

Abstract
Background
Autonomic imbalances including parasympathetic withdrawal and sympathetic overactivity are cardinal features of heart failure regardless of etiology; however, mechanisms underlying these imbalances remain unknown. Animal model studies of heart and visceral organ hypertrophy predict that nerve growth factor levels should be elevated in heart failure; whether this is so in human heart failure, though, remains unclear. We tested the hypotheses that neurons in cardiac ganglia are hypertrophied in human, canine, and rat heart failure and that nerve growth factor, which we hypothesize is elevated in the failing heart, contributes to this neuronal hypertrophy.
Methods and Results
Somal morphology of neurons from human (579.54±14.34 versus 327.45±9.17 μm2; P<0.01) and canine hearts (767.80±18.37 versus 650.23±9.84 μm2; P<0.01) failing secondary to ischemia and neurons from spontaneously hypertensive rat hearts (327.98±3.15 versus 271.29±2.79 μm2; P<0.01) failing secondary to hypertension reveal significant hypertrophy of neurons in cardiac ganglia compared with controls. Western blot analysis shows that nerve growth factor levels in the explanted, failing human heart are 250% greater than levels in healthy donor hearts. Neurons from cardiac ganglia cultured with nerve growth factor are significantly larger and have greater dendritic arborization than neurons in control cultures.
Conclusions
Hypertrophied neurons are significantly less excitable than smaller ones; thus, hypertrophy of vagal postganglionic neurons in cardiac ganglia would help to explain the parasympathetic withdrawal that accompanies heart failure. Furthermore, our observations suggest that nerve growth factor, which is elevated in the failing human heart, causes hypertrophy of neurons in cardiac ganglia.
Keywords: autonomic, cardiac ganglia, heart failure, nerve growth factor, nervous system
Introduction
Regardless of etiology, whether ischemia, increased afterload, infection, or idiopathic, heart failure is associated with autonomic imbalances including sympathetic overactivity and parasympathetic withdrawal.1–6 Such imbalances contribute to the myocardial hypertrophy associated with heart failure and accelerate the process leading to failure of the heart.2,6 Furthermore, parasympathetic activity protects patients from most cardiac arrhythmias including ventricular tachycardias and ventricular fibrillation as well as from sudden cardiac death;7–8 sympathetic activity and increased cardiac catecholamines, however, predispose patients to arrhythmogenesis and sudden death.9 Autonomic imbalances in heart failure, then, contribute to the compromised hemodynamics of the failing heart and the morbidity and mortality associated with heart failure; despite this, the neural mechanisms underlying autonomic imbalances in heart failure remain largely unknown. Electrophysiological studies in canines with pacing‐induced heart failure, though, have shown that cardiac ganglia are the primary site of defective neurotransmission of vagal impulses in heart failure.5,10 Stimulation of preganglionic vagal fibers or postganglionic neurons in cardiac ganglia, with and without ganglionic blockade, reveals that changes in preganglionic parasympathetic neurons in the brainstem or postganglionic neuronal effector sites, including nodal or myocardial tissue, do not significantly contribute to the parasympathetic withdrawal in heart failure. Rather, the vagal withdrawal occurs because of changes within cardiac ganglia.5,10
Myocardial hypertrophy, ischemia, infarction, and infection as well as stress and aging are all associated with increased message or protein levels of nerve growth factor in the myocardium.11–23 Whether nerve growth factor levels are similarly elevated in patients with heart failure is suggested;24 however, this remains largely unresolved. Interestingly, experimental overexpression of nerve growth factor in mice leads to myocardial hypertrophy, sympathetic ganglia hypertrophy, and increased catecholamine spillover.13 Furthermore, cardiac nerve growth factor levels are elevated in spontaneously hypertensive rats, which develop cardiac hypertrophy secondary to hypertension, compared with their Wistar‐Kyoto, normotensive controls.11–12 Chronic administration of nerve growth factor to newborn Wistar‐Kyoto rats leads to hypertrophy and hyperplasia of cardiac sympathetic neurons and cardiac catecholamine content that exceeds levels found in spontaneously hypertensive rats.25 Hypertrophy of the bladder or colon, as in Hirschsprung's disease, is also associated with elevated nerve growth factor levels, hypertrophy of autonomic neurons innervating the bladder or colon, and increased catecholamine synthesis and release.26–27 If nerve growth factor levels are similarly elevated in hypertrophied human hearts, then neurons in cardiac ganglia may hypertrophy as well. Compared with smaller neurons, hypertrophied neurons have decreased input resistance, due to an increased number of leak channels, and greater membrane capacitance due to an increased surface area; thus, hypertrophied neurons require more current to reach threshold.28–31 Hypertrophy of neurons in cardiac ganglia would, therefore, render them significantly less excitable and help to explain the parasympathetic withdrawal associated with heart failure.
In the present study, we sought to test the following hypotheses: one, neurons in cardiac ganglia from hypertrophied or failing human, canine, and rat hearts are hypertrophied compared with those from normal controls; two, nerve growth factor levels are elevated in explanted, failing human hearts compared with healthy donor hearts; and three, parasympathetic neurons from cardiac ganglia cultured with nerve growth factor, mimicking presumptive conditions in the failing heart, hypertrophy.
Materials and Methods
Human, Canine, and Rat Heart Tissue
Human heart tissue procurement was in accordance with the Institutional Review Board and use of animals with the Institutional Animal Care and Use Committee at Loyola University Medical Center. Human heart tissue concentrated with cardiac ganglia (Figure 1; Table 1) was removed from hearts of patients without apparent cardiovascular pathology (n=5: 401 neurons sampled) and from those with autopsy‐confirmed heart failure secondary to coronary artery disease and ischemia (n=5: 411 neurons); the tissue was immersion‐fixed in 10% w/v buffered formalin (Sigma, St Louis, MO). In previous experiments, ischemic heart failure was induced in canines (weight, 20 to 27 kg; n=4: 364 neurons) by multiple, sequential intracoronary embolizations (n=4: 398 neurons).33 Atrial tissue from regions homologous to those in humans was removed from formalin‐fixed hearts and processed for histology (heart weight, 211.57±7.75 versus 150.95±12.82 g; P<0.01). Similar regions from 6‐month‐old spontaneously hypertensive rats (n=7: 773 neurons) and their age‐matched normotensive Wistar‐Kyoto controls (n=7: 717 neurons) were removed and processed. Hearts from spontaneously hypertensive rats were markedly hypertrophied compared with hearts from their Wistar‐Kyoto controls (heart/body weight ratio, 0.54 versus 0.38; P<0.05).
Figure 1.
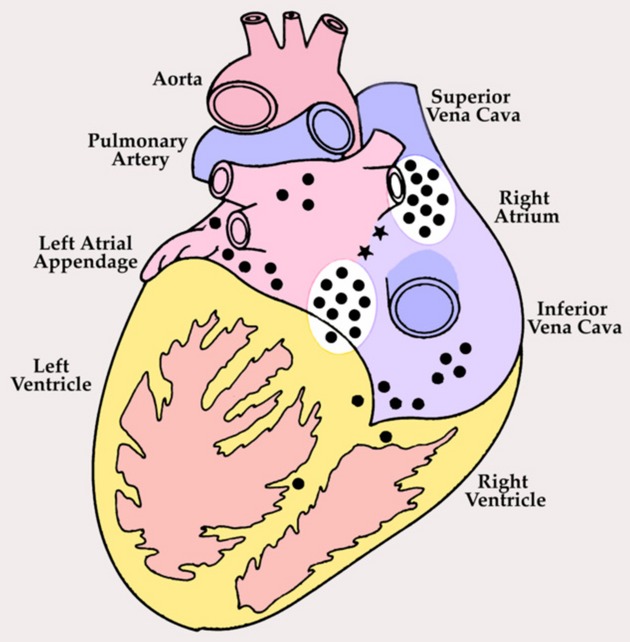
Posterior view of adult human heart showing distribution of cardiac ganglia on the surface (•) and in the interatrial septum (*). Cardiac ganglia adjacent to the sinoatrial and atrioventricular nodes (white ovals) were harvested for morphological studies from normal human hearts and those with ischemic heart failure; homologous regions from canines with ischemic heart failure and spontaneously hypertensive rats with heart failure secondary to hypertension were also harvested. Modified from Singh et al.32
Table 1.
Clinical Data for Controls and Patients With Ischemic Heart Failure
| Age and Sex | Heart Weight (g) | Left Ventricular Thickness (cm) | Pathology Findings |
|---|---|---|---|
| 51F | 400 | 1.4 | Gastric carcinoma |
| 65M | 400 | 1.3 | Adenocarcinoma |
| 71M | 310 | 1.6 | Hodgkin's lymphoma; pneumonia |
| 78F | 360 | 1.5 | Acute pulmonary emboli and edema |
| 45M | 790 | 1.5 | CAD; carcinoma |
| 70F | 830 | 1.2 | CAD; myocardial infarction* |
| 71M | 750 | 1.4 | CAD; myocardial infarction* |
| 75M | 780 | 1.4 | CAD; myocardial infarction* |
CAD indicates coronary artery disease.
Previous.
Morphology and Image Analysis
Unbiased, quantitative stereology was used to measure somal profiles of neurons in cardiac ganglia. Serial cryostat (canine) or paraffin‐embedded (human and rat) tissue sections, 10 μm thick, were placed on poly‐l‐lysine (Sigma) subbed slides and stained with hematoxylin and eosin Y (Sigma). The first tissue section was chosen randomly, and thereafter every 12th section examined. Every neuron in a section in which the nucleus and nucleolus were clearly visible was digitally captured at a magnification of 1000×, and somal cross‐sectional area determined using NIH ImageJ (http://rsbweb.nih.gov/ij/); image files were coded so that the investigator was “blind” to the origin of the tissue. Mixed‐model analysis using restricted‐likelihood method of fit was used to determine if the morphology of neurons in failing hearts versus normal hearts was significantly different; all data are reported as mean±standard error of the mean.
Western Blot Analysis
Left ventricular tissue from normal donor hearts was obtained from aborted transplants (n=4: 28.8±3.9 years; all male) and explanted human hearts with heart failure secondary to coronary artery disease and ischemia during separate transplant procedures (n=4: 51.8±4.3 years; 3 males); all patients were New York Heart Association class IV with end‐stage heart failure and left ventricle ejection fraction ≤35%. Tissue samples were flash‐frozen in liquid nitrogen and stored at −70°C; after thawing, the heart tissue was homogenized in Tris‐buffered saline (TBS: pH 7.5; Sigma) and cells lysed using a mammalian tissue lysis buffer (Sigma). Tissue sample homogenates were centrifuged at 5000g for 10 minutes at 4°C and protein concentrations determined using a bicinchoninic acid protein assay (Pierce) with bovine serum albumin (BSA) as the standard (Sigma). Thereafter, equal concentrations of protein were loaded in each well (100 μg) of a precast 10% sodium dodecyl sulfate polyacrylamide gel and current applied for 1 hour in a Mini‐Protean electrophoreses module containing running buffer (Bio‐Rad). A Mini‐Trans‐Blot cell containing transfer buffer was used to transfer proteins onto nitrocellulose paper (Bio‐Rad) with 35 mA of current for 1 hour. The nitrocellulose paper was washed three times in TBS and blocked using 5% nonfat dry milk and 3% BSA in TBS with 0.1% Tween 20 (TBS‐T; Sigma). Rabbit‐derived primary antibodies to nerve growth factor (1:1500; Chemicon) were incubated with the nitrocellulose paper in TBS‐T for 1 hour and then washed. Goat‐derived secondary antibodies linked to horseradish peroxidase (1:12000; Chemicon) were applied for 1 hour and the resulting blot blocked and washed. High‐resolution X‐ray film (Fuji Medical Systems) scans of horseradish peroxidase–linked chemiluminescence (Pierce, Rockford, IL) and NIH ImageJ were used to determine the density of chemiluminescence representing nerve growth factor; westerns were run in triplicates and data‐pooled.
Cell Culture
Mixed cell cultures of neonate rat atrial tissue were prepared by rinsing atrial tissue in Hank's solution (Sigma) containing penicillin and streptomycin (5 mL; Sigma); the tissue was first cut and then digested with trypsin (0.4 mg/mL; Sigma) and collagenase (1.2 mg/mL; Sigma) for 1 hour at 37°C. Resulting tissue fragments were triturated and then centrifuged at 300g for 10 minutes. The supernatant was discarded and pellet resuspended in minimum essential medium (Gibco) supplemented with 10% fetal bovine serum (Atlanta Biologicals). Cells were plated at ≈5000 cells/cm2 and incubated with either nerve growth factor (2 μg/mL; Chemicon [n=5: 181 neurons], without nerve growth factor [n=5: 259 neurons]), or antibodies to nerve growth factor (2 μL/mL; Chemicon [n=5: 271 neurons]), at 37°C in a 5% CO2 and 95% air mixture for 5 days; culture media were replaced with like media on the third day. Neurons from each culture were digitally captured at 400×, and somal area determined using “blinded” image analysis. Mixed‐model analysis using restricted‐likelihood method of fit was used to evaluate statistical differences in the somal area of neurons incubated with nerve growth factor versus that of those without nerve growth factor.
Results
Neuronal Hypertrophy in Heart Failure
Hearts of patients with heart failure secondary to coronary artery disease and ischemia were markedly enlarged compared with normal hearts (788±16.5 versus 368±21.4 g [mean±SEM]; P<0.01) and, as expected, the left ventricular wall thickness was similar in failing and normal hearts (both, 1.5±0.1 cm). Neurons in cardiac ganglia from failing human hearts are markedly hypertrophied compared with neurons from hearts of patients without cardiovascular pathology (579.54±14.34 versus 327.45±9.17 μm2; P<0.01); see Figure 2. Notice the significant rightward shift in the distribution of somal area for neurons from failing hearts versus those from healthy controls. The largest neuron observed in failing hearts is nearly twice the area of the largest neuron in normal hearts (2239.62 versus 1327.13 μm2). In addition to being hypertrophied, neurons in failing hearts are markedly edematous in appearance with notably lighter cytoplasm reflecting, in part, reduced staining of neuronal Nissl substance (Figure 3).
Figure 2.
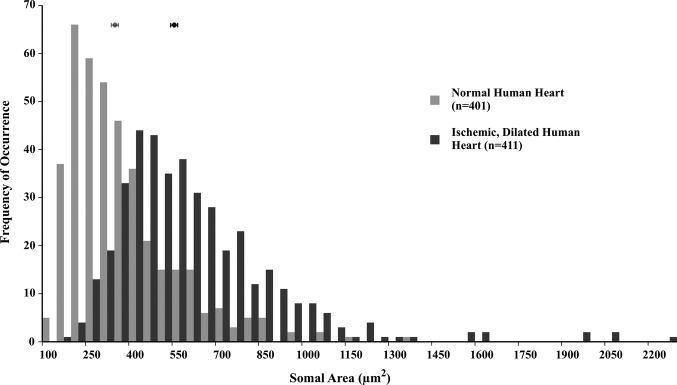
Somal area of neurons in cardiac ganglia from normal human hearts (light gray) and those with ischemic heart failure (dark gray). Each bin represents 50 μm2 and shows the frequency of occurrence of neurons in that range; the means±standard error of the means are shown above each histogram. The distribution histogram for neurons from ischemic, failing human hearts is shifted significantly toward larger sizes compared with that for neurons from normal human hearts (579.54±14.34 versus 327.45±9.17 μm2; P<0.01).
Figure 3.

Representative neurons in cardiac ganglia of normal human hearts (top) and those with ischemic heart failure (bottom). Image contrast and levels were identically adjusted in all photomicrographs. Neurons in ischemic, failing human hearts are conspicuously larger and appear more pale and edematous compared with neurons in normal hearts. Scale bars=20 μm.
Neurons from canine hearts with ischemic heart failure induced by sequential latex microsphere injections into the coronary vasculature are significantly hypertrophied compared with neurons from sham‐operated normal controls (767.80±18.37 versus 650.23±9.84 μm2; P<0.01); see Figure 4. The largest neuron observed in failing canine hearts is almost twice the area of the largest neuron in controls hearts (2170.48 versus 1344.96 μm2). Histologically and similar to observations in failing human hearts, neurons in failing canine hearts are edematous and pale in appearance compared with neurons from normal controls.
Figure 4.
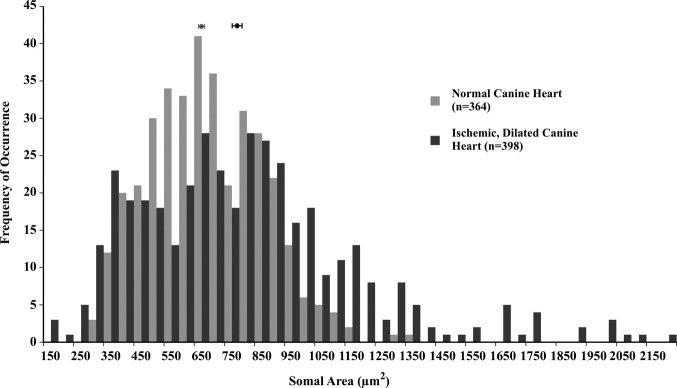
Somal area of neurons in cardiac ganglia from normal canine hearts (light gray) and those with ischemic heart failure (dark gray); each bin represents 50 μm2. The distribution histogram for neurons from ischemic, failing canine hearts is shifted significantly toward larger sizes compared with that for neurons from normal canine hearts (767.80±18.37 versus 650.23±9.84 μm2; P<0.01).
In the failing human and canine hearts examined in this study cardiac hypertrophy and failure occurred secondary to ischemia, leading to ischemic heart failure. In spontaneously hypertensive rats, however, hearts hypertrophied secondary to idiopathic hypertension and increased afterload, leading to nonischemic heart failure. Neurons in hypertrophied hearts of spontaneously hypertensive rats are, nonetheless, also hypertrophied compared with neurons from their normotensive Wistar‐Kyoto controls (327.98±3.15 versus 271.29±2.79 μm2; P<0.01); see Figure 5. The largest neuron observed in spontaneously hypertensive rat hearts was 708.32 μm2, whereas that in Wistar‐Kyoto rats was 575.72 μm2. Thus, regardless of etiology, whether coronary artery disease and ischemia or secondary to hypertension and increased afterload, neurons in failing hearts are hypertrophied compared with neurons from matched controls.
Figure 5.
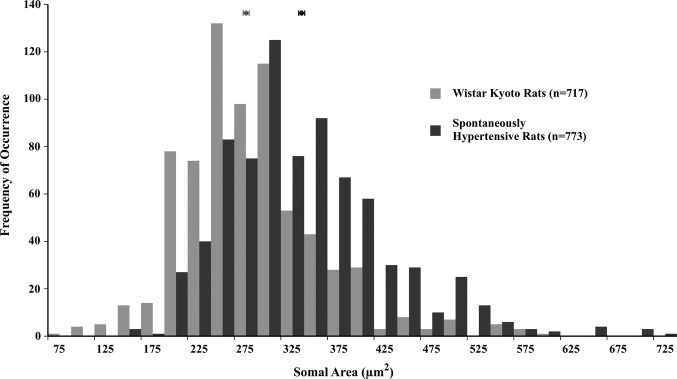
Somal area of neurons in cardiac ganglia from normotensive Wistar‐Kyoto rats (light gray) and spontaneously hypertensive rats (dark gray); each bin represents 25 μm2. The distribution histogram for neurons from spontaneously hypertensive rats with heart failure secondary to hypertension is shifted significantly toward larger sizes compared with that for neurons from their normotensive Wistar‐Kyoto controls (327.98±3.15 versus 271.29±2.79 μm2; P<0.01).
Nerve Growth Factor in Failing Human Hearts
Western blot analysis of left ventricular tissue shows increased levels of nerve growth factor in explanted, failing human hearts compared with tissue from normal, donor hearts (Figure 6). Density measurements of x‐ray film exposed to horseradish peroxidase–linked chemiluminescence show that failing human hearts had over 250% greater levels of nerve growth factor than tissue from donor controls. In 1 failing human heart, the levels of nerve growth factor were more than 500% the average level in donor hearts. In arbitrary density units, the density of the nerve growth factor representing bands was 1829±418 (n=4) in normal hearts and 4745±1397 (n=4) in failing human hearts. Immunocontrols for antibody specificity included omission and preincubation controls for the primary and omission controls for the secondary. The immunostained band migrated at ≈146 kD, the molecular weight for nerve growth factor, further establishing specificity of the Western immunoprotocol.
Figure 6.
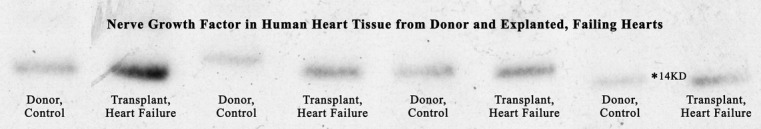
Western blot analysis reveals that nerve growth factor protein levels in the explanted, failing human heart are greater than 250% of those in normal donor hearts; in one failing heart the levels of nerve growth factor are greater than 500% of those in normal controls. The bands representing nerve growth factor migrated at approximately 146 kD,* the molecular weight for nerve growth factor.
Nerve Growth Factor in Cell Culture
Neonate rat heart cell cultures incubated with nerve growth factor for 5 days show marked neuronal hypertrophy compared with neurons cultured without nerve growth factor (1005.78±50.48 versus 711.63±29.71 μm2; P<0.01); see Figure 7. Qualitatively, the dendritic arborization of neurons cultured with nerve growth factor was notably more extensive than the arborization of neurons from control cultures. Cultures incubated with antibodies to nerve growth factor showed no somal or dendritic hypertrophy and in fact were atrophied compared with control cultures (327.07±12.88 μm2; P<0.01). The mixed cell cultures were immunostained for neuronal markers, neurofilament‐100, and choline acetyltransferase to confirm the phenotype of cells analyzed; further, the imaged cells were the only ones with extensive dendritic arborization, which is indicative of neurons.
Figure 7.
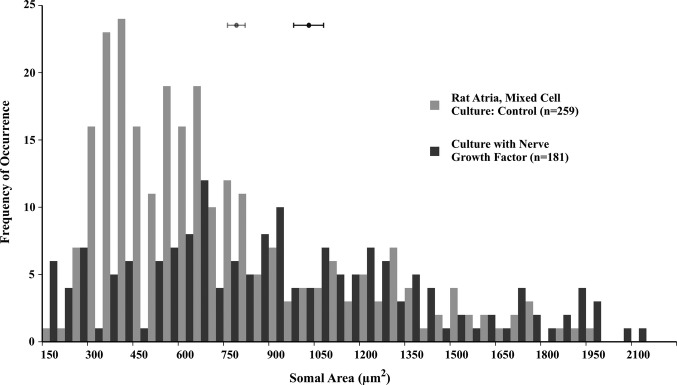
Somal area of neurons in rat atria, mixed cell cultures (light gray), and similar cultures incubated with nerve growth factor (2 mg/mL, dark gray); each bin represents 50 μm2. The distribution histogram for rat atrial neurons cultured with nerve growth factor is shifted significantly toward larger sizes compared with that for neurons from normal control cultures (1005.78±50.48 versus 711.63±29.71 μm2; P<0.01).
Discussion
Neuronal Hypertrophy and Vagal Withdrawal
Neuronal hypertrophy has significant consequences for the excitability of a neuron and the generation of action potentials.28–31 Hypertrophy of neurons in cardiac ganglia would render them significantly less excitable28–31 and, thus, would help to explain the parasympathetic withdrawal that occurs in human heart failure. According to Ohm's law, ∆V=IRin, the current required to change the voltage across the membrane varies inversely with the input, or membrane, resistance; thus, lower input resistance means more current is required to change the membrane voltage.29 Input resistance, in turn, is inversely proportional to the square of the radius of the cell: Rin=Rm/4πa2, where Rm is the unit or specific membrane resistance, which depends only on the density and conductance of leak channels.29 With neuronal hypertrophy, as the membrane surface area increases, the density of leak channels remains the same, and, therefore, the total number of leak channels increases; as a result, input resistance decreases.29 Hypertrophied neurons also have increased membrane capacitance due to increased membrane surface area; thus, more charge is required to change the membrane voltage: ∆V=∆Q/C, where Q is charge and C capacitance.29 The internal capacitance of the cell, in turn, is directly related to the surface area of the cell: Cin=Cm·4πa2, where Cm is the unit or specific capacitance, which for biological membranes is 1 μF/cm2.29 As a consequence of decreased input resistance, hypertrophied neurons have a shorter length constant (λ=√Rm/Ri), because the membrane excitation is essentially short‐circuited through open leak channels. As a consequence of the increased capacitance, they have a longer time constant (τ=RmC) and, thus, a given patch of membrane takes longer to reach threshold.29 A useful analogy is that of a bucket (the neuron) and water (the current): as the number of holes in the bucket increases, the input resistance decreases and water simply dissipates through open channels, and more is required to fill the bucket or, by analogy, more current is required for the neuron to reach threshold. As the size of the bucket increases, the membrane capacitance increases, and more water and time are required to fill the bucket or, again, for the neuron to reach threshold.
Although electrophysiological studies correlating hypertrophy of neurons in cardiac ganglia with membrane capacitance have yet to be done, direct electrophysiological measurements of membrane capacitance in hypertrophied sympathetic neurons, due to nerve growth factor overexpression, reveal an increase in membrane capacitance of 102% in phasic neurons and 44% in tonic neurons.34 Based on our human somal morphology data, we calculated that membrane capacitance in hypertrophied neurons from failing human hearts should be increased by 75% and that input resistance should be decreased by 57% compared with neurons from normal hearts. Neurons from cardiac ganglia cultured with nerve growth factor have greater dendritic arborization; given that failing hearts have increased nerve growth factor levels, we would suspect parasympathetic neurons in failing hearts to have greater arborization as well. Because the vast majority of neuronal surface area is dendritic arborization,35 capacitance increases and input resistance decreases in neurons from failing hearts are likely to be much greater than those based on calculations considering the soma alone. In mice overexpressing nerve growth factor, electrophysiological measurements of hypertrophied cardiomyocytes showed that even with just a 12% increase in membrane capacitance, the action potential duration, at 90% of repolarization, is increased by 142%.14 In our laboratory, in situ electrophysiology of neurons in canine cardiac ganglia revealed that tonic patterns of neuronal activity correlate with smaller neurons and greater input resistance, whereas phasic patterns correlate with larger neurons and lower input resistance; the largest neurons were unexcitable.35 Hypertrophied neurons in failing human hearts may, therefore, convert from tonic to phasic patterns or, possibly, fail to reach threshold. The net effect of morphological changes in cardiac ganglia in heart failure will be significantly decreased excitability, possible changes in firing pattern, and, ultimately, reduced vagal tone and decreased acetylcholine release onto the myocardium.
The only other description of somal hypertrophy of neurons in cardiac ganglia with myocardial hypertrophy, apart from this one, is a qualitative time‐course study by Meerson and Krokhina.36 After aortic banding in rabbits and canines, they found that neurons in cardiac ganglia began to enlarge within days, 2 or more months after aortic banding neurons became notably hypertrophied, and 2 years after banding, neurons in cardiac ganglia remained noticeably hypertrophied.36 Similar to our observations, Meerson and Krokhina reported an edematous and pale appearance to the hypertrophied neurons in hypertrophied hearts.36 Recently, canines with pacing‐induced heart failure were shown to have increased diameter of parasympathetic nerve bundles in the left atrium.37 These observations are consistent with ours of somal hypertrophy; however, the authors did not consider the size principle29 or somal hypertrophy and suggest that increased fiber diameter leads to increased vagal activity in heart failure.37 This is contrary to many lines of evidence and Bibevski and Dunlap have shown directly that cardiac ganglia are the site of defective vagal neurotransmission in heart failure.3–5,10 In a separate study, high‐frequency stimulation of atrioventricular ganglia and surrounding myocardium in canines led to increased expression of nerve growth factor, likely because of local pacing‐induced failure of atrial tissue that then, much like in heart failure, led to hypertrophy of neurons in cardiac ganglia;38 these authors also did not consider the size principle29 and suggest that increasing nerve growth factor levels and the resulting neuronal hypertrophy will actually increase vagal activity.38
Nerve Growth Factor and Hypertrophy
Our Western blot analysis is the first direct demonstration of nerve growth factor changes in the myocardium of the failing human heart. Our findings are consistent with the vast majority of the literature examining nerve growth factor in myocardial hypertrophy and failure, aging, and pathophysiological stress.11–21,23 Other lines of evidence including changes in endothelin‐1 in heart failure, studies of myocardial ischemia and infarction, and studies of bladder and gut hypertrophy also corroborate our findings.16,22,26–27,26–40 Certain discrepancies exist in the literature, though, based on studies of norepinephrine‐induced heart failure in rats and indirect, transcardiac plasma measurements of nerve growth factor in patients with heart failure24,41–42; plausible explanations are discussed. Recently, Wistar rats with pressure‐induced right ventricular hypertrophy were shown to have significantly greater concentrations of nerve growth factor in the right ventricle compared with controls.19 Trypanosoma cruzi–induced myocarditis in rats, a model for Chagas' also leads to significant increases in nerve growth factor in the myocardium.18 Based on glyoxylic acid histofluorescence the authors suggest sympathetic denervation of the myocardium;18 most studies, though, correlate nerve growth factor overexpression with increased sympathetic innervation.13,17,25,42 However, glyoxylic acid histofluorescence is an insensitive technique compared with immunodetection but is used in many studies to qualitatively stain for catecholamines in nerve fibers and extrapolate the histofluorescence to the density of sympathetic nerve fibers.24,41 Given that catecholamine uptake is severely compromised in heart failure, though spillover is significantly increased,43 catecholamine histofluorescence will not reflect sympathetic nerve fiber density but rather would reflect diminished catecholamine content of the nerve fiber. Further, markers such as tyrosine hydroxylase or catecholamines are ambiguous as sympathetic markers because some 70% of “parasympathetic” neurons in cardiac ganglia express tyrosine hydroxylase and catecholamine immunoreactivity.44–45
Measurements of nerve growth factor in rats show significant correlation of nerve growth factor level with increases in age and heart weight but no such correlations with increases in skeletal muscle weight.15 On the basis of these observations, Stuerenburg and Kunze15 postulated that increases in nerve growth factor in excess of age‐related changes would lead to pathological cardiac hypertrophy caused by increased sympathetic drive and cardiac catecholamines. Their hypothesis is consistent with our findings; we further propose that nerve growth factor increases in cardiac hypertrophy and failure lead to morphological, electrical, and neurochemical changes in vagal postganglionic neurons of cardiac ganglia. Such changes further tip the autonomic imbalance toward sympathetic overactivity due to parasympathetic withdrawal. Restraint stress in rats increases cardiac and vascular nerve growth factor levels and endotoxemic stress from gastric perforation leads to increases in cardiac endothelin‐1, nerve growth factor, and, subsequently, sympathetic markers;20,23 anti‐inflammatory steroidal therapy during sepsis reduces both endothelin‐1 and nerve growth factor levels toward normal.23 Endothelin‐1 is known to be elevated in human heart failure and correlates with New York Heart Association functional classification and ventricular dysfunction.39,46–47 Importantly for this discussion, endothelin‐1 upregulates nerve growth factor;42 thus, elevated endothelin‐1 in heart failure would predict elevated levels of nerve growth factor as well.
Thirty minutes after ischemia and reperfusion, nerve growth factor message and protein levels increase in the peri‐ischemic region and from 2 to 120 hours message levels are increased more than 4‐fold.16 After an infarct, myocardial tissue in the peri‐infarct region has significantly higher levels of nerve growth factor message and protein, whereas remote to the infarct, nerve growth factor expression is unchanged.46 In mice, blocking nerve growth factor postinfarction increases apoptosis, reduces neovascularization, and worsens left ventricular function, presumably because of decreased sympathetic drive.22 Alternatively, administration of nerve growth factor after an infarct decreases apoptosis, increases neovascularization, and improves left ventricular function.22 Postinfarct, the heterogeneity of nerve growth factor expression over the myocardium leads to heterogeneous sympathetic innervation, dispersion of refractoriness of the myocardium, and, thus, predisposition to reentrant arrhythmias. Given that ischemia and, often, multiple infarcts secondary to coronary artery disease are consistent with the pathophysiology of ischemic heart failure, we would expect increased levels of nerve growth factor in failing, ischemic hearts. The progression from cardiac dilation and hypertrophy to failure is a positive‐feedback, downward spiral. Depending on where in the spiral the patient may be, nerve growth factor–induced sympathetic activity is beneficial early, as increased sympathetic drive helps to meet demands for tissue perfusion, but detrimental later, as increased sympathetic tone and catecholamine spillover along with parasympathetic withdrawal drive the heart further and further into failure.1,6–9,19,43,47
The only other study to examine nerve growth factor levels in human heart failure is one in which transcardiac arteriovenous blood nerve growth factor levels were measured and reportedly diminished.24 Nerve growth factor is present in vanishingly low concentrations in the myocardium, and we suspect that measurements of arteriovenous differences may be a poor reflection of myocardial levels; our study and other recent studies bring these previous findings24 into question.18–19,38,48 In the same study and two similar ones, rats chronically infused with catecholamines are found to have decreased levels of nerve growth factor in the myocardium.24,43–44 Chronic administration of catecholamines in otherwise healthy rats, however, would lead to negative feedback reductions in nerve growth factor levels and does not model in vivo neurotrophic cascades of heart failure. Given that sympathetic overactivity and catecholamine spillover along with parasympathetic withdrawal are the final common pathways in heart failure, exogenously administered catecholamines will certainly lead to myocardial hypertrophy and failure. In human heart failure, though, the neurotrophic cascade likely begins with increases in endothelin‐1, triggered by oxidative, inflammatory, hemodynamic, or infectious stressors that then upregulate nerve growth factor; subsequently, nerve growth factor increases sympathetic drive and myocardial catecholamine spillover along with decreased vagal tone due to nerve growth factor induced morphological, electrical, and neurochemical changes within cardiac ganglia.9,13,18–19,25,40 Knocking down nerve growth factor levels by passive immunization or via viral vectors should normalize endogenous levels and ameliorate the autonomic imbalance induced arrhythmogenesis, myocardial hypertrophy, and failure of the heart.6,13,19,26,49
Disclosures
None.
Acknowledgments
We thank Steven Pogwizd, John M. Lee, and Vassyl A. Lonchyna for providing canine, autopsy, and transplant tissue, respectively, for this study. We also thank Vanessa Raschke for assistance with the mixed‐model statistical analysis.
References
- 1.Oliveira JS. A natural human model of intrinsic heart nervous system denervation: Chagas' cardiopathy. Am Heart J. 1985; 110:1092-1098 [DOI] [PubMed] [Google Scholar]
- 2.Binkley PF, Nunziata E, Hass GJ, Nelson SD, Cody RJ. Parasympathetic withdrawal is an integral component of autonomic imbalance in congestive heart failure: demonstration in human subjects and verification in a paced canine model of ventricular failure. J Am Coll Cardiol. 1991; 18:464-472 [DOI] [PubMed] [Google Scholar]
- 3.Ajiki K, Murakawa Y, Yanagisawa‐Miwa A, Usui M, Yamashita T, Oikawa N, Inoue H. Autonomic nervous system activity in idiopathic dilated cardiomyopathy and in hypertrophic cardiomyopathy. Am J Cardiol. 1993; 71:1316-1320 [DOI] [PubMed] [Google Scholar]
- 4.Meredith IT, Eisenhofer G, Lambert GW, Dewar EM, Jennings GL, Esler MD. Cardiac sympathetic nervous activity in congestive heart failure. Evidence for increased neuronal norepinephrine release and preserved neuronal uptake. Circulation. 1993; 88:136-145 [DOI] [PubMed] [Google Scholar]
- 5.Bibevski S, Dunlap ME. Ganglionic mechanisms contribute to diminished vagal control in heart failure. Circulation. 1999; 99:2958-2963 [DOI] [PubMed] [Google Scholar]
- 6.Zhang Y, Popovic ZB, Bibevski S, Fakhry I, Sica DA, Van Wagoner DR, Mazgalev TN. Chronic vagus nerve stimulation improves autonomic control and attenuates systemic inflammation and heart failure progression in a canine high‐rate pacing model. Circ Heart Fail. 2009; 2:692-699 [DOI] [PubMed] [Google Scholar]
- 7.Waxman MB, Cameron D, Wald RW. In: Levy MN, Schwartz PJ. (eds.). Vagal activity and ventricular tachyarrhythmias. Vagal Control of the Heart: Experimental Basis and Clinical Implications. 1994New York: Futura Publishers; 579-612 [Google Scholar]
- 8.Ferrari GM, Vanoli E, Schwartz PJ. In: Levy MN, Schwartz PJ. (eds.). Vagal activity and ventricular fibrillation. Vagal Control of the Heart: Experimental Basis and Clinical Implications. 1994New York: Futura Publishers; 613-636 [Google Scholar]
- 9.Pozzati A, Pancaldi LG, Di Pasquale G, Pinelli G, Bugiardini R. Transient sympathovagal imbalance triggers “ischemic” sudden death in patients undergoing electrocardiographic Holter monitoring. J Am Coll Cardiol. 1996; 27:847-852 [DOI] [PubMed] [Google Scholar]
- 10.Dunlap ME, Bibevski S, Rosenberry TL, Ernsberger P. Mechanisms of altered vagal control in heart failure: influence of muscarinic receptors and acetylcholinesterase activity. Am J Physiol Heart Circ Physiol. 2003; 285:H1632-H1640 [DOI] [PubMed] [Google Scholar]
- 11.Ueyama T, Hamada M, Hano T, Nishio I, Masuyama Y, Furukawa S. Increased nerve growth factor levels in spontaneously hypertensive rats. J Hypertens. 1992; 10:215-219 [DOI] [PubMed] [Google Scholar]
- 12.Zettler C, Rush RA. Elevated concentrations of nerve growth factor in heart and mesenteric arteries of spontaneously hypertensive rats. Brain Res. 1993; 614:15-20 [DOI] [PubMed] [Google Scholar]
- 13.Hassankhani A, Steinhelper ME, Soonpaa MH, Katz EB, Taylor DA, Andrade‐Rozental A, Factor SM, Steinberg JJ, Field LJ, Federoff HJ. Overexpression of NGF within the heart of transgenic mice causes hyperinnervation, cardiac enlargement, and hyperplasia of ectopic cells. Dev Biol. 1995; 169:309-321 [DOI] [PubMed] [Google Scholar]
- 14.Heath BM, Xia J, Dong E, An RH, Brooks A, Liang C, Federoff HJ, Kass RS. Overexpression of nerve growth factor in the heart alters ion channel activity and beta‐adrenergic signalling in an adult transgenic mouse. J Physiol. 1998; 512:779-791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stuerenburg HJ, Kunze K. Tissue concentrations of nerve growth factor in aging rat heart and skeletal muscle. Muscle Nerve. 1998; 21:404-406 [DOI] [PubMed] [Google Scholar]
- 16.Hiltunen JO, Laurikainen A, Vakeva A, Meri S, Saarma M. Nerve growth factor and brain‐derived neurotrophic factor mRNAs are regulated in distinct cell populations of rat heart after ischaemia and reperfusion. J Pathol. 2001; 194:247-253 [DOI] [PubMed] [Google Scholar]
- 17.Kiriazis H, Du XJ, Feng X, Hotchkin E, Marshall T, Finch S, Gao XM, Lambert G, Choate JK, Kaye DM. Preserved left ventricular structure and function in mice with cardiac sympathetic hyperinnervation. Am J Physiol Heart Circ Physiol. 2005; 289:H1359-H1365 [DOI] [PubMed] [Google Scholar]
- 18.Martinelli PM, Camargos ER, Azevedo AA, Chiari E, Morel G, Machado CR. Cardiac NGF and GDNF expression during Trypanosoma cruzi infection in rats. Auton Neurosci. 2006; 130:32-40 [DOI] [PubMed] [Google Scholar]
- 19.Kimura K, Ieda M, Kanazawa H, Yagi T, Tsunoda M, Ninomiya S, Kurosawa H, Yoshimi K, Mochizuki H, Yamazaki K, Ogawa S, Fukuda K. Cardiac sympathetic rejuvenation: a link between nerve function and cardiac hypertrophy. Circ Res. 2007; 100:1755-1764 [DOI] [PubMed] [Google Scholar]
- 20.Manni L, Di Fausto V, Fiore M, Aloe L. Repeated restraint and nerve growth factor administration in male and female mice: effect on sympathetic and cardiovascular mediators of the stress response. Curr Neurovasc Res. 2008; 5:1-12 [DOI] [PubMed] [Google Scholar]
- 21.Lujan HL, Chen Y, Dicarlo SE. Paraplegia increased cardiac NGF content, sympathetic tonus, and the susceptibility to ischemia‐induced ventricular tachycardia in conscious rats. Am J Physiol Heart Circ Physiol. 2009; 296:H1364-H1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meloni M, Caporali A, Graiani G, Lagrasta C, Katare R, Van Linthout S, Spillmann F, Campesi I, Madeddu P, Quaini F, Emanueli C. Nerve growth factor promotes cardiac repair following myocardial infarction. Circ Res. 2010; 106:1275-1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tsai MS, Chung SD, Liang JT, Ko YH, Hsu WM, Lai HS, Chang KC. Enhanced expression of cardiac nerve growth factor and nerve sprouting markers in rats following gastric perforation: the association with cardiac sympathovagal balance. Shock. 2010; 33:170-178 [DOI] [PubMed] [Google Scholar]
- 24.Kaye DM, Vaddadi G, Gruskin SL, Du XJ, Esler MD. Reduced myocardial nerve growth factor expression in human and experimental heart failure. Circ Res. 2000; 86:E80-E84 [DOI] [PubMed] [Google Scholar]
- 25.Zettler C, Head RJ, Rush RA. Chronic nerve growth factor treatment of normotensive rats. Brain Res. 1991; 538:251-262 [DOI] [PubMed] [Google Scholar]
- 26.Steers WD, Kolbeck S, Creedon D, Tuttle JB. Nerve growth factor in the urinary bladder of the adult regulates neuronal form and function. J Clin Invest. 1991; 88:1709-1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuroda T, Ueda M, Nakano M, Saeki M. Altered production of nerve growth factor in aganglionic intestines. J Pediatr Surg. 1994; 29:288-292 [DOI] [PubMed] [Google Scholar]
- 28.Henneman E, Somjen G, Carpenter DO. Functional significance of cell size in spinal motorneurons. J Neurophysiol. 1965; 28:560-580 [DOI] [PubMed] [Google Scholar]
- 29.Koester J. In: Kandel ER, Schwartz JH, Jessell TM. (eds.). Local signaling: passive electrical properties of the neuron. Principles of Neural Science. 2000New York: McGraw‐Hill; 136-143 [Google Scholar]
- 30.Steinback CD, Salmanpour A, Breskovic T, Dujic Z, Shoemaker JK. Sympathetic neural activation: an ordered affair. J Physiol. 2010; 588:4825-4836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arora RC, Cardinal R, Smith FM, Ardell JL, Dell'Italia LJ, Armour JA. Intrinsic cardiac nervous system in tachycardia induced heart failure. Am J Physiol Regul Integr Comp Physiol. 2003; 285:R1212-R1223 [DOI] [PubMed] [Google Scholar]
- 32.Singh S, Johnson PI, Lee RE, Orfei E, Lonchyna VA, Sullivan HJ, Montoya A, Tran H, Wehrmacher WH, Wurster RD. Topography of cardiac ganglia in the adult human heart. J Thorac Cardiovas Surg. 1996; 112:943-953 [DOI] [PubMed] [Google Scholar]
- 33.Pogwizd SM. Focal mechanisms underlying ventricular tachycardia during prolonged ischemic cardiomyopathy. Circulation. 1994; 90:1441-1458 [DOI] [PubMed] [Google Scholar]
- 34.Davis BM, Wang HS, Albers KM, Carlson SL, Goodness TP, McKinnon D. Effects of NGF overexpression on anatomical and physiological properties of sympathetic postganglionic neurons. Brain Res. 1996; 724:47-54 [DOI] [PubMed] [Google Scholar]
- 35.Xi X, Randall WC, Wurster RD. Morphology of intracellularly labeled canine intracardiac ganglion cells. J Comp Neurol. 1991; 314:396-402 [DOI] [PubMed] [Google Scholar]
- 36.Meerson FZ, Krokhina EM. Neurons of intramural ganglia on the heart in its hyperfunction and hypertrophy. Acta Anat. 1965; 62:161-175 [DOI] [PubMed] [Google Scholar]
- 37.Ng J, Villuendas R, Cokic I, Schliamser JE, Gordon D, Koduri H, Benefield B, Simon J, Murthy SN, Lomasney JW, Wasserstrom JA, Goldberger JJ, Aistrup GL, Arora R. Autonomic remodeling in the left atrium and pulmonary veins in heart failure: creation of a dynamic substrate for atrial fibrillation. Circ Arrhythm Electrophysiol. 2011; 4:388-396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rana OR, Saygili E, Gemein C, Zink MD, Buhr A, Mischke K, Nolte KW, Weis J, Weber C, Marx N, Schauerte P. Chronic electrical neuronal stimulation increases cardiac parasympathetic tone by eliciting neurotrophic effects. Circ Res. 2011; 108:1209-1219 [DOI] [PubMed] [Google Scholar]
- 39.Serneri GG, Modesti PA, Boddi M, Cecioni I, Paniccia R, Coppo M, Galanti G, Simonetti I, Vanni S, Papa L, Bandinelli B, Migliorini A, Modesti A, Maccherini M, Sani G, Toscano M. Cardiac growth factors in human hypertrophy. Relations with myocardial contractility and wall stress. Circ Res. 1999; 85:57-67 [DOI] [PubMed] [Google Scholar]
- 40.Ieda M, Fukuda K, Hisaka Y, Kimura K, Kawaguchi H, Fujita J, Shimoda K, Takeshita E, Okano H, Kurihara Y, Kurihara H, Ishida J, Fukamizu A, Federoff HJ, Ogawa S. Endothelin‐1 regulates cardiac sympathetic innervation in the rodent heart by controlling nerve growth factor expression. J Clin Invest. 2004; 113:876-884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Qin F, Vulapalli RS, Stevens SY, Liang CS. Loss of cardiac sympathetic neurotransmitters in heart failure and NE infusion is associated with reduced NGF. Am J Physiol Heart Circ Physiol. 2002; 282:H363-H371 [DOI] [PubMed] [Google Scholar]
- 42.Kimura K, Kanazawa H, Ieda M, Kawaguchi‐Manabe H, Miyake Y, Yagi T, Arai T, Sano M, Fukuda K. Norepinephrine‐induced nerve growth factor depletion causes cardiac sympathetic denervation in severe heart failure. Auton Neurosci. 2010; 156:27-35 [DOI] [PubMed] [Google Scholar]
- 43.Eisenhofer G, Friberg P, Rundqvist B, Quyyumi AA, Lambert G, Kaye DM, Kopin IJ, Goldstein DS, Esler MD. Cardiac sympathetic nerve function in congestive heart failure. Circulation. 1996; 93:1667-1676 [DOI] [PubMed] [Google Scholar]
- 44.Singh S, Johnson PI, Javed A, Gray TS, Lonchyna VA, Wurster RD. Monoamine‐ and histamine‐synthesizing enzymes and neurotransmitters within neurons of adult human cardiac ganglia. Circulation. 1999; 99:411-419 [DOI] [PubMed] [Google Scholar]
- 45.Hoover DB, Isaacs ER, Jacques F, Hoard JL, Page P, Armour JA. Localization of multiple neurotransmitters in surgically derived specimens of human atrial ganglia. Neuroscience. 2009; 164:1170-1179 [DOI] [PubMed] [Google Scholar]
- 46.McMurray JJ, Ray SG, Abdullah I, Dargie HJ, Morton JJ. Plasma endothelin in chronic heart failure. Circulation. 1992; 85:1374-1379 [DOI] [PubMed] [Google Scholar]
- 47.Tang WH, Shrestha K, Martin MG, Borowski AG, Jasper S, Yandle TG, Richards AM, Klein AL, Troughton RW. Clinical significance of endogenous vasoactive neurohormones in chronic systolic heart failure. J Card Fail. 2010; 16:635-640 [DOI] [PubMed] [Google Scholar]
- 48.Kristen AV, Kreusser MM, Lehmann L, Kinscherf R, Katus HA, Haass M, Backs J. Preserved norepinephrine reuptake but reduced sympathetic nerve endings in hypertrophic volume‐overloaded rat hearts. J Card Fail. 2006; 12:577-583 [DOI] [PubMed] [Google Scholar]
- 49.Gorin PD, Johnson EM., Jr Effects of long‐term nerve growth factor deprivation on the nervous system of the adult rat: an experimental autoimmune approach. Brain Res. 1980; 198:27-42 [DOI] [PubMed] [Google Scholar]