

Abstract
Background
C3H/HeJ (C3H) mice are extremely resistant to atherosclerosis, especially males. To understand the underlying genetic basis, we performed quantitative trait locus (QTL) analysis on a male F2 (the second generation from an intercross between 2 inbred strains) cohort derived from an intercross between C3H and C57BL/6 (B6) apolipoprotein E–deficient (Apoe−/−) mice.
Methods and Results
Two hundred forty‐six male F2 mice were started on a Western diet at 8 weeks of age and kept on the diet for 5 weeks. Atherosclerotic lesions in the aortic root and fasting plasma lipid levels were measured. One hundred thirty‐four microsatellite markers across the entire genome were genotyped. Four significant QTLs on chromosomes (Chr) 2, 4, 9, and 15 and 4 suggestive loci on Chr1, Chr4, and Chr7 were identified for atherosclerotic lesions. Unexpectedly, the C3H allele was associated with increased lesion formation for 2 of the 4 significant QTLs. Six loci for high‐density lipoprotein (HDL), 6 for non‐HDL cholesterol, and 3 for triglycerides were also identified. The QTL for atherosclerosis on Chr9 replicated Ath29, originally mapped in a female F2 cohort derived from B6 and C3H Apoe−/− mice. This locus coincided with a QTL for HDL, and there was a moderate, but statistically significant, correlation between atherosclerotic lesion sizes and plasma HDL cholesterol levels in F2 mice.
Conclusions
These data indicate that most atherosclerosis susceptibility loci are distinct from those for plasma lipids except for the Chr9 locus, which exerts effect through interactions with HDL.
Keywords: atherosclerosis, cholesterol, mapping, quantitative trait loci, sex
Introduction
Atherosclerosis is a complex inflammatory disease of large and medium‐sized arteries, resulting from interactions between genetic and environmental factors.1 The mouse is the leading mammalian model organism for finding genes involved in atherosclerosis and many other complex diseases.2 A number of candidate genes suspected of contributing to the development of atherosclerosis have been tested through construction and analysis of gene knockout or transgenic mice.3 Inbred mouse strains that display phenotypic differences in atherosclerosis or related traits have been used to conduct quantitative trait locus (QTL) analysis for finding new genes and pathways that give rise to the traits. To date, >18 mouse crosses from 12 inbred strains have been constructed to identify loci for atherosclerosis, leading to the identification of 43 QTLs for aortic plaques (http://www.informatics.jax.org/searches/allele_form.shtml). C3H/HeJ (C3H) and C57BL/6 (B6) are the most phenotypically divergent inbred mouse strains in terms of variation in atherosclerotic lesion sizes in the aortic root.4 C3H mice develop much smaller lesions than B6 mice when fed an atherogenic diet or deficient in apolipoprotein E (Apoe–/–),5.6 In a female F2 cohort derived from B6.Apoe−/− and C3H.Apoe−/− mice, we identified a major locus on chromosome (Chr) 9, named Ath29 (initially named Ath22), that had a major effect on atherosclerotic lesion formation in the aortic root.7 This locus was subsequently replicated in 2 separate intercrosses that developed fatty streak or advanced lesions.8
Previous studies have shown that atherosclerosis susceptibility loci mapped from a female population are often different from those mapped from a male population even though they were derived from the same cross.9–13 In this study, we sought to identify QTLs for atherosclerosis in a male F2 cohort generated from B6.Apoe–/– and C3H.Apoe–/– mice. Male mice are more resistant to atherosclerosis than their female counterparts, partially due to higher HDL cholesterol levels.14–16 Thus, the potential genetic link of atherosclerosis with plasma lipids was also explored through the cohort.
Materials and Methods
Mice
B6.Apoe−/− mice were purchased from the Jackson Laboratories, and C3H.Apoe−/− mice were created in our laboratory. The creation of a male F2 population from the 2 Apoe−/− mouse strains was done as recently reported.17 Briefly, B6.Apoe−/− mice were mated with C3H.Apoe−/− mice to generate F1s, which were intercrossed by brother–sister mating to generate 246 male F2s. Mice were weaned onto a chow diet at 3 weeks of age. At 8 weeks of age, mice were switched onto a Western diet containing 21% fat, 34.1% sucrose, 0.15% cholesterol, and 19.5% casein (TD 88137; Harlan Laboratories) and maintained on the diet for 5 weeks. Mice were fasted overnight before being killed, and blood was collected via retro‐orbital venous plexus puncture with the animals under isoflurane anesthesia. After being bled, mice were killed to allow collection of the heart and tail samples. All procedures were carried out in accordance with the National Institutes of Health guidelines and approved by the institutional Animal Care and Use Committee.
Aortic Lesion Analysis
Atherosclerotic lesions in aortic root were measured as previously reported.7 Briefly, mice were killed via cervical dislocation after isoflurane anesthesia. The vasculature of the animals was perfusion‐fixed with 10% formalin through the left ventricle of the heart. The aortic root and adjacent heart were excised en bloc and embedded in optimal cutting temperature compound. Serial 10‐μm‐thick cryosections from the middle portion of the ventricle to the aortic arch were collected and mounted on slides. In the region from the appearance to the disappearance of the aortic valves, every other section was collected. In all other regions, every fifth section was collected. Sections were stained with oil red O and hematoxylin and counterstained with fast green. Atherosclerotic lesion areas were measured using an ocular lens with a square‐micrometer grid on a light microscope. The lesion areas on all sections were summed and then normalized to 44 sections for each mouse, and this number was used for statistical analysis.
Plasma Lipid Analysis
The measurements of total cholesterol, HDL cholesterol, and triglycerides were performed as reported previously.18 Briefly, 6 μL of plasma samples (for total cholesterol measurements, plasma was diluted 1:5 in distilled water), lipid standards, and controls was loaded onto a 96‐well plate in duplicate and then mixed with 150 μL of cholesterol or triglyceride reagents. After an 8‐minute incubation at 37°C, the absorbance at 500 nm was read on a Molecular Devices plate reader. Non‐HDL cholesterol was calculated as the difference between total and HDL cholesterol.
Genotyping
DNA was isolated from the tails of mice by using the standard phenol–chloroform extraction and ethanol precipitation method. One hundred thirty‐four microsatellite markers on all 19 autosomes and the X chromosome at an interval of approximately 11 cM were screened for all of the F2 progeny individually by polymerase chain reaction. Parental and F1 DNA was also typed as controls for each marker so as to readily identify the parental origin of alleles at the marker in F2s.
Statistical Analysis
QTL analysis was performed using J/qtl for atherosclerotic lesion size and plasma lipid levels in 246 male F2s that were genotyped for 134 micosatellite markers across the entire genome (the data have been deposited in the Mouse Genome Informatics database), as we previously reported.7,17,19 J/qtl is a Java graphic user interface for the popular QTL data analysis software R/qtl (http://churchill.jax.org/software/jqtl.shtml). One thousand permutations of trait values were run to define the genomewide logarithm of odds (LOD) score thresholds for significant and suggestive linkage to each trait. Loci that exceeded the 95th percentile of the permutation distribution were defined as significant (P<0.05), and those exceeding the 37th percentile were suggestive (P<0.63), as recommended by the Complex Trait Consortium.20 Permutation tests are the standard statistical approach for obtaining threshold values that are adjusted for multiple testing.21 In these permutation tests, genome scans are repeatedly carried out on shuffled versions of the data to estimate an LOD threshold that is appropriate for the given data set.
ANOVA was performed to determine the statistical significance of differences in phenotypic values of F2 mice among different genotypes at a specific marker. The mode of inheritance for a QTL was determined based on relative trait values of F2 mice among 3 different genotypes at the nearest marker: if a heterozygous allele (BC) produced the same phenotypic effect as a homozygous allele (BB or CC), the QTL effect was called “dominant” and the allele with a larger effect was called the “high allele”; if a heterozygous allele produced a phenotypic effect somewhere between the effect of the 2 homozygous alleles (BB and CC), it was called “additive”; if an allele only expressed a phenotypic effect in the homozygous condition, it was called “recessive”; and if a heterozygous allele produced a phenotypic effect that exceeded the effect of the 2 homozygous alleles, it was called “heterosis.”
Prioritization of Positional Candidate Genes
The Sanger SNP database (http://www.sanger.ac.uk/cgi-bin/modelorgs/mousegenomes/snps.pl) was used to prioritize candidate genes for significant atherosclerosis QTLs that had been mapped in ≥2 crosses generated from different parental strains. Probable candidate genes were defined as those with ≥1 single nucleotide polymorphisms (SNPs) in coding or upstream promoter regions that were shared by the parental strains carrying the “high” allele but were different from the parental strains carrying the “low” allele at a QTL.
Results
Trait Value Frequency Distributions
Values of atherosclerotic lesion sizes in the aortic root of 246 male F2 mice were distributed in a Pareto manner: the number of F2 mice with a lesion size of ≤10 000 μm2 is the largest and then decreases as lesion sizes increase (Figure 1). After being transformed using natural logs (LN), the values of atherosclerotic lesion sizes approach a normal distribution. The values of log‐transformed HDL cholesterol, non‐HDL cholesterol, and log‐transformed triglyceride concentrations in F2 mice are approximately normally distributed. These data were then analyzed using J/qtl software to detect significant and suggestive QTLs affecting the traits.
Figure 1.

Frequency distributions of atherosclerotic lesion sizes and plasma lipid levels in 246 male F2 mice after being fed a Western diet for 5 weeks. The F2 progeny was generated from an intercross between B6.Apoe−/− and C3H.Apoe−/− mice. Note that the unit for untransformed atherosclerotic lesion sizes is “μm2×1000,” whereas the unit for LN‐transformed atherosclerotic lesion sizes is “μm2.” LN indicates natural logs.
Atherosclerotic Lesions
We first performed a genomewide scan using untransformed atherosclerotic lesion size data to detect main‐effect loci with the nonparametric mode and found 4 significant QTLs, located on Chr2, Chr4, Chr9, and Chr15, and 2 suggestive QTLs, with 1 on distal Chr4 and 1 on Chr7 (Figure 2). Details of the QTLs found, including locus name, LOD score, peak location, 95% CI, genomewide significance P value, high allele, and mode of inheritance, are presented in Table 1. Unexpectedly, the C3H allele was associated with increased lesion sizes for 2 of the 4 significant QTLs, including the Chr2 and Chr15 QTLs (Table 2). The Chr2 locus peaked at 100.5 cM and had a significant LOD score of 3.59 and a genomewide P value of 0.041. This locus was overlapping in the CI with Ath28, mapped in an Akr.Apoe−/−×DBA.Apoe−/− intercross.9 The Chr15 locus peaked at 37.8 cM and had a highly significant LOD score of 5.92. This QTL replicated Ath33, a locus identified in a B6.Apoe−/−×C3H.Apoe−/− intercross fed a Western diet.13 The interval mapping plot for Chr4 revealed 2 distinct peaks, each with an LOD score exceeding the suggestive LOD score threshold of 2.05 (Figure 3). The proximal peak occurred at 31.6 cM with a significant LOD score of 3.81, and the distal peak appeared at 77.6 cM with a suggestive LOD score of 2.14. The proximal QTL was partially overlapping with Ath8, a suggestive locus mapped in an SM/J×NZB/BlNJ intercross,22 and the distal QTL replicated Athsq1, a locus identified in an MOLF/Ei×B6. Ldlr−/− backcross.11 Both loci exhibited a dominant effect from the B6 allele on lesion formation. The Chr9 QTL had a highly significant LOD score of 5.40 and a genomewide P value of <0.0001. This QTL replicated Ath29, originally identified in a female cohort derived from B6.Apoe−/− and C3H.Apoe−/− mice.7 The suggestive QTL on Chr7 replicated Ath31, identified in a B6.Apoe−/−×C3H.Apoe−/− intercross.13
Figure 2.
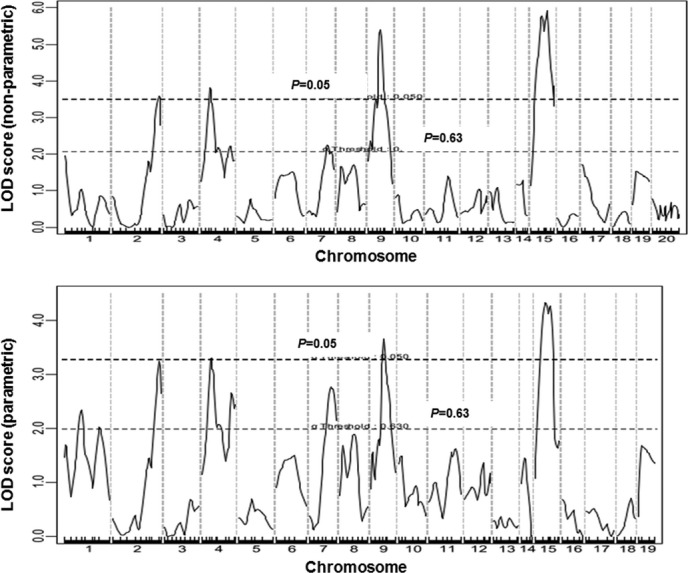
Genomewide QTL analysis to search for loci influencing atherosclerotic lesion sizes in male F2 mice. Chromosomes 1 through 20 are represented numerically on the x axis. The relative width of the space allotted for each chromosome reflects the number of microsatellite markers typed for that chromosome. The y axis represents the LOD score. Two horizontal dashed lines denote genomewide thresholds for suggestive (P=0.63) and significant (P=0.05) linkage. The top panel shows a genomewide scan for atherosclerotic lesions using the nonparametric mode, and the bottom panel shows an autosome scan for atherosclerotic lesions using the parametric mode. For the latter scan, the X chromosome was not included due to a strong biased influence from the chromosome. QTL indicates quantitative trait locus; LOD, logarithm of odds.
Table 1.
Significant and Suggestive QTLs for Atherosclerosis and Plasma Lipid Levels in Male F2 Mice Derived from B6.Apoe−/− and C3H.Apoe−/− Mice
| Locus Name | Chr | Trait | LOD* | Peak, cM | 95% CI* | P Value* | High Allele* | Mode of Inheritance* |
|---|---|---|---|---|---|---|---|---|
| Ath28 | 2 | Lesion (nonparametric) | 3.59 | 100.5 | 90.2 to 103.8 | 0.041 | C3H | Recessive |
| Ath8 | 4 | Lesion (nonparametric) | 3.81 | 31.6 | 23.6 to 77.6 | 0.022 | B6 | Dominant |
| Athsq1 | 4 | Lesion (nonparametric) | 2.14 | 77.6 | 63 to 82.6 | … | B6 | Dominant |
| Ath31 | 7 | Lesion (nonparametric) | 2.25 | 59.1 | 47.1 to 73.0 | 0.489 | B6 | Dominant |
| Ath29 | 9 | Lesion (nonparametric) | 5.40 | 44.2 | 40.2 to 48.2 | 0.0001 | B6 | Additive |
| Ath33 | 15 | Lesion (nonparametric) | 5.92 | 37.8 | 21.8 to 43.8 | 0.0001 | C3H | Additive |
| Ath30 | 1 | Lesion (parametric) | 2.34 | 39.2 | 3.7 to 91.7 | 0.385 | B6 | Dominant |
| Ath1 | 1 | Lesion (parametric) | 2.10 | 78 | 66 to 93 | … | B6 | Dominant |
| Ath28 | 2 | Lesion (parametric) | 3.24 | 100.5 | 88.2 to 103.8 | 0.054 | C3H | Recessive |
| Ath8 | 4 | Lesion (parametric) | 3.30 | 33.6 | 25.6 to 82.6 | 0.047 | B6 | Heterosis |
| Athsq1 | 4 | Lesion (parametric) | 2.65 | 75.6 | 67.6 to 82.6 | … | B6 | Dominant |
| Ath31 | 7 | Lesion (parametric) | 2.77 | 61.1 | 51.1 to 73.0 | 0.175 | B6 | Dominant |
| Ath29 | 9 | Lesion (parametric) | 3.65 | 46.2 | 40.2 to 58.2 | 0.019 | B6 | Additive |
| Ath33 | 15 | Lesion (parametric) | 4.33 | 25.8 | 15.2 to 39.8 | 0.005 | C3H | Additive |
| Hdlq5 | 1 | HDL | 7.30 | 67.71 | 65.67 to 73.67 | 0.0001 | C3H | Additive |
| Hdlq16 | 8 | HDL | 2.11 | 17.7 | 13.7 to 45.7 | 0.617 | Heterosis | |
| Hdlq17 | 9 | HDL | 3.04 | 20.2 | 0 to 34 | … | C3H | Additive |
| Hdlq54 | 9 | HDL | 3.55 | 43.91 | 18.24 to 48.24 | 0.054 | C3H | Additive |
| Hdlq18 | 12 | HDL | 2.81 | 12.04 | 10.04 to 44.04 | 0.228 | C3H | Heterosis |
| Lipq2 | 13 | HDL | 3.05 | 64.72 | 21.99 to 64.72 | 0.145 | C3H | Dominant |
| Cq1 | 1 | Non‐HDL | 4.92 | 75.67 | 67.71 to 85.67 | 0.002 | C3H | Additive |
| Chol8 | 4 | Non‐HDL | 2.51 | 13.55 | 13.55 to 57.55 | 0.323 | B6 | Additive |
| Nhdlq12 | 12 | Non‐HDL | 6.59 | 54.04 | 30.04 to 58.04 | 0.0001 | B6 | Dominant |
| Chldq8 | 14 | Non‐HDL | 2.24 | 29.37 | 19.37 to 35.37 | 0.488 | C3H | Additive |
| Nhdlq9 | 15 | Non‐HDL | 2.43 | 51.82 | 3.82 to 53.94 | 0.367 | C3H | Recessive |
| Nhdlq2 | X | Non‐HDL | 2.22 | 24.59 | 20.59 to 58.59 | 0.511 | ||
| Tglq1 | 1 | Triglyceride | 6.42 | 77.67 | 73.67 to 85.67 | 0.0001 | C3H | Additive |
| Tgq10 | 2 | Triglyceride | 2.62 | 49.35 | 24.23 to 58.23 | 0.305 | C3H | Dominant |
| Tgq28 | 16 | Triglyceride | 2.51 | 9.66 | 9.66 to 35.66 | 0.355 | B6 | Dominant |
QTL indicates quantitative trait locus; Chr, chromosome; LOD, logarithm of odds.
LOD scores were obtained from genomewide QTL analysis using J/qtl software. The significant LOD scores are highlighted in bold. The suggestive and significant LOD score thresholds were determined by 1000 permutation tests for each trait. Suggestive and significant LOD scores were 2.045 and 3.362 for atherosclerotic lesion size (nonparametric), 1.986 and 3.277 for natural log–transformed lesion sizes (parametric), 2.123 and 3.559 for HDL cholesterol, 2.087 and 3.49 for non‐HDL cholesterol, and 2.12 and 3.432 for triglycerides, respectively.
The 95% CI in cM defined by a whole genome QTL scan.
The P values reported represent the level of genomewide significance as they were generated by J/qtl basedon genomewide permutation tests.
High allele—the allele with a larger allelic effect at the peak marker of a QTL.
Mode of inheritance was defined according to allelic effect at the nearest marker of a QTL: dominant, a heterozygous allele produced the same phenotypic effect as a homozygous allele; additive, a heterozygous allele produced a phenotypic effect between the effect of the 2 homozygous alleles; recessive, an allele only expressed a phenotypic effect in the homozygous condition; and heterosis, a heterozygous allele produced a phenotypic effect exceeding the effect of the 2 homozygous alleles.
Table 2.
Effects of B6 (B) and C3H (C) Alleles in Different QTLs on Atherosclerosis and Plasma Lipids in the Intercross Between B6.Apoe−/− and C3H.Apoe−/− Mouse Strains
| Locus Namea | Chr | Trait | LOD | Peak, cM | Closest Marker | BB | BC | CC | P Value |
|---|---|---|---|---|---|---|---|---|---|
| Ath28 | 2 | Lesion (nonparametric) | 3.59 | 100.5 | D2Mit148 | 12 595±12 897 (n=73) | 10 848±10 165 (n=99) | 28 271±36 479 (n=56) | 1.8E−06 |
| Ath8 | 4 | Lesion (nonparametric) | 3.81 | 31.6 | D4Mit139 | 15 296±13 347 (n=59) | 16 945±22 166 (n=104) | 12 520±24 876 (n=73) | 0.396 |
| Athsq1 | 4 | Lesion (nonparametric) | 2.14 | 77.6 | D4Mit33 | 15 861±24 952 (n=53) | 17 479±23 433 (n=118) | 10 616±12 244 (n=59) | 0.136 |
| Ath31 | 7 | Lesion (nonparametric) | 2.25 | 59.1 | D7Mit330 | 17 873±27 165 (n=56) | 16 152±20 439 (n=118) | 11 153±16 419 (n=60) | 0.198 |
| Ath29 | 9 | Lesion (nonparametric) | 5.40 | 44.2 | D9Mit236 | 22 162±22 430 (n=54) | 15 020±20 472 (n=120) | 10 123±20 992 (n=63) | 9.26E−03 |
| Ath33 | 15 | Lesion (nonparametric) | 5.92 | 37.8 | D15Mit188 | 9386±11 194 (n=48) | 14 341±23 011 (n=120) | 24 495±25 337 (n=50) | 1.95E−03 |
| Ath30 | 1 | Lesion (parametric) | 2.34 | 39.2 | D1MIT161 | 16 369±17 243 (n=53) | 16 027±22 747 (n=121) | 13 748±22 869 (n=58) | 0.765 |
| Ath1 | 1 | Lesion (parametric) | 2.1 | 78 | D1Mit270 | 17 752±24 695 (n=64) | 16 318±23 341 (n=102 | 11 913±14 121 (n=67) | 0.261 |
| Ath28 | 2 | Lesion (parametric) | 3.24 | 100.5 | D2Mit148 | 12 595±12 897 (n=74) | 10 848±10 165 (n=99) | 28 271±36 479 (n=56) | 1.8E−06 |
| Ath8 | 4 | Lesion (parametric) | 3.30 | 33.6 | D4Mit178 | 14 252±12 034 (n=56) | 19 297±28 313 (n=107) | 10 376±12 777 (n=69) | 2.41E−02 |
| Athsq1 | 4 | Lesion (parametric) | 2.65 | 75.6 | D4Mit33 | 15 861±24 952 (n=53) | 17479±23433 (n=118) | 10 616±12 244 (n=59) | 0.136 |
| Ath31 | 7 | Lesion (parametric) | 2.77 | 61.1 | D7Mit330 | 17 873±27 165 (n=56) | 16152±20439 (n=118) | 11 153±16 419 (n=60) | 0.198 |
| Ath29 | 9 | Lesion (parametric) | 3.65 | 46.2 | D9Mit236 | 22 162±22 430 (n=54) | 15 020±20 472 (n=120 | 10 123±20 992 (n=63) | 9.26E−03 |
| Ath33 | 15 | Lesion (parametric) | 4.33 | 25.8 | D15Mit143 | 9107±10943 (n=47) | 14 181±21 364 (n=127) | 22 794±25 419 (n=62) | 2.41E−03 |
| Hdlq5 | 1 | HDL | 7.30 | 67.71 | D1Mit425 | 64.8±45.3 (n=63) | 107.1±61.3 (n=113) | 114.1±66.7 (n=55) | 2.9E−06 |
| Hdlq16 | 8 | HDL | 2.11 | 17.7 | D8Mit191 | 81.0±53.3 (n=56) | 107.9±66.8 (n=134) | 83.5±49.3 (n=43) | 6.5E−03 |
| Hdlq17 | 9 | HDL | 3.04 | 0 to 34 | D9Mit297 | 81.1±53.8 (n=65) | 95.0±62.8 (n=105) | 116.2±63.8 (n=64) | 4.80E−03 |
| Hdlq54 | 9 | HDL | 3.55 | 43.91 | D9Mit236 | 73.2±48.7 (n=55) | 95.8±60.6 (n=120) | 120.2±66.7 (n=61) | 1.8E−04 |
| Hdlq18 | 12 | HDL | 2.81 | 12.04 | D12Mit84 | 77.7±52.5 (n=60) | 112.3±65.5 (n=98) | 92.8±60.6 (n=73) | 2.1E−03 |
| Lipq2 | 13 | HDL | 3.05 | 64.72 | D13Mit151 | 73.1±49.1 (n=55) | 101.8±60.0 (n=116) | 108.4±70.2 (n=64) | 3.7E−03 |
| Cq1 | 1 | Non‐HDL | 4.92 | 75.67 | D1Mit270 | 696.2±341.6 (n=61) | 859.7±363.9 (n=101) | 976.9±332.6 (n=69) | 4.3E−05 |
| Chol8 | 4 | Non‐HDL | 2.51 | 13.55 | D4Mit192 | 938.6±353.6 (n=48) | 873.3±361.4 (n=106) | 730.4±344.9 (n=75) | 3.1E−03 |
| Nhdlq12 | 12 | Non‐HDL | 6.59 | 54.04 | D12mit277 | 865.6±291.4 (n=49) | 909.7±373.4 (n=148) | 575.3±265.7 (n=38) | 1.1E−06 |
| Chldq8 | 14 | Non‐HDL | 2.24 | 29.37 | D14MIT155 | 743.9±336.3 (n=68) | 869.0±359.5 (n=109) | 941.3±377.4 (n=54) | 8.1E−03 |
| Nhdlq9 | 15 | Non‐HDL | 2.43 | 51.82 | D15Mit161 | 830.6±327.7 (n=54) | 798.9±364.9 (n=123) | 977.1±358.8 (n=56) | 7.9E−03 |
| Nhdlq2 | X | Non‐HDL | 2.22 | 24.59 | DXMit81 | 804.2±366.2 (n=110) | 875.9±358.2 (n=116) | 1.4E−01 | |
| Tglq1 | 1 | Triglyceride | 6.42 | 77.67 | D1Mit270 | 111.6±26.2 (n=61) | 135.5±41.6 (n=102) | 148.5±48.0 (n=69) | 1.9E−06 |
| Tgq10 | 2 | Triglyceride | 2.62 | 49.35 | D2Mit126 | 117.4±34.5 (n=57) | 139.3±46.9 (n=124) | 135.7±35.6 (n=53) | 4.6E−03 |
| Tgq28 | 16 | Triglyceride | 2.51 | 9.66 | D16Mit165 | 133.5±48.7 (n=68) | 140.9±42.7 (n=97) | 119.0±31.5 (n=62) | 3.7E−03 |
Data are mean±SD. The units for these measurements are μm2/section for atherosclerotic lesions and mg/dL for plasma lipid levels. The number in the brackets represents the number of progeny with a specific genotype at a peak marker. ANOVA was used to determine the significance level (P value) of differences for a specific phenotype among progeny with different genotypes at a specific marker. The significant LOD scores were highlighted in bold. BB indicates homozygous for B6 alleles at the linked peak marker; CC, homozygous for C3H alleles; BC, heterozygous for B6 and C3H alleles at the peak marker; QTL, quantitative trait locus; Chr, chromosome; LOD, logarithm of odds.
Figure 3.
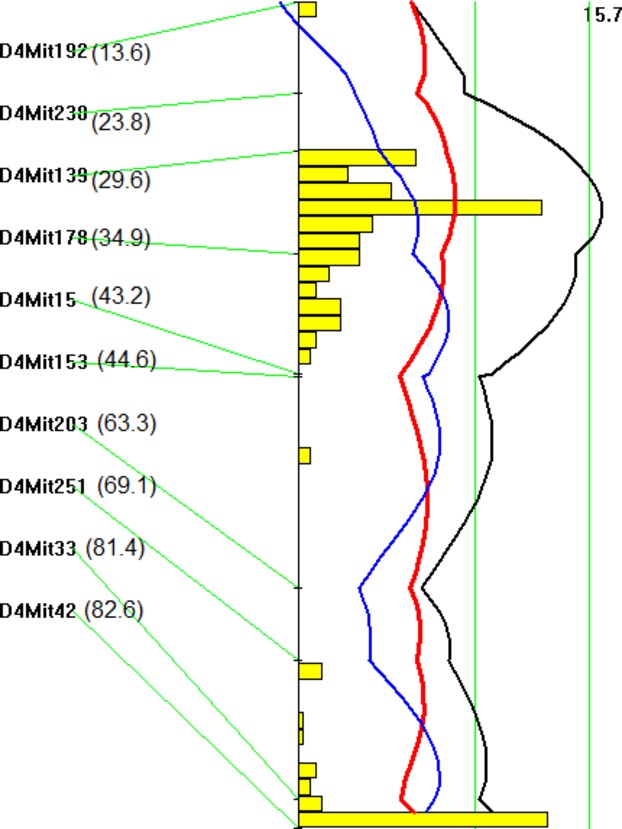
Interval mapping plot for atherosclerotic lesions on chromosome 4. The plot was created with the interval mapping function of Map Manager QTX, which includes a bootstrap test shown as a histogram estimating the CI of a QTL. Two straight vertical lines on the plot represent the genomewide significance thresholds for suggestive and significant linkage. The black line denotes the LOD score calculated at 1‐cM intervals. The blue line represents the effect of the B6 allele, and the red line represents the effect of the C3H allele. The histogram indicates the existence of 2 QTLs on the chromosome. The number in the bracket denotes the mapping distance from the centromere of a specific marker in cM. LOD indicates logarithm of odds; QTL, quantitative trait locus.
We then localized QTLs using the LN‐transformed atherosclerotic lesion size data and identified 2 additional suggestive QTLs on Chr1 (Figure 2). The proximal QTL peaked at 39.2 cM and had a suggestive LOD score of 2.34. This locus replicated Ath30, identified in a B6.Apoe−/−×C3H.Apoe−/− intercross.13 The distal QTL peaked at 78 cM and had a suggestive LOD score of 2.10. This locus replicated Ath1, initially detected in recombinant inbred strains derived from B6 and C3H mice and subsequently replicated in several crosses.13,15,23–24 Both QTLs affected lesion sizes in a dominant manner from the B6 allele (Table 2).
Plasma Lipid Levels
Genomewide scans revealed that plasma HDL, non‐HDL cholesterol, and triglyceride levels were each controlled by multiple QTLs (Figure 4, Table 1). For HDL, 2 significant QTLs, located on Chr1 and Chr9, and 4 suggestive QTLs, located on Chr8, Chr9, Chr12, and Chr13, were identified. The significant QTL on Chr1 replicated Hdlq5, which had been mapped in numerous crosses.25 The interval mapping graph for Chr9 showed 2 distinct QTLs with each surpassing the suggestive LOD score threshold of 2.087 (Figure 5). The distal locus peaked at 43.9 cM and had a significant LOD score of 3.55. This QTL coincided precisely with the atherosclerosis susceptibility locus on Chr9. The proximal QTL on Chr9 and the suggestive QTLs on Chr8 and Chr12 corresponded to Hdlq17, Hdlq16, and Hdlq18, respectively, previously mapped in a B6×129 F2 population.26 The QTL on Chr13 was partially overlapping in the confidence interval with Lipq2, identified in B6.C‐H25C×BALB/cJ F2 mice.27 For non‐HDL cholesterol, 2 significant QTLs, located on Chr1 and Chr12, and 4 suggestive QTLs, on Chr4, Chr14, Chr15, and ChrX, were identified. The Chr1 QTL replicated Cq1, a locus identified in a number of crosses.28 The Chr12 QTL replicated Nhdlq12, previously mapped in female mice derived from an intercross between B6.Apoe−/− and C3H.Apoe−/− mice.29 The suggestive QTLs on Chr4, Chr14, Chr15, and ChrX replicated Chol8, Chldq8, Nhdlq9, and Nhdlq2, respectively (http://www.informatics.jax.org/phenotypes.shtml). Plasma triglyceride levels were controlled by 1 significant QTL on Chr1 and 2 suggestive QTLs on Chr2 and Chr16, corresponding to Tglq1, Tgq10, and Tgq28, respectively (http://www.informatics.jax.org/phenotypes.shtml).
Figure 4.
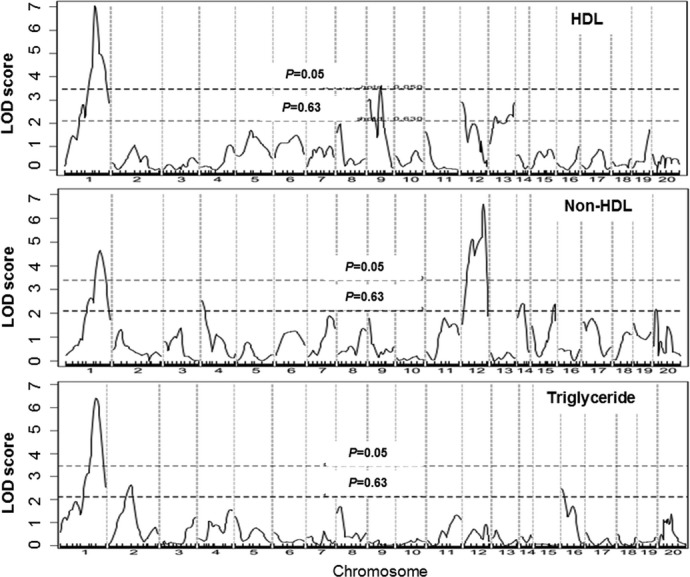
Genomewide scans for HDL, non‐HDL cholesterol, and triglyceride levels in the F2 population. Chromosomes 1 through 20 are represented numerically on the x axis, and the y axis represents the LOD score. Two horizontal dashed lines denote genomewide thresholds for suggestive and significant linkage. LOD indicates logarithm of odds; HDL, high‐density lipoprotein.
Figure 5.
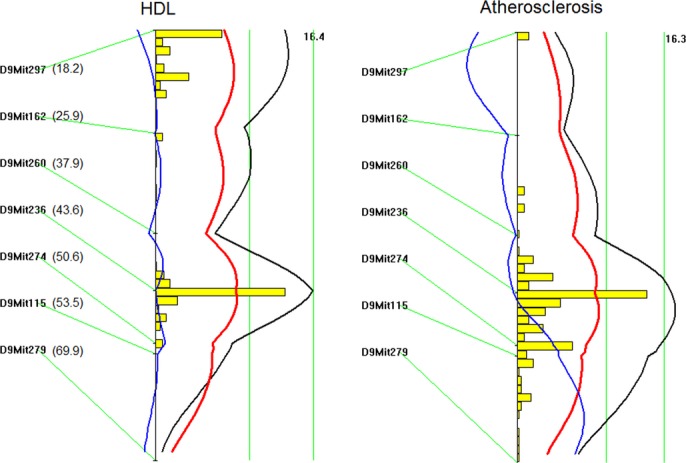
LOD score plots for HDL cholesterol levels (left) and atherosclerotic lesion size (right) on chromosome 9. Plots were created with the interval mapping function of Map Manager QTX. The histogram shown in the plots indicates the confidence interval of a QTL. The distal QTL for HDL coincided with the QTL for atherosclerotic lesions on chromosome 9. The number in the bracket denotes the cM value of a specific marker. LOD indicates logarithm of odds; QTL, quantitative trait locus; HDL, high‐density lipoprotein.
Relationships Between Plasma Lipids and Atherosclerosis
The associations of atherosclerotic lesion sizes with plasma lipid levels were analyzed using the F2 population (Figure 6). A significant inverse correlation between lesion sizes and plasma HDL cholesterol levels was observed (r=−0.19, P=0.0041). LN‐transformed lesion sizes showed an improved inverse association with plasma HDL cholesterol levels (r=−0.327, P=4.07E−7). F2 mice with higher HDL cholesterol levels tended to develop smaller atherosclerotic lesions. No significant correlation was observed between non‐HDL cholesterol levels and lesion sizes, although there was a trend toward statistical significance between untransformed lesion sizes and non‐HDL cholesterol levels (r=0.12, P=0.063).
Figure 6.

Scatterplots showing relationships of atherosclerotic lesion sizes with HDL and non‐HDL cholesterol levels in the F2 population. Each point represents an individual value of an F2 mouse. The correlation coefficient (r) and significance (P) are shown. Plasma levels of HDL but not non‐HDL cholesterol were significantly correlated with the sizes of atherosclerotic lesions. Top row: untransformed atherosclerotic lesion sizes in “μm2×1000”; bottom row: LN‐transformed atherosclerotic lesion sizes in “μm2”. LN indicates natural logs; HDL, high‐density lipoprotein.
Prioritization of Positional Candidate Genes for Atherosclerosis QTLs
The C3H allele was responsible for increased lesion formation for 2 of the 4 significant QTLs: Ath28 on Chr2 and Ath33 on Chr15. As Ath28 has been mapped in 2 separate intercrosses, including a previously reported AKR.Apoe−/−×DBA.Apoe−/− cross,9 we conducted a haplotype analysis using the Sanger SNP database to prioritize positional candidate genes for the QTL. A few candidate genes underneath the linkage peak of Ath28 were identified, including Rbm38 (173 Mb), Cdh4 (179 Mb), Ss18l1 (179.7 Mb), Hrh3 (179.8 Mb), Osbpl2 (179.8 Mb), and Lama5 (179.9 Mb) (Table 3). These candidates contain ≥1 nonsynonymous SNPs in coding regions or SNPs in the upstream regulatory region that are shared by the low allele strains (B6 and AKR) but are different from the high allele strains (C3H and DBA) at the QTL. These genes were further examined for associations with relevant human diseases using a public accessible genomewide association study database (http://www.genome.gov/GWAStudies/). Rbm38 has been shown to be associated with variation in the magnitude of statin‐mediated reduction in total and LDL cholesterol30 and Cdh4 with sudden cardiac arrest in patients with coronary heart disease.31 Lama5 was associated with colorectal cancer in European ancestry individuals.32
Table 3.
Haplotype Analysis to Prioritize Candidate Genes for Atherosclerosis QTLs on Chromosomes 2 and 15
| Gene | Chromosome | Position | Low Allele | High Allele | Consequence | ||
|---|---|---|---|---|---|---|---|
| B6 | AKR/J | C3H/HeJ | DBA/2J | ||||
| Rbm38 | 2 | 172847290 | G | G | c | c | 5′_UTR |
| Rbm38 | 2 | 172847496 | G | G | A | A | 5′_UTR |
| Cdh4 | 2 | 179177190 | C | C | T | T | 5′_UTR |
| Cdh4 | 2 | 179179412 | G | G | A | A | NON_SYNONYMOUS_CODING |
| Ss18l1 | 2 | 179777235 | C | C | G | G | 5′_UTR |
| Hrh3 | 2 | 179835661 | C | C | T | T | NON_SYNONYMOUS_CODING |
| Osbpl2 | 2 | 179854116 | G | G | A | A | 5′_UTR |
| Lama5 | 2 | 179913809 | C | C | A | A | NON_SYNONYMOUS_CODING |
| Lama5 | 2 | 179915098 | T | T | G | G | NON_SYNONYMOUS_CODING |
| Lama5 | 2 | 179920402 | C | C | T | T | NON_SYNONYMOUS_CODING |
| Lama5 | 2 | 179920521 | G | G | T | T | NON_SYNONYMOUS_CODING |
| Lama5 | 2 | 179921374 | T | T | C | C | NON_SYNONYMOUS_CODING |
| Lama5 | 2 | 179925043 | C | C | T | T | NON_SYNONYMOUS_CODING |
| Lama5 | 2 | 179928191 | A | A | G | G | 5′_UTR |
| Lama5 | 2 | 179928517 | G | G | A | A | 5′_UTR |
| Lama5 | 2 | 179929976 | C | C | T | T | NON_SYNONYMOUS_CODING |
| Lama5 | 2 | 179933048 | G | G | A | A | 5′_UTR |
| Lama5 | 2 | 179933067 | G | G | T | T | NON_SYNONYMOUS_CODING |
| Lama5 | 2 | 179933747 | T | T | A | A | 5′_UTR |
| Lama5 | 2 | 179933873 | G | G | A | A | NON_SYNONYMOUS_CODING |
| Gene | Chromosome | Position | B6 | C3H/HeJ | 129 | Consequence | |
|---|---|---|---|---|---|---|---|
| Rhpn1 | 15 | 75543724 | A | G | G | NON_SYNONYMOUS_CODING | |
| Apol11b | 15 | 77468437 | A | c | C | NON_SYNONYMOUS_CODING | |
| Cacng2 | 15 | 77950358 | G | A | A | 5′_UTR | |
| Ift27 | 15 | 78004380 | C | A | A | 5′_UTR | |
| Ncf4 | 15 | 78075365 | T | C | C | 5′_UTR | |
| Ncf4 | 15 | 78081428 | G | A | A | NON_SYNONYMOUS_CODING | |
| Csf2rb2 | 15 | 78115054 | G | t | t | NON_SYNONYMOUS_CODING | |
| Csf2rb2 | 15 | 78115310 | T | C | c | NON_SYNONYMOUS_CODING | |
| Csf2rb2 | 15 | 78115528 | C | t | t | NON_SYNONYMOUS_CODING | |
| Csf2rb2 | 15 | 78115654 | A | G | G | NON_SYNONYMOUS_CODING | |
| Csf2rb2 | 15 | 78115727 | T | C | C | NON_SYNONYMOUS_CODING | |
| Csf2rb2 | 15 | 78117449 | A | G | G | NON_SYNONYMOUS_CODING | |
| Csf2rb2 | 15 | 78119364 | T | A | A | NON_SYNONYMOUS_CODING | |
| Csf2rb2 | 15 | 78122957 | C | G | g | NON_SYNONYMOUS_CODING | |
| Csf2rb2 | 15 | 78122983 | G | a | A | NON_SYNONYMOUS_CODING | |
| Csf2rb2 | 15 | 78123018 | T | g | G | NON_SYNONYMOUS_CODING | |
| Csf2rb2 | 15 | 78123283 | G | A | A | NON_SYNONYMOUS_CODING | |
| Csf2rb2 | 15 | 78127509 | C | G | G | NON_SYNONYMOUS_CODING | |
| Mpst | 15 | 78240804 | A | G | G | NON_SYNONYMOUS_CODING | |
| Sstr3 | 15 | 78370921 | C | T | T | NON_SYNONYMOUS_CODING | |
| Sstr3 | 15 | 78370941 | G | A | A | NON_SYNONYMOUS_CODING | |
| Sstr3 | 15 | 78371011 | C | G | G | 5′_UTR | |
| Sstr3 | 15 | 78374301 | G | A | A | 5′_UTR | |
| Sstr3 | 15 | 78374693 | A | G | G | 5′_UTR | |
| Sstr3 | 15 | 78374764 | T | C | C | 5′_UTR | |
| Gm6723 | 15 | 78384992 | C | T | T | NON_SYNONYMOUS_CODING | |
| Gm6723 | 15 | 78385151 | G | C | C | NON_SYNONYMOUS_CODING | |
| Mfng | 15 | 78603821 | A | G | G | 5′_UTR | |
| Card10 | 15 | 78633071 | G | A | A | 5′_UTR | |
| Card10 | 15 | 78633319 | T | C | C | 5′_UTR | |
| Cdc42ep1 | 15 | 78677764 | G | A | A | 5′_UTR | |
| Cdc42ep1 | 15 | 78679988 | T | C | C | NON_SYNONYMOUS_CODING | |
| Cdc42ep1 | 15 | 78680104 | G | A | A | NON_SYNONYMOUS_CODING | |
| Cdc42ep1 | 15 | 78680263 | G | A | A | NON_SYNONYMOUS_CODING | |
| Lgals2 | 15 | 78681495 | T | C | C | NON_SYNONYMOUS_CODING | |
| Gcat | 15 | 78873505 | A | G | G | NON_SYNONYMOUS_CODING | |
| Micall1 | 15 | 78957630 | C | T | T | NON_SYNONYMOUS_CODING | |
| Pick1 | 15 | 79060137 | A | G | G | 5′_UTR | |
| Pick1 | 15 | 79079229 | A | G | G | NON_SYNONYMOUS_CODING | |
| Pick1 | 15 | 79086211 | A | C | C | NON_SYNONYMOUS_CODING | |
| Pick1 | 15 | 79086466 | G | T | T | NON_SYNONYMOUS_CODING | |
| Pick1 | 15 | 79086508 | A | g | G | NON_SYNONYMOUS_CODING | |
| Pick1 | 15 | 79086512 | C | g | G | NON_SYNONYMOUS_CODING | |
| Tmem184b | 15 | 79209126 | C | A | A | 5′_UTR | |
| Csnk1e | 15 | 79269395 | A | G | G | 5′_UTR | |
| Csnk1e | 15 | 79271799 | C | T | T | 5′_UTR | |
| Nptxr | 15 | 79624881 | T | G | G | NON_SYNONYMOUS_CODING | |
| Apobec3 | 15 | 79722929 | C | A | A | 5′_UTR | |
| Apobec3 | 15 | 79725453 | A | T | T | 5′_UTR | |
| Apobec3 | 15 | 79725492 | T | A | A | 5′_UTR | |
| Apobec3 | 15 | 79725494 | T | G | G | 5′_UTR | |
| Apobec3 | 15 | 79725534 | A | G | G | 5′_UTR | |
| Apobec3 | 15 | 79725547 | G | A | A | 5′_UTR | |
| Apobec3 | 15 | 79725550 | T | A | A | 5′_UTR | |
| Apobec3 | 15 | 79725566 | G | A | A | 5′_UTR | |
| Apobec3 | 15 | 79725868 | G | A | A | 5′_UTR | |
| Apobec3 | 15 | 79725887 | G | C | C | NON_SYNONYMOUS_CODING | |
| Apobec3 | 15 | 79725897 | A | T | T | NON_SYNONYMOUS_CODING | |
| Apobec3 | 15 | 79725900 | G | A | A | NON_SYNONYMOUS_CODING | |
| Apobec3 | 15 | 79728257 | A | G | G | NON_SYNONYMOUS_CODING | |
| Apobec3 | 15 | 79728260 | G | C | C | NON_SYNONYMOUS_CODING | |
| Apobec3 | 15 | 79728323 | G | A | A | NON_SYNONYMOUS_CODING | |
| Apobec3 | 15 | 79728327 | A | G | G | NON_SYNONYMOUS_CODING | |
| Apobec3 | 15 | 79728339 | C | A | A | NON_SYNONYMOUS_CODING | |
| Apobec3 | 15 | 79728604 | G | A | A | NON_SYNONYMOUS_CODING | |
| Apobec3 | 15 | 79729463 | T | C | C | NON_SYNONYMOUS_CODING | |
| Apobec3 | 15 | 79729537 | G | C | C | NON_SYNONYMOUS_CODING | |
| Apobec3 | 15 | 79735878 | A | G | G | NON_SYNONYMOUS_CODING | |
| Apobec3 | 15 | 79735881 | G | C | C | NON_SYNONYMOUS_CODING | |
| Apobec3 | 15 | 79735885 | C | T | T | NON_SYNONYMOUS_CODING | |
| Apobec3 | 15 | 79735953 | C | G | G | NON_SYNONYMOUS_CODING | |
| Apobec3 | 15 | 79736838 | C | T | T | NON_SYNONYMOUS_CODING | |
| Apobec3 | 15 | 79737380 | G | A | A | NON_SYNONYMOUS_CODING | |
| Cbx7 | 15 | 79749323 | C | T | T | NON_SYNONYMOUS_CODING | |
| Cbx7 | 15 | 79749453 | G | C | C | NON_SYNONYMOUS_CODING | |
| Cbx7 | 15 | 79764235 | T | G | G | NON_SYNONYMOUS_CODING | |
| Cbx7 | 15 | 79764259 | A | T | T | STOP_GAINED | |
| Cbx7 | 15 | 79764307 | A | T | T | NON_SYNONYMOUS_CODING | |
| Cbx7 | 15 | 79801396 | G | T | T | 5′_UTR | |
| Cbx7 | 15 | 79801461 | A | C | C | 5′_UTR | |
| Pdgfb | 15 | 79830813 | C | T | T | NON_SYNONYMOUS_CODING | |
| Pdgfb | 15 | 79844499 | T | G | G | 5′_UTR | |
| Pdgfb | 15 | 79844550 | A | C | C | 5′_UTR | |
| Pdgfb | 15 | 79844807 | G | T | T | 5′_UTR | |
| Pdgfb | 15 | 79844895 | G | A | A | 5′_UTR |
Analysis was performed using the Sanger SNP database (http://www.sanger.ac.uk/cgi-bin/modelorgs/mousegenomes/snps.pl). QTL indicates quantitative trait locus.
Ath33 on Chr15 has been mapped in 3 independent intercrosses, including 2 B6.Apoe−/−×C3H.Apoe−/− intercrosses and 1 B6.Apoe−/−×129.Apoe−/− intercross.13,23 C3H and 129 are the high allele strains and B6 is the low allele strain for the QTL. Positional candidate genes, such as Rhpn1 (75.5 Mb), Apol11b (77.4 Mb), Csf2rb2 (78.1 Mb), Cdc42ep1 (78.6 Mb), Apobec3 (79.7 Mb), and Pdgfb (79.8 Mb), contain ≥1 nonsynonymous SNPs that are shared by the high allele strains but different from the low allele strain. Cacng2 (77.9 Mb) and Card10 (78.6 Mb) are positional genes possessing SNPs in the upstream promoter region that are shared by the high allele strains but different from the low allele strain. However, none of these candidate genes have been reported for associations with atherosclerotic arterial disease in recent genomewide association studies.
Discussion
Male mice are more resistant to atherosclerosis than their female counterparts for almost all inbred strains examined.14,33–34 Higher HDL cholesterol levels have been considered to be a major contributor to the resistance of male mice to atherosclerosis.14 In this study, we performed QTL analysis using an F2 cohort from the most phenotypically divergent Apoe−/− mouse strains to explore potential genetic connections between atherosclerosis and plasma lipids. Four significant and 4 suggestive QTLs for atherosclerotic lesion sizes and 15 QTLs for plasma HDL, non‐HDL cholesterol, and triglyceride levels have been identified. Atherosclerosis susceptibility loci are distinct from those for plasma lipids except for the Chr9 locus, which exerts effects through interactions with HDL.
Ath29 is a significant QTL for atherosclerosis initially identified by our group from a female F2 cohort derived from B6.Apoe−/− and C3H.Apoe−/− mice.7 It is located on mouse Chr9 at 42 cM with the B6 allele contributing to increased lesion sizes. The present study replicated this QTL in a male F2 population and further revealed its coincidence with an HDL QTL, Hdlq54.32,35 The colocalization of loci for 2 different traits suggests a possibility that these traits are controlled by a same gene. Elovl5 (77.7 Mb), Elovl4 (83.7 Mb), and Me1 (86.5 Mb) are positional candidate genes involved in fatty acid synthesis or elongation, and Cyb5r4 (86.9 Mb) is a positional candidate gene involved in fatty acid catabolism and oxidative stress.36 These genes are polymorphic among the parental strains (B6 versus C3H and 129) of 3 intercrosses that have led to detection of the QTL.23,35 Tcf12 (71.7 Mb) is also a promising candidate gene with multiple nonsynonymous SNPs between the B6 and C3H strains. It encodes the transcription factor 12 (also called HTF4 or HEB), a member of the helix‐loop‐helix protein family.37 The product of Tcf12 has diverse functions, including downregulating E‐cadherin expression, enhancing cell migration and invasion,38 and activating T‐lymphocyte differentiation,39 all of which have a role in atherosclerosis.
An interesting finding of this study is the detection of atherosclerosis susceptibility QTLs on Chr2 and Chr15 in which the C3H allele contributed to increased lesion sizes. F2 mice homozygous for the C3H allele at the loci had larger lesion sizes than those homozygous for the B6 allele. This finding was unexpected given the extremity of resistance that C3H.Apoe−/− mice exhibit during the development of atherosclerosis. However, this may not be surprising given the fact that QTL mapping detects loci based on the phenotypic variance among different genotypes at genetic markers within an F2 or N2 (second backcross generation) population but not on the phenotypic variance between the 2 parental strains. Both F2s and N2s are genetically and phenotypically diverse compared with both parents and thus can lead to revelations of cryptic genetic effects. Atherosclerosis susceptibility loci derived from the resistant strains of crosses have been previously detected in at least 3 independent crosses, including a B6×FVB/N Ldlr−/− intercross, B6×FVB/N Apoe−/− intercross, and a B×H Apoe−/− intercross.10,12–13 The Chr2 QTL replicated Ath28, originally identified in a DBA/2.Apoe−/−×AKR.Apoe−/− intercross9 and recently mapped in a B6.Apoe−/−×129.Apoe−/− intercross.23 The CI of the Chr2 QTL (90 to 104 cM) is corresponding to human Chr20q13, a region associated with sudden cardiac arrest in patients with coronary artery disease and inflammatory and autoimmunal diseases.31,40–41 The 2 strongest candidate genes for this QTL are Lama5 and Cdh4. Lama5 encodes laminin, α5, which, together with collagen IV, nidogen/entactin, and heparan sulfate proteoglycans, constitutes the structural component of vascular basement membrane.42 Cdh4 encodes cadherin 4, a calcium‐dependent transmembrane adhesion molecule. The protein products of Lama5 and Cdh4 have a broad range of functions, including direct cell–cell cohesion, regulation of cell migration, proliferation, survival, and cell signaling.43–44
The Chr15 QTL replicated Ath33, previously mapped in a B6.Apoe−/−×C3H.Apoe−/− cross and a B6.Apoe−/−×129.Apoe−/− intercross.13,23 The CI (22 to 44 cM) of this locus corresponds to human Chr8q24 and Chr22q13, regions that are associated with carotid intima‐media thickness,45 sudden cardiac arrest in patients with coronary heart disease,31 Crohn disease,46 and inflammatory bowel disease.47 The strongest candidate gene for the Chr15 QTL is Pdgfb, which encodes the platelet‐derived growth factor‐β polypeptide. Blood cells, especially monocytes and platelets, are a major source of platelet‐derived growth factor‐β after being activated.48 The absence of Pdgfb in circulating cells promotes inflammatory responses and early atherosclerosis and delays fibrous cap formation in Apoe−/− mice.49–50
QTLs on distal Chr1 contributed to major variations in plasma HDL, non‐HDL cholesterol, and triglyceride levels of the cross. This finding is consistent with our previous observation in a female cohort derived from an intercross between B6.Apoe−/− and C3H.Apoe−/− mice.29 Apoa2 is a well‐characterized QTL gene in the distal Chr1 region accounting for variations in plasma lipid levels of mice.51 Soat1 is also a QTL gene in the region that has been confirmed to contribute to variations in plasma levels of lipids, especially HDL cholesterol.52 A significant QTL for non‐HDL cholesterol was mapped to Chr12 at 54 cM in the current cross. This QTL was partially overlapping in the CI with Nhdlq12, mapped in a B6.Apoe−/−×C3H.Apoe−/− intercross.29 One prominent candidate gene for this QTL is Cyp46a1, encoding a member of the cytochrome P450 superfamily of monooxygenases that converts cholesterol to 24S‐hydroxycholesterol.53 The oxysterol products activate the liver X receptor NR1H2 and are subsequently metabolized to esters by sterol o‐acyltransferase 1.
A moderate but statistically significant reverse correlation was observed between plasma HDL cholesterol levels and atherosclerotic lesion sizes in this cross. This finding supports the concept that HDL is protective against atherosclerosis. However, such a correlation was not observed in female F2 mice derived from B6.Apoe−/− and C3H.Apoe−/− mice.7 The HDL cholesterol levels in the male F2s were ≈3× higher and exhibited larger variance compared with the female F2s (96.6±61.7 versus 34.0±27.4 mg/dL), which might explain why statistical associations with atherosclerosis were observed in the males but not in the females. We have also observed a significant reverse correlation between plasma HDL cholesterol levels and atherosclerotic lesion sizes in a B6.Apoe−/−×BALB/c.Apoe−/− intercross, whose HDL levels are higher and display a large variance.54 No significant correlation between plasma levels of non‐HDL cholesterol and sizes of atherosclerotic lesions was observed in this cross or in previous crosses.7,13,29,54 This finding suggests that in the absence of Apoe, other genetic factors than those involved in regulating plasma lipids play a key role in atherosclerosis. In the previous female F2 cohort derived from B6.Apoe−/− and C3H.Apoe−/− mice, we only detected 2 QTLs for atherosclerosis and 5 QTLs for plasma lipid levels.7 In contrast, in the present male F2 cohort, we have identified 8 QTLs for atherosclerosis and 15 QTLs for plasma lipid levels (Table 4) despite the fact that the 2 intercrosses were derived from the same parental strains. There were several factors that could contribute to the discrepancy in the results between the 2 crosses: first, the previous female F2 mice were fed a Western diet for 12 weeks starting at 6 weeks of age, while the present male F2 mice were fed the diet for 5 weeks starting at 8 weeks of age. Thus, female F2 mice should have developed larger and more advanced lesions than male F2 mice. Genetic factors could exert effect differently on different stages of atherosclerosis. Second, as discussed earlier, the HDL cholesterol level in the male F2s was much higher and exhibited larger variance compared with the female F2s, which was conducive to the identification of QTLs for the trait. Third, the sex of the mice could exert a significant influence on genetic factors on atherosclerosis, as previously illustrated.9–10,13 Finally, as the 2 crosses were constructed in different time, environmental factors could influence the results.
Table 4.
QTLs for Atherosclerosis and Plasma Lipids Identified in the Present Male Versus the Previously Reported Female F2 Cohort Derived From B6.Apoe−/− and C3H.Apoe−/− Mice
| Locus Name | Chr | Trait | LOD (Male) | LOD (Female) |
|---|---|---|---|---|
| Ath30 | 1 | Lesion | 2.34 | … |
| Ath1 | 1 | Lesion | 2.10 | … |
| Ath28 | 2 | Lesion | 3.59 | … |
| Ath8 | 4 | Lesion | 3.30 | … |
| Athsq1 | 4 | Lesion | 2.65 | … |
| Ath31 | 7 | Lesion | 2.77 | … |
| Ath29 | 9 | Lesion | 3.65 | 4.1 |
| Ath19 | 11 | Lesion | … | 2.4 |
| Ath33 | 15 | Lesion | 4.33 | … |
| Hdlq5 | 1 | HDL | 7.30 | 3.0 |
| Hdlq21 | 3 | HDL | … | 2.3 |
| Hdlq16 | 8 | HDL | 2.11 | … |
| Hdlq17 | 9 | HDL | 3.04 | … |
| Hdlq54 | 9 | HDL | 3.55 | … |
| Hdlq18 | 12 | HDL | 2.81 | … |
| Lipq2 | 13 | HDL | 3.05 | … |
| Cq1 | 1 | Non‐HDL | 4.92 | 6.3 |
| Chol8 | 4 | Non‐HDL | 2.51 | … |
| Chldq3 | 5 | Non‐HDL | … | 2.2 |
| Nhdlq11 | 9 | Non‐HDL | … | 2.5 |
| Nhdlq12 | 12 | Non‐HDL | 6.59 | … |
| Chldq8 | 14 | Non‐HDL | 2.24 | … |
| Nhdlq9 | 15 | Non‐HDL | 2.43 | … |
| Nhdlq2 | X | Non‐HDL | 2.22 | … |
| Tglq1 | 1 | Triglyceride | 6.42 | 3.8 |
| Tgq10 | 2 | Triglyceride | 2.62 | … |
| Trigg2 | 8 | Triglyceride | … | 3.2 |
| Tgq28 | 16 | Triglyceride | 2.51 | … |
QTL indicates quantitative trait locus; Chr, chromosome; LOD, logarithm of odds; male: the present male F2 cohort derived from B6.Apoe−/− and C3H.Apoe−/− mice; female: the female F2 cohort derived from B6.Apoe−/− and C3H.Apoe−/− mice previously reported by our group.7 The significant LOD scores were highlighted in bold.
In summary, we have identified a number of QTLs affecting atherosclerosis and plasma lipid levels using a male F2 cohort derived from B6.Apoe−/− and C3H.Apoe–/– mice. Atherosclerosis susceptibility loci are independent of those for plasma lipids except for the Chr9 QTL, which appears to exert effects through interactions with HDL. Through haplotype analysis, we have narrowed the list of candidate genes for major atherosclerosis susceptibility loci.
Sources of Funding
This work was supported by National Institutes of Health grant HL82881. The original data of this cross associated with this article have been submitted to the Mouse Genome Informatics database.
Disclosures
None.
References
- 1.Stylianou IM, Bauer RC, Reilly MP, Rader DJ. Genetic basis of atherosclerosis: insights from mice and humans. Circ Res. 2012; 110:337-355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peters LL, Robledo RF, Bult CJ, Churchill GA, Paigen BJ, Svenson KL. The mouse as a model for human biology: a resource guide for complex trait analysis. Nat Rev Genet. 2007; 8:58-69 [DOI] [PubMed] [Google Scholar]
- 3.Wang X, Ishimori N, Korstanje R, Rollins J, Paigen B. Identifying novel genes for atherosclerosis through mouse‐human comparative genetics. Am J Hum Genet. 2005; 77:1-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paigen B, Morrow A, Brandon C, Mitchell D, Holmes P. Variation in susceptibility to atherosclerosis among inbred strains of mice. Atherosclerosis. 1985; 57:65-73 [DOI] [PubMed] [Google Scholar]
- 5.Paigen B, Morrow A, Holmes PA, Mitchell D, Williams RA. Quantitative assessment of atherosclerotic lesions in mice. Atherosclerosis. 1987; 68:231-240 [DOI] [PubMed] [Google Scholar]
- 6.Shi W, Wang NJ, Shih DM, Sun VZ, Wang X, Lusis AJ. Determinants of atherosclerosis susceptibility in the C3H and C57BL/6 mouse model: evidence for involvement of endothelial cells but not blood cells or cholesterol metabolism. Circ Res. 2000; 86:1078-1084 [DOI] [PubMed] [Google Scholar]
- 7.Su Z, Li Y, James JC, McDuffie M, Matsumoto AH, Helm GA, Weber JL, Lusis AJ, Shi W. Quantitative trait locus analysis of atherosclerosis in an intercross between C57BL/6 and C3H mice carrying the mutant apolipoprotein E gene. Genetics. 2006; 172:1799-1807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang SS, Shi W, Wang X, Velky L, Greenlee S, Wang MT, Drake TA, Lusis AJ. Mapping, genetic isolation, and characterization of genetic loci that determine resistance to atherosclerosis in C3H mice. Arterioscler Thromb Vasc Biol. 2007; 27:2671-2676 [DOI] [PubMed] [Google Scholar]
- 9.Smith JD, Bhasin JM, Baglione J, Settle M, Xu Y, Barnard J. Atherosclerosis susceptibility loci identified from a strain intercross of apolipoprotein E‐deficient mice via a high‐density genome scan. Arterioscler Thromb Vasc Biol. 2006; 26:597-603 [DOI] [PubMed] [Google Scholar]
- 10.Teupser D, Tan M, Persky AD, Breslow JL. Atherosclerosis quantitative trait loci are sex‐ and lineage‐dependent in an intercross of C57BL/6 and FVB/N low‐density lipoprotein receptor‐/‐ mice. Proc Natl Acad Sci USA. 2006; 103:123-128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Welch CL, Bretschger S, Latib N, Bezouevski M, Guo Y, Pleskac N, Liang CP, Barlow C, Dansky H, Breslow JL, Tall AR. Localization of atherosclerosis susceptibility loci to chromosomes 4 and 6 using the Ldlr knockout mouse model. Proc Natl Acad Sci USA. 2001; 98:7946-7951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dansky HM, Shu P, Donavan M, Montagno J, Nagle DL, Smutko JS, Roy N, Whiteing S, Barrios J, McBride TJ, Smith JD, Duyk G, Breslow JL, Moore KJ. A phenotype‐sensitizing Apoe‐deficient genetic background reveals novel atherosclerosis predisposition loci in the mouse. Genetics. 2002; 160:1599-1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang SS, Schadt EE, Wang H, Wang X, Ingram‐Drake L, Shi W, Drake TA, Lusis AJ. Identification of pathways for atherosclerosis in mice: integration of quantitative trait locus analysis and global gene expression data. Circ Res. 2007; 101:e11-e30 [DOI] [PubMed] [Google Scholar]
- 14.Paigen B, Holmes PA, Mitchell D, Albee D. Comparison of atherosclerotic lesions and HDL‐lipid levels in male, female, and testosterone‐treated female mice from strains C57BL/6, BALB/c, and C3H. Atherosclerosis. 1987; 64:215-221 [DOI] [PubMed] [Google Scholar]
- 15.Paigen B, Mitchell D, Reue K, Morrow A, Lusis AJ, LeBoeuf RC. Ath‐1, a gene determining atherosclerosis susceptibility and high density lipoprotein levels in mice. Proc Natl Acad Sci USA. 1987; 84:3763-3767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qiao JH, Xie PZ, Fishbein MC, Kreuzer J, Drake TA, Demer LL, Lusis AJ. Pathology of atheromatous lesions in inbred and genetically engineered mice. Genetic determination of arterial calcification. Arterioscler Thromb. 1994; 14:1480-1497 [DOI] [PubMed] [Google Scholar]
- 17.Yuan Z, Pei H, Roberts DJ, Zhang Z, Rowlan JS, Matsumoto AH, Shi W. Quantitative trait locus analysis of neointimal formation in an intercross between C57BL/6 and C3H/HeJ apolipoprotein E‐deficient mice. Circ Cardiovasc Genet. 2009; 2:220-228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tian J, Pei H, James JC, Li Y, Matsumoto AH, Helm GA, Shi W. Circulating adhesion molecules in apoE‐deficient mouse strains with different atherosclerosis susceptibility. Biochem Biophys Res Commun. 2005; 329:1102-1107 [DOI] [PubMed] [Google Scholar]
- 19.Su Z, Li Y, James JC, Matsumoto AH, Helm GA, Lusis AJ, Shi W. Genetic linkage of hyperglycemia, body weight and serum amyloid‐P in an intercross between C57BL/6 and C3H apolipoprotein E‐deficient mice. Hum Mol Genet. 2006; 15:1650-1658 [DOI] [PubMed] [Google Scholar]
- 20.Abiola O, Angel JM, Avner P, Bachmanov AA, Belknap JK, Bennett B, Blankenhorn EP, Blizard DA, Bolivar V, Brockmann GA, Buck KJ, Bureau JF, Casley WL, Chesler EJ, Cheverud JM, Churchill GA, Cook M, Crabbe JC, Crusio WE, Darvasi A, de Haan G, Dermant P, Doerge RW, Elliot RW, Farber CR, Flaherty L, Flint J, Gershenfeld H, Gibson JP, Gu J, Gu W, Himmelbauer H, Hitzemann R, Hsu HC, Hunter K, Iraqi FF, Jansen RC, Johnson TE, Jones BC, Kempermann G, Lammert F, Lu L, Manly KF, Matthews DB, Medrano JF, Mehrabian M, Mittlemann G, Mock BA, Mogil JS, Montagutelli X, Morahan G, Mountz JD, Nagase H, Nowakowski RS, O'Hara BF, Osadchuk AV, Paigen B, Palmer AA, Peirce JL, Pomp D, Rosemann M, Rosen GD, Schalkwyk LC, Seltzer Z, Settle S, Shimomura K, Shou S, Sikela JM, Siracusa LD, Spearow JL, Teuscher C, Threadgill DW, Toth LA, Toye AA, Vadasz C, Van Zant G, Wakeland E, Williams RW, Zhang HG, Zou FComplex Trait Consortium The nature and identification of quantitative trait loci: a community's view. Nat Rev Genet. 2003; 4:911-916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doerge RW, Churchill GA. Permutation tests for multiple loci affecting a quantitative character. Genetics. 1996; 142:285-294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Korstanje R, Eriksson P, Samnegard A, Olsson PG, Forsman‐Semb K, Sen S, Churchill GA, Rollins J, Harris S, Hamsten A, Paigen B. Locating Ath8, a locus for murine atherosclerosis susceptibility and testing several of its candidate genes in mice and humans. Atherosclerosis. 2004; 177:443-450 [DOI] [PubMed] [Google Scholar]
- 23.Tomita H, Zhilicheva S, Kim S, Maeda N. Aortic arch curvature and atherosclerosis have overlapping quantitative trait loci in a cross between 129S6/SvEvTac and C57BL/6J apolipoprotein E‐null mice. Circ Res. 2010; 106:1052-1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Z, Rowlan JS, Wang Q, Shi W. Genetic analysis of atherosclerosis and glucose homeostasis in an intercross between C57BL/6 and BALB/cJ apolipoprotein E‐deficient mice. Circ Cardiovasc Genet. 2012; 5:190-201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wang X, Paigen B. Genetics of variation in HDL cholesterol in humans and mice. Circ Res. 2005; 96:27-42 [DOI] [PubMed] [Google Scholar]
- 26.Ishimori N, Li R, Kelmenson PM, Korstanje R, Walsh KA, Churchill GA, Forsman‐Semb K, Paigen B. Quantitative trait loci analysis for plasma HDL‐cholesterol concentrations and atherosclerosis susceptibility between inbred mouse strains C57BL/6J and 129S1/SvImJ. Arterioscler Thromb Vasc Biol. 2004; 24:161-166 [DOI] [PubMed] [Google Scholar]
- 27.Welch CL, Bretschger S, Wen PZ, Mehrabian M, Latib N, Fruchart‐Najib J, Fruchart JC, Myrick C, Lusis AJ. Novel QTLs for HDL levels identified in mice by controlling for Apoa2 allelic effects: confirmation of a chromosome 6 locus in a congenic strain. Physiol Genomics. 2004; 17:48-59 [DOI] [PubMed] [Google Scholar]
- 28.Wang X, Paigen B. Genome‐wide search for new genes controlling plasma lipid concentrations in mice and humans. Curr Opin Lipidol. 2005; 16:127-137 [DOI] [PubMed] [Google Scholar]
- 29.Li Q, Li Y, Zhang Z, Gilbert TR, Matsumoto AH, Dobrin SE, Shi W. Quantitative trait locus analysis of carotid atherosclerosis in an intercross between C57BL/6 and C3H apolipoprotein E‐deficient mice. Stroke. 2008; 39:166-173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barber MJ, Mangravite LM, Hyde CL, Chasman DI, Smith JD, McCarty CA, Li X, Wilke RA, Rieder MJ, Williams PT, Ridker PM, Chatterjee A, Rotter JI, Nickerson DA, Stephens M, Krauss RM. Genome‐wide association of lipid‐lowering response to statins in combined study populations. PLoS ONE. 2010; 5:e9763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aouizerat BE, Vittinghoff E, Musone SL, Pawlikowska L, Kwok PY, Olgin JE, Tseng ZH. GWAS for discovery and replication of genetic loci associated with sudden cardiac arrest in patients with coronary artery disease. BMC Cardiovasc Disord. 2011; 11:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Houlston RS, Cheadle J, Dobbins SE, Tenesa A, Jones AM, Howarth K, Spain SL, Broderick P, Domingo E, Farrington S, Prendergast JG, Pittman AM, Theodoratou E, Smith CG, Olver B, Walther A, Barnetson RA, Churchman M, Jaeger EE, Penegar S, Barclay E, Martin L, Gorman M, Mager R, Johnstone E, Midgley R, Niittymaki I, Tuupanen S, Colley J, Idziaszczyk SCOGENT Consortium Thomas HJ, Lucassen AM, Evans DG, Maher ERCORGI Consortium, COIN Collaborative Group, COINB Collaborative Group Maughan T, Dimas A, Dermitzakis E, Cazier JB, Aaltonen LA, Pharoah P, Kerr DJ, Carvajal‐Carmona LG, Campbell H, Dunlop MG, Tomlinson IP. Meta‐analysis of three genome‐wide association studies identifies susceptibility loci for colorectal cancer at 1q41, 3q26.2, 12q13.13 and 20q13.33. Nat Genet. 2010; 42:973-977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith DD, Tan X, Tawfik O, Milne G, Stechschulte DJ, Dileepan KN. Increased aortic atherosclerotic plaque development in female apolipoprotein E‐null mice is associated with elevated thromboxane A2 and decreased prostacyclin production. J Physiol Pharmacol. 2010; 61:309-316 [PMC free article] [PubMed] [Google Scholar]
- 34.Teupser D, Persky AD, Breslow JL. Induction of atherosclerosis by low‐fat, semisynthetic diets in LDL receptor‐deficient C57BL/6J and FVB/NJ mice: comparison of lesions of the aortic root, brachiocephalic artery, and whole aorta (en face measurement). Arterioscler Thromb Vasc Biol. 2003; 23:1907-1913 [DOI] [PubMed] [Google Scholar]
- 35.Su Z, Wang X, Tsaih SW, Zhang A, Cox A, Sheehan S, Paigen B. Genetic basis of HDL variation in 129/SvImJ and C57BL/6J mice: importance of testing candidate genes in targeted mutant mice. J Lipid Res. 2009; 50:116-125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu M, Wang W, Frontera JR, Neely MC, Lu J, Aires D, Hsu FF, Turk J, Swerdlow RH, Carlson SE, Zhu H. Ncb5or deficiency increases fatty acid catabolism and oxidative stress. J Biol Chem. 2011; 286:11141-11154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y, Babin J, Feldhaus AL, Singh H, Sharp PA, Bina M. HTF4: a new human helix‐loop‐helix protein. Nucleic Acids Res. 1991; 19:4555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen WS, Chen CC, Chen LL, Lee CC, Huang TS. Secreted heat shock protein 90alpha induces NF‐kappaB‐mediated TCF12 expression to down‐regulate E‐cadherin and enhance cell migration and invasion in colorectal cancer cells. J Biol Chem. 2013; 288:9001-9010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Braunstein M, Anderson MK. HEB in the spotlight: transcriptional regulation of T‐cell specification, commitment, and developmental plasticity. Clin Dev Immunol. 2012; 2012:678705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kugathasan S, Baldassano RN, Bradfield JP, Sleiman PM, Imielinski M, Guthery SL, Cucchiara S, Kim CE, Frackelton EC, Annaiah K, Glessner JT, Santa E, Willson T, Eckert AW, Bonkowski E, Shaner JL, Smith RM, Otieno FG, Peterson N, Abrams DJ, Chiavacci RM, Grundmeier R, Mamula P, Tomer G, Piccoli DA, Monos DS, Annese V, Denson LA, Grant SF, Hakonarson H. Loci on 20q13 and 21q22 are associated with pediatric‐onset inflammatory bowel disease. Nat Genet. 2008; 40:1211-1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.UK IBD Genetics Consortium, Barrett JC, Lee JC, Lees CW, Prescott NJ, Anderson CA, Phillips A, Wesley E, Parnell K, Zhang H, Drummond H, Nimmo ER, Massey D, Blaszczyk K, Elliott T, Cotterill L, Dallal H, Lobo AJ, Mowat C, Sanderson JD, Jewell DP, Newman WG, Edwards C, Ahmad T, Mansfield JC, Satsangi J, Parkes M, Mathew CGWellcome Trust Case Control Consortium 2 Donnelly P, Peltonen L, Blackwell JM, Bramon E, Brown MA, Casas JP, Corvin A, Craddock N, Deloukas P, Duncanson A, Jankowski J, Markus HS, Mathew CG, McCarthy MI, Palmer CN, Plomin R, Rautanen A, Sawcer SJ, Samani N, Trembath RC, Viswanathan AC, Wood N, Spencer CC, Barrett JC, Bellenguez C, Davison D, Freeman C, Strange A, Donnelly P, Langford C, Hunt SE, Edkins S, Gwilliam R, Blackburn H, Bumpstead SJ, Dronov S, Gillman M, Gray E, Hammond N, Jayakumar A, McCann OT, Liddle J, Perez ML, Potter SC, Ravindrarajah R, Ricketts M, Waller M, Weston P, Widaa S, Whittaker P, Deloukas P, Peltonen L, Mathew CG, Blackwell JM, Brown MA, Corvin A, McCarthy MI, Spencer CC, Attwood AP, Stephens J, Sambrook J, Ouwehand WH, McArdle WL, Ring SM, Strachan DP. Genome‐wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat Genet. 2009; 41:1330-1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sasaki T, Fassler R, Hohenester E. Laminin: the crux of basement membrane assembly. J Cell Biol. 2004; 164:959-963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aszodi A, Legate KR, Nakchbandi I, Fassler R. What mouse mutants teach us about extracellular matrix function. Annu Rev Cell Dev Biol. 2006; 22:591-621 [DOI] [PubMed] [Google Scholar]
- 44.Jeanes A, Gottardi CJ, Yap AS. Cadherins and cancer: how does cadherin dysfunction promote tumor progression? Oncogene. 2008; 27:6920-6929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bis JC, Kavousi M, Franceschini N, Isaacs A, Abecasis GR, Schminke U, Post WS, Smith AV, Cupples LA, Markus HS, Schmidt R, Huffman JE, Lehtimaki T, Baumert J, Munzel T, Heckbert SR, Dehghan A, North K, Oostra B, Bevan S, Stoegerer EM, Hayward C, Raitakari O, Meisinger C, Schillert A, Sanna S, Volzke H, Cheng YC, Thorsson B, Fox CS, Rice K, Rivadeneira F, Nambi V, Halperin E, Petrovic KE, Peltonen L, Wichmann HE, Schnabel RB, Dorr M, Parsa A, Aspelund T, Demissie S, Kathiresan S, Reilly MP, Taylor K, Uitterlinden A, Couper DJ, Sitzer M, Kahonen M, Illig T, Wild PS, Orru M, Ludemann J, Shuldiner AR, Eiriksdottir G, White CC, Rotter JI, Hofman A, Seissler J, Zeller T, Usala G, Ernst F, Launer LJ, Sr D'Agostino RB, O'Leary DH, Ballantyne C, Thiery J, Ziegler A, Lakatta EG, Chilukoti RK, Harris TB, Wolf PA, Psaty BM, Polak JF, Li X, Rathmann W, Uda M, Boerwinkle E, Klopp N, Schmidt H, Wilson JF, Viikari J, Koenig W, Blankenberg S, Newman AB, Witteman J, Heiss G, Duijn C, Scuteri A, Homuth G, Mitchell BD, Gudnason V, O'Donnell CJCARDIoGRAM Consortium Meta‐analysis of genome‐wide association studies from the CHARGE consortium identifies common variants associated with carotid intima media thickness and plaque. Nat Genet. 2011; 43:940-947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Franke A, McGovern DP, Barrett JC, Wang K, Radford‐Smith GL, Ahmad T, Lees CW, Balschun T, Lee J, Roberts R, Anderson CA, Bis JC, Bumpstead S, Ellinghaus D, Festen EM, Georges M, Green T, Haritunians T, Jostins L, Latiano A, Mathew CG, Montgomery GW, Prescott NJ, Raychaudhuri S, Rotter JI, Schumm P, Sharma Y, Simms LA, Taylor KD, Whiteman D, Wijmenga C, Baldassano RN, Barclay M, Bayless TM, Brand S, Buning C, Cohen A, Colombel JF, Cottone M, Stronati L, Denson T, De Vos M, D'Inca R, Dubinsky M, Edwards C, Florin T, Franchimont D, Gearry R, Glas J, Van Gossum A, Guthery SL, Halfvarson J, Verspaget HW, Hugot JP, Karban A, Laukens D, Lawrance I, Lemann M, Levine A, Libioulle C, Louis E, Mowat C, Newman W, Panes J, Phillips A, Proctor DD, Regueiro M, Russell R, Rutgeerts P, Sanderson J, Sans M, Seibold F, Steinhart AH, Stokkers PC, Torkvist L, Kullak‐Ublick G, Wilson D, Walters T, Targan SR, Brant SR, Rioux JD, D'Amato M, Weersma RK, Kugathasan S, Griffiths AM, Mansfield JC, Vermeire S, Duerr RH, Silverberg MS, Satsangi J, Schreiber S, Cho JH, Annese V, Hakonarson H, Daly MJ, Parkes M. Genome‐wide meta‐analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat Genet. 2010; 42:1118-1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, Essers J, Mitrovic M, Ning K, Cleynen I, Theatre E, Spain SL, Raychaudhuri S, Goyette P, Wei Z, Abraham C, Achkar JP, Ahmad T, Amininejad L, Ananthakrishnan AN, Andersen V, Andrews JM, Baidoo L, Balschun T, Bampton PA, Bitton A, Boucher G, Brand S, Buning C, Cohain A, Cichon S, D'Amato M, De Jong D, Devaney KL, Dubinsky M, Edwards C, Ellinghaus D, Ferguson LR, Franchimont D, Fransen K, Gearry R, Georges M, Gieger C, Glas J, Haritunians T, Hart A, Hawkey C, Hedl M, Hu X, Karlsen TH, Kupcinskas L, Kugathasan S, Latiano A, Laukens D, Lawrance IC, Lees CW, Louis E, Mahy G, Mansfield J, Morgan AR, Mowat C, Newman W, Palmieri O, Ponsioen CY, Potocnik U, Prescott NJ, Regueiro M, Rotter JI, Russell RK, Sanderson JD, Sans M, Satsangi J, Schreiber S, Simms LA, Sventoraityte J, Targan SR, Taylor KD, Tremelling M, Verspaget HW, De Vos M, Wijmenga C, Wilson DC, Winkelmann J, Xavier RJ, Zeissig S, Zhang B, Zhang CK, Zhao HInternational IBD Genetics Consortium (IIBDGC) Silverberg MS, Annese V, Hakonarson H, Brant SR, Radford‐Smith G, Mathew CG, Rioux JD, Schadt EE, Daly MJ, Franke A, Parkes M, Vermeire S, Barrett JC, Cho JH. Host‐microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012; 491:119-124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raines EW. PDGF and cardiovascular disease. Cytokine Growth Factor Rev. 2004; 15:237-254 [DOI] [PubMed] [Google Scholar]
- 49.Tang J, Kozaki K, Farr AG, Martin PJ, Lindahl P, Betsholtz C, Raines EW. The absence of platelet‐derived growth factor‐B in circulating cells promotes immune and inflammatory responses in atherosclerosis‐prone ApoE‐/‐ mice. Am J Pathol. 2005; 167:901-912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kozaki K, Kaminski WE, Tang J, Hollenbach S, Lindahl P, Sullivan C, Yu JC, Abe K, Martin PJ, Ross R, Betsholtz C, Giese NA, Raines EW. Blockade of platelet‐derived growth factor or its receptors transiently delays but does not prevent fibrous cap formation in ApoE null mice. Am J Pathol. 2002; 161:1395-1407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang X, Korstanje R, Higgins D, Paigen B. Haplotype analysis in multiple crosses to identify a QTL gene. Genome Res. 2004; 14:1767-1772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lu Z, Yuan Z, Miyoshi T, Wang Q, Su Z, Chang CC, Shi W. Identification of Soat1 as a quantitative trait locus gene on mouse chromosome 1 contributing to hyperlipidemia. PLoS ONE. 2011; 6:e25344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lorbek G, Lewinska M, Rozman D. Cytochrome P450s in the synthesis of cholesterol and bile acids–from mouse models to human diseases. FEBS J. 2012; 279:1516-1533 [DOI] [PubMed] [Google Scholar]
- 54.Rowlan JS, Zhang Z, Wang Q, Fang Y, Shi W. New quantitative trait loci for carotid atherosclerosis identified in an intercross derived from apolipoprotein E‐deficient mouse strains. Physiol Genomics. 2013; 45:332-342 [DOI] [PMC free article] [PubMed] [Google Scholar]