the fluorogenic substrate is useful for kinetic assessment, MazFSa cleaves mRNA transcripts as its endogenous substrate. Thus, a method for the analysis of MazFSa(His)6 endoribonuclease activity against mRNA was developed. An RNA substrate was generated by in vitro transcription as described in the Materials and Methods. The resultant transcript from this template contained three MazFSa cleavage sites, including one preferentially cleaved site (site 1, UACAU), a sub-optimal site at which cleavage was not detected (site 2, UACGU) and a second sub-optimal site that was cleaved (site 3, UACAU) (Fig. 2, Left). The primary cleavage site was approximately 66 bases from the 5′ end of the full length transcript, complicating analysis of the cleavage products by gel electrophoresis (Fig. 2, Left). To improve the resolution of the cleavage products on gels, the original template was engineered by site-directed mutagenesis to remove sites 1 and 2 and optimize site 3, such that the new substrate contained only one cleavage site of the optimal sequence UACAU (Fig. 2, Right). Cleavage of this substrate by MazFSa resulted in easily distinguishable products (Fig. 2, Right).
Fig. 2.

Cleavage of an mRNA transcript substrate by MazFSa(His)6. A transcript was produced as described in Materials and Methods. Incubation of the full length transcript (1070 nucleotides [nts]) with MazFSa(His)6 resulted in the cleavage products shown in the gel and depicted in the schematic below the gel. Left: Cleavage of the 3-site substrate (50 nM) produced 4 cleavage products within 240 s incubation with MazFSa(His)6 (50 nM). The products from MazFSa(His)6 cleavage at site 1 (UACAU) and site 3 (UACGU) are depicted below the gel. MazFSa(His)6 did not cleave at site 2 (UACGU) within 240 s. Right: Cleavage of the 1-site substrate under the same conditions results in two well-separated products. As shown below the gel, in the 1-site substrate the original cleavage sites 1 and 2 were removed and site 3 was changed to the optimal MazFSa recognition sequence, UACAU. Products were separated by denaturing gel electrophoresis and visualized by ethidium bromide staining.
The sensitivity of this substrate for MazFSa(His)6 activity was tested with lower and perhaps more physiologically relevant concentrations of MazFSa. A time course experiment with 1 nM MazFSa(His)6 and 25 nM RNA resulted in rapid processing, with approximately 50% cleavage of the substrate in 5 minutes (Fig. 3). To our knowledge, this is the most sensitive assay for in vitro assessment of the ribonuclease activity of purified MazFSa. Furthermore, MazFSa(His)6 activity was inhibited by (His)6MazESa, (+E lane, Fig. 3) demonstrating the specificity of the cleavage of this substrate.
Fig. 3.
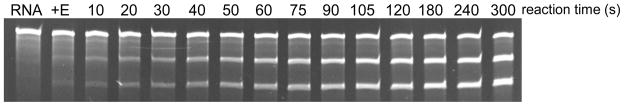
Time course of MazFSa endoribonuclease activity on an mRNA transcript. MazFSa(His)6 (1 nM) was incubated at room temperature with the single site substrate (from Fig. 2, Right; 25 nM). Activity is detected within 10 s and >50% processing is achieved by 5 min. To inhibit MazFSa activity, (His)6MazESa was incubated with MazFSa(His)6 for 15 min prior to reaction with the single site mRNA substrate for 60 seconds (+E lane). Products were separated by denaturing gel electrophoresis and visualized by ethidium bromide staining.
Radiometric assay for MazFSa activity
As a prelude to experiments with endogenous MazFSa from S. aureus lysate, the activity of MazFSa(His)6 was assessed with a 32P-labeled version of the single site substrate (from Fig. 2, Right) in a time course experiment. As shown in Fig. 4, the sensitivity of the assay allows for clear differentiation in the activity of increasing concentrations of MazFSa. In this experiment, minimal cleavage was detected by 1 nM MazFSa(His)6 after 11 min incubation, moderate processing was observed by 5 nM MazFSa(His)6, and there was significant processing by 10 nM MazFSa(His)6 (Fig. 4).
Fig. 4.
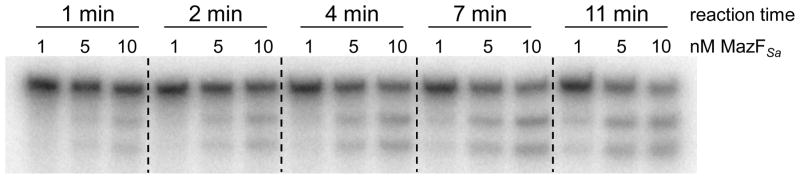
Radiometric assay for MazFSa activity. MazFSa(His)6 (1, 5, or 10 nM) was incubated at room temperature with 32P-labeled single-site substrate (100 nM). The resultant products were separated by denaturing gel electrophoresis and visualized by phosphorimaging.
Detection of endogenous MazFSa activity in S. aureus cell lysate
The highly sensitive and specific cleavage of the radiolabeled substrate allowed for detection of endogenous MazFSa activity in S. aureus cell lysate. Lysate was prepared from the S. aureus strain NRS26, in which the mazEFSa genes and transcript were previously detected [17]. The radiolabeled substrate was added to the lysate 7, 15, 35 and 65 min post-lysis. As shown in Fig. 5, cleavage of the RNA substrate increased with longer post-lysis incubation times, consistent with MazESa being susceptible to proteolysis by endogenous proteases in the lysate, resulting in the release of active MazFSa. Addition of recombinant (His)6MazESa to lysate 65 min post-lysis inhibited MazFSa activity, demonstrating the specificity of the RNA cleavage by endogenous MazFSa (far right lane in Fig. 5). This is the first detection of the enzymatic activity of endogenous MazFSa.
Fig. 5.
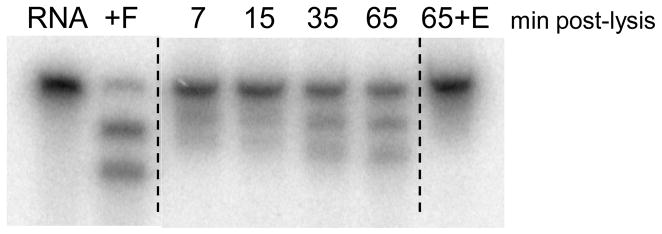
Detection of endogenous MazFSa activity in S. aureus cell lysate. Radiolabeled RNA transcript (100 nM) was added to S. aureus lysate 7, 15, 35 or 65 min post-lysis and incubated at room temperature for 10 min. CONTROLS: Far Left -- RNA only (R) and RNA processed in vitro by 10 nM MazFSa(His)6 (+F); Far Right -- S. aureus lysate 65 min post-lysis was incubated with 200 nM (His)6MazESa prior to reaction with radiolabeled RNA for 10 min (65+E). Products were separated by denaturing gel electrophoresis and visualized by phosphorimaging.
Discussion
Toxin-antitoxin systems are an ingenious method to maintain a plasmid in a bacterial host. Although their role on the bacterial chromosome is unclear, at the biochemical level Type II TA systems operate via the same mechanism whether plasmid or chromosomal; that is, a stable toxin protein is inhibited by a proteolytically labile (and co-transcribed/translated) antitoxin protein. Important work has shown that the genes for TA systems are present in a variety of pathogenic bacteria [17, 26–30]. Fewer publications have reported on the detection of TA transcripts in such bacteria [17, 26, 27] and fewer still on the detection of the actual TA proteins [18, 21]. While the presence of the DNA, mRNA, and proteins is clearly informative, it does not tell the whole story, especially for TA systems. Specifically, transcription and translation does not mean that the protein toxin is enzymatically active, given the co-transcription and co-translation of the proteic antitoxin.
Here we describe an assay that allows, for the first time, for the detection of MazFSa enzymatic activity from its endogenous source. The principles applied for the design of this substrate and this assay should be readily transferable to the study of other toxins from TA pairs, especially those that are ribonucleases. There are many questions about the roles of TA systems in basic bacteriology [9, 10, 31–34], their function in response to stress [35, 36] and their potential as novel antibacterial targets [11–14, 16]. The tools described herein for the direct detection of endogenous toxin activity should significantly facilitate the answering of those questions.
Acknowledgments
We thank the National Institutes of Health (R01GM068385) for support of our research. J.J.W. was partially supported by NIH Cell and Molecular Biology Training grant T32 GM007283.
Abbreviations
- TA
Toxin-Antitoxin
- PSK
Post-segregational killing
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ogura T, Hiraga S. Mini-F plasmid genes that couple host cell division to plasmid proliferation. Proc Natl Acad Sci U S A. 1983;80:4784–8. doi: 10.1073/pnas.80.15.4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gerdes K, Rasmussen PB, Molin S. Unique type of plasmid maintenance function: postsegregational killing of plasmid-free cells. Proc Natl Acad Sci U S A. 1986;83:3116–20. doi: 10.1073/pnas.83.10.3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hayes CS, Sauer RT. Cleavage of the A site mRNA codon during ribosome pausing provides a mechanism for translational quality control. Mol Cell. 2003;12:903–11. doi: 10.1016/s1097-2765(03)00385-x. [DOI] [PubMed] [Google Scholar]
- 4.Jensen RB, Gerdes K. Programmed cell death in bacteria: proteic plasmid stabilization systems. Mol Microbiol. 1995;17:205–10. doi: 10.1111/j.1365-2958.1995.mmi_17020205.x. [DOI] [PubMed] [Google Scholar]
- 5.Lehnherr H, Yarmolinsky MB. Addiction protein Phd of plasmid prophage P1 is a substrate of the ClpXP serine protease of Escherichia coli. Proc Natl Acad Sci U S A. 1995;92:3274–7. doi: 10.1073/pnas.92.8.3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tsuchimoto S, Nishimura Y, Ohtsubo E. The stable maintenance system pem of plasmid R100: degradation of PemI protein may allow PemK protein to inhibit cell growth. J Bacteriol. 1992;174:4205–11. doi: 10.1128/jb.174.13.4205-4211.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brzozowska I, Zielenkiewicz U. Plasmid. 2013. Regulation of toxin-antitoxin systems by proteolysis. [DOI] [PubMed] [Google Scholar]
- 8.Pandey DP, Gerdes K. Toxin-antitoxin loci are highly abundant in free-living but lost from host-associated prokaryotes. Nucleic Acids Res. 2005;33:966–76. doi: 10.1093/nar/gki201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Van Melderen L. Toxin-antitoxin systems: why so many, what for? Curr Opin Microbiol. 2010;13:781–5. doi: 10.1016/j.mib.2010.10.006. [DOI] [PubMed] [Google Scholar]
- 10.Tsilibaris V, Maenhaut-Michel G, Mine N, Van Melderen L. What is the benefit to Escherichia coli of having multiple toxin-antitoxin systems in its genome? J Bacteriol. 2007;189:6101–8. doi: 10.1128/JB.00527-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alonso JC, Balsa D, Cherny I, Espinosa M, Francuski D, Gazit E, Gerdes K, Hitchin E, Martin MT, Nieto C, Overweg K, Pellicer T, Saenger W, Welfle W, Welfle H, Wells J. In: Enzyme-mediated resistance to antibiotics: mechanisms, dissemination, and prospects for inhibition. Bonomo RA, Tomalsky ME, editors. American Society for Microbiology Press; Washington DC: 2007. pp. 313–329. [Google Scholar]
- 12.Engelberg-Kulka H, Sat B, Reches M, Amitai S, Hazan R. Bacterial programmed cell death systems as targets for antibiotics. Trends Microbiol. 2004;12:66–71. doi: 10.1016/j.tim.2003.12.008. [DOI] [PubMed] [Google Scholar]
- 13.DeNap JC, Hergenrother PJ. Bacterial death comes full circle: targeting plasmid replication in drug-resistant bacteria. Org Biomol Chem. 2005;3:959–66. doi: 10.1039/b500182j. [DOI] [PubMed] [Google Scholar]
- 14.Park SJ, Son WS, Lee BJ. Structural overview of toxin-antitoxin systems in infectious bacteria: A target for developing antimicrobial agents. Biochim Biophys Acta. 2013;1834:1155–67. doi: 10.1016/j.bbapap.2013.02.027. [DOI] [PubMed] [Google Scholar]
- 15.Williams JJ, Hergenrother PJ. Exposing plasmids as the Achilles’ heel of drug-resistant bacteria. Curr Opin Chem Biol. 2008;12:389–99. doi: 10.1016/j.cbpa.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Williams JJ, Hergenrother PJ. Artificial activation of toxin-antitoxin systems as an antibacterial strategy. Trends Microbiol. 2012;20:291–8. doi: 10.1016/j.tim.2012.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Williams JJ, Halvorsen EM, Dwyer EM, DiFazio RM, Hergenrother PJ. Toxin-antitoxin (TA) systems are prevalent and transcribed in clinical isolates of Pseudomonas aeruginosa and methicillin-resistant Staphylococcus aureus. FEMS Microbiol Lett. 2011;322:41–50. doi: 10.1111/j.1574-6968.2011.02330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fu Z, Donegan NP, Memmi G, Cheung AL. Characterization of MazFSa, an endoribonuclease from Staphylococcus aureus. J Bacteriol. 2007;189:8871–9. doi: 10.1128/JB.01272-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhu L, Inoue K, Yoshizumi S, Kobayashi H, Zhang Y, Ouyang M, Kato F, Sugai M, Inouye M. Staphylococcus aureus MazF specifically cleaves a pentad sequence, UACAU, which is unusually abundant in the mRNA for pathogenic adhesive factor SraP. J Bacteriol. 2009;191:3248–55. doi: 10.1128/JB.01815-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fu Z, Tamber S, Memmi G, Donegan NP, Cheung AL. Overexpression of MazFSa in Staphylococcus aureus induces bacteriostasis by selectively targeting mRNAs for cleavage. J Bacteriol. 2009;191:2051–9. doi: 10.1128/JB.00907-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Donegan NP, Cheung AL. Regulation of the mazEF toxin-antitoxin module in Staphylococcus aureus and its impact on sigB expression. J Bacteriol. 2009;191:2795–805. doi: 10.1128/JB.01713-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Donegan NP, Thompson ET, Fu Z, Cheung AL. Proteolytic regulation of toxin-antitoxin systems by ClpPC in Staphylococcus aureus. J Bacteriol. 2010;192:1416–22. doi: 10.1128/JB.00233-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Robichon C, Luo J, Causey TB, Benner JS, Samuelson JC. Engineering Escherichia coli BL21(DE3) derivative strains to minimize E. coli protein contamination after purification by immobilized metal affinity chromatography. Appl Environ Microbiol. 2011;77:4634–46. doi: 10.1128/AEM.00119-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang NR, Hergenrother PJ. A continuous fluorometric assay for the assessment of MazF ribonuclease activity. Anal Biochem. 2007;371:173–83. doi: 10.1016/j.ab.2007.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Boynton TO, McMurry JL, Shimkets LJ. Characterization of Myxococcus xanthus MazF and implications for a new point of regulation. Mol Microbiol. 2013;87:1267–76. doi: 10.1111/mmi.12165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moritz EM, Hergenrother PJ. Toxin-antitoxin systems are ubiquitous and plasmid-encoded in vancomycin-resistant enterococci. Proc Natl Acad Sci USA. 2007;104:311–6. doi: 10.1073/pnas.0601168104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Halvorsen EM, Williams JJ, Bhimani AJ, Billings EA, Hergenrother PJ. Txe, an endoribonuclease of the enterococcal Axe-Txe toxin-antitoxin system, cleaves mRNA and inhibits protein synthesis. Microbiology. 2011;157:387–97. doi: 10.1099/mic.0.045492-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosvoll TC, Pedersen T, Sletvold H, Johnsen PJ, Sollid JE, Simonsen GS, Jensen LB, Nielsen KM, Sundsfjord A. PCR-based plasmid typing in Enterococcus faecium strains reveals widely distributed pRE25-, pRUM-, pIP501- and pHTbeta-related replicons associated with glycopeptide resistance and stabilizing toxin-antitoxin systems. FEMS Immunol Med Microbiol. 2010;58:254–68. doi: 10.1111/j.1574-695X.2009.00633.x. [DOI] [PubMed] [Google Scholar]
- 29.Nieto C, Sadowy E, de la Campa AG, Hryniewicz W, Espinosa M. The relBE2Spn toxin-antitoxin system of Streptococcus pneumoniae: role in antibiotic tolerance and functional conservation in clinical isolates. PLoS ONE. 2010;5:e11289. doi: 10.1371/journal.pone.0011289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bjorkeng E, Rasmussen G, Sundsfjord A, Sjoberg L, Hegstad K, Soderquist B. Clustering of polyclonal VanB-type vancomycin-resistant Enterococcus faecium in a low-endemic area was associated with CC17-genogroup strains harbouring transferable vanB2-Tn5382 and pRUM-like repA containing plasmids with axe-txe plasmid addiction systems. APMIS. 2011;119:247–58. doi: 10.1111/j.1600-0463.2011.02724.x. [DOI] [PubMed] [Google Scholar]
- 31.Magnuson RD. Hypothetical functions of toxin-antitoxin systems. J Bacteriol. 2007;189:6089–92. doi: 10.1128/JB.00958-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hayes CS, Sauer RT. Toxin-antitoxin pairs in bacteria: killers or stress regulators? Cell. 2003;112:2–4. doi: 10.1016/s0092-8674(02)01282-5. [DOI] [PubMed] [Google Scholar]
- 33.Yamaguchi Y, Park JH, Inouye M. Toxin-antitoxin systems in bacteria and archaea. Annu Rev Genet. 2011;45:61–79. doi: 10.1146/annurev-genet-110410-132412. [DOI] [PubMed] [Google Scholar]
- 34.Hayes F, Van Melderen L. Toxins-antitoxins: diversity, evolution and function. Crit Rev Biochem Mol Biol. 2011;46:386–408. doi: 10.3109/10409238.2011.600437. [DOI] [PubMed] [Google Scholar]
- 35.Gerdes K, Christensen SK, Lobner-Olesen A. Prokaryotic toxin-antitoxin stress response loci. Nat Rev Microbiol. 2005;3:371–82. doi: 10.1038/nrmicro1147. [DOI] [PubMed] [Google Scholar]
- 36.Wang X, Wood TK. Toxin-antitoxin systems influence biofilm and persister cell formation and the general stress response. Appl Environ Microbiol. 2011;77:5577–83. doi: 10.1128/AEM.05068-11. [DOI] [PMC free article] [PubMed] [Google Scholar]