

Abstract
Background
Egr‐1 is implicated in the pathogenesis of myocardial ischemia–reperfusion injury. The aim of this study was to ascertain the effectiveness of intracoronary delivery of DNAzyme targeting the transcription factor Egr‐1 at reperfusion following experimental myocardial ischemia.
Methods and Results
Functional DNAzyme targeting Egr‐1 or a size‐matched scrambled control were delivered via the intracoronary route immediately on reperfusion after 60 minutes' balloon occlusion of the left anterior descending coronary artery in a pig model of myocardial I/R injury (n=7 per treatment group). Heart function and extent of myocardial infarction were determined following intervention by echocardiography and cardiac magnetic resonance imaging, respectively. Hearts were removed and examined for molecular and histological markers of inflammation and apoptosis. Administration of functional DNAzyme led to an overall decrease in the expression of inflammatory markers including intracellular adhesion molecule‐1, tissue factor, and complement 3, with associated decreases in the extent of neutrophil infiltration, oxidative damage, and subsequent apoptosis within the infarct border zone. Functional significance was indicated by an increase in salvaged left ventricular myocardium (P=0.012), ejection fraction (P=0.002), and fractional area change (P=0.039) in the functional DNAzyme–treated group compared with the control.
Conclusions
Egr‐1 silencing through intracoronary delivery of a targeting DNAzyme at the time of reperfusion following acute myocardial ischemia decreases myocardial inflammation and apoptosis leading to improved cardiac function.
Keywords: acute myocardial ischemia reperfusion, DNAzyme, Egr‐1
Introduction
The deleterious molecular processes initiated by both ischemia and reperfusion are thought to be key elements in contributing negatively to outcomes despite optimal pharmacoinvasive treatment of myocardial infarction (MI). There is a body of growing literature that implicates the transcription factor early growth response‐1 (Egr‐1) as playing a pivotal role in ischemia as well as ischemia–reperfusion (I/R) injury in the heart, gut, kidneys, and lungs.1 The importance of Egr‐1 in the pathogenesis of myocardial injury and its role as a potential therapeutic target is accentuated by 2 characteristics: first, it is quiescent under basal conditions and is rapidly expressed following ischemia and I/R injury; and second, it is a master regulator gene, positioned upstream of genes governing key facets of I/R injury including inflammation, apoptosis. and thrombosis such as ICAM‐1, PAI‐1, TF, MCP‐1, MIP 2, and p53.2 Silencing Egr‐1 therefore represents an exciting opportunity to interrupt gene signaling and potentially attenuate myocardial damage. DNA enzymes (DNAzymes) are novel catalytic oligonucleotides that can be engineered to silence genes by selectively binding and inhibiting mRNA expression. They have been applied therapeutically against plasminogen activator inhibitor‐I (PAI‐I), c‐Jun, and vitamin D–upregulated protein 1 to reduce myocardial injury following ischemia and I/R.3–4 DNAzymes targeting Egr‐1 (DZF) have been shown to ablate the pathological response in a range of experimental models of disease including cancer, I/R, and vascular injury. We have recently demonstrated that DZF can be delivered preemptively via the intracoronary route in pigs to block Egr‐1 expression and reduce heart attack size as measured at 3 hours postreperfusion.1 The present study extends this work by assessing the effects of DZF delivered via the intracoronary route at the time of reperfusion in a preclinical large animal model at a clinically relevant time point. We examine the impact on MI size and heart function using state‐of‐the‐art imaging and characterize the effect of Egr‐1 silencing on signaling pathways critical to myocardial I/R injury.
Methods
DNAzyme Preparation
DNAzymes were commercially synthesized as described previously.1 DZF targets Egr‐1, while DZF Scr is the size‐matched control molecule with a preserved 10–23 catalytic core but possessing scrambled flanking binding arm sequences. DNAzyme (1 mg) was suspended in 2 mL of PBS containing 300 μL of FuGENE6 (Roche Diagnostics) and 1 mmol/L MgCl2 and delivered via the intracoronary route immediately on reperfusion.
Porcine Model of Myocardial I/R
Procedures were performed in accordance with the Royal North Shore Hospital Animal Care Ethics Committee guidelines. Male pigs (n=7 per group) were subjected to I/R by endoluminal occlusion of the left anterior descending coronary artery distal to the first major diagonal for 60 minutes, followed by reperfusion and recovery for 48 hours as described previously.1 Immediately following balloon deflation at the time of reperfusion, pigs received an intracoronary bolus of either DZF or DZF Scr via the guiding catheter. A third group was included in this study as a control to establish baseline parameters for subsequent analyses and underwent a sham operation in which the catheter, guide wire, and balloon were advanced into position as described earlier, but the balloon was not inflated. On termination of the study, hearts were excised, and apices were removed and immediately frozen in liquid nitrogen for gene and protein expression studies as detailed later.
Quantitative Real‐Time RT‐PCR
Total RNA was extracted from heart apices using a Total RNA isolation kit (Sigma) as per manufacturer's instructions. Primers specific for genes of interest (Table 1) were designed using Primer3 software and synthesized by Sigma‐Aldrich. Quantitative real‐time RT‐PCR (qPCR) was performed on generated cDNA with iQ SYBR Green Supermix (BioRad) as per manufacturer's instructions on a Corbett Rotor‐gene 6000 thermocycler (Qiagen) as described previously.5 Data are expressed as the average expression of the gene of interest relative to the housekeeping gene from 4 hearts per DNAzyme‐treated group from of 4 replicate extractions per heart.
Table 1.
Primer Sequences Used in qRT‐PCR Analysis
| Gene | Primer Sequence | GeneBank Reference |
|---|---|---|
| Egr‐1 | 5′‐CAGTACCCACCTCCTGCCTA‐3′ | AJ238156 |
| 5′‐GAGAGTGGAGTGAGCGAAGG‐3′ | ||
| ICAM‐1 | 5′‐TTCCTTGGATGGACTGTTCC‐3′ | AF156712 |
| 5′‐CGTCTGCCAGCATTATCTCA‐3′ | ||
| VCAM‐1 | 5′‐ATCCAAGCTGCTCCAAAAGA‐3′ | NM213891 |
| 5′‐GGCCCTGTGGATGGTATATG‐3′ | ||
| Tissue factor | 5′‐GAAGCTGCTCGCACCTTAGT‐3′ | AY504424 |
| 5′‐CGACTTTCGGGACTCTTCTG‐3′ | ||
| C3 | 5′‐ACAAATTGACCCAGCGTAGG‐3′ | AF154933 |
| 5′‐GCACGTCCTTGCTGTACTGA‐3′ | ||
| C3aR | 5′‐GCAGGTTCCTATGCAAGCTC‐3′ | NM004054 |
| 5′‐CACATCACAAAAGCCACCAC‐3′ | ||
| p53 | 5′‐CCTCACCATCATCACACTGG‐3′ | AF098067 |
| 5′‐GGCTTCTTCTTTTGCACTGG‐3′ | ||
| PTEN | 5′‐ACCAGGACCAGAGGAAACCT‐3′ | U93051 |
| 5′‐GCTAGCCTCTGGATTTGACG‐3′ | ||
| GAPDH | 5′‐GGGCATGAACCATGAGAAGT‐3′ | AF017079 |
| 5′‐AAGCAGGGATGATGTTCTGG‐3′ |
qRT‐PCR indicates quantitative real‐time polymerase chain reaction; Egr‐1, early growth response‐1; ICAM‐1, intercellular adhesion molecule 1; VCAM‐1, vascular cell adhesion molecule 1; C3aR, complement 3 receptor; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; PTEN, phosphatase and tensin homolog.
Western Blot Analysis
Western blotting was performed on tissue homogenates obtained from the heart apices using specific antibodies as previously described.5
Immunohistochemistry
Tissue inflammation within the infarct border region of hearts following myocardial I/R injury was assessed through the use of immunohistochemistry as previously described2 with corresponding hematoxylin and eosin–stained sections analyzed for neutrophils and the extent of apoptosis determined through the use of an ApopTag® Plus kit as per the manufacturer's instructions (Millipore). Intensity of interleukin (IL)‐8 staining, neutrophils, and apoptotic cells were counted within 6 randomly selected high‐powered fields of view within the infarct border zone from 4 hearts per treated group.
Enzyme Immunoassay
Heart tissue levels of F2‐isoprostanes were measured in homogenates using a commercially available kit as per the manufacturer's instructions (Cayman Chemical).
CMR Imaging
CMR imaging was performed 48 hours postreperfusion on a 3.0‐T GE 750 Discovery scanner (GE Healthcare). Steady‐state free‐precession cine sequences were performed with field of view (FOV) of 350 to 400 mm, repetition time (TR)/echo time (TE) of 3.2/1.6 ms, flip angle of 60°, matrix of 224×192, 20 segments, and 6‐mm slices. T2‐weighted double inversion‐recovery FSE sequence parameters were FOV of 380 to 400 mm, TR of 2 R‐R intervals, TE of 70 ms, matrix of 256×192, and slice thickness of 6 mm. Both T2 and cine acquisitions were acquired in short‐axis orientation and the left ventricle was encompassed by contiguous slices. Late gadolinium enhancement (LGE) imaging was performed 7 to 15 minutes after contrast administration (0.2 mmol/kg body weight gadolinium‐diethylenetriamine‐pentaacetic acid) with inversion time of 180 to 210 ms, FOV of 350 to 400 mm, TR/TE of 4.6/1.3 ms, flip angle of 20°, matrix of 256×192, and slice thickness of 6 mm. Infarct size was defined as an area of late enhancement on LGE images of the MI. Persistent microvascular obstruction (MVO) was defined as late hypoenhancement within a hyper‐enhanced region on LGE images. Cine CMR was used to derive LV volumes, mass, and global function. On T2‐weighted images, infarct‐related edema was considered present when the signal intensity (SI) of the myocardium was >2 SDs of the mean SI of the contralateral remote region. Area at risk (AAR) extent was obtained by manually tracing the hyperintense region on T2‐weighted, short‐axis images. On DE imaging, MI was considered present if the SI of hyperenhanced myocardium was >5 SDs of the mean SI of the remote region, and MVO was defined as a hypoenhanced region within infarcted myocardium. MI size and MVO extent were obtained by manually drawing the regions of interest and expressed as LV percentage. Salvage index was calculated as (AAR−LGE)/AAR×100.6
Echocardiography
Two‐dimensional echocardiography was performed using a Toshiba Xario (model UICW‐680A) with a 3‐MHz probe. LV systolic function was estimated by measuring the fractional area change as described previously.7
Statistics
After confirming normality of data comparison among 3 sample groups (sham/DZF/DZF Scr), data were analyzed by 1‐way ANOVA with Tukey's post‐test. For analysis between 2 sample groups (DZF and DZF Scr), data were analyzed by unpaired t test using Prism software (version 4; GraphPad), all data are expressed as mean±SEM with significance defined as P≤0.05. Standard linear regression analysis was used to explore the relationship between AAR and LGE in both the Scr and DZF groups.
Results
Egr‐1 mRNA and Protein Expression Is Inhibited at 48 Hours by DNAzyme Delivered via the Intracoronary Route at the Time of Reperfusion
Representative Western blot images of protein expression in pig hearts following DNAzyme treatment are shown in Figure 1. Evaluation of Egr‐1 mRNA expression via qRT‐PCR (Figure 2A) in heart tissue 48 hours postreperfusion revealed a significant upregulation of Egr‐1 in pigs receiving DZF Scr compared with sham‐operated controls. Conversely, DZF inhibited Egr‐1 mRNA expression. Similarly, quantification of protein expression from Western blot analysis (Figure 2B) revealed that downregulation of Egr‐1 mRNA expression in the DZF group was paralleled by a trend to decrease in Egr‐1 protein accumulation, whereas Egr‐1 protein expression in hearts from pigs receiving DZF Scr remained significantly elevated compared with sham controls. Taken together, these results indicate that DNAzyme targeting Egr‐1, delivered at the time of reperfusion via the intracoronary route, efficiently inhibits expression of Egr‐1 mRNA and protein and that this inhibition of Egr‐1 via DNAzyme delivery remains effectual for at least 48 hours following reperfusion.
Figure 1.
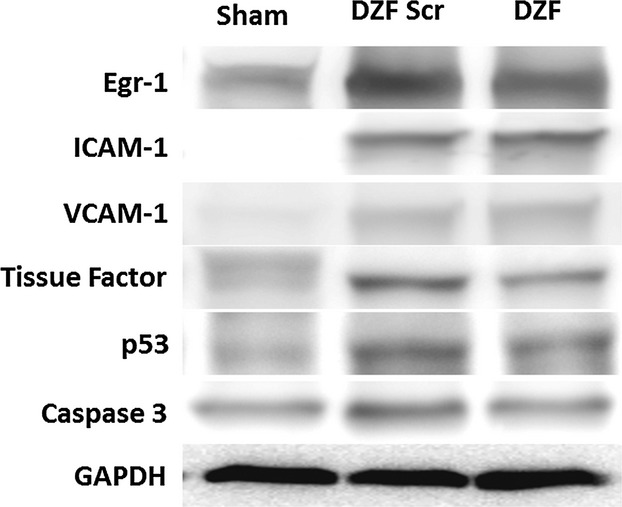
Representative Western blots demonstrating protein expression in hearts from pigs subjected to myocardial I/R injury. Pigs were administered functional (DZF) or dysfunctional (DZF Scr) DNAzyme at the time of reperfusion. Blots shown are representative of protein accumulation within n=5 hearts per DNAzyme‐treated group. I/R indicates ischemia–reperfusion; DZF, functional DNAzyme; DZF Scr, scrambled DNAzyme; Egr‐1, early growth response‐1; ICAM‐1, intercellular adhesion molecule 1; VCAM‐1, vascular cell adhesion molecule 1; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; ELISA, enzyme‐linked immunosorbent assay; PTEN, phosphatase and tensin homolog; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
Figure 2.
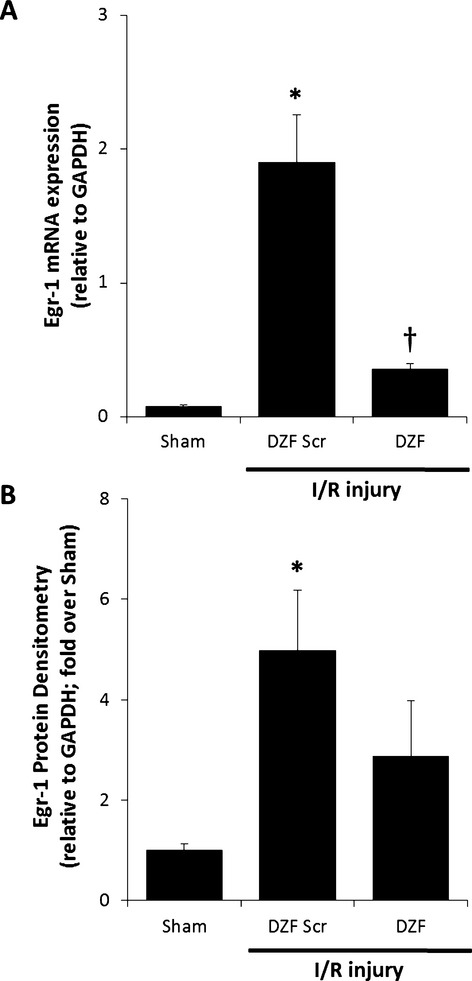
EGR‐1 expression within myocardial tissue following I/R injury. A, Egr‐1 mRNA expression representative of 4 replicate mRNA extractions from 4 hearts per DNAzyme‐treated group. B, Egr‐1 protein densitometry normalized to GAPDH expression from 5 hearts per DNAzyme‐treated group. *Significantly different compared with sham control; †significantly different compared with DZF Scr group (P ≤ 0.05). EGR‐1 indicates early growth response‐1; I/R, ischemia–reperfusion; DZF Scr, scrambled DNAzyme; DZF, functional DNAzyme.
DNAzyme Targeting Egr‐1 Inhibits the I/R‐Mediated Inflammatory Response and Reduces Oxidative Damage to the Myocardium
Intracoronary administration of DZF at the time of reperfusion following myocardial ischemia resulted in the decreased expression of Egr‐1–regulated inflammatory mediators. Evaluation by qRT‐PCR (Figure 3A) revealed that hearts in the DZF Scr cohort exhibited an upregulation in ICAM‐1, VCAM‐1, and tissue factor mRNA compared with the sham control group, although these did not reach statistical significance at the 48‐hour time point measured. There was, however, a significant increase in ICAM‐1 and tissue factor protein levels within the DZF Scr cohort, whereas within the DZF‐treated group, protein expression of these inflammatory mediators remained within background levels exhibited in the sham group (Figures 1 and 3B). The increased expression of I/R‐induced inflammatory mediators in hearts from the DZF Scr–treated group was concurrent with heightened inflammation as well as an increase in oxidative injury. Activation of the complement‐mediated inflammatory response was confirmed through qRT‐PCR analysis of C3 and the C3 receptor, C3aR (Figure 4A), with administration of intracoronary DZF but not DZF Scr at the time of reperfusion attenuating this expression. To determine the extent of I/R‐induced inflammation, tissue sections were assessed for IL‐8 expression (Figure 4B and 4C). Hearts that had received DZF had significantly lower IL‐8 expression and a significant decrease in the extent of neutrophil infiltration within the affected heart tissue (Figure 4D and 4E) compared with the DZF Scr–treated group.
Figure 3.

Inflammatory markers within myocardial tissue following I/R injury. A, ICAM‐1, VCAM‐1, and tissue factor mRNA expression relative to GAPDH expression representative of 4 replicate mRNA extractions from 4 hearts per DNAzyme‐treated group. B, Quantitation of protein densitometry from Western blots probing for ICAM‐1, VCAM‐1, and tissue factor (relative to GAPDH) from 5 hearts per DNAzyme‐treated group. *Significantly different compared with sham control; †significantly different compared with DZF Scr group (P≤0.05). I/R indicates ischemia–reperfusion; DZF Scr, scrambled DNAzyme; DZF, functional DNAzyme.
Figure 4.

Oxidative injury and the inflammatory response following myocardial I/R injury. A, mRNA expression of complement 3 and the complement 3 receptor (C3aR) assessed via qRT‐PCR (relative to GAPDH expression) from 4 hearts per DNAzyme‐treated group (with an average of 4 replicates per heart included in analysis). B, Representative immunohistochemistry of IL‐8 staining and (C) quantification of IL‐8 within heart tissue from DZF and DZF Scr–treated groups. D, Representative H&E‐stained sections demonstrating neutrophils and (E) quantification of neutrophil infiltration within heart tissue. F, ELISA analysis of F2‐isoprostane levels from 5 hearts per DNAzyme‐treated group (with an average of 2 replicates per heart used for this analysis). *Significantly different compared with sham control; †significantly different compared with DZF Scr group (P≤0.05). I/R indicates ischemia–reperfusion; qRT‐PCR, quantitative real‐time polymerase chain reaction; IL‐8, interleukin‐8; DZF, functional DNAzyme; DZF Scr, scrambled DNAzyme; H&E, hematoxylin and eosin.
To assess whether the reduction in the extent of inflammation in the DZF‐treated group was coupled with decreased oxidative damage, homogenates prepared from the isolated hearts were analyzed for the extent of F2‐isoprostanes (Figure 4F), an indicator of lipid peroxidation. Compared with the DZF Scr group, tissue from hearts administered DZF contained significantly decreased content of F2‐isoprostanes. These results indicate that by silencing Egr‐1, inflammation severity and subsequent myocardial tissue damage are reduced.
DNAzyme Targeting Egr‐1 Inhibits I/R‐Induced Myocardial Apoptosis
I/R injury resulted in heart tissue exhibiting an increase in proapoptotic markers of cell death (Figures 1, and 5A, 5B). DZF but not DZF Scr treatment resulted in inhibition of I/R‐induced p53 and PTEN mRNA that was coupled with a trend toward lower p53 and active caspase 3 protein expression, although this did not reach statistical significance. To ascertain if reduced levels of these proapoptotic markers translated into a reduction in the extent of cells undergoing apoptosis, terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling assay staining of histological sections was performed (Figure 5C). Consistent with the decrease in p53 and active caspase 3 expression, treatment with DZF resulted in a significant reduction myocyte apoptosis within the infarct border region of hearts compared with sections of hearts treated with DZF Scr.
Figure 5.
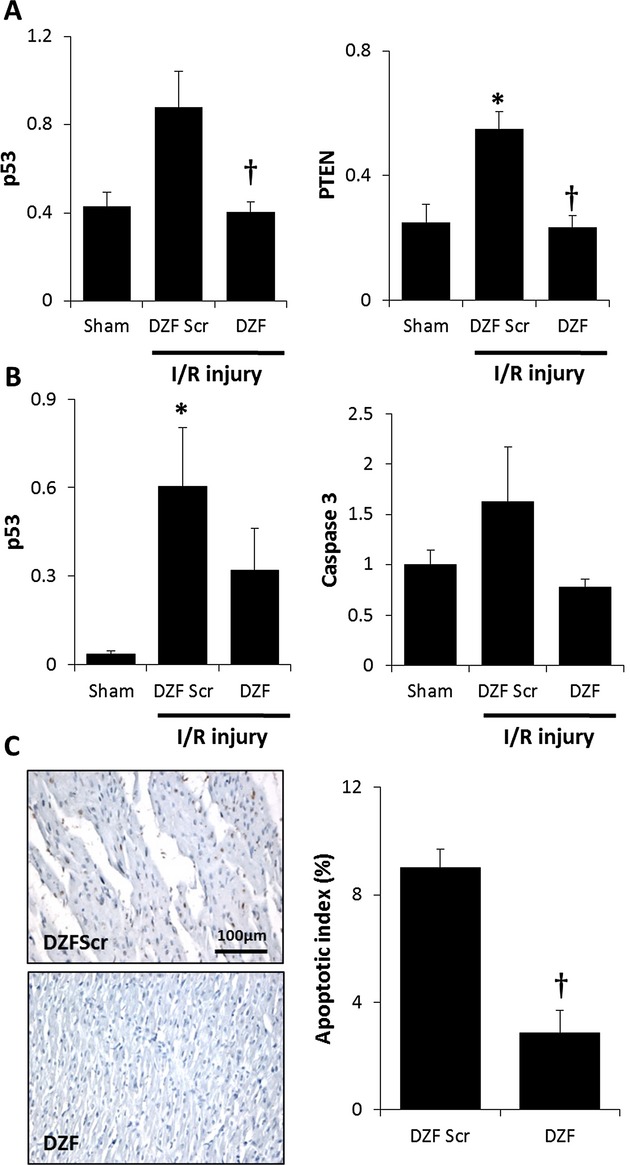
Extent of apoptosis following myocardial I/R injury. A, p53 and PTEN and mRNA expression (relative to GAPDH expression) representative of 4 replicate mRNA extractions from 4 hearts per DNAzyme‐treated group. B, Quantitation of protein densitometry from Western blots probing for p53 and caspase 3 (relative to GAPDH, ‐fold over sham) from 5 hearts per DNAzyme‐treated group. C, Representative terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling assay micrographs of DZF and DZF Scr heart sections. Data are the average of 3 hearts (from 6 high‐powered field readings per heart included in this analysis). *Significantly different compared with sham control; †significantly different compared with DZF Scr group (P≤0.05). I/R indicates ischemia–reperfusion; DZF, functional DNAzyme; DZF Scr, scrambled DNAzyme.
DNAzyme Targeting Egr‐1 Improves Myocardial Salvage and Cardiac Function Following Myocardial I/R Injury
Treatment had no significant effect on AAR or on LV and right ventricular volumes at either end diastole or systole between the treatment groups (Table 2). Infarct size was 14.4 g (16.9±1.5% of LV mass) in the DZF treatment group compared with 19.6 g (25.3±3.1% of LV mass) in the scrambled control (NS; P=0.195). Myocardial salvage index (Figure 6A and 6B) was calculated using with the formula (AAR−LGE)/AAR×100, to adjust for varying coronary anatomy, as described in the Methods section. DZF treatment improved myocardial salvage by 84% (Figure 6C; P=0<0.05). The relationship between AAR and LGE is shown graphically for the 2 groups in Figure 6D (DZF Scr treatment group R2=0.742; and DZF treatment group R2=0.329).
Table 2.
CMR Measurement of Heart Function Following I/R Injury
| Parameter | DZF Scr | DZF | P Value |
|---|---|---|---|
| HR, bpm | 83.8±6 | 83.6±5 | 0.989 |
| LV mass, g | 76.4±6 | 84.9±11 | 0.491 |
| LVEDV, mL | 83.7±5 | 99.4±9 | 0.131 |
| LVESV, mL | 58.9±3 | 58.8±6 | 0.978 |
| LV stroke volume, mL | 24.7±2 | 40.8±4 | 0.005 |
| LV EF, % | 29.4±1 | 41±3 | 0.002 |
| Cardiac output, L/min | 2.09±0.3 | 3.38±0.3 | 0.011 |
| RV EF, % | 40.7±1 | 42.7±3 | 0.519 |
| AAR, g | 28.3±2 | 35.9±4 | 0.07 |
| AAR/LV mass, % | 37.3±1 | 44.3±4 | 0.122 |
| LGE, g | 19.6±3 | 14.4±2 | 0.195 |
| LGE/LV mass, % | 25.3±3 | 16.9±1 | 0.038 |
| LGE/AAR, % | 67.4±7 | 40.1±6 | 0.012 |
Pigs receiving either functional DNAzyme (DZF) or a size‐matched scrambled control (DZF) Scr at the time of reperfusion were assessed at 48 hours postreperfusion for functional parameters, including heart rate (HR), left ventricular (LV) volume, ejection fraction (EF), and cardiac output. Data represents values from 7 pigs per DNAzyme‐treated group. CMR indicates cardiac magnetic resonance; I/R, ischemia–reperfusion; LVEDV, LV end‐diastolic volume; LVESV, LV end‐systolic volume; RV, right ventricular; AAR, area at risk; LGE, late gadolinium enhancement.
Figure 6.
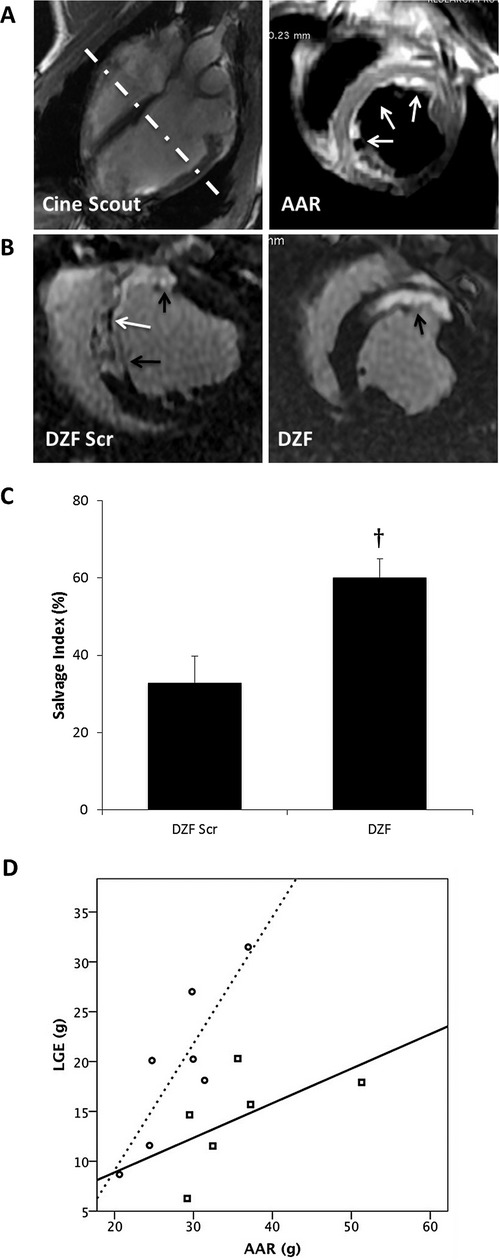
CMR assessment of myocardial salvage and heart function following I/R injury. A, Representative cine 4‐chamber scout image (left) with hashed line demonstrating orientation of ensuing short‐axis slice obtained using double inversion recovery with TE=80 (right). The region of high T2 signal intensity (white arrows) reflects the edematous myocardium defined as AAR. B, Representative LGE sequences measured at 48 hours postreperfusion from pigs treated with either DZF Scr or DZF demonstrating LGE (black arrows) and microvascular obstruction (white arrow). C, Quantification of myocardial salvage demonstrating percentage of (AAR–LGE)/AAR% (salvage index). Data represent analysis from n=7 hearts per DNAzyme‐treated group. †Significantly different compared with DZF Scr group (P≤0.05). D, Comparative scatterplot demonstrating the effect of treatment on infarct size (LGE) as a function of AAR between DZF Scr (open circles/dotted line) and DZF (open squares/filled line)‐treated groups. CMR indicates cardiac magnetic resonance; I/R, ischemia–reperfusion; TE, echo time; AAR, area at risk; LGE, late gadolinium enhancement; DZF Scr, scrambled DNAzyme; DZF, functional DNAzyme.
Importantly, the improved myocardial salvage in the DZF‐treated group was associated with a significant increase in LV ejection fraction and cardiac output as assessed by CMR (Table 2). Assessment of LV function using echocardiography revealed results consistent with the CMR data (Figure 7). All pigs experienced a significant reduction in LV systolic function during the ischemic period, as expected. Administration of DZF via the intracoronary route at the time of reperfusion resulted in improvement in LV function when measured at 48 hours postreperfusion, compared with administration of the DZF Scr control.
Figure 7.
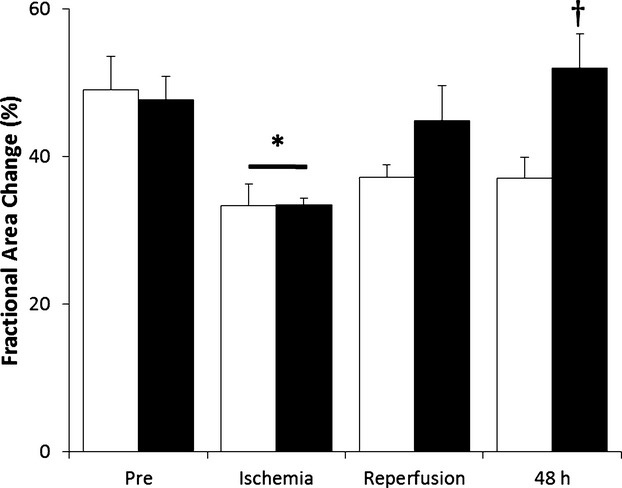
Heart LV function following I/R injury. Echocardiography was performed on pigs receiving DZF Scr (white bars) or DZF (black bars) and images acquired in the parasternal short axis at mid papillary level prior to ischemia (Pre), during ischemia, at reperfusion, and at 48 hours postreperfusion. LV systolic function was estimated by measuring the fractional area change. Data represent measurements from 7 pigs per treatment group. *Significantly different compared with respective measurements recorded prior to ischemia; †significantly different compared with DZF Scr group (P≤0.05). LV indicates left ventricular; I/R, ischemia–reperfusion; DZF Scr, scrambled DNAzyme; DZF, functional DNAzyme.
Discussion
The present study demonstrated effective inhibition of Egr‐1 through delivery of a DNAzyme via the intracoronary route at the time of reperfusion in a large‐animal model of I/R injury. We found that, in association with reduced inflammation and oxidative damage at the cellular level, DZF resulted in a substantial increase in salvaged myocardium and improved LV function. The restoration of blood flow and subsequent reoxygenation of ischemic tissue cause the generation of ROS that, in turn, stimulates the release of cytokines and expression of adhesion molecules on the surface of damaged cells within the reperfused tissue.8 This forms a key part of the potentially injurious processes of myocardial inflammation and apoptosis. The transcription factor Egr‐1 acts as both a trigger and a convergence point for many signaling cascades and is implicated in the regulation and expression of a variety of genes involved in seemingly disparate physiological mechanisms, including cellular growth, differentiation, inflammation, and apoptosis.1 Egr‐1–null mice have markedly less activation of mediators of vascular injury and improved survival compared with wild‐type mice exposed to pulmonary I/R. Following I/R injury, Egr‐1 mediates the expression of ICAM‐1, VCAM‐1, and tissue factor, within the myocardium and associated vasculature.9 An increase in the expression of ICAM‐1 acts as a leukocyte chemoattractant, whereas increased cellular VCAM‐1 expression aids in leukocyte migration from the vasculature to sites of tissue damage. Moreover, tissue factor initiates the blood coagulation cascade, causing increased accumulation of fibrin, which is further exacerbated by the Egr‐1–controlled induction of PAI‐1, which acts to suppress fibrinolysis.10 Consequently, through the induction of these key processes, Egr‐1 holds a critical role in myocardial damage occurring as a result of I/R injury.
Activation of the complement cascade is also recognized as a key effector mechanism in mediating the severity of myocardial injury following I/R.11–12 C3 plays a central role in the activation of the complement system, ultimately inducing the deposition of the membrane attack complex on damaged cells. Depending on the severity of membrane attack complex deposition, affected cells are either lysed directly or, as is the case for endothelial cells, activated to promote neutrophil chemotaxis through the secretion of IL‐8.13 Previously, it has been shown that Egr‐1 plays a pivotal role in the sustained expression of IL‐8.5 Here, we have demonstrated that myocardial I/R injury results in increases in C3 and C3aR expression that are coupled with increases in tissue expression of IL‐8. Furthermore, silencing of Egr‐1 resulted in a reduction of C3, C3aR, and subsequent IL‐8 expression that was concurrent with the decreased recruitment of infiltrating neutrophils within the border zone of the infarcted heart tissue. Additionally, Egr‐1 regulation of ICAM‐1 expression has been shown to mediate the induction in IL‐1β 9 and tumor necrosis factor‐α 14 in models of I/R injury, with expression of these proinflammatory mediators further stimulating the production of IL‐8 from endothelial cells.11 Taken together, these data support a role for Egr‐1 within the inflammatory mechanisms evident in this model.
A recent study using a pig model of acute myocardial I/R injury similar to our own concluded that apoptosis is a reperfusion‐triggered phenomenon, with evident increases in proinflammatory cytokines likely contributing to the stimulation of caspase‐dependent apoptotic cell death.15 It has also been previously demonstrated that activity of the pro‐apoptotic molecule p53 is increased following I/R injury, resulting directly in an increase in the hypoxia‐induced apoptosis of cardiac myocytes.16 Egr‐1 upregulates synthesis of p53 mRNA and resultant protein by binding directly to the 5′‐CGCCTACGC‐3′ sequence in the promoter region of the p53 tumor suppressor gene, 17 supporting a role for Egr‐1 in the induction of the p53 apoptotic pathway. Similarly, the PTEN gene contains a functional 5′‐GCGGCGGCG‐3′ Egr‐1 binding site within the 5′‐untranslated region, and activation of Egr‐1 results in the upregulation of PTEN and subsequent increase in apoptosis 18 with inhibition of Egr‐1 conversely shown to abolish PTEN expression and rescue cells from apoptosis.19 In addition, PTEN is also activated by p53, indicating that Egr‐1 may not only target PTEN directly but also indirectly through the subsequent binding of p53 within the PTEN promoter.20 In this study, we have shown that inhibition of Egr‐1 at the time of reperfusion following myocardial ischemia effectively reduces the proapoptotic processes of p53 and PTEN, as well as the mechanistic activity of caspase 3, resulting in significantly decreased apoptotic cells within the infarct border zone of the heart tissue following I/R injury.
The culmination of the aforementioned protection against the detrimental molecular and cellular events afforded through Egr‐1–targeted DNAzyme delivery is an overall increase in parameters indicative of improved heart function. As measured with both CMR and echocardiography, DZF administration via the intracoronary route at the time of reperfusion resulted in significant improvement in the percentage of salvaged myocardium and myocardial function. Taken together, these data demonstrate for the first time that the targeting of Egr‐1 through the use of DNAzymes delivered via a clinically relevant route and time following myocardial infarction is able to rescue heart tissue and protect heart function from the damaging effects of I/R injury.
Sources of Funding
The research detailed here was funded by a National Health and Medical Research Council (NHMRC) grant (APP632551) and a North Shore Heart Research Foundation grant awarded to R.B.
Disclosures
None.
Acknowledgments
The authors wish to thank Rafael Dye for anesthetic services, Joseph Alcorace for technical assistance, Janos Tomka for technical advice, the radiologists and radiographers at Royal North Shore Hospital, Sydney, for their assistance, and the staff at the Kearns Facility for animal husbandry.
References
- 1.Bhindi R, Fahmy RG, McMahon AC, Khachigian LM, Lowe HC. Intracoronary delivery of DNAzymes targeting human EGR‐1 reduces infarct size following myocardial ischaemia reperfusion. J Pathol. 2012; 227:157-164 [DOI] [PubMed] [Google Scholar]
- 2.Khachigian LM. Early growth response‐1 in cardiovascular pathobiology. Circ Res. 2006; 98:186-191 [DOI] [PubMed] [Google Scholar]
- 3.Xiang G, Schuster MD, Seki T, Witkowski P, Eshghi S, Itescu S. Downregulated expression of plasminogen activator inhibitor‐1 augments myocardial neovascularization and reduces cardiomyocyte apoptosis after acute myocardial infarction. J Am Coll Cardiol. 2005; 46:536-541 [DOI] [PubMed] [Google Scholar]
- 4.Xiang G, Seki T, Schuster MD, Witkowski P, Boyle AJ, See F, Martens TP, Kocher A, Sondermeijer H, Krum H, Itescu S. Catalytic degradation of vitamin D up‐regulated protein 1 mRNA enhances cardiomyocyte survival and prevents left ventricular remodeling after myocardial ischemia. J Biol Chem. 2005; 280:39394-39402 [DOI] [PubMed] [Google Scholar]
- 5.Hoffmann E, Ashouri J, Wolter S, Doerrie A, Dittrich‐Breiholz O, Schneider H, Wagner EF, Troppmair J, Mackman N, Kracht M. Transcriptional regulation of EGR‐1 by the interleukin‐1‐JNK‐MKK7‐c‐Jun pathway. J Biol Chem. 2008; 283:12120-12128 [DOI] [PubMed] [Google Scholar]
- 6.Lonborg J, Vejlstrup N, Mathiasen AB, Thomsen C, Jensen JS, Engstrom T. Myocardial area at risk and salvage measured by T2‐weighted cardiovascular magnetic resonance: reproducibility and comparison of two T2‐weighted protocols. J Cardiovasc Magn Reson. 2011; 13:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nozaki S, DeMaria AN, Helmer GA, Hammond HK. Detection of regional left ventricular dysfunction in early pacing‐induced heart failure using ultrasonic integrated backscatter. Circulation. 1995; 92:2676-2682 [DOI] [PubMed] [Google Scholar]
- 8.Diepenhorst GM, van Gulik TM, Hack CE. Complement‐mediated ischemia‐reperfusion injury: lessons learned from animal and clinical studies. Ann Surg. 2009; 249:889-899 [DOI] [PubMed] [Google Scholar]
- 9.Yan SF, Fujita T, Lu J, Okada K, Shan Zou Y, Mackman N, Pinsky DJ, Stern DM. Egr‐1, a master switch coordinating upregulation of divergent gene families underlying ischemic stress. Nat Med. 2000; 6:1355-1361 [DOI] [PubMed] [Google Scholar]
- 10.Okada M, Fujita T, Sakaguchi T, Olson KE, Collins T, Stern DM, Yan SF, Pinsky DJ. Extinguishing Egr‐1‐dependent inflammatory and thrombotic cascades after lung transplantation. FASEB J. 2001; 15:2757-2759 [DOI] [PubMed] [Google Scholar]
- 11.Walsh MC, Bourcier T, Takahashi K, Shi L, Busche MN, Rother RP, Solomon SD, Ezekowitz RA, Stahl GL. Mannose‐binding lectin is a regulator of inflammation that accompanies myocardial ischemia and reperfusion injury. J Immunol. 2005; 175:541-546 [DOI] [PubMed] [Google Scholar]
- 12.Zhang H, Qin G, Liang G, Li J, Chiu I, Barrington RA, Lui D. Suppression of complement regulatory protein C1 inhibitor in vascular endothelial activation by inhibiting vascular cell adhesion molecule‐1 action. Biochem Biophys Res Commun. 2007; 358:1120-1127 [DOI] [PubMed] [Google Scholar]
- 13.Kilgore KS, Flory CM, Miller BF, Evans VM, Warren JS. The membrane attack complex of complement induces interleukin‐8 and monocyte chemoattractant protein‐1 secretion from human umbilical vein endothelial cells. Am J Pathol. 1996; 149:953-961 [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou Y, Zhang Y, Gao F, Guo F, Wnag J, Cai W, Chen Y, Zheng J, Shi G. N‐n‐butyl haloperidol iodide protects cardiac microvascular endothelial cells from hypoxia/reoxygenation injury by down‐regulating Egr‐1 expression. Cell Physiol Biochem. 2010; 26:839-848 [DOI] [PubMed] [Google Scholar]
- 15.Vilahur G, Juan‐Babot O, Pena E, Onate B, Casani L, Badimon L. Molecular and cellular mechanisms involved in cardiac remodeling after acute myocardial infarction. J Mol Cell Cardiol. 2011; 50:522-533 [DOI] [PubMed] [Google Scholar]
- 16.Long X, Boluyt MO, Hipolito ML, Lundberg MS, Zheng JS, O'Neill L, Cirielli C, Lakatta EG, Crow MT. p53 and the hypoxia‐induced apoptosis of cultured neonatal rat cardiac myocytes. J Clin Invest. 1997; 99:2635-2643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nair P, Muthukkumar S, Sells SF, Han SS, Sukhatme VP, Rangnekar VM. Early growth response‐1‐dependent apoptosis is mediated by p53. J Biol Chem. 1997; 272:20131-20138 [DOI] [PubMed] [Google Scholar]
- 18.Virolle T, Adamson ED, Baron V, Birle D, Mercola D, Mustelin T, de Belle I. The Egr‐1 transcription factor directly activates PTEN during irradiation‐induced signalling. Nat Cell Biol. 2001; 3:1124-1128 [DOI] [PubMed] [Google Scholar]
- 19.Okamura H, Yoshida K, Morimoto H, Haneji T. PTEN expression elicited by EGR‐1 transcription factor in calyculin A‐induced apoptotic cells. J Cell Biochem. 2005; 94:117-125 [DOI] [PubMed] [Google Scholar]
- 20.Thiel G, Cibelli G. Regulation of life and death by the zinc finger transcription factor Egr‐1. J Cell Physiol. 2002; 193:287-292 [DOI] [PubMed] [Google Scholar]