

Abstract
Background
Peripheral arterial disease (PAD) is characterized by myofiber degeneration and loss of function in muscles of the lower limbs. Human enterovirus (HEV) infection has been implicated in the pathogenesis of a number of muscle diseases. However, its association with PAD has not been studied. In this study, we tested the hypothesis that infectious HEV is present in skeletal muscle of patients with PAD and is associated with severity of disease.
Methods and Results
Gastrocnemius biopsies from 37 patients with PAD and 14 controls were examined for the presence of HEV RNA, viral capsid protein, viral RNA copy number, and viral infectivity. HEV RNA was detected in 54% of the biopsies from patients with PAD but was not detected in muscle biopsies from control patients. This difference in prevalence among PAD and control patients was significant at P<0.001. Viral RNA copy numbers were increased significantly at the later stages of disease; Fontaine Stage IV (105.50 copies/mg muscle wet weight, at P<0.005) and Stage III (104.87 copies/mg, at P<0.010) compared to Stage II (102.50 copies/mg). Viral replication was confirmed by the presence of the negative‐strand of viral RNA in all specimens positive for HEV RNA. Cultures of HeLa and human skeletal muscle cells treated with muscle homogenates showed HEV replication and the presence of HEV capsid protein.
Conclusion
Our data identified infectious HEV in the gastrocnemius of PAD patients but not in controls. Viral copy number and prevalence of infection were higher in the later stages of disease. Our data point to the need for further studies to determine the contribution of HEV infection to the pathophysiology of PAD.
Keywords: coxsackievirus, human enterovirus, peripheral arterial disease, skeletal muscle degeneration
Introduction
Peripheral arterial disease (PAD) affects over 8.5 million, mostly elderly, individuals in the Unites States.1 According to the Fontaine classification system,2 clinical stages of PAD are comprised of Stage I (asymptomatic PAD), Stage II (claudication—the most common clinical manifestation of symptomatic PAD), Stage III (ischemic rest pain), and Stage IV (tissue loss). Work from our and other laboratories has demonstrated a myopathy in the ischemic limbs of patients with PAD.3–5
Infection with the human enterovirus (HEV) has been implicated in the pathogenesis of a number of skeletal and cardiac muscle diseases.6–12 HEV RNA or its structural protein VP1 has been localized in the cytoplasm of myofibers in the skeletal muscle of patients with idiopathic dilated cardiomyopathy,12 chronic fatigue syndrome,6 chronic inflammatory muscle diseases,7 fibromyalgia,7 and in the myocardium of patients with myocarditis,8–9 dilated cardiomyopathy,8–9,11 or idiopathic dilated cardiomyopathy.10 These studies support an association between enterovirus and diseases of the skeletal muscle and demonstrate that the prevalence of enterovirus is highest in skeletal muscle with more pronounced myopathic changes such as polymyositis, dermatomyositis, granulomatous myositis, and inclusions body myositis.7 The myopathy of the ischemic limbs of patients with PAD has biochemical abnormalities that are similar to those seen in patients with fibromyalgia. Additionally, in advanced stages of PAD the myopathy demonstrates ultrastructural changes that can be similar, and in many cases worse, than those seen in chronic inflammatory myopathies.3,5 The presence of these similarities between PAD and other diseases of skeletal muscles, raises the question of whether HEV is present in the leg muscles of PAD patients and whether that is associated with the pathophysiology of the disease. In this study we tested the hypothesis that infectious HEV is present in skeletal muscle of patients with PAD and correlates with a stage of disease. We analyzed biopsy specimens of the gastrocnemius from PAD and control patients for HEV RNA, viral capsid protein, viral RNA copy number, and viral infectivity.
Methods
Muscle and Serum Specimens
The institutional review board at the University of Nebraska Medical Center (UNMC) and the VA Nebraska and Western Iowa Medical Center approved the experimental protocols and consent forms. After informed consent, muscle and serum samples were obtained from PAD and control patients. Gastrocnemius samples weighing ≈250 mg were obtained from the anteromedial aspect of the muscle belly, 10 cm distal to the tibial tuberosity. All biopsies were obtained with a 6 mm Bergstrom needle and were frozen for future analysis.
PAD group
We recruited 37 patients undergoing lower extremity operations for symptomatic PAD (Table1). For every patient, the diagnosis of PAD was established on the basis of medical history, physical examination, significantly decreased ankle‐brachial index (ABI<0.9), and computerized or standard arteriography demonstrating stenosis and/or occlusions in the arteries supplying the lower extremities. PAD patients presenting with ischemic, nonhealing ulcers, and/or gangrene were categorized as stage IV PAD patients. PAD patients presenting with ischemic rest pain and no evidence of tissue loss were categorized as stage III PAD patients. Those presenting with intermittent claudication, who had no symptoms of ischemic rest pain and/or evidence of tissue loss were categorized as stage II PAD patients.
Table 1.
Demographics for Control and Peripheral Arterial Disease (PAD) Patients Recruited for this Study
| Control Patients | PAD Patients | |||||
|---|---|---|---|---|---|---|
| *Stage II | Stage III | Stage IV | Combined | P Value* | ||
| Number of patients | 14 | 11 | 9 | 17 | 37 | — |
| Gender male/female, % | 86/14 | 91/9 | 100/0 | 94/6 | 95/5 | 0.29 |
| Age, y* | 63.4±2.0 | 61.6±2.3 | 59.7±1.9 | 63.5±2.2 | 62.0±1.3 | 0.59 |
| Race, w/h/a* | 86/7/7 | 100/0/0 | 100/0/0 | 88/6/6 | 94/3/3 | 0.33 |
| Smoking, n/f/c* | 43/21/36 | 9/18/73 | 11/33/56 | 24/29/47 | 16/27/57 | 0.13 |
| CAD | 35.7% | 45.5% | 33.3% | 75.0% | 55.6% | 0.21 |
| Obesity | 28.6% | 27.3% | 44.4% | 17.7% | 27.0% | 0.92 |
| DM | 21.4% | 18.2% | 33.3% | 35.3% | 29.7% | 0.55 |
| Dyslipidemia | 50.0% | 63.6% | 66.7% | 58.8% | 62.2% | 0.53 |
| HTN | 57.1% | 81.8% | 66.7% | 82.4% | 78.4% | 0.17 |
| Family history | 21.4% | 9.1% | 22.2% | 17.6% | 16.2% | 0.69 |
| ABI* | 1.12±0.04 | 0.34±0.05 | 0.24±0.05 | 0.17±0.02 | 0.54±0.02 | <0.001 |
CAD indicates coronary artery disease; DM, diabetes mellitus; HTN, hypertension; ABI, ankle brachial index.
Fontaine Stage.
P values are for differences between Control and all PAD patients combined; Chi‐Square or Fisher's exact tests for categorical variables; Independent t Test for continuous variables.
Values are mean±SEM.
Percent of patients in each category; w, Caucasian; h, Hispanic; a, African American.
Percent of patients in each category; n, never; f, former; c, current.
Control group
We recruited 14 patients with normal blood flow to their lower limbs undergoing lower extremity operations for indications other than PAD (Table1). These patients had no history of PAD symptoms and all had normal lower extremity pulses at examination. All controls were selected to have normal ABIs at rest and after stress, and all led sedentary lifestyles.
Cells and Virus Stocks
HeLa cells (ATCC) were used for HEV propagation, HEV detection, and infectivity experiments.4,13–14 Primary human skeletal muscle (HSkm) cells, cultured in MyoTonic Growth Medium (Cook MyoSite), also were used to study virus infectivity. Control HEV strain, Coxsackievirus B1 (CVB1, ATCC) was propagated in HeLa cells, and its stocks were titrated by an assay based on the 50% tissue culture infective dose (TCID50).14 CVB1 was used for viral infectivity testing14 and subgroup‐genotyping.15
Muscle Homogenates and Cell Lysates
Muscle samples (~100 mg wet weight) were homogenized in tissue buffer (PBS/proteinase inhibitor) with a 2 mL‐Dounce homogenizer (Wheaton), and stored in aliquots at −84°C. Cell lysates were prepared in RIPA buffer (Thermo Scientific) and stored in aliquots at −84°C as described elsewhere. Muscle homogenates were frozen/thawed 3‐times and clarified by centrifugation. Tissue pellets were stored in RNAlater (Ambion) at −84°C for RNA isolation.
To determine virus infectivity,13 the homogenates prepared from all muscle biopsies or the supernatants of cell lysates were used to infect HeLa cell cultures. At 48 to 72 hours post inoculation (hpi) the treated cells were frozen/thawed and clarified by centrifugation, and supernatant was collected (VS1; viral supernatant “1”). Next, fresh HeLa cells were treated with a 1:2 dilution of the VS1. At 48 to 72 hpi, supernatants of the treated HeLa cells (VS2; viral supernatant “2”) were prepared as described for VS1. Treated cell cultures were inspected for cytopathic effect (CPE; an indication of HEV replication) by light microscopy. For the detection of CPE, HeLa cells were fixed and stained with crystal violet.
Prior to analysis, supernatants of muscle homogenates or cell lysates were treated with RNase A/T cocktail (Ambion) to digest exogenous free RNA in the sample, leaving encapsidated viral RNA intact, filtered through 0.45‐μm filters to remove fine cell debris, and stored in aliquots at −84°C.
Analysis of Muscle Homogenates, Sera, and Cell Lysates by RT‐PCR, RT‐qPCR and Western Blot
RT‐PCR analyses were performed to detect HEV RNA in the muscle homogenates, sera, or cell lysates. A primer pair, EV1 (5′‐CGGCCCCTGAATGCGGC‐3′) and EV2 (5′‐CACCGGATGGCCAATCCA‐3′) was used for HEV RNA detection15 as these primers are specific for sequences located within a highly conserved region of the 5′‐NTR of HEV. Other primers used in this study are presented in Table S1. The numbering system is based on the published sequence for CVB1 (GeneBank Accession No. M16560.1).
By utilizing real‐time quantitative PCR (RT‐qPCR),13–14 positive‐sense viral RNA copy numbers present in muscle homogenates or cell lysates were determined. A standard curve for the determination of viral RNA copy numbers was developed with a PCR‐fragment (193 bp long), amplified by RT‐PCR using a primer pair EV1/EV2, and cloned into a pGEM‐T easy vector (Promega) (Figure S3A).
For the detection of the negative‐strand of viral RNA,14 total RNAs were isolated from samples that were not treated with RNAse and then mixed with biotinylated strand‐specific primer EV1 (for the negative‐strand). The annealing was carried out in an ABI 2720 thermal cycler (Applied Biosystems) by incubating the RNA/Primer mixture at 75°C for 5 minutes followed by gradual cooling to room temperature. Viral RNAs annealed with the biotinylated primer were mixed with Streptavidin MagneSphere Paramagnetic particles (Promega) and separated using the MagneSphere Magnetic Separation Stand (Promega) per the manufacture's instruction. The biotinylated primer/RNA complex was then washed with nuclease‐free water. The separation/washing steps were performed 3 to 4 times. After suspending the complex, RT‐qPCR was carried out as described above. Synthesized cDNA was then treated with RNase H to remove the RNA from the cDNA/RNA complex. PCR with a primer pair EV1/EVU2 (5′‐CCCCTGAATGCGGCTAAT‐3′)15 was carried out to amplify the negative‐strand viral RNA.
HEV capsid protein VP1 was detected by Western blot with the primary monoclonal HEV VP1 antibody (5‐D8/1, Dako).14,16
Statistical Analysis
Differences between demographic values of controls and all PAD patients combined were tested by Chi‐square analysis for categorical variables and an independent t test for continuous variables. Fisher's exact tests were used to analyze categorical variables yielding small cell counts by Chi‐square analysis, and to compare the HEV prevalence in control and PAD‐stage groups. Kruskal–Wallis nonparametric statistics were used to test the effect of PAD stage on HEV RNA copy number. All analyses were completed using SAS statistical software version 9.3 (SAS Institute Inc), with P<0.05 considered significant.
For a more extensive description of methods, please refer to the “Supplemental Materials.”
Results
Study and Control Groups
Thirty‐seven patients with PAD and undergoing lower extremity operations (28 bypass procedures and 9 amputations) were recruited. Eleven were diagnosed as Fontaine Stage II (ABI; 0.34±0.05, Mean±SEM), 9 were diagnosed as Fontaine Stage III (ABI; 0.24±0.05), and 17 were diagnosed as Fontaine Stage IV (ABI; 0.17±0.02). For the control group, 14 patients undergoing lower extremity operations for the treatment of superficial varicose veins were recruited (ABI; 1.15±0.03). These patients had no history of PAD symptoms and all had absence of PAD based on physical exam and noninvasive testing. The demographics of PAD and control subjects are presented in Table 1. No significant differences were found between the control and the PAD groups except for ABI (P<0.001).
HEV RNA is Present in Biopsies of PAD Gastrocnemius
Total RNAs of the muscle biopsies were analyzed for the presence of HEV RNA by RT‐PCR (Figure 1A), using the HEV detection specific primer pair, EV1 and EV2 (EV1/EV2, Figure 1C). RT‐PCR analysis showed that 20 of 37 PAD samples (54%) had detectable HEV RNA while HEV RNA was not detected in the control group. The percentage of muscle samples positive for HEV RNA varied with disease stage (Table 2); 36% (4/11) for Stage II, 66% (6/9) for Stage III, and 58% (10/17) for Stage IV patients. Negative‐strand viral RNA, indicative of viral replication, was found in those muscle specimens that had detectable HEV RNA but not in specimens that lacked detectable HEV RNA (Figure 1B). In addition, HEV RNA was not detected in any of the serum samples from controls or PAD patients (data not shown). The nucleotide sequence of HEV RNA showed sequence variation, indicating no cross‐contamination (Figure S1). Furthermore, utilizing RT‐PCR with HEV‐specific subgroup primer pairs, HEV RNA detected in the muscle samples was found to be a member of the HEV‐B subgroup (Figure S2), which are currently under serotyping.
Figure 1.
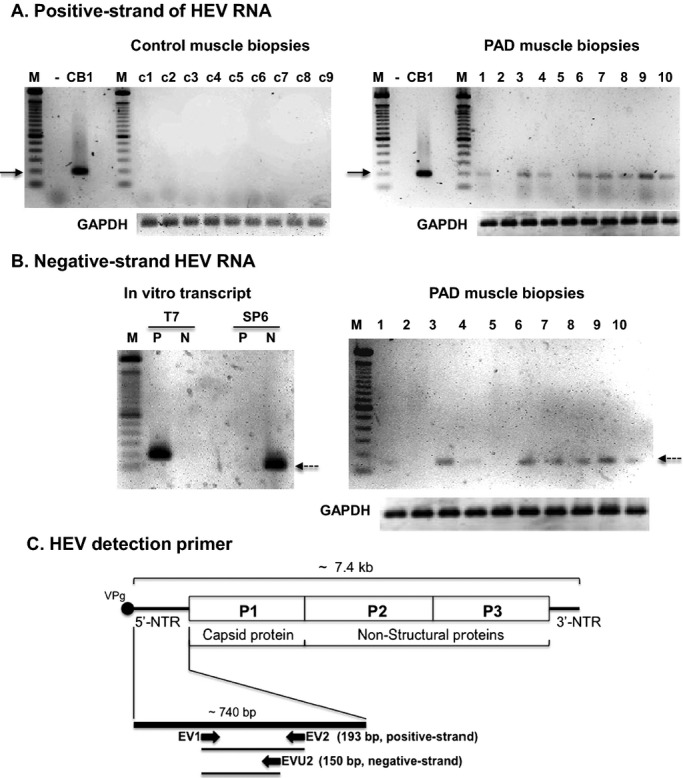
HEV RNA is present in muscle biopsies of PAD gastrocnemius. Total RNA from muscle homogenates of 9 controls and 10 PAD patients comprising those with disease Stage II (samples 2, 5, 7, and 9), Stage III (samples 1, 4, and 10) and Stage IV (samples 3, 6, and 8) were analyzed by RT‐PCR (T7 RNA polymerase) for HEV RNA (positive‐strand genomic RNA) (A). All controls had no detectable HEV RNA (left panel) while 8 of the 10 PAD specimens were positive for HEV RNA (right panel). RNAs of the 10 PAD patients also were analyzed by RT‐PCR (SP6 RNA polymerase) for negative‐strand viral RNA (indicating viral replication) (B). All PAD specimens that were positive for HEV RNA (specimens 1, 3, 4, 6 to 10) had detectable negative‐strand RNA and those PAD specimens negative for HEV RNA (2 and 5) had no detectable negative‐strand RNA. For a control, T7 RNA polymerase‐transcribed positive‐strand HEV RNA (P) and SP6 RNA polymerase‐transcribed negative‐strand viral RNA (SP6) were synthesized from a linearized pGEM‐T harboring CVB1EV1/2 fragment (data not shown). A 100 bp ladder is seen in lane “M”. GAPDH was used as an internal housekeeping gene. Solid arrows indicate 200 bp DNA and dashed arrows indicate negative‐strand viral RNA. (C), HEV RNA detection primers, EV1, EV2 and EVU2 are shown. The EV1/EV2 primer pair was used for detection of positive‐strand HEV RNA (193 bp). The EV1/EVU2 primer pair was used for detection of negative‐strand viral RNA (150 bp). HEV indicates human enterovirus; PAD, peripheral arterial disease; GAPDH, glyceraldehyde 3‐phosphate dehydrogenase; RT‐PCR, reverse‐transcription polymerase chain reaction; VP1, viral capsid protein.
Table 2.
Prevalence of HEV Infection in PAD Gastrocnemius and Viral RNA Copy Number in the HEV Positive Specimens
| Control | Fontaine Stage | P Value | |||
|---|---|---|---|---|---|
| Stage II | Stage III | Stage IV | |||
| Total number | 14 | 11 | 9 | 17 | — |
| HEV positive*, count/% | 0/0% | 4/36% | 6/66% | 10/58% | 0.001* |
| HEV RNA copy number, log10/mg | N/A | 2.5 (2.25 to 3.75)* | 4.87 (3.25 to 5.75) | 5.5 (4.75 to 6.25) | 0.002* |
HEV indicates human enterovirus; PAD, peripheral arterial disease.
Detection of HEV RNA by RT‐PCR and VP1 by Western blot.
Fisher's exact tests: PAD patients grouped by Fontaine Stage differed significantly from the controls: Stage II (P=0.026), Stage III (P=0.001) and Stage IV (P<0.001).
HEV RNA copy number expressed as median (ranges).
Kruskal–Wallis test; Stage II differed significantly from Stage III (P=0.010) and Stage IV P<0.005). A P value of 0.016 was set as significant in these post hoc comparisons.
HEV in PAD Muscle is Infectious but Lacks Cytolytic Activity
Next, we determined whether the HEV RNA, detected in the muscle biopsies by RT‐PCR, represents infectious virus. HeLa cells (an indicator cell line for HEV replication) were treated with homogenates of all muscle biopsies. Cell lysates VS1 (HeLa cells treated with muscle homogenates) or VS2 (HeLa cells treated with VS1) appeared to have no CPE (upper panels in Figure 2A and 2B), showing no difference from untreated HeLa cells (TC, Figure 2C). In contrast, cell lysis was observed within 24 hours after treatment of HeLa cells with the control virus, CVB1, or supernatants from the cells infected with CVB1 (Figure 2C). These data led us to investigate whether HEV RNA was still present in cell cultures treated with muscle homogenates.
Figure 2.
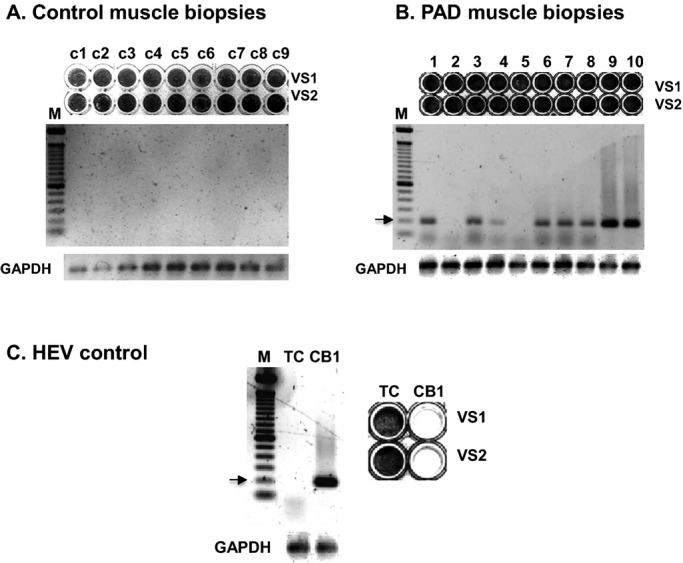
HEV RNA was detected in the absence of a cytopathic effect (CPE). VS1 and VS2 prepared from HeLa cells treated with muscle homogenates of controls (c1 to c9) (A) and PAD patients (B) with Stage II (2, 5, 7, and 9), Stage III (1, 4, and 10) and Stage IV (3, 6, and 8) disease did not produce a CPE (upper panels) and yet HEV RNA was detected in 8 of the 10 VS2 preparations by RT‐PCR (lower panels). (C) CPE was observed in cultures infected with the cytolytic CVB1 virus, as a positive control. CPE was not observed in uninfected cell cultures serving as negative controls (TC). For the detection of CPE, HeLa cell cultures were fixed and stained with crystal violet. A 100 bp DNA ladder (lane M) was used as a sizing reference. The arrows indicate 200 bp DNA. HEV indicates human enterovirus; VS, viral supernatant; PAD, peripheral arterial disease; RT‐PCR, reverse‐transcription polymerase chain reaction; CVB1, coxsackievirus B1.
Viral RNA was readily detectable in both VS1 (data not shown) and VS2 (lower panel in Figure 2A and 2B), derived from PAD muscle homogenates previously determined as HEV RNA positive (Figure 1). However, no viral RNA was detected in VS1 or VS2, derived from PAD muscle homogenates determined as HEV RNA‐negative. As previously noted in the methods section (also in Supplemental Materials), VS1 and VS2 were treated with RNase A/T1 cocktail to remove free RNA before analysis. Therefore, the detection of HEV RNA in VS1 and VS2 cell lysates indicates the presence of infectious encapsidated HEV rather than free, viral RNA.
Subsequently, HEV‐positive muscle homogenates or cell lysates were analyzed for viral capsid protein VP1 by Western Blot analysis. Viral VP1 protein was detected at varying levels in the homogenates with nonspecific labeling of host proteins, possibly due to cross‐reactivity of the anti‐VP1 antibody (data not shown). On the other hand, HEV VP1 protein was readily detected in VS2 cell lysates as a single band, in the absence of the nonspecific labeling (Figure 3). Also, HEV RNA and VP1 were readily detected in primary human skeletal muscle cells treated with PAD muscle homogenates positive for HEV RNA (data not shown). Hence, these data, together with the data from RT‐PCR, strongly suggest that infectious HEV is present in skeletal muscles of PAD patients. Such noncytolytic HEV has been reported for cardiac muscle diseases.14,17
Figure 3.
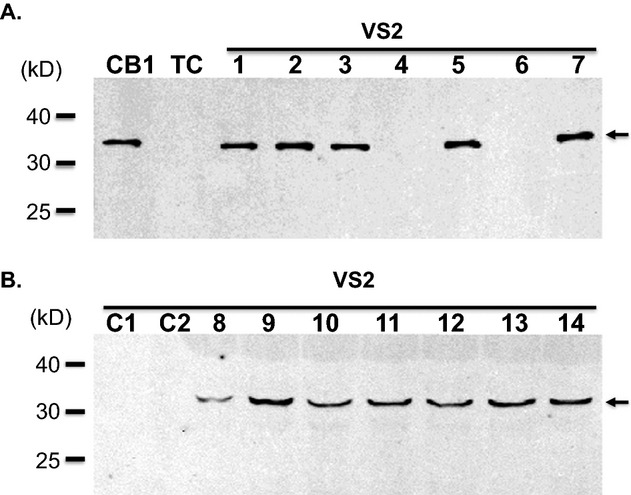
Viral capsid protein (VP) was detected by Western blot analysis. VP1 was detected in VS2 from HeLa cells treated with HEV RNA‐positive muscle homogenates from PAD patients (lanes 1 to 3, 5 and 7 to 14) but was not detected in VS2 from HeLa cells treated with HEV RNA‐negative homogenates from PAD patients (lanes 4 and 6) or in VS2 from HeLa Cells treated with control homogenates (A, B). Arrows indicate VP1. CVB1 infected HeLa cell lysate (positive control), lane “CB1”; Uninfected HeLa cell lysate (negative control), lane “TC.” Patient samples: Controls, (lanes C1 and C2); PAD Stage II, (lanes 4 and 11 to 13); PAD Stage III, (lanes 2 and 8 to 10); PAD Stage IV, (lanes 1, 3, 5 to 7 and 14). VS indicates viral supernatant; HEV, human enterovirus; PAD, peripheral arterial disease; CVB1, coxsackievirus B1.
HEV‐RNA Copy Numbers were Higher in the Later Stages of Disease
HEV found in PAD muscles does not induce CPE in HeLa cells (upper panels in Figure 2A and 2B), consequently the virus could not be quantified on the basis of plaque forming or TCID50 units. Instead, HEV RNA copy number in PAD muscle was measured by RT‐qPCR (Figure 4A). Viral RNA copy numbers varied with disease stage (Table 2). Overall, higher copy numbers were observed in Stage III and IV (median copy number 4.87 and 5.50 [log10 copy number/mg muscle wet weight], respectively), compared to Stage II (median copy number 2.50 [log10 copy number/mg wet weight]).
Figure 4.
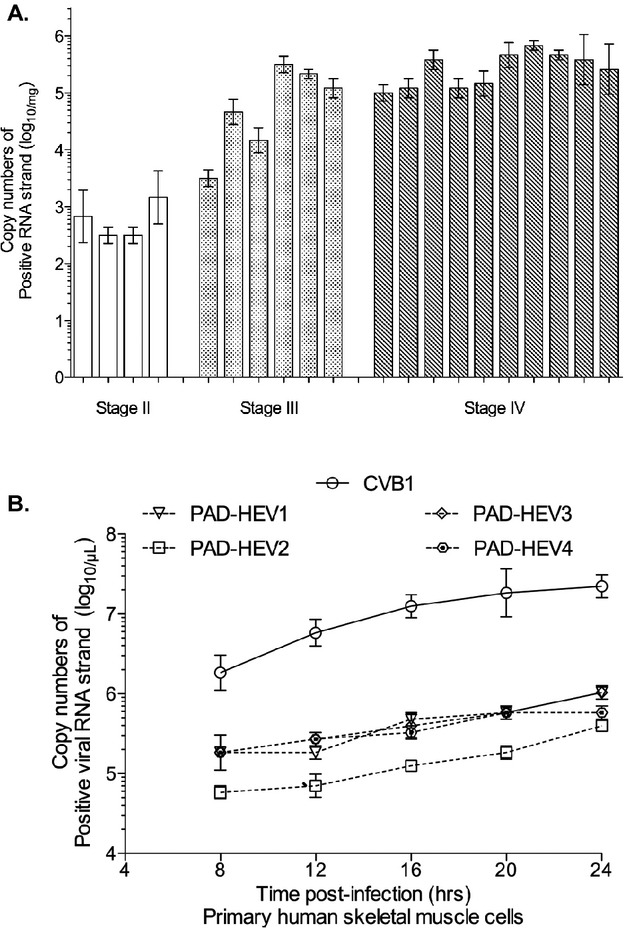
Viral RNA copy numbers were determined by RT‐qPCR. (A) Viral RNA copy numbers were compared among the Stage groups. (B) Single‐step growth curves of PAD‐HEV were examined at the various time points in the primary HSkm cells. Viral RNA copy numbers were determined using in vitro transcripts (see more detail in Supplemental Materials). PAD‐HEV1 (Stage III), PAD‐HEV2 (Stage II), PAD‐HEV3 and 4 (Stage IV). RT‐qPCR indicates reverse‐transcription real‐time quantitative polymerase chain reaction; PAD, peripheral arterial disease; HEV, human enterovirus; HSkm, primary human skeletal muscle; CVB1, coxsackievirus B1.
PAD Muscle HEV Replicates in Vitro in Primary Human Skeletal Muscle Cells and in Situ
Replication of HEV present in PAD muscle was analyzed in a single step growth curve (Figure 4B).13–14 Primary HSkm cells were treated either with supernatants of PAD muscle that were positive for HEV (PAD‐HEV) or with control virus CVB1. No lysis (CPE) was observed in the HSkm cell cultures infected with PAD‐HEV or CVB1 throughout the experimental period extending 24 hpi. Lysis was evident at 20 hpi in HeLa cell cultures infected with CVB1 (data not shown). Consequently, PAD‐HEV could not be quantified on the basis of plaque forming or TCID50 units. Instead, the copy number of HEV RNA at various time points was determined by RT‐qPCR. Overall the RNA copy numbers of both PAD‐HEV and CVB1 were lower in HSkm cells compared to their numbers in HeLa cells (Figure 4B). By 24 hpi, the RNA copy number of PAD‐HEV in the HSKm cell cultures was about 2 log‐fold lower than that of CVB1. Although PAD‐HEV generated fewer copy numbers and replicated slower than CVB1, it nevertheless replicated efficiently in primary HSKm cells. These data indicate persistent replication of PAD‐HEV in skeletal muscle of patients with PAD.
Discussion
Persistent HEV infections have been implicated in the pathogenesis of a number of disorders of skeletal and cardiac muscle.6–12 Here we present, for the first time, data demonstrating persistent HEV in the leg muscles of PAD patients. HEV was present in 54% of skeletal muscle samples from PAD patients, but was not detected in the muscles of healthy controls. HEV detected in PAD muscle both replicates and is infectious. These findings are compelling and raise the possibility that HEV infection is associated with initiation and/or severity of PAD. Although our study design does not permit a determination of the mechanism by which HEV infection of PAD skeletal muscles may lead to myopathy, a review of the literature identified 3 likely pathways. First, infectious HEV may produce myofiber damage by competing with native mRNA for ribosomal eIF4E, a translation initiation factor that directs mRNAs to the ribosomal start site, thus suppressing synthesis of native proteins.18–20 Second, viral infection may cause an accumulation of unfolded proteins in the endoplasmic reticulum (ER), interfering with normal ER protein assembly and trafficking, a condition termed ER stress. Unresolved ER stress caused by persistent viral infection will lead to cell death.21–22 Finally, myofiber infection by HEV may lead to the release of skeletal muscle antigens that can induce an autoimmune response which may further damage the skeletal muscles.12 These mechanisms, based on infection of myofibers per se, are supported by the presence of virus‐like particles in the sarcoplasm of patients with idiopathic dilated cardiomyopathy.12 In regard to PAD, it will be necessary to determine (eg, by in situ hybridization or viral capsid labeling) whether the virus is present in myofibers of infected muscle. However, it is possible that HEV simply coexists in patients with PAD and is not involved in the pathogenesis of PAD.
The absence of detectable virus in serum and the nonlytic phenotype of HEV in PAD gastrocnemius are consistent with chronic, persistent infection of PAD skeletal muscle. In patients with sporadic idiopathic dilated cardiomyopathy, skeletal muscle hosts persistent enteroviral infection.12 Consequently, myocardial damage may be caused by recurrent heart infection by virus harbored in skeletal muscle or by enterovirus‐dependent myopathy that releases antigens shared by skeletal muscle and myocardium, inducing an autoimmune response against the heart. In another report,23 the investigators concluded from their study of persistent, limited heart infection with enterovirus that viral RNA persisted in the myocardium but was not detected in peripheral blood mononuclear cells. The authors point out that attempts to isolate virus from blood often fail because enterovirus viremias are short in duration and occur early in infection. Thus, the characteristics of persistent versus acute enterovirus infection include the presence of virus in skeletal muscle as host during persistent infection and the absence of a viremia (seen during acute infection). Spread of persistent enterovirus does not require a lytic phase that can produce viremia. Nonlytic virus may be transmitted to other sites of infection by exploiting microvesiculation, a process by which cells produce and release microvesicles that mediate cell‐to‐cell communication.24 This process can be induced by many cellular events (eg, cell death, hypoxia, stress, and viral infection) and would permit direct transmission of viral particles from infected cells to nearby uninfected cells (eg, myofiber‐to‐myofiber, or to more distant uninfected cells) avoiding release of virions into the extracellular environment. Fiber‐to‐fiber transmission of microvesiculated virions may favor skeletal muscle as depot of a slowly expanding population of viral particles. Our study design does not permit determination of the clinical and functional impairment produced by persistent viral infection in the gastrocnemius of PAD patients. However, in a recent study of patients with prior viral myocarditis, Kuhl et al25 demonstrated persistence of enteroviral genome in almost one‐third of the samples 1 year after acute infection. Persistence of viral RNA in the myocardium of patients was associated with a progressive impairment of heart function, whereas spontaneous viral elimination was associated with a significant improvement in the function of the left ventricle.
Our data indicate a higher prevalence of HEV infection in the gastrocnemius of PAD patients (54%) compared to the affected tissues of patients with other chronic diseases of skeletal and cardiac muscle, including chronic fatigue syndrome (20.8% to 26.4%),26–27 chronic inflammatory muscle diseases (20%),7 fibromyalgia (13%),7 dilated cardiomyopathy (37.5% to 46%),8–9,11 or idiopathic dilated cardiomyopathy.10,12 Again, our study design does not permit identification of a mechanism for these differences; however, chronic progression of end‐organ (skeletal muscle) damage seen in PAD may provide an expanding tissue involvement over a longer period of time, producing increased accumulation of viral particles. Another possible mechanism is related to the presence of oxidative stress in PAD muscle.28 It is possible that the levels of oxidative stress in PAD muscle support increased viral replication. Oxidative stress has been shown to enhance replication of a number of viruses including HEV,29–30 HCV,21 and West Nile virus.22 A preliminary study in our laboratory has shown that HEV replicates at higher rates in HSkm cells with increased oxidative stress induced by tunicamycin30 than in normal cells (data not shown).
The prevalence of HEV infection of the gastrocnemius increased from 0% in control patients to 36%, 66% and 58% in PAD patients with disease stage II, III, and IV, respectively. Similarly, viral copy number increased from 2.5 to 4.87 and 5.5 (log10/mg) in disease stage II, III, and IV, respectively. Again, a mechanism that involves increased oxidative stress may be responsible for the higher prevalence of HEV RNA‐positive specimens and increased viral copy number per specimen among patients with more advanced PAD.21–22,30 Increased oxidative stress characteristic of PAD muscle28 may induce increased sensitivity to HEV infection and increased viral replication in the later stages of disease. It is also possible that a longer period of viral growth and expansion in patients with more advanced, and likely longer lasting, disease may account for these observations.
The principal limitation of this study is that the findings are based on correlational analyses of the association between HEV and PAD and, as such, do not identify cause and effect linkages. These correlations, nonetheless, establish the rationale for mechanistic studies that can determine whether HEV infection contributes to initiation and/or severity of PAD. Another concern is that the hemodynamic limitation of most patients in our study may be considered severe on the basis of their low ABIs and raises the possibility that our findings may not apply in the same manner to patients with milder hemodynamic limitation. We also note that biopsies were not taken from skeletal muscles other than the gastrocnemius. Consequently, we were not able to determine whether virus detected in PAD but not in control patients was localized to muscles of the affected leg, only. Finally, our study design identified control and PAD patients who had sedentary life styles, but did not allow precise quantification of differences in physical activity among our study groups. Different levels of inactivity among patients could produce differences in the prevalence of HEV infection in their gastrocnemius.
To the best of our knowledge, this is the first reported study of HEV infection of skeletal muscle in the ischemic limbs of PAD patients. HEV was found in the gastrocnemius of PAD patients but not in controls and both viral copy number and prevalence of infection increased with disease severity. Our data point to the need for further studies to determine the contribution of HEV infection to the pathophysiology of PAD.
Sources of Funding
This work was supported in part by a grant from the NIH (R01 AG034995), the Alexander S. Onassis Public Benefit Foundation and the Charles and Mary Heider Fund for Excellence in Vascular Surgery. Furthermore, this material is the result of work supported with resources and the use of facilities at the Veterans Affair Nebraska‐Western Iowa Health Care System.
Disclosures
None.
References
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MBAmerican Heart Association Statistics C, Stroke Statistics S Heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation. 2013; 127:e6-e245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pentecost MJ, Criqui MH, Dorros G, Goldstone J, Johnston KW, Martin EC, Ring EJ, Spies JBSpecial Writing Group of the Councils on Cardiovascular Radiology AC‐T, Vascular Surgery CC, Epidemiology, Prevention AHA Guidelines for peripheral percutaneous transluminal angioplasty of the abdominal aorta and lower extremity vessels. A statement for health professionals from a Special Writing Group of the Councils on Cardiovascular Radiology, Arteriosclerosis, Cardio‐Thoracic and Vascular Surgery, Clinical Cardiology, and Epidemiology and prevention, the American Heart Association. J Vasc Interv Radiol. 2003; 14:S495-S515 [DOI] [PubMed] [Google Scholar]
- 3.Pipinos II, Judge AR, Selsby JT, Zhu Z, Swanson SA, Nella AA, Dodd SL. The myopathy of peripheral arterial occlusive disease: part 1. Functional and histomorphological changes and evidence for mitochondrial dysfunction. Vasc Endovascular Surg. 2007; 41:481-489 [DOI] [PubMed] [Google Scholar]
- 4.Pipinos II, Judge AR, Selsby JT, Zhu Z, Swanson SA, Nella AA, Dodd SL. The myopathy of peripheral arterial occlusive disease: part 2. Oxidative stress, neuropathy, and shift in muscle fiber type. Vasc Endovascular Surg. 2008; 42:101-112 [DOI] [PubMed] [Google Scholar]
- 5.Brass EP, Hiatt WR. Acquired skeletal muscle metabolic myopathy in atherosclerotic peripheral arterial disease. Vasc Med. 2000; 5:55-59 [DOI] [PubMed] [Google Scholar]
- 6.Chia JK. The role of enterovirus in chronic fatigue syndrome. J Clin Pathol. 2005; 58:1126-1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Douche‐Aourik F, Berlier W, Feasson L, Bourlet T, Harrath R, Omar S, Grattard F, Denis C, Pozzetto B. Detection of enterovirus in human skeletal muscle from patients with chronic inflammatory muscle disease or fibromyalgia and healthy subjects. J Med Virol. 2003; 71:540-547 [DOI] [PubMed] [Google Scholar]
- 8.Calabrese F, Rigo E, Milanesi O, Boffa GM, Angelini A, Valente M, Thiene G. Molecular diagnosis of myocarditis and dilated cardiomyopathy in children: clinicopathologic features and prognostic implications. Diagn Mol Pathol. 2002; 11:212-221 [DOI] [PubMed] [Google Scholar]
- 9.Gasparri MG, Pipinos II, Kralovich KA, Margolin DA. Retrograde jejunogastric intussusception. South Med J. 2000; 93:499-500 [PubMed] [Google Scholar]
- 10.Pauschinger M, Kuhl U, Dorner A, Schieferecke K, Petschauer S, Rauch U, Schwimmbeck PL, Kandolf R, Schultheiss HP. Detection of enteroviral RNA in endomyocardial biopsies in inflammatory cardiomyopathy and idiopathic dilated cardiomyopathy. Z Kardiol. 1998; 87:443-452 [DOI] [PubMed] [Google Scholar]
- 11.Archard LC, Khan MA, Soteriou BA, Zhang H, Why HJ, Robinson NM, Richardson PJ. Characterization of Coxsackie B virus RNA in myocardium from patients with dilated cardiomyopathy by nucleotide sequencing of reverse transcription‐nested polymerase chain reaction products. Hum Pathol. 1998; 29:578-584 [DOI] [PubMed] [Google Scholar]
- 12.Arbustini E, Grasso M, Porcu E, Bellini O, Diegoli M, Fasani R, Banchieri N, Pilotto A, Morbini P, Dal Bello B, Campana C, Gavazzi A, Vigano M. Enteroviral RNA and virus‐like particles in the skeletal muscle of patients with idiopathic dilated cardiomyopathy. Am J Cardiol. 1997; 80:1188-1193 [DOI] [PubMed] [Google Scholar]
- 13.Kim KS, Chapman NM, Tracy S. Replication of Coxsackievirus B3 in primary cell cultures generates novel viral genome deletions. J Virol. 2008; 82:2033-2037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim KS, Tracy S, Tapprich W, Bailey J, Lee CK, Kim K, Barry WH, Chapman NM. 5′‐terminal deletions occur in Coxsackievirus B3 during replication in murine hearts and cardiac myocyte cultures and correlate with encapsidation of negative‐strand viral RNA. J Virol. 2005; 79:7024-7041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Iturriza‐Gomara M, Megson B, Gray J. Molecular detection and characterization of human enteroviruses directly from clinical samples using RT‐PCR and DNA sequencing. J Med Virol. 2006; 78:243-253 [DOI] [PubMed] [Google Scholar]
- 16.Trabelsi A, Grattard F, Nejmeddine M, Aouni M, Bourlet T, Pozzetto B. Evaluation of an enterovirus group‐specific anti‐VP1 monoclonal antibody, 5‐D8/1, in comparison with neutralization and PCR for rapid identification of enteroviruses in cell culture. J Clin Microbiol. 1995; 33:2454-2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chapman NM, Kim KS. Persistent Coxsackievirus infection: enterovirus persistence in chronic myocarditis and dilated cardiomyopathy. Curr Top Microbiol Immunol. 2008; 323:275-292 [DOI] [PubMed] [Google Scholar]
- 18.Heim A, Stille‐Seigener M, Pring‐Akerblom P, Grumbach I, Brehm C, Kreuzer H, Figulla HR. Recombinant interferons β and γ have a higher antiviral activity than interferon‐α in coxsackievirus B3‐infected carrier state cultures of human myocardial fibroblasts. J Interferon Cytokine Res. 1996; 16:283-287 [DOI] [PubMed] [Google Scholar]
- 19.Oberste MS, Maher K, Williams AJ, Dybdahl‐Sissoko N, Brown BA, Gookin MS, Penaranda S, Mishrik N, Uddin M, Pallansch MA. Species‐specific RT‐PCR amplification of human enteroviruses: a tool for rapid species identification of uncharacterized enteroviruses. J Gen Virol. 2006; 87:119-128 [DOI] [PubMed] [Google Scholar]
- 20.Rozovics JM, Chase AJ, Cathcart AL, Chou W, Gershon PD, Palusa S, Wilusz J, Semler BL. Picornavirus modification of a host mRNA decay protein. MBio. 2012; 3:e00412-e00431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tardif KD, Waris G, Siddiqui A. Hepatitis C virus, ER stress, and oxidative stress. Trends Microbiol. 2005; 13:159-163 [DOI] [PubMed] [Google Scholar]
- 22.Ambrose RL, Mackenzie JM. West Nile virus differentially modulates the unfolded protein response to facilitate replication and immune evasion. J Virol. 2011; 85:2723-2732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heim A, Grumbach I, Hake S, Muller G, Pring‐Akerblom P, Mall G, Figulla HR. Enterovirus heart disease of adults: a persistent, limited organ infection in the presence of neutralizing antibodies. J Med Virol. 1997; 53:196-204 [DOI] [PubMed] [Google Scholar]
- 24.Inal JM, Jorfi S. Coxsackievirus B transmission and possible new roles for extracellular vesicles. Biochem Soc Trans. 2013; 41:299-302 [DOI] [PubMed] [Google Scholar]
- 25.Kuhl U, Pauschinger M, Seeberg B, Lassner D, Noutsias M, Poller W, Schultheiss HP. Viral persistence in the myocardium is associated with progressive cardiac dysfunction. Circulation. 2005; 112:1965-1970 [DOI] [PubMed] [Google Scholar]
- 26.Gow JW, Behan WM, Simpson K, McGarry F, Keir S, Behan PO. Studies on enterovirus in patients with chronic fatigue syndrome. Clin Infect Dis. 1994; 18suppl 1:S126-S129 [DOI] [PubMed] [Google Scholar]
- 27.Lane RJ, Soteriou BA, Zhang H, Archard LC. Enterovirus related metabolic myopathy: a postviral fatigue syndrome. J Neurol Neurosurg Psychiatry. 2003; 74:1382-1386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pipinos II, Judge AR, Zhu Z, Selsby JT, Swanson SA, Johanning JM, Baxter BT, Lynch TG, Dodd SL. Mitochondrial defects and oxidative damage in patients with peripheral arterial disease. Free Radical Biol Med. 2006; 41:262-269 [DOI] [PubMed] [Google Scholar]
- 29.Tung WH, Hsieh HL, Lee IT, Yang CM. Enterovirus 71 induces integrin β1/EGFR‐Rac1‐dependent oxidative stress in SK‐N‐SH cells: role of HO‐1/CO in viral replication. J Cell Physiol. 2011; 226:3316-3329 [DOI] [PubMed] [Google Scholar]
- 30.Zhang HM, Ye X, Su Y, Yuan J, Liu Z, Stein DA, Yang D. Coxsackievirus B3 infection activates the unfolded protein response and induces apoptosis through downregulation of p58IPK and activation of CHOP and SREBP1. J Virol. 2010; 84:8446-8459 [DOI] [PMC free article] [PubMed] [Google Scholar]