

Abstract
Background
Dilated cardiomyopathy (DCM) is a leading cause of chronic morbidity and mortality in muscular dystrophy (MD) patients. Current pharmacological treatments are not yet able to counteract chronic myocardial wastage, thus novel therapies are being intensely explored. MicroRNAs have been implicated as fine regulators of cardiomyopathic progression. Previously, miR‐669a downregulation has been linked to the severe DCM progression displayed by Sgcb‐null dystrophic mice. However, the impact of long‐term overexpression of miR‐669a on muscle structure and functionality of the dystrophic heart is yet unknown.
Methods and Results
Here, we demonstrate that intraventricular delivery of adeno‐associated viral (AAV) vectors induces long‐term (18 months) miR‐669a overexpression and improves survival of Sgcb‐null mice. Treated hearts display significant decrease in hypertrophic remodeling, fibrosis, and cardiomyocyte apoptosis. Moreover, miR‐669a treatment increases sarcomere organization, reduces ventricular atrial natriuretic peptide (ANP) levels, and ameliorates gene/miRNA profile of DCM markers. Furthermore, long‐term miR‐669a overexpression significantly reduces adverse remodeling and enhances systolic fractional shortening of the left ventricle in treated dystrophic mice, without significant detrimental consequences on skeletal muscle wastage.
Conclusions
Our findings provide the first evidence of long‐term beneficial impact of AAV‐mediated miRNA therapy in a transgenic model of severe, chronic MD‐associated DCM.
Keywords: cardiomyopathy, microRNAs, miRNA therapy, muscular dystrophy
Introduction
Muscular dystrophies (MDs) constitute a heterogeneous group of hereditary disorders, mainly characterized by chronic muscle degeneration.1 MDs encompass, among others, dystrophinopathies (eg, Duchenne MD [DMD]) and sarcoglycanopathies (eg, recessive limb girdle MD [LGMD2]) forms. Dystrophin and sarcoglycans are components of the dystrophin‐associated glycoprotein complex (DGC).2 Genetic disruption of DGC components leads to pathological calcium leakage and chronic muscle fiber wastage.3 Beside skeletal muscle impairment, chronic cardiomyopathy occurs in almost all DMD patients who survive beyond their third decade,4 and in ≈30% of LGMD2 patients.5 MD‐associated cardiomyopathy is characterized by arrhythmias, myocardial ischemia, cardiomyocyte necrosis, and fibrotic infiltrations. Affected myocardium undergoes hypertrophic remodeling and ventricular dilation, resulting in progressive functional deterioration (dilated cardiomyopathy [DCM]).6
Currently, gene therapy is regarded as a promising tool for reducing or counteracting cardiomyopathy progression in MDs.7 However, gene therapy approaches must carefully address the choice of animal models, vector types, and expressed genes.8
Animal models of MD‐associated cardiomyopathy should confront its complex and chronic manifestations. Mdx mice genetically mimic DMD, but exhibit cardiomyopathic features prevalently after 20 months of age.9 Conversely, Sgcb‐null mice, which genetically model LGMD2E sarcoglycanopathy, already show necrotic foci in 9‐week‐old hearts and prominent signs of ischemic‐like lesions, arrhythmia, and DCM from 20 weeks of age.10 Notably, cardiomyopathy in Sgcb‐null mice features severe, chronic progression, and variable worsening with age.
Because of its internal structure and well‐protected position, cardiac muscle therapy still poses challenges to vector delivery and fiber transduction.11 Adeno‐associated viral (AAV) vectors appear to be a promising tool for efficient gene delivery in dystrophic hearts.12 Direct delivery of AAV‐micro‐dystrophin into the cardiac cavity of newborn mdx mice resulted in micro‐dystrophin expression and DGC restoration in extensive myocardial areas.13 AAV‐mediated micro‐dystrophin delivery has been recently tested in the myocardium of 21‐month‐old mdx mice, partially mitigating electrocardiogram (ECG) abnormalities, but not fibrosis and most hemodynamic parameters.14 Albeit promising in short term studies, the potential of AAV‐based therapies in chronic cardiac dysfunctions remains largely unexplored in the long term.
Suitable candidates for heart gene therapy can be identified in missing genes or cardiomyogenic factors, but also noncoding RNAs, such as microRNAs (miRNAs).15 MicroRNAs are small, untranslated RNAs able to finely tune expression or translational levels of downstream genes in a wide range of processes, including cardiomyopathy and dysfunctional heart remodeling.16 Cardiac‐specific deletion of Dicer, an essential endoribonuclease for miRNA processing, led to DCM and heart failure.17 Altered levels of miR‐1 and miR‐133a were implicated in cardiomyopathic and arrhythmogenic processes,18–19 whereas miR‐208a upregulation was associated with adverse clinical outcome in DCM patients.20 Moreover, miR‐669a downregulation has been recently shown to couple calcium leakage and abnormal MyoD expression in chronic DCM of Sgcb‐null mice.21 However, assessment of long‐term effects of microRNA therapies in in vivo models of chronic cardiomyopathy is still lacking.
In the present study, we investigated the long‐term (18 months) effects of AAV‐mediated intraventricular miR‐669a delivery in Sgcb‐null mice. Therapeutic relevance of this approach was assessed by measuring histologic, molecular, and functional properties of MD‐associated end‐stage chronic cardiomyopathy. We show that miR669a expression is maintained up to 18 months postinjection and results in increased mice viability and marked reduction of adverse hypertrophic remodeling and myocardial fibrosis. Furthermore, such miRNA therapy consistently alleviates cardiomyopathic progression and increases left ventricular performance, as evidenced by protein, gene and microRNA markers, and by functional parameters. In addition, miR‐669a delivery fails in triggering off‐target effects in the skeletal muscle of treated mice. In summary, our study provides the first evidence of long‐term, beneficial effects of AAV‐mediated in vivo miRNA therapy of MD‐associated advanced chronic cardiomyopathy.
Materials and Methods
Animal Procedures
All animal protocols were conducted in compliance with Ethical Committee Guidelines of KU Leuven (project 095/2012) and Belgian legislation. Sgcb‐null mice were generated by the group of K.P. Campbell10 (University of Iowa). Recombinant AAV vectors were produced as previously described.21 109 AAV2/9 particles in 10 μL PBS (sham, 10 μL PBS only) were delivered into pups (sham, n=15; miR‐dsRed, n=15; miR‐669a, n=15; randomized groups) through intraventricular puncture through the chest wall within 24 hours after birth. Survival was monitored during 18 months. At 18 months postinjection, all surviving mice (sham, n=3; miR‐dsRed, n=3; miR‐669a, n=7) were analyzed using echocardiography and subsequently euthanized for histologic and molecular analyses.
Histologic Analyses
Cardiac and skeletal muscle OCT‐embedded transversal cryosections were stained via Artisan Staining kit (Dako) for Masson's trichromic staining after fixation in paraformaldehyde (4% paraformaldehyde [PFA] in PBS, 30 minutes at room temperature) and Bouin's solution (8 hours at room temperature), according to manufacturer's protocol. Sirius Red staining was performed on formalin‐embedded transverse, 5‐μm‐thick sections using Direct Red 80 reagent (Sigma) as previously described,22 and pictures were taken with polarized light and appropriate filter at Leica DM5500B microscope. X‐Gal staining was performed by overnight incubation at 37°C of gluteraldehyde‐fixed cryosections with ready‐to‐use X‐Gal staining solution (Fermentas), followed by subsequent eosin (Sigma) counterstaining. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay was performed on cyosections, after fixation first in PFA, then in ethanol/acetic acid, by Apoptag kit (Millipore), according to manufacturer's instructions. Bright field and fluorescence images were acquired at 10× magnification via Eclipse Ti inverted microscope (Nikon) and quantifications were conducted on >10 sections per mouse via ImageJ software. As regards transmission electron microscopy, immersion fixation was performed by incubating the ventricle biopsies in PBS supplemented with 4% PFA and 0.5% glutaraldehyde at 4°C for 4 hours. The samples were washed with PBS, carefully minced, postfixed in 1% osmium tetraoxide in collidine buffer for 1 hour at 4°C, then dehydrated through ice‐cold gradient EtOH series (25%, 50%, 75%, 80%, 90% and 100%) for 15 minutes each. Tissue samples were then routinely embedded in Epon, ultrathin‐sectioned (75 to 90 nm) and placed on nickel grids. Grids were stained with a saturated solution of uranyl acetate followed by Reynold's lead citrate, then examined by EM10 Zeiss Electron Microscope.
Immunostaining and Western Blot Analysis
Immunostaining reactions were performed on PFA‐fixed, TritonX100‐permeabilized (Sigma) transverse, 10‐μm‐thick cryosections by overnight incubation at 4°C with primary antibody in PBS supplemented with 5% w/v BSA (Sigma). Primary antibodies and dilutions were as follows: chicken antilaminin (Abcam), 1:300; mouse antisarcomeric α‐actinin (Abcam), 1:300; rabbit anti‐Cx43 (SantaCruz), 1:300. All secondary Alexa Fluor donkey antibodies (Life Technologies) were diluted 1:500 in PBS supplemented with 5% w/v BSA. Fluorescence images were acquired at 20× magnification via Eclipse Ti inverted microscope (Nikon). Western blot (WB) analysis was performed on 10 μg protein extracts on parallel gels and primary antibody was incubated with nitrocellulose blots overnight at 4°C in Tris‐buffered saline (TBS) supplemented with 5% w/v skim milk powder (Sigma). Primary antibodies and dilutions were as follows: rabbit anti‐Cx43 (SantaCruz), 1:500; rabbit antiatrial natriuretic peptide (ANP) (Millipore), 1:500. All secondary horseradish peroxidase (HRP) ‐conjugated goat antibodies (SantaCruz) were diluted 1:5000 in TBS supplemented with 5% w/v skim milk powder. WB imaging was performed on GelDoc chemiluminescence detection system (BioRad) and protein levels were quantified by relative densitometry, normalizing versus background and Gapdh, through QuantityOne software (BioRad).
Gene/miRNA Expression Analyses
RNA was extracted from ≈10 mg cardiac tissue of all surviving mice using RNA mini kit (Life Technologies). Reverse transcription was performed using Superscript III kit (Life Technologies) and gene expression was assessed via SybrGreen (Life Technologies) qPCR, after normalization versus Pgk values. Primers; MyoD, Fw CCACTCCGGGACATAGACTTGACA, Rev TCTGGTGAGTCGAAACACGGATCA; Agtr1a, Fw AGGAGCTGGATGGATTTGTGGGTT, Rev ACAAACAAGGTTCCTTGCCCTTGG; Il6, Fw TCCAGTTGCCTTCTTGGGACTGAT, Rev TAAGCCTCCGACTTGTGAAGTGGT; Il1b, Fw AGCAGCTATGGCAACTGTTCCTGA, Rev CAAAGGTTTGGAAGCAGCCCTTCA; Pgk, Fw CAAAATGTCGCTTTCCAACAAG, Rev AACGTTGAAGTCCACCCTCATC. Total miRNA fraction was extracted from ≈10 mg cardiac tissue of all surviving mice through miRNA isolation kit (Life Technologies). Reverse transcription and Taqman‐based qPCR were conducted using miRNA‐specific probes (Life Technologies). Expression quantification was normalized versus miR‐16 levels.
Functional Assessment
Mice were anesthetized using Isoflurane (1.5% to 2%, Ecuphar). Subsequently, the animal was positioned in a supine position on a heating pad and chest hair was removed using a hair removing cream (Veet). Body temperature was kept constant by feeding the signal of a rectal probe back to the heating pad while heart and respiratory rates were continuously monitored. Transthoracic echocardiography was performed using a high frequency ultrasound system dedicated to small animal imaging (VisualSonics Vevo 2100, VisualSonics Inc) using a MS 400 linear array transducer (18 to 38 MHz). Parasternal short axis images at the level of the papillary muscles were recorded as well as a parasternal long axis image. All images were analyzed offline using dedicated software (Vevo 2100 version 1.3, VisualSonics). Left ventricle (LV) diameter at end‐diastole (LVIDd) and end‐systole (LVIDs), and posterior wall thickness (PWT) were measured on the short axis images as well as apex‐to‐base dimension of the LV on the long axis image. From these measurements, fractional shortening (FS) was calculated.
Skeletal Muscle Analyses
Transverse 7‐μm‐thick cryosections of soleus, tibialis anterior, and diaphragm muscles, embedded in Tragacanth Gum, were rehydrated with PBS and analysed for the presence of satellite cells as previously described.23 Briefly, after blocking, muscle sections were stained with primary Pax7 (Hybridoma Bank, Iowa University) and laminin (Sigma) antibodies and appropriate secondary antibodies (Molecular Probes) to identify satellite cells under the basal lamina of muscle fibers. Sections were mounted on polylysine‐coated slides with fluorescent mounting medium (DAKO) containing DAPI (10 μg/mL). Fluorescence images were taken using a Zeiss Axiophoto microscope (Carl Zeiss) and Metamorph image capture software (Metamorph Production). Numbers of satellite cells per muscle fiber in representative transverse sections were counted and presented as percentages of satellite cells.
Statistical Analysis
Survival curves were analyzed with Mantel‐Cox and Gehan‐Breslow‐Wilcoxon tests. Variation significance was scored when P<0.05 in both tests (*in figures and text). All data comparison analyses among sham, miR‐dsRed ,and miR‐669a were analysed through Kruskal‐Wallis test. When Kruskal‐Wallis test showed P<0.05 significance among the three groups, Mann‐Whitney test was used to compare miR‐669a versus sham and miR‐669a versus miR‐dsRed. Significance was scored when P<0.05 in both analyses (**in figures and text). All statistical tests mentioned above were performed via Prism software (GraphPad).
Results
In Vivo miR‐669a Therapy Ameliorates Survival and is Maintained Long‐Term
To determine long‐term efficacy and maintenance of in vivo miR‐669a therapy, AAV2/9 vectors bearing nuclear LacZ cDNA (nLacZ) as tracer and, alternatively, two copies of anti‐dsRed miRNA (miR‐dsRed) or miR‐669a, as control scramble and therapeutic miRNAs respectively, were generated and concentrated as reported (Figure S1,21). Newborn Sgcb‐null dystrophic mice were randomized into three groups and injected with PBS (sham, n=15), AAV2/9‐miR‐dsRed (miR‐dsRed, n=15), or AAV2/9‐miR‐669a (miR‐669a, n=15) intraventricularly, and survival was monitored for 18 months. Analysis of fatalities over time indicated that only miR‐669a treatment, but not miR‐dsRed, did significantly improve mice survival, as compared to sham (Figure 1A).
Figure 1.

AAV‐mediated miR‐669a expression improves survival and cardiac tissue structure in Sgcb‐null mice. A, Analysis of survival percentages over time shows a significant difference between miR‐669a and control experimental groups (**P<0.05 according to Mantel‐Cox and Gehan‐Breslow‐Wilcoxon tests). B, qPCR data of miR‐669a expression fold change as compared to sham. C, Immunostaining of Laminin (green, nuclei counterstaining in blue) reveals hypertrophy‐typical enlarged and unorganized cardiomyocytes in sham and miR‐dsRed. Conversely, miR‐669a‐treated myocardium shows a more regular pattern of smaller cardiomyocytes among experimental groups. D, Fiber percentage distribution among cross‐sectional area (CSA) classes reveals a shift of miR‐669a‐treated myocardium toward lower CSA values. E, Masson's trichromic staining reveals prominent fibrotic scars or infiltrations (in blue) in sham and miR‐dsRed, dramatically reduced in miR‐669a condition. F, Electron microscopy imaging shows sarcomere organization impairment (arrows) in sham and miR‐dsRed, whereas sarcomeres partially recover proper organization after miR‐669a treatment (arrowheads). Inset, high magnification of a spiral polyribosome in close relationship with nascent myofibrils. G, Immunostaining of connexin‐43 (green) and sarcomeric α‐actinin (red) reveals improvement of Cx43‐marked gap junctions in miR‐669a group, as compared to controls. H, Protein level quantification of Cx43 and ANP indicates Cx43 upregulation and ANP downregulation in miR‐669a‐treated myocardia (protein levels normalized vs miR‐669a for dsRed, vs sham for Cx43 and ANP). Error bars depict standard deviation; *P<0.05; **P<0.05 vs sham and P<0.05 vs miR‐dsRed; scale bars, 100 μm (1 μm in F). Sham, n=3; miR‐dsRed, n=3; miR‐669a, n=7. AAV, indicates adeno‐associated viral; qPCR, quantitative polymerase chain reaction; Cx43, connexin‐43; ANP, atrial natriuretic peptide.
In order to check for long‐term maintenance of injected vectors and expression of target miRNAs and tracers, the myocardium of all mice surviving at 18 months postinjection was analyzed for miR‐669a and nLacZ expression. Quantitative PCR (qPCR) data showed a significant increase in miR‐669a levels only in miR‐669a mice, as compared to sham and miR‐dsRed (Figure 1B). X‐Gal histological staining revealed virtually ubiquitous nLacZ expression throughout the myocardium both in miR‐dsRed and in miR‐669a mice (Figure S2).
Thus, AAV‐mediated miR669a therapy is maintained for at least 18 months and miR‐669a‐treated, Sgcb‐null mice show significant amelioration of survival rate over time, as compared to controls.
miR‐669a Expression Ameliorates Myocardial Hypertrophy and Fibrosis
Hypertrophic remodeling and myocardial fibrosis are hallmarks of MD‐associated DCM. In order to assess miRNA therapy effects on ventricular hypertrophy, we analyzed morphological pattern and cross‐sectional area (CSA) values of cardiomyocytes, as previously reported.24 Immunostaining on myocardial sections revealed a heterogeneously unorganized pattern of enlarged cardiomyocytes in sham and miR‐dsRed, whereas cardiomyocytes appeared smaller and more compactly organized in miR‐669a (Figure 1C). Accordingly, quantitative analyses showed that treatment with miR‐669a significantly decreased average cardiomyocyte CSA (Figure S3) and dramatically shifted CSA value distribution toward lower classes (Figure 1D), as compared to sham and miR‐dsRed.
We next asked whether treatment with miR‐669a induced beneficial effects on myocardial fibrosis and cardiomyocyte apoptosis. Fibrotic areas were revealed by histological staining and quantified as percentage of total section area. Quantitative analysis of intramyocardial fibrotic areas using Masson's trichromic staining indicated a significant reduction in miR‐669a mice, as compared to sham and miR‐dsRed (Figure 1E and Figure S4). Comparable results were quantifiable after Sirius Red staining of collagen deposits (Figure S5). Pyknotic nuclei quantification by TUNEL assay, moreover, revealed a significant reduction in apoptotic nuclei in miR‐669a mice, as compared to sham and miR‐dsRed (Figure S6).
Ventricular myocardium under chronic DCM conditions is also characterized by impairment of sarcomeric structures25 and gap junctions,26 and by increased local release of ANP.27 Because of the beneficial effects of miR‐669 therapy on hypertrophy, fibrosis, and apoptosis, we sought to determine whether miR‐669a treatment triggered long‐term effects also on sarcomere organization, connexin‐43 (Cx43) pattern, and ANP levels. Transmission electron microscopy imaging revealed disrupted and variably orientated myofibrils in sham and miR‐dsRed myocardia, whereas sarcomere organization was partially restored and de novo‐forming myofibrils were visible after miR‐669a treatment (Figure 1F). Immunostaining revealed ameliorated Cx43 pattern at intercalated disks in miR‐669a mice, as compared to sham and miR‐dsRed (Figure 1G). Furthermore, protein analysis revealed that an increase in Cx43 quantity and a decrease in ANP levels were also significantly quantifiable in the ventricular myocardium of miR‐669a mice, as compared to sham and miR‐dsRed (Figure 1H and Figure S7).
Hence, miR‐669a therapy ameliorated hypertrophic remodeling, fibrosis, and structural organization of end‐stage Sgcb‐null myocardium.
miR‐669a Expression Alters Cardiomyopathic Gene/miRNA Signature
Given the long‐term effects on survival and histological properties, we then asked whether AAV‐mediated miR‐669a therapy was also sufficient to perturb transcription levels of gene and miRNA markers of cardiomyopathy.
We examined myocardial levels of MyoD, abnormally reactivated in Sgcb‐null cardiac cells and a direct target of miR‐669a,21 and of Agtr1a, Il6 and Il1b, which were previously shown to be upregulated in hypertrophic cardiomyopathy.28–30 Quantitative PCR data indicated significant downregulation of all gene markers in miR‐669a mice, as compared to sham and miR‐dsRed (Figure 2A).
Figure 2.
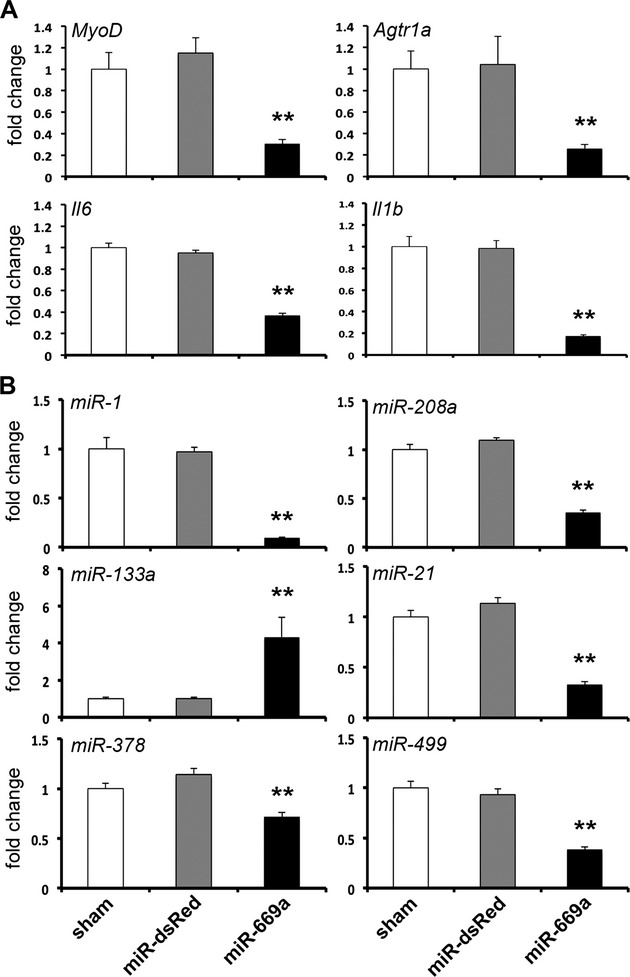
Effects of long‐term miR‐669a‐based therapy on gene/miRNA profile of Sgcb‐null hearts. A, qPCR data indicate significant decrease of MyoD, Agtr1a, Il6 and Il1b expression levels in miR‐669a‐treated myocardium (fold change normalized vs sham). B, Expression levels of miR‐1, miR‐208a, miR‐21, miR‐378 and miR‐499 significantly decrease, while miR‐133a levels increase, in miR‐669a‐treated myocardium (fold change normalized vs sham). Error bars depict standard deviation; **P<0.05 vs sham and P<0.05 vs miR‐dsRed. Sham, n=3; miR‐dsRed, n=3; miR‐669a, n=7. miRNA indicates MicroRNA; qPCR, quantitative polymerase chain reaction.
Furthermore, we investigated potential alterations in expression levels of miRNAs implicated in hypertrophy, cardiac degeneration, and DCM. Beside miR‐1, miR‐133a, and miR‐208a, we quantified expression levels of miR‐21, miR‐378, and miR‐499, previously reported to be upregulated during cardiomyopathy or cardiomyocyte stress.31–33 Quantitative PCR data showed significant downregulation of miR‐1 and miR‐208a and upregulation of miR‐133a in the myocardium of miR‐669a mice, as compared to sham and miR‐dsRed (Figure 2B). In addition, miR‐21, miR‐378, and miR‐499 were also significantly downregulated (Figure 2B).
Thus, miR‐669a sustained expression was sufficient to trigger long‐term perturbation of many gene/miRNA markers of cardiomyopathy.
Long‐Term miR‐669a Expression Alleviates Cardiac Functional Impairment
DCM progression affects not only histologic and contractile, but also functional properties of the myocardium, resulting in dilation of ventricular chambers,34 augmentation of posterior wall thickness,35 and reduction of systolic fractional shortening.36 We therefore investigated whether miR‐669a treatment affected functional cardiac properties in all surviving mice at 18 months postinjection, using transthoracic echocardiography. Possible effects on DCM‐related cardiac dysfunction were measured by quantifying long axis of LV, posterior wall thickness, ventricle diameter, and fractional shortening.
Treatment with miR669a, but not with miR‐dsRed, significantly reduced the end‐diastolic long axis dimension of the LV (Lax‐endo, Figure 3A). Moreover, posterior wall thickness showed a significant decrease in miR‐669a mice, as compared to sham and miR‐dsRed (PWT, Figure 3A). Importantly, long‐term miR‐669a expression significantly decreased LV internal diameter both in diastole and in systole (LVIDd and LVIDs respectively, Figure 3B) and, as a consequence, increased the systolic fractional shortening, as compared to sham and miR‐dsRed (Figure 3C).
Figure 3.
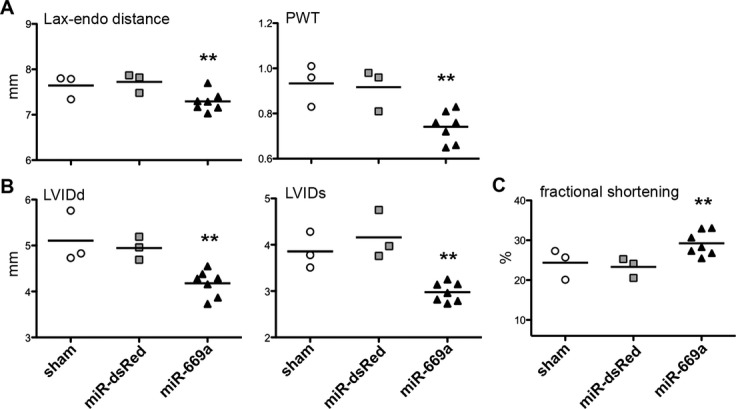
Long‐term miR‐669a‐based therapy alleviates MD‐associated dilated cardiomyopathy. Echocardiography‐based measurements show a significant improvement in distance between mitral valve and endocardial apex border (Lax‐endo) and posterior wall thickness (PWT) (A) decrease in left ventricle internal diameter both in diastole and in systole (LVIDd and LVIDs respectively, (B) and increase in the fractional shortening of the left ventricle in miR‐669a group, as compared to sham and miR‐dsRed (C). Bars, average value; each data point refers to one mouse surviving at 18 months post‐injection. **P<0.05 vs sham and P<0.05 vs miR‐dsRed; sham, n=3; miR‐dsRed, n=3; miR‐669a, n=7. MD indicates muscular dystrophy.
Hence, echocardiography‐based quantifications demonstrated significant alleviation of DCM dysfunctional phenotype in miR‐669a‐treated mice at 18 months postinjection.
AAV‐Mediated miRNA Therapy Does not Present Off‐Target Detrimental Effects in the Skeletal Muscle
Because of the dramatic negative regulation exerted by miR‐669a on MyoD21 and the tropism of AAV serotype 2/9 also for skeletal muscle,37 we next sought to assess potential long‐term effects of AAV‐mediated miRNA therapy on skeletal muscle structure of injected dystrophic mice. In addition, satellite cell (SC) number was also investigated, as an indication of regenerative capability of the skeletal muscle. Hind limb and diaphragm muscles of all surviving mice were therefore analyzed for fibrosis extension, fiber diameter, and SC percentage per fiber.
X‐Gal histological staining showed virtually absent nLacZ expression in the skeletal muscles of sham, miR‐dsRed, and miR‐669a mice (Figure S8A). Masson's trichromic staining revealed deep fibrotic infiltrations in gastrocnemius muscles in all three experimental conditions, without significant differences (Figure S8B). Moreover, no significant differences were quantifiable in average CSA (Figure S9) or CSA value distribution of gastrocnemius fibers among sham, miR‐dsRed, and miR‐669a conditions (Figure 4A). Similar results were also obtained by analyzing the quadriceps muscles (data not shown). In addition, no significant differences were found when quantifying the SC percentage/fiber in diaphragm, tibialis anterior, and soleus muscles among the three experimental conditions (Figure 4B).
Figure 4.
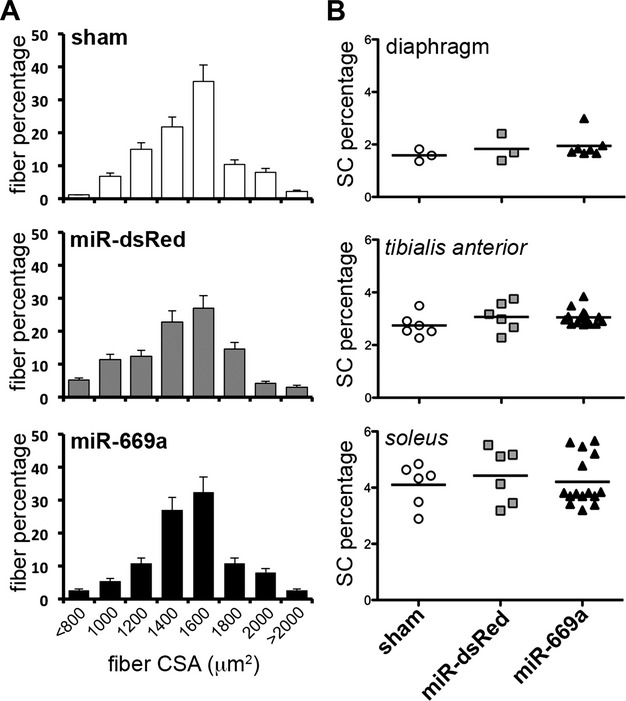
Intraventricular miR‐669a delivery does not alter skeletal muscle properties in the long term. A, Fiber percentage distribution among cross‐sectional area (CSA) classes reveals no significant shifts among the experimental groups. Error bars depict standard deviation. B, Analysis of SC percentage per fiber in diaphragm, tibialis anterior and soleus skeletal muscles reveals no significant differences among sham, miR‐dsRed and miR‐669a groups. Bar, average value; each data point refers to one skeletal muscle of mice surviving at 18 months postinjection. **P<0.05 vs sham and P<0.05 vs miR‐dsRed; sham, n=3; miR‐dsRed, n=3; miR‐669a, n=7. SC indicates satellite cell.
Thus, AAV‐mediated intraventricular delivery of both miR‐dsRed and miR‐669a did not result in significant deleterious effects on morphology and regenerative potential of hind limb and diaphragm muscles of treated mice as compared to sham conditions.
Discussion
MD‐associated DCM is currently treatable only by symptom alleviation, as effective cardioprotective or reparative intervention is yet lacking. Taken together, our results provide the first evidence of long‐term beneficial effects of a miRNA therapy in a murine model of MD‐associated DCM. In fact, AAV‐mediated miR‐669a expression is maintained for at least 18 months in Sgcb‐null hearts, significantly improves myocardial structure and function, and reduces adverse left ventricular remodeling, thereby increasing survival rate of dystrophic, severely cardiomyopathic mice.
In our study, Sgcb‐null dystrophic mice constituted a suitable model to assess long‐term effects and efficacy of AAV‐mediated miRNA therapy. In fact, spontaneous, early onset, and severe, chronic progression of DCM constitute a challenging yet clinically relevant model to test novel therapeutic approaches. Interestingly, miR‐669a‐mediated effects were consistently quantifiable at 18 months postinjection, corresponding to an end‐stage point with regards to both life expectancy and severity of cardiomyopathy in these mice.
In our approach, AAV2/9 vectors were tested. AAV vectors are gathering increasing interest in the cardiac gene therapy field, given the susceptibility of cardiomyocytes to AAV transduction.38 In addition, the 2/9 serotype recently showed high tropism for the cardiac muscle also in nonhuman primates.39 A possible drawback of AAV‐based therapies resides in variable immune response against viral capsids or occasionally against AAV‐transduced host cells.40–41 The main limitation of the present study is that AAV particles were injected before 24 hours after birth, escaping possible immune responses by the developing host immune system. In this regard, detailed studies are yet required to determine immunological sustainability over time of such an AAV therapy in juvenile or adult dystrophic mice.
Given the small size of miRNA molecules, many alternatives to viral vectors are currently tested for cardiac miRNA delivery or blockade, including injection of chemically modified oligonucleotides.42 Oligonucleotide‐based approaches could bypass the obstacles posed by immune response and viral genome integration, although durability and need of repeated injections might still hamper applications on MD‐associated DCM cases.43 Our results indicate that a single AAV injection is able to partially counteract cardiomyopathic progression for up to 18 months in a small animal model of MD‐associated DCM.
In our experimental conditions, histological, protein, and molecular assays showed significant miR‐669a‐dependent reduction of myocardial hypertrophy, apoptosis, and fibrosis. In accordance with previously reported data,21 miR‐669a overexpression was able to diminish aberrant MyoD levels in the dystrophic myocardium also in the long term. MyoD downregulation is likely linked to a decrease in aberrant myogenic fate and in related apoptotic events during remodeling of dystrophic myocardium, thus partially explaining the beneficial effects on treated hearts. Furthermore, we observed potentially beneficial variations also in transcriptional levels of cardiac miRNAs that are involved in cardiomyopathy and cardiac dysfunction. Notably, miR‐669a‐injected hearts displayed decreased miR‐1 and miR‐208a, and increased miR‐133a levels. Interestingly, miR‐1 was recently reported to suppress Cx43 in a model of myocarditis.44 This could additionally explain Cx43 upregulation in miR‐669a‐treated hearts. Furthermore, miR‐208a downregulation correlates with decreased remodeling, as miR‐208a‐null mice fail to undergo hypertrophy and fibrosis in response to several forms of cardiac stress.45 Conversely, miR‐133a upregulation can counteract Caspase9‐mediated apoptosis upon myocardial injury.46 Moreover, miR‐133a downregulation has been recently shown to further worsen prohypertrophic calcium signaling and pathological remodeling in the heart.47 However, changes in global miRNA levels could theoretically lead to adverse side events (eg, high miR‐133a levels have been associated with retinal degeneration).48 Although far from the scope of this work, a systemic investigation in off‐target organs is in fact required to address possible detrimental effects of such miRNA therapies.
Importantly, intraventricular miR‐669a delivery ameliorated structure and function of DCM hearts, apparently without inducing off‐target detrimental effects in the skeletal muscle. Moreover, after miR‐669a treatment, neither the dystrophic muscle wastage, nor the SC percentage was different from control groups. Interestingly, nuclei positive to nlacZ staining were virtually absent in skeletal muscles of AAV‐injected mice. Given MyoD‐dependent effects on the delicate balance between quiescence and apoptosis in SCs,49 it might be possible that rare miR669a‐transduced SCs were eventually withdrawn from the muscles after repeated cycles of de/regeneration, typical of MD progression.
In summary, this study provides the first evidence of long‐term (18 months) beneficial effects of AAV‐mediated miR‐669a therapy in a murine model of MD‐associated chronic DCM. Further refinement of administration routes and combination with cell therapy will potentially increase therapeutic effectiveness of miRNA‐based strategies for MD cardiomyopathy.
Sources of Funding
The Translational Cardiomyology laboratory is supported by CARE‐MI FP7, AFM, CARIPLO, FWO, GOA, IUAP and OT grants. Dr Quattrocelli is supported by a FWO PostDoctoral Fellowship. Dr Morgan is funded by a Wellcome Trust University Award. Dr Boldrin is funded by the BBSRC. Dr Toelen is funded by the KOOR Fonds of the University Hospitals Leuven. The authors would like to thank also Paolo Luban and Rondoufonds voor Duchenne Onderzoek for kind donations.
Disclosures
None.
Acknowledgments
AAV‐based viral vectors were produced by the Leuven Viral Vector Core. We appreciated Danny Huylebroeck, Karin Sipido, and Catherine Verfaillie for critical discussion. The authors would like to thank Christina Vochten and Vicky Raets for professional secretarial service.
References
- 1.Emery AE. The muscular dystrophies. Lancet. 2002; 359:687-695 [DOI] [PubMed] [Google Scholar]
- 2.Lapidos KA, Kakkar R, McNally EM. The dystrophin glycoprotein complex: signaling strength and integrity for the sarcolemma. Circ Res. 2004; 94:1023-1031 [DOI] [PubMed] [Google Scholar]
- 3.Gissel H. The role of Ca2+ in muscle cell damage. Ann N Y Acad Sci. 2005; 1066:166-180 [DOI] [PubMed] [Google Scholar]
- 4.McNally EM. New approaches in the therapy of cardiomyopathy in muscular dystrophy. Annu Rev Med. 2007; 58:75-88 [DOI] [PubMed] [Google Scholar]
- 5.Melacini P, Fanin M, Duggan DJ, Freda MP, Berardinelli A, Danieli GA, Barchitta A, Hoffman EP, Dalla Volta S, Angelini C. Heart involvement in muscular dystrophies due to sarcoglycan gene mutations. Muscle Nerve. 1999; 22:473-479 [DOI] [PubMed] [Google Scholar]
- 6.Verhaert D, Richards K, Rafael‐Fortney JA, Raman SV. Cardiac involvement in patients with muscular dystrophies: magnetic resonance imaging phenotype and genotypic considerations. Circ Cardiovasc Imaging. 2011; 4:67-76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lai Y, Duan D. Progress in gene therapy of dystrophic heart disease. Gene Ther. 2012; 19:678-685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Duan D. Challenges and opportunities in dystrophin‐deficient cardiomyopathy gene therapy. Hum Mol Genet. 2006; 15‐ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lefaucheur JP, Pastoret C, Sebille A. Phenotype of dystrophinopathy in old mdx mice. Anat Rec. 1995; 242:70-76 [DOI] [PubMed] [Google Scholar]
- 10.Durbeej M, Cohn RD, Hrstka RF, Moore SA, Allamand V, Davidson BL, Williamson RA, Campbell KP. Disruption of the beta‐sarcoglycan gene reveals pathogenetic complexity of limb‐girdle muscular dystrophy type 2E. Mol Cell. 2000; 5:141-151 [DOI] [PubMed] [Google Scholar]
- 11.Hedman M, Hartikainen J, Yla‐Herttuala S. Progress and prospects: hurdles to cardiovascular gene therapy clinical trials. Gene Ther. 2011; 18:743-749 [DOI] [PubMed] [Google Scholar]
- 12.Vandendriessche T, Thorrez L, Acosta‐Sanchez A, Petrus I, Wang L, Ma L, DE Wale L, Iwasaki Y, Gillijns V, Wilson JM, Collen D, Chuah MK. Efficacy and safety of adeno‐associated viral vectors based on serotype 8 and 9 vs. Lentiviral vectors for hemophilia B gene therapy. J Thromb Haemost. 2007; 5:16-24 [DOI] [PubMed] [Google Scholar]
- 13.Yue Y, Li Z, Harper SQ, Davisson RL, Chamberlain JS, Duan D. Microdystrophin gene therapy of cardiomyopathy restores dystrophin‐glycoprotein complex and improves sarcolemma integrity in the mdx mouse heart. Circulation. 2003; 108:1626-1632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bostick B, Shin JH, Yue Y, Wasala NB, Lai Y, Duan D. AAV micro‐dystrophin gene therapy alleviates stress‐induced cardiac death but not myocardial fibrosis in >21‐m‐old mdx mice, an end‐stage model of duchenne muscular dystrophy cardiomyopathy. J Mol Cell Cardiol. 2012; 53:217-222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kairouz V, Lipskaia L, Hajjar RJ, Chemaly ER. Molecular targets in heart failure gene therapy: current controversies and translational perspectives. Ann N Y Acad Sci. 2012; 1254:42-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orenes‐Pinero E, Montoro‐Garcia S, Patel JV, Valdes M, Marin F, Lip GY. Role of microRNAs in cardiac remodelling: new insights and future perspectives. Int J Cardiol. 2012. ‐‐10.1016/j.ijcard.2012.09.120 [DOI] [PubMed] [Google Scholar]
- 17.Chen JF, Murchison EP, Tang R, Callis TE, Tatsuguchi M, Deng Z, Rojas M, Hammond SM, Schneider MD, Selzman CH, Meissner G, Patterson C, Hannon GJ, Wang DZ. Targeted deletion of dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc Natl Acad Sci USA. 2008; 105:2111-2116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bagnall RD, Tsoutsman T, Shephard RE, Ritchie W, Semsarian C. Global microRNA profiling of the mouse ventricles during development of severe hypertrophic cardiomyopathy and heart failure. PLoS One. 2012; 7:e44744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Belevych AE, Sansom SE, Terentyeva R, Ho HT, Nishijima Y, Martin MM, Jindal HK, Rochira JA, Kunitomo Y, Abdellatif M, Carnes CA, Elton TS, Gyorke S, Terentyev D. MicroRNA‐1 and ‐133 increase arrhythmogenesis in heart failure by dissociating phosphatase activity from RYR2 complex. PLoS One. 2011; 6:e28324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Satoh M, Minami Y, Takahashi Y, Tabuchi T, Nakamura M. Expression of microrna‐208 is associated with adverse clinical outcomes in human dilated cardiomyopathy. J Card Fail. 2010; 16:404-410 [DOI] [PubMed] [Google Scholar]
- 21.Crippa S, Cassano M, Messina G, Galli D, Galvez BG, Curk T, Altomare C, Ronzoni F, Toelen J, Gijsbers R, Debyser Z, Janssens S, Zupan B, Zaza A, Cossu G, Sampaolesi M. MiR669a and miR669q prevent skeletal muscle differentiation in postnatal cardiac progenitors. J Cell Biol. 2011; 193:1197-1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Whittaker P, Kloner RA, Boughner DR, Pickering JG. Quantitative assessment of myocardial collagen with picrosirius red staining and circularly polarized light. Basic Res Cardiol. 1994; 89:397-410 [DOI] [PubMed] [Google Scholar]
- 23.Boldrin L, Neal A, Zammit PS, Muntoni F, Morgan JE. Donor satellite cell engraftment is significantly augmented when the host niche is preserved and endogenous satellite cells are incapacitated. Stem Cells. 2012; 30:1971-1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Russo SB, Baicu CF, Van Laer A, Geng T, Kasiganesan H, Zile MR, Cowart LA. Ceramide synthase 5 mediates lipid‐induced autophagy and hypertrophy in cardiomyocytes. J Clin Invest. 2012; 122:3919-3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lefta M, Campbell KS, Feng HZ, Jin JP, Esser KA. Development of dilated cardiomyopathy in Bmal1‐deficient mice. Am J Physiol Heart Circ Physiol. 2012; 303:H475-H485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li J, Goossens S, van Hengel J, Gao E, Cheng L, Tyberghein K, Shang X, De Rycke R, van Roy F, Radice GL. Loss of alphaT‐catenin alters the hybrid adhering junctions in the heart and leads to dilated cardiomyopathy and ventricular arrhythmia following acute ischemia. J Cell Sci. 2012; 125:1058-1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kasama S, Furuya M, Toyama T, Ichikawa S, Kurabayashi M. Effect of atrial natriuretic peptide on left ventricular remodelling in patients with acute myocardial infarction. Eur Heart J. 2008; 29:1485-1494 [DOI] [PubMed] [Google Scholar]
- 28.Masuda T, Muto S, Fujisawa G, Iwazu Y, Kimura M, Kobayashi T, Nonaka‐Sarukawa M, Sasaki N, Watanabe Y, Shinohara M, Murakami T, Shimada K, Kobayashi E, Kusano E. Heart angiotensin II‐induced cardiomyocyte hypertrophy suppresses coronary angiogenesis and progresses diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol. 2012; 302:H1871-H1883 [DOI] [PubMed] [Google Scholar]
- 29.Bobbert P, Jenke A, Bobbert T, Kuhl U, Rauch U, Lassner D, Scheibenbogen C, Poller W, Schultheiss HP, Skurk C. High leptin and resistin expression in chronic heart failure: adverse outcome in patients with dilated and inflammatory cardiomyopathy. Eur J Heart Fail. 2012; 14:1265-1275 [DOI] [PubMed] [Google Scholar]
- 30.Ock S, Ahn J, Lee SH, Park H, Son JW, Oh JG, Yang DK, Lee WS, Kim HS, Rho J, Oh GT, Abel ED, Park WJ, Min JK, Kim J. Receptor activator of nuclear factor‐kappab ligand is a novel inducer of myocardial inflammation. Cardiovasc Res. 2012; 94:105-114 [DOI] [PubMed] [Google Scholar]
- 31.Yang KC, Ku YC, Lovett M, Nerbonne JM. Combined deep microRNA and mRNA sequencing identifies protective transcriptomal signature of enhanced PI3Kalpha signaling in cardiac hypertrophy. J Mol Cell Cardiol. 2012; 53:101-112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knezevic I, Patel A, Sundaresan NR, Gupta MP, Solaro RJ, Nagalingam RS, Gupta M. A novel cardiomyocyte‐enriched microRNA, miR‐378, targets insulin‐like growth factor 1 receptor: implications in postnatal cardiac remodeling and cell survival. J Biol Chem. 2012; 287:12913-12926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matkovich SJ, Hu Y, Eschenbacher WH, Dorn LE, Dorn GW., 2nd Direct and indirect involvement of microRNA‐499 in clinical and experimental cardiomyopathy. Circ Res. 2012; 111:521-531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Politano L, Nigro G. Treatment of dystrophinopathic cardiomyopathy: review of the literature and personal results. Acta Myol. 2012; 31:24-30 [PMC free article] [PubMed] [Google Scholar]
- 35.Carvalho JS, Silva CM, Shinebourne EA, Redington AN. Prognostic value of posterior wall thickness in childhood dilated cardiomyopathy and myocarditis. Eur Heart J. 1996; 17:1233-1238 [DOI] [PubMed] [Google Scholar]
- 36.Chun JL, O'Brien R, Berry SE. Cardiac dysfunction and pathology in the dystrophin and utrophin‐deficient mouse during development of dilated cardiomyopathy. Neuromuscul Disord. 2012; 22:368-379 [DOI] [PubMed] [Google Scholar]
- 37.Koo T, Malerba A, Athanasopoulos T, Trollet C, Boldrin L, Ferry A, Popplewell L, Foster H, Foster K, Dickson G. Delivery of AAV2/9‐microdystrophin genes incorporating helix 1 of the coiled‐coil motif in the C‐terminal domain of dystrophin improves muscle pathology and restores the level of alpha1‐syntrophin and alpha‐dystrobrevin in skeletal muscles of mdx mice. Hum Gene Ther. 2011; 22:1379-1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lovric J, Mano M, Zentilin L, Eulalio A, Zacchigna S, Giacca M. Terminal differentiation of cardiac and skeletal myocytes induces permissivity to AAV transduction by relieving inhibition imposed by DNA damage response proteins. Mol Ther. 2012; 20:2087-2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tarantal AF, Lee CC. Long‐term luciferase expression monitored by bioluminescence imaging after adeno‐associated virus‐mediated fetal gene delivery in rhesus monkeys (Macaca Mulatta). Hum Gene Ther. 2010; 21:143-148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mingozzi F, High KA. Immune responses to AAV in clinical trials. Curr Gene Ther. 2011; 11:321-330 [DOI] [PubMed] [Google Scholar]
- 41.Vulin A, Barthelemy I, Goyenvalle A, Thibaud JL, Beley C, Griffith G, Benchaouir R, le Hir M, Unterfinger Y, Lorain S, Dreyfus P, Voit T, Carlier P, Blot S, Garcia L. Muscle function recovery in golden retriever muscular dystrophy after AAV1‐U7 exon skipping. Mol Ther. 2012; 20:2120-2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Montgomery RL, Hullinger TG, Semus HM, Dickinson BA, Seto AG, Lynch JM, Stack C, Latimer PA, Olson EN, van Rooij E. Therapeutic inhibition of miR‐208a improves cardiac function and survival during heart failure. Circulation. 2011; 124:1537-1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Prakash TP. An overview of sugar‐modified oligonucleotides for antisense therapeutics. Chem Biodivers. 2011; 8:1616-1641 [DOI] [PubMed] [Google Scholar]
- 44.Xu HF, Ding YJ, Shen YW, Xue AM, Xu HM, Luo CL, Li BX, Liu YL, Zhao ZQ. MicroRNA‐ 1 represses Cx43 expression in viral myocarditis. Mol Cell Biochem. 2012; 362:141-148 [DOI] [PubMed] [Google Scholar]
- 45.van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress‐dependent cardiac growth and gene expression by a microRNA. Science. 2007; 316:575-579 [DOI] [PubMed] [Google Scholar]
- 46.He B, Xiao J, Ren AJ, Zhang YF, Zhang H, Chen M, Xie B, Gao XG, Wang YW. Role of miR‐1 and miR‐133a in myocardial ischemic postconditioning. J Biomed Sci. 2011; 18:22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Drawnel FM, Wachten D, Molkentin JD, Maillet M, Aronsen JM, Swift F, Sjaastad I, Liu N, Catalucci D, Mikoshiba K, Hisatsune C, Okkenhaug H, Andrews SR, Bootman MD, Roderick HL. Mutual antagonism between IP3RII and miRNA‐133a regulates calcium signals and cardiac hypertrophy. J Cell Biol. 2012; 199:783-798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Loscher CJ, Hokamp K, Kenna PF, Ivens AC, Humphries P, Palfi A, Farrar GJ. Altered retinal microRNA expression profile in a mouse model of retinitis pigmentosa. Genome Biol. 2007; 8:R248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hirai H, Verma M, Watanabe S, Tastad C, Asakura Y, Asakura A. MyoD regulates apoptosis of myoblasts through microRNA‐mediated down‐regulation of Pax3. J Cell Biol. 2010; 191:347-365 [DOI] [PMC free article] [PubMed] [Google Scholar]