

Abstract
Background
Vascular smooth muscle cell (VSMC) senescence and apoptosis are involved in atherosclerotic plaque vulnerability. Arginase‐II (Arg‐II) has been shown to promote vascular dysfunction and plaque vulnerability phenotypes in mice through uncoupling of endothelial nitric oxide synthase and activation of macrophage inflammation. The function of Arg‐II in VSMCs with respect to plaque vulnerability is unknown. This study investigated the functions of Arg‐II in VSMCs linking to plaque vulnerability.
Methods and Results
In vitro studies were performed on VSMCs isolated from human umbilical veins, whereas in vivo studies were performed on atherosclerosis‐prone apolipoprotein E‐deficient (ApoE−/−) mice. In nonsenescent VSMCs, overexpressing wild‐type Arg‐II or an l‐arginine ureahydrolase inactive Arg‐II mutant (H160F) caused similar effects on mitochondrial dysfunction, cell apoptosis, and senescence, which were abrogated by silencing p66Shc or p53. The activation of p66Shc but not p53 by Arg‐II was dependent on extracellular signal‐regulated kinases (ERKs) and sequential activation of 40S ribosomal protein S6 kinase 1 (S6K1)—c‐Jun N‐terminal kinases (JNKs). In senescent VSMCs, Arg‐II and S6K1, ERK‐p66Shc, and p53 signaling levels were increased. Silencing Arg‐II reduced all these signalings and cell senescence/apoptosis. Conversely, silencing p66Shc reduced ERK and S6K1 signaling and Arg‐II levels and cell senescence/apoptosis. Furthermore, genetic ablation of Arg‐II in ApoE−/− mice reduced the aforementioned signaling and apoptotic VSMCs in the plaque of aortic roots.
Conclusions
Arg‐II, independently of its l‐arginine ureahydrolase activity, promotes mitochondrial dysfunction leading to VSMC senescence/apoptosis through complex positive crosstalk among S6K1‐JNK, ERK, p66Shc, and p53, contributing to atherosclerotic vulnerability phenotypes in mice.
Keywords: apoptosis, arginase, p53, p66Shc, vascular smooth muscle cells
Introduction
The rupture of an unstable plaque with thrombotic occlusion of coronary arteries is a fatal complication of atherosclerosis, leading to acute coronary syndromes, that is, unstable angina, myocardial infarction, and sudden cardiac death.1–2 Unstable plaque is characterized by a thin fibrous cap overlying a large necrotic lipid core containing infiltrated inflammatory cells with lower vascular smooth muscle cell (VSMC) content and extracellular matrix.1–2 Senescence and apoptosis of VSMCs have been shown to be important contributors to plaque vulnerability.3–5 The senescent VSMCs reveal impaired plaque‐repairing capacity, whereas cell apoptosis causes loss of cellularity in the plaque, conferring plaque vulnerability. In advanced atherosclerotic lesions obtained from patients, senescent VSMCs are found in fibrous caps and intima and exhibit increased oxidative stress.5 Senescence is characterized by the irreversible loss of the proliferative capability of the cells, which show typical morphological features such as large flattened shape accompanied by an increase in several senescent markers including enhanced senescence‐associated β‐galactosidase (SA‐β‐gal) activity, activation of p53 and increased oxidative stress, shortened telomeres, and elevated levels of a series of cyclin‐dependent kinase inhibitors, including p16ink4, p21cip1, or p27kip1 proteins.6 In addition to senescence, VSMC apoptosis has also been identified in advanced atherosclerotic plaques and is increased in unstable versus stable lesions.7 Cellular apoptosis is the process of programmed cell death that is characterized by distinct morphological and biochemical alterations, including mitochondrial dysfunction, activation of caspase‐3, DNA damage such as DNA strand breaks, which can be detected by the terminal dUTP nick end‐labeling (TUNEL) method, and externalization of phosphatidylserine residues on the outer plasma membrane of apoptotic cells, which can be assayed by annexin V staining.8
Compelling evidence suggests that the overwhelming production of reactive oxygen species (ROS) promotes both cellular senescence and apoptosis and accelerates atherogenesis.9–11 Studies in past years have demonstrated that mitochondria are the important source of intracellular ROS, including O2•– and H2O2, which ultimately cause cell senescence and apoptosis.12–14 Among the mechanisms, the mitochondrial redox enzyme p66Shc has been shown to play an important role in mitochondrial ROS generation. On phosphorylation at serine 36 (p66Shc‐S36), p66Shc translocates into mitochondria, where it directly generates H2O2 by transferring electron from cytochrome c to oxygen, promoting cellular senescence and apoptosis.15–16 Phosphorylation of p66Shc‐S36 has been reported to be mediated by protein kinases including protein kinase C‐β (PKC‐β), extracellular signal‐regulated kinases (ERKs), and c‐Jun N‐terminal kinases (JNKs).16–18 These mechanisms are implicated in organism aging and aging‐associated pathologies, including oxidative stress, cellular senescence and apoptosis, and vascular disease.11,19
Interestingly, arginase‐II (Arg‐II), an l‐arginine ureahydrolase metabolizing l‐arginine to urea and l‐ornithine, is mainly localized in the mitochondria of cells and has been shown to exert similar biological functions as p66Shc. Both Arg‐II and p66Shc increase mitochondrial ROS production, accelerate endothelial senescence and dysfunction, and promote atherogenesis in mouse models.11,19–21 Our recent studies demonstrated that Arg‐II promotes mitochondrial ROS generation in macrophages20 and positively regulates 40S ribosomal protein S6 kinase 1 (S6K1) signaling, leading to induction of endothelial nitric oxide synthase (eNOS) uncoupling, that is, enhanced ROS and decreased NO production from eNOS in endothelial cells.21 S6K1 is a serine/threonine kinase that phosphorylates the 40S ribosomal protein S6, the first identified S6K1 substrate, resulting in enhanced protein translation.22 The activation of S6K1 relies on the coordinated phosphorylation of Thr389 and Thr229 by mammalian target of rapamycin (mTOR) and phosphoinositide‐dependent kinase‐1, respectively.23–24 It is a key mediator of mTOR complex 1 (mTORC1), which senses and integrates a variety of environmental cues to regulate many major cellular functions, including cell growth, cell survival, and cell motility.25 Exaggerated persistent activation of mTORC1‐S6K1 signaling is implicated in many aging‐associated pathological conditions including cancer, obesity, neurodegeneration, and cardiovascular disease.25–27
It has been reported that Arg‐II is involved in hypoxia‐induced VSMC proliferation,28 which does not seem to support the observation of a more stable plaque phenotype in atherosclerosis‐prone apolipoprotein E (ApoE)–deficient mice with Arg‐II deficiency (ApoE−/−Arg‐II−/−) compared with ApoE−/−Arg‐II+/+ control mice, that is, a decreased number of proinflammatory macrophages in the plaque, smaller size of necrotic core, and decreased amounts of various proinflammatory cytokines and MMPs in the plaques.20 Given that Arg‐II enhances mitochondrial ROS production,20 which is an important mechanism of both cellular senescence and apoptosis,9 in the present study, we investigated the functional role of Arg‐II in in vitro cultured human VSMCs and in a mouse model in the context of atherosclerotic plaque‐vulnerable phenotypes and explored a novel function of Arg‐II, that is, an l‐arginine ureahydrolase activity–independent effect on activation of p53 and various signaling pathways that activate p66Shc in VSMCs, leading to mitochondrial dysfunction, cell senescence, and cell apoptosis.
Methods
Materials
If not specifically indicated, all chemicals including those used for immunoblotting, mouse anti‐tubulin monoclonal antibody, and 2′,7′‐dichlorofluorescein (H2DCF, #35845) were obtained from Sigma (Buchs, Switzerland). Nω‐hydroxy‐nor‐l‐arginine (nor‐NOHA; #399275), S‐12‐bromoethyl‐l‐cystine HCl (BEC; #197900), Gö6976 (#365250), and PD98059 (#513000) were from Calbiochem (VWR, Geneva, Switzerland). SP600125 (#1496) was from Tocris (Anawa, Zürich, Switzerland). Rabbit antibodies against phospho‐S6‐S235/236 (#2211s), S6 (#2217s), phospho‐p53‐S15 (#9284s), phospho‐ERK‐T202/Y204 (#9101s), and mouse anti‐p53 (#2524s) for immunofluorescence staining were from Cell Signaling (Allschwil, Switzerland); mouse anti‐eNOS (#610297), mouse anti‐S6K1 (#611260) for immunoblotting, and mouse anti‐ERK (#610123) were from BD Transduction Laboratories (Allschwil, Switzerland); rabbit anti‐Arg‐II (sc‐20151) and rabbit anti‐p53 (sc‐6243) for immunoblotting were from Santa‐Cruz (Nunningen, Switzerland). PhosphoDetect mouse anti‐p66Shc (p66Shc‐Ser36; #566807) was purchased from Millipore (Zug, Switzerland). Rabbit anti‐p66Shc (#06‐203) was from Upstate (LucernaChem, Lucerne, Switzerland). Rabbit and mouse anti‐α‐smooth muscle actin (α‐SMA; #ab5694 and #ab7817, respectively), rabbit anti–proliferating cell nuclear antigen (PCNA; #ab18197) and mouse anti‐S6K1 (#ab119252) for immunofluorescence staining were from Abcam (Cambridge, UK). Alexa Fluor 680–conjugated anti‐mouse IgG (#A21057), Alexa Fluor 488–conjugated goat anti‐mouse IgG, Alexa Fluor 594–conjugated goat anti‐rabbit F(ab)2, dihydroethidium (DHE), MitoSox, and 4′6‐diamidino‐2‐phenyl‐indole, dihydrochloride (DAPI) were from Molecular Probes/Invitrogen (Lucerne, Switzerland); IRDye800‐conjugated anti‐rabbit IgG (#926‐32211) was from LI‐COR Biosciences (Bad Homburg, Germany); the membrane‐permeable 4,5‐diaminofluoresceine acetate (DAF‐2DA) was from VWR International SA (Dietikon, Switzerland); and X‐gal was from Promega (Dübendorf, Switzerland). All cell culture media and materials were purchased from Gibco BRL (Lucerne, Switzerland).
Cell Culture
VSMCs were isolated from human umbilical veins and cultured in Dulbecco's Modified Eagle Medium (DMEM) culture medium containing 10% fetal calf serum (FCS), 5.5 mmol/L glucose, 100 U/mL penicillin, 0.17 mmol/L streptomycin, and 2 mmol/L l‐glutamine. Cells of passages 2 to 4 (P2 to P4) were used as “young” cells. Some of the cells were further split in a ratio of 1:3 continuously over a period of several weeks until replicative senescence was reached, as assessed by SA‐β‐gal staining (P20 to P23, which were referred to as senescent cells). VSMCs isolated from 18 individual human umbilical cords were used in this study. The “n” for number of experiments conducted with VSMCs in the figures represents the number of repeated sets of individual experiments, not the number of human samples.
Animals
The ApoE−/− mice were from Jackson Laboratory. The Arg‐II−/− mice were kindly provided by Dr William O'Brien29 and backcrossed to C57BL/6J for more than 8 generations. Genotyping was performed by polymerase chain reaction as previously described.29 The ApoE−/− mice and Arg‐II−/− mice, both on a C57BL/6J background, were interbred to obtain ApoE−/−Arg‐II−/− mice as described previously.20 To accelerate the atherosclerotic lesion formation, 10‐week‐old male ApoE−/−Arg‐II+/+ and ApoE−/−Arg‐II−/− mice were fed a high‐fat diet (HF; energy content: 55% fat, 21% protein, and 24% carbohydrate; Harlan Teklad TD 93075) for 10 weeks. At 20 weeks of age, animals were anesthetized with ketamine (100 mg/kg IP) and xylazine (10 mg/kg IP), the entire aortas from the heart to the iliac bifurcation were removed and dissected free from fat and adhering perivascular tissue for immunoblotting analysis. For immunostaining, the aortic roots (ARs) were snap‐frozen in optimal cutting temperature compound. The 7‐μm‐thick cryosections of the ARs were stained with an In Situ Cell Death Detection Kit, TMR red (#12156792910; Roche Applied Science), for apoptotic cells followed by immunostaining of VSMCs with anti‐α‐SMA. Four mice of each genotype were included in experiments. Housing and animal experimentation were approved by the Swiss Federal Veterinary Office.
Immunofluorescence Staining of ARs and VSMCs
AR cryosections (7 μm) were fixed with 4% paraformaldehyde and blocked with 2% bovine serum albumin in phosphate‐buffered saline (PBS) for 30 minutes. To assess the changes in Arg‐II, p66Shc, p53, and PCNA in VSMCs in aortas, costaining of these proteins with α‐SMA was performed. Mouse anti‐α‐SMA was used for costaining with rabbit antibodies against Arg‐II, p66Shc, p53‐S15, and PCNA, whereas rabbit anti‐α‐SMA was used for costaining with mouse anti‐p53 and anti‐S6K1. The sections were first incubated for 2 hours with both anti‐α‐SMA and the corresponding primary antibody. After washing, the sections were incubated with Alexa Fluor 488–conjugated goat anti‐mouse IgG and Alexa Fluor 594–conjugated goat anti‐rabbit F(ab)2 at room temperature for 1 hour. The sections were finally counterstained with 300 nmol/L DAPI for 2 minutes. The immunofluorescence signals were visualized under a Leica DIM6000 confocal microscope. The same procedure was applied for immunofluorescence staining of the VSMCs that were cultured on a coverslip, except that the cells were stained with a single first antibody.
Recombinant Adenovirus
Generation of recombinant adenovirus (rAd)–expressing shRNA targeting human p66Shc and p53 driven by the U6 promoter (rAd/U6‐hp66ShcshRNA and ‐hp53shRNA, respectively) and wild‐type p66Shc and a dominant‐negative mutant of p66Shc (p66Shc‐S36A)30 driven by the cytomegalovirus (CMV) promoter (rAd/CMV‐p66Shc and ‐p66Shc‐S36A, respectively) was carried out with Gateway Technology (Invitrogen Life Technologies) according to the manufacturer's instructions.31 Expression plasmids encoding p66Shc and p66Shc‐S36A (pcDNA3.1‐His‐p66Shc and ‐p66Shc‐S36A, respectively)32 were purchased from Addgene (Cambridge, MA). The targeting sequences for shRNA are indicated in boldface with only the sense strand shown: GAATGAGTCTCTGTCATCG for the human p66Shc located within p66Shc‐specific CH2 domain33; GCGCACAGAGGAAGAGAATCT for human p53. rAd/U6‐LacZshRNA, rAd/U6‐Arg‐IIshRNA, rAd/U6‐S6K1shRNA, rAd/CMV empty vector, rAd/CMV‐Arg‐II, rAd/CMV‐Arg‐II‐H160F (an l‐arginine ureahydrolase inactive mutant of Arg‐II), and rAd/CMV‐HA‐S6K1ca (an active S6K1 mutant), generated as previously described.21
Adenoviral Transduction of VSMC
Transduction of VSMCs by rAd was performed as previously described.34 Cells were transduced with the rAd at titers of 50 to 100 multiplicities of infection and cultured in complete medium for 2 to 4 days before experiments as indicated in the figures.
Immunoblotting
Cell or tissue lysate preparation, SDS‐PAGE, and transfer of SDS gels to Immobilon‐P membranes (Millipore) were performed as previously described.35 The resultant membrane was first incubated with the corresponding primary antibody at room temperature for 2 hours with gentle agitation after blocking with 5% skim milk. The blot was then further incubated with a corresponding anti‐mouse (Alexa Fluor 680–conjugated) or anti‐rabbit (IRDye 800‐conjugated) secondary antibodies. Signals were visualized using an Odyssey Infrared Imaging System (LI‐COR Biosciences). Quantification of the signals was performed using NIH Image 1.47 software (US National Institutes of Health).
Arginase Activity Assay
Arginase activity was measured by colorimetric determination of urea formed from l‐arginine as previously described.36 Briefly, cells were lysed in lysis buffer containing 10 mmol/L Tris‐HCl (pH 7.4), 0.4% Triton X‐100, 10 μg/mL leupeptin, and 0.1 mmol/L phenylmethylsulfonyl fluoride (PMSF). Samples were centrifuged at 13 000 rpm at 4°C for 10 minutes, and protein concentration of the supernatant was determined by the Lowry method (Bio‐Rad). For measurement of arginase activity, equal amount of the cell lysate in 50 μL (2 μg total protein for young cells overexpressing LacZ, Arg‐II, or Arg‐II‐H160F; 30 μg total protein for comparison between young and senescent VSMCs) was added to 50 μL of Tris‐HCl (10 mmol/L [pH 7.4]) containing 5 mmol/L MnCl2. Arginase was then activated by heating the mixture at 56°C for 10 minutes. The hydrolysis reaction of l‐arginine by arginase was conducted by incubating the mixture containing activated arginase with 100 μL of l‐arginine (100 mmol/L [pH 9.6]) at 37°C for 1 hour. For colorimetric determination of urea, 1 mL of chromogenic reagent consisting of 1 volume of 3% 2,3‐butanedione monoxine and 29 volumes of the acid solution mixture (H2SO4:H3PO4:H2O 1:3:7) was added, and the mixture was then heated at 100°C for 30 minutes. After placing the samples in the dark for 10 minutes at room temperature, the urea concentration was determined spectrophotometrically by the absorbance at 492 nm. The amount of the urea produced was used as an index for arginase activity.
Detection of Nitric Oxide (NO), Cytoplasmic O2•−, Mitochondrial O2•−, and H2O2
NO, cytoplasmic O2•−, mitochondrial O2•−, and H2O2 detection was performed by using DAF‐2DA, DHE, MitoSox, and H2DCF, respectively, as previously described.20,35 For detection of NO, cells were gently washed twice with Ca2+‐free PBS and incubated in a modified Krebs–Ringer bicarbonate solution (NaCl, 118 mmol/L; KCl, 4.7 mmol/L; CaCl2, 2.5 mmol/L; MgSO4, 1.2 mmol/L; KH2PO4, 1.2 mmol/L; NaHCO3, 25 mmol/L; EDTA, 0.026 mmol/L; and glucose, 5.5 mmol/L) containing 5 μmol/L of DAF‐2DA for 30 minutes. For detection of cytoplasmic or mitochondrial superoxide, cells were incubated with 5 μmol/L DHE for 20 minutes or 5 μmol/L MitoSox for 10 minutes, respectively. For detection of H2O2, cells were washed with PBS and incubated with 10 μmol/L H2DCF in PBS for 25 minutes. The cells were then washed 3 times and fixed with 3.7% of paraformaldehyde, and images were obtained with Zeiss fluorescence microscopy. The intensity of the fluorescence was quantified by NIH Image J 1.47 software and normalized by cell number.
Mitochondrial Membrane Potential
Mitochrondrial membrane (Δψm) was assessed using a JC‐1 Mitochondrial Membrane Potential Detection Kit (#10009172; Cayman) according to the manufacturer's instructions. In healthy cells with high mitochondrial Δψm, JC‐1 spontaneously forms complexes known as J‐aggregates with intense red fluorescence that was detected at 600‐nm emission after excitation at 550 nm. On the other hand, in apoptotic or unhealthy cells with low Δψm, JC‐1 remains in the monomeric form, which shows only green fluorescence that was detected at 535‐nm emission after excitation at 485 nm. Quantification of the signals was performed using NIH Image 1.47 software.
SA‐β‐gal staining
SA‐β‐gal staining was performed as previously described.35 Briefly, cells were washed twice with PBS followed by fixation with a 3.7% formaldehyde solution for 10 minutes. After washing, cells were then incubated with the SA‐β‐gal staining solution (1 mg/mL X‐gal, 40 mmol/L citric acid, 5 mmol/L potassium ferrocyanide, 5 mmol/L potassium ferricyanide, 150 mmol/L sodium chloride, 2 mmol/L magnesium chloride dissolved in phosphate buffer) at pH 6.0 overnight at 37°C in a CO2‐free atmosphere. The stained senescent cells were detected by conventional microscopy. Senescent cells were determined by the ratio of SA‐β‐gal‐positive cells to the total cell count.
Detection of Apoptotic Cells
Apoptosis of cultured VSMCs was detected with an Annexin‐V‐FLUOS Staining Kit (#1988549; Roche Applied Science), whereas detection of apoptotic cells in atherosclerotic plaque was carried out by staining of the 7‐μm‐thick cryosections of the aortic roots with the In Situ Cell Death Detection Kit, TMR red (TUNEL; #12156792910; Roche Applied Science) according to the manufacturer's instructions. Quantification of the signals was performed using NIH Image 1.47 software.
Assessment of Cell Proliferation
Proliferation of cultured VSMCs was assessed by monitoring the proliferation marker PCNA37 by immunoblotting and/or immunostaining.
Statistical Analysis
Statistical analysis was performed as described previously.20 In brief, the Kolmogorov–Smirnov test was used to first determine whether the data deviated from Gaussian distributions. For normally distributed values, statistical analysis was performed with the Student t test for unpaired observations or analysis of variance (ANOVA) with Bonferroni's posttest, and data are given as means±SEMs. For nonnormally distributed values, nonparametric statistical analysis was performed with the Mann–Whitney test or the Kruskal–Wallis test with a Dunn's multiple comparison posttest, and data are expressed as medians with 25th and 75th percentiles. P≤0.05 was considered a statistically significant difference. The number of treatment groups in each set of experiment and the “n” (number of repeated sets of in vitro experiments conducted with human VSMCs or the number of animals of each genotype used) is indicated in each figure.
Results
Effects of Overexpression of Arg‐II in Young VSMCs
Arg‐II overexpression induces oxidative stress and mitochondrial dysfunction in VSMCs
In the young VSMCs, overexpression of wild‐type Arg‐II and of the l‐arginine ureahydrolase inactive mutant H160F21 was confirmed by immunoblotting with the antibody that reacts with both wild‐type and mutant Arg‐II, and the activity of the wild‐type and mutant enzymes was confirmed by the arginase activity assay (Figure 1A). Overexpression of Arg‐II in the cells enhances cytosolic O2•− (assessed by DHE staining), mitochondrial O2•− production (assessed by MitoSox staining), and H2O2 generation (assessed by H2DCF staining) (Figure 1B). Interestingly, the inactive Arg‐II mutant (H160F) with abolished l‐arginine ureahydrolase activity, as shown in Figure 1A, was unable to stimulate O2•− production but was still able to induce H2O2 (Figure 1B). The results demonstrate that Arg‐II exerts both an l‐arginine ureahydrolase activity–dependent effect (generation of O2•−) and an enzymatic activity–independent effect, that is, generation of H2O2 in VSMCs. In addition, mitochondrial membrane potential (Δψm) as assessed by JC‐1 staining (red fluorescent JC‐1 signal is indicative of healthy cells with high Δψm, whereas green fluorescent JC‐1 signal is indicative of unhealthy cells with low Δψm) was significantly attenuated in the VSMCs overexpressing Arg‐II or the inactive H160F mutant (Figure 1C). These results demonstrate that Arg‐II decreases mitochondrial Δψm and induces H2O2 production in VSMCs independently of its l‐arginine ureahydrolase activity. For the following study, we mainly focused on the novel enzymatic activity–independent effects of Arg‐II. Moreover, there was no NO production as assessed by DAF‐2DA (Figure 2A) and no detectable expression of eNOS or inducible nitric oxide synthase (iNOS) (Figure 2B), demonstrating that Arg‐II's effects in VSMCs are independent of NOS/NO.
Figure 1.
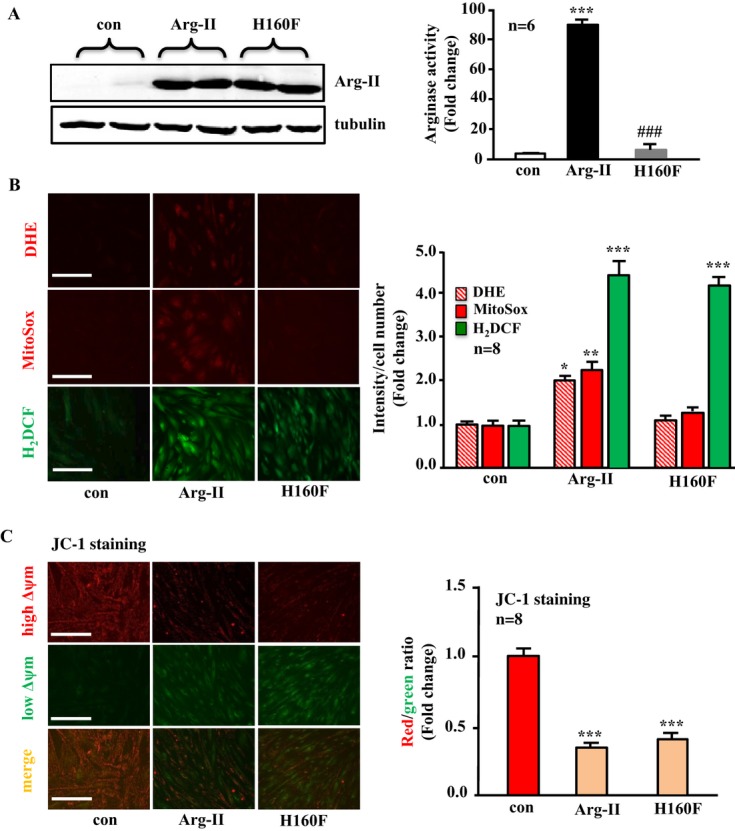
Arginase‐II (Arg‐II) overexpression enhances cytoplasmic and mitochondrial O2•− generation in an l‐arginine ureahydrolase activity‐dependent manner, promotes production of H2O2, and decreases mitochondrial membrane potential (Δψm) independently of its enzymatic activity in young VSMCs. Young VSMCs were transduced with empty vector rAd/CMV as control (con), rAd/CMV‐Arg‐II (Arg‐II), or rAd/CMV‐Arg‐II‐H160F (H160F, an inactive Arg‐II mutant). Seventy‐two hours posttransduction, the cells were subjected to (A) immunoblotting analysis for detection of overexpressed Arg‐II taking tubulin as a loading control (left) and arginase activity assay (right). B, DHE and MitoSox staining for detection of cytoplasmic and mitochondrial O2•−, respectively, and H2DCF staining for detection of H2O2 production. C, Δψm assessment using a JC‐1 Mitochondrial Membrane Potential Detection Kit. Red fluorescence JC‐1 signal is indicative of healthy cells with high Δψm, whereas green fluorescence JC‐1 signal is indicative of unhealthy cells with low Δψm. Quantification of the signals is shown in the corresponding right‐sided panels (n=6 or 8 as indicated in the graphs). **P<0.01, ***P<0.001 vs control group, ###P<0.001 vs Arg‐II. Scale bar=0.2 mm. VSMC indicates vascular smooth muscle cell; rAd, recombinant adenovirus; DHE, dihydroethidium; H2DCF, 2′,7′‐dichlorofluorescein; CMV, cytomegalovirus.
Figure 2.
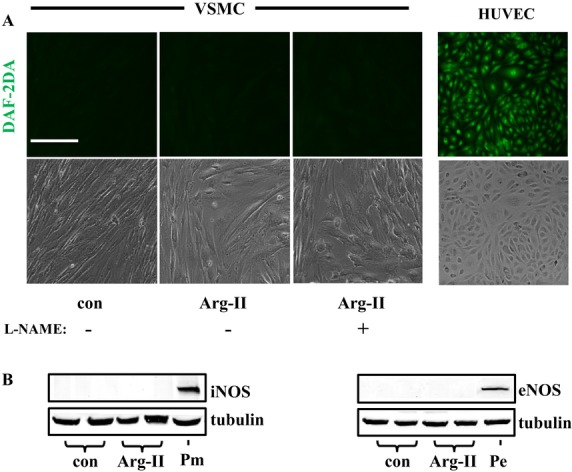
NOS/NO is not involved in the actions of Arg‐II in VSMCs. VSMCs were transduced with empty vector rAd/CMV as control or rAd/CMV‐Arg‐II. Seventy‐two hours posttransduction, cells were treated with 5 mmol/L L‐NG‐Nitroarginine methyl ester (L‐NAME) for 1.5 hours and then subjected to (A) DAF‐2DA staining for nitric oxide (NO) production. Human umbilical veins endothelial cells (HUVECs) were used as a positive control for NO staining. B, Immunoblotting analysis of iNOS and eNOS (P, positive control). Macrophage (Pm) was used as a positive control for iNOS (left), whereas HUVECs (Pe) was used as a positive control for eNOS (right). Scale bar=0.2 mm. These experiments were repeated 3 times. NOS indicates nitric oxide synthase; VSMC, vascular smooth muscle cell; con, control; rAd, recombinant adenovirus; DAF‐2DA, 4,5‐diaminofluoresceine acetate; iNOS, inducible nitric oxide synthase; eNOS, endothelial nitric oxide synthase; Arg‐II, arginase‐II.
Arg‐II overexpression promotes VSMC senescence and apoptosis independently of its l‐arginine ureahydrolase activity but enhances VSMC proliferation in an enzymatic activity–dependent manner
Overexpression of Arg‐II in the young VSMCs promoted cell senescence as demonstrated by the increase in the percentage of the cells positive for SA‐β‐gal staining (Figure 3A). Moreover, more apoptotic cells as demonstrated by positive Annexin‐V‐FLUOS staining were observed in cells overexpressing Arg‐II (Figure 3B). In addition, p53‐S15 levels were increased by overexpressing Arg‐II, although total p53 levels were not altered (Figure 3C). In line with its effects in promoting H2O2 production and mitochondrial dysfunction, the inactive Arg‐II mutant H160F exerted similar effects in inducing cellular senescence, apoptosis, and p53‐S15 similar to those in wild‐type Arg‐II (Figure 3A through 3C). In contrast, only the wild type, not the inactive mutant H160F, showed a stimulatory effect on cell proliferation as monitored by the proliferation marker PCNA with immunoblotting (Figure 3C) or by immunostaining (Figure 3D). These data demonstrate that Arg‐II promotes VSMC senescence and apoptosis through an l‐arginine ureahydrolase activity–independent mechanism, whereas it stimulates VSMC proliferation in an enzymatic activity–dependent manner (please also refer to the scheme in Figure 17A).
Figure 3.
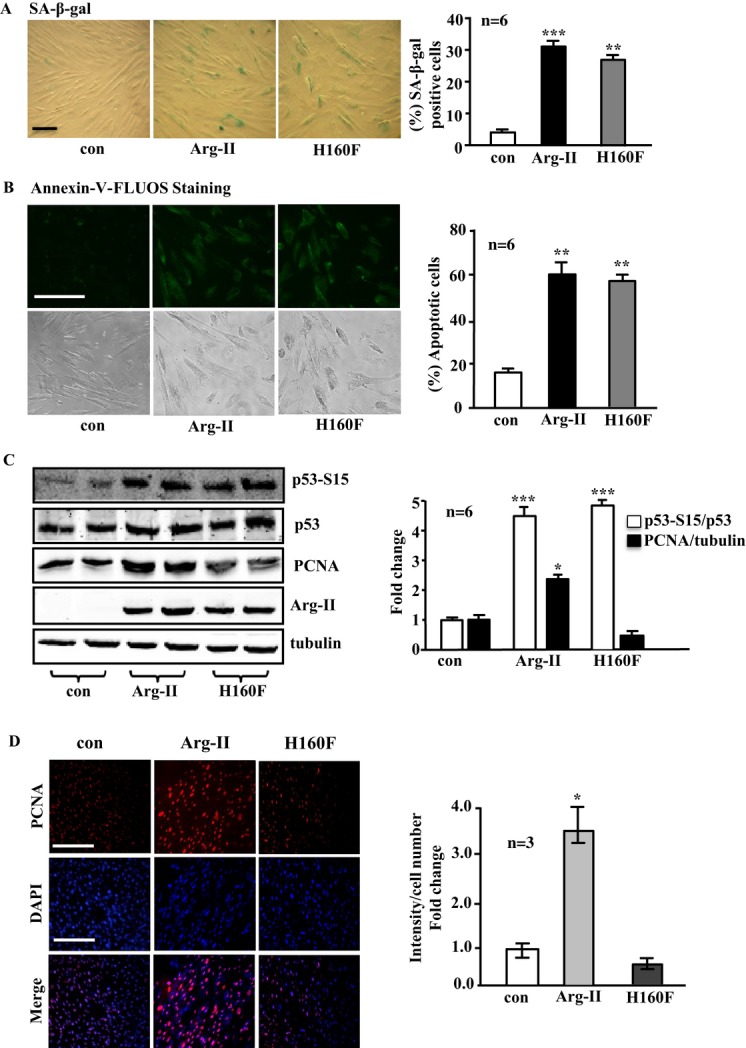
Overexpression of Arg‐II in young VSMCs causes cell senescence and apoptosis independently of its enzymatic activity, whereas it enhances cell proliferation in an enzymatic activity–dependent manner. The young VSMCs were as described in Figure 1. A, SA‐β‐gal staining for senescent cells. B, Annexin‐V‐FLUOS staining for detection of apoptotic cells. C, Immunoblotting analysis of p53‐S15, p53, PCNA, and Arg‐II levels. Tubulin served as a loading control. D, Representative images showing immunofluorescence staining of PCNA level (red) in VSMCs followed by counterstaining with DAPI (blue). The merged images are also shown. Bar graphs show quantifications of the corresponding signals (n=6 or 3). *P<0.05, **P<0.01, ***P<0.001 vs control. Scale bar=0.2 mm. Arg‐II indicates arginase‐II; VSMC, vascular smooth muscle cell; con, control; SA‐β‐gal, senescence‐associated β‐galactosidase; PCNA, proliferating cell nuclear antigen; DAPI, 4′6‐diamidino‐2‐phenyl‐indole, dihydrochloride.
Arg‐II overexpression activates p66Shc through S6K1 in VSMC independently of its enzymatic activity
To elucidate the mechanisms of Arg‐II in inducing VSMC mitochondrial dysfunction, cell senescence, and apoptosis, the effects of Arg‐II on the mTORC1‐S6K1 signaling pathway and the mitochondrial redox protein p66Shc were investigated. The cells overexpressing Arg‐II reveal enhanced levels of S6K1‐T389, S6‐S235/236, and p66Shc‐S36, as well as of total p66Shc (Figure 4A). Similar effects were obtained with the inactive Arg‐II mutant H160F (Figure 4A), demonstrating an Arg‐II enzymatic activity–independent effect on activation of S6K1 and p66Shc. Furthermore, treatment of the young VSMCs with the arginase inhibitors BEC (200 μmol/L) or nor‐NOHA (50 μmol/L) overnight did not affect the stimulating effect of Arg‐II overexpression on S6K1 and p66Shc activation (Figure 4B). These results further support our conclusion on the enzymatic activity–independent effect of Arg‐II on S6K1 and p66Shc. Moreover, silencing S6K1 abrogated the Arg‐II‐induced increase in p66Shc‐S36 and total p66Shc levels, whereas elevated p53‐S15 on Arg‐II overexpression was not affected by silencing S6K1 (Figure 5). These results demonstrate that Arg‐II activates p66Shc through S6K1 signaling, whereas activation of p53 by Arg‐II is independent of S6K1 in the VSMCs (please also refer to the scheme in Figure 17A).
Figure 4.
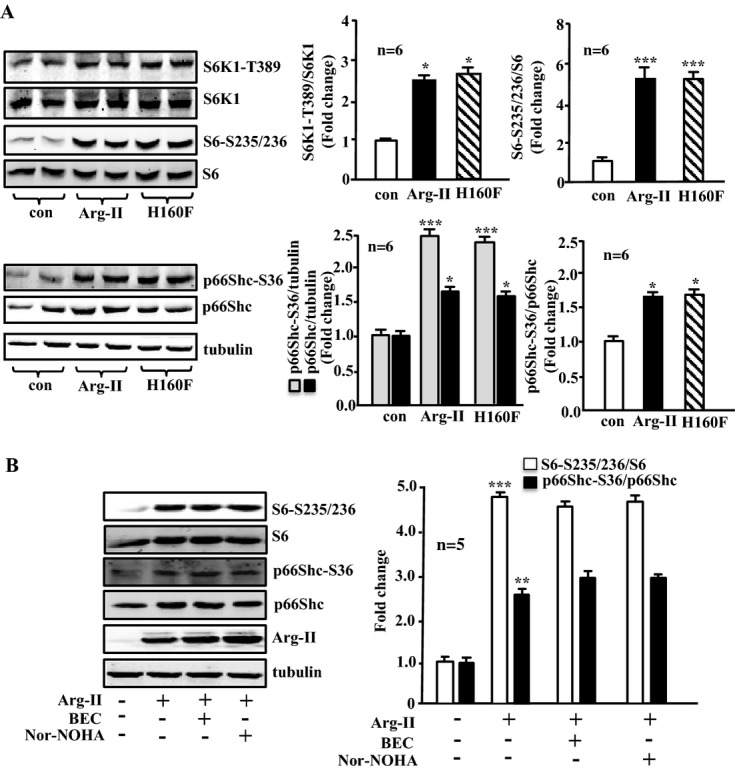
Overexpression of Arg‐II in young VSMCs activates S6K1 and p66Shc independently of its enzymatic activity. A, Young VSMCs were transduced as in Figure 1. Shown is immunoblotting analysis of phosphorylated S6K1‐T389 and total S6K1, phosphorylated S6‐S235/236 and total S6, and phosphorylated p66Shc‐S36 and total p66Shc levels. Tubulin served as a loading control. B, Young VSMCs were transduced with empty vector rAd/CMV as control (Arg‐II: −) or rAd/CMV‐Arg‐II (Arg‐II: +). Cells were treated with or without the arginase inhibitor BEC (200 μmol/L) or nor‐NOHA (50 μmol/L) overnight during serum starvation before cell lysate preparation 72 hours posttransduction. Shown is the immunoblotting analysis of phosphorylated S6‐S235/236 and total S6, phosphorylated p66Shc‐S36, and total p66Shc levels and Arg‐II level. Tubulin served as a loading control. Bar graphs show quantifications of the signals (n=6 in A, n=5 in B). *P<0.05, **P<0.01, ***P<0.001 vs control. Arg‐II indicates arginase‐II; con, control; VSMC, vascular smooth muscle cell; rAd, recombinant adenovirus; BEC, S‐12‐bromoethyl‐l‐cystine; Nor‐NOHA, Nω‐hydroxy‐nor‐l‐arginine.
Figure 5.
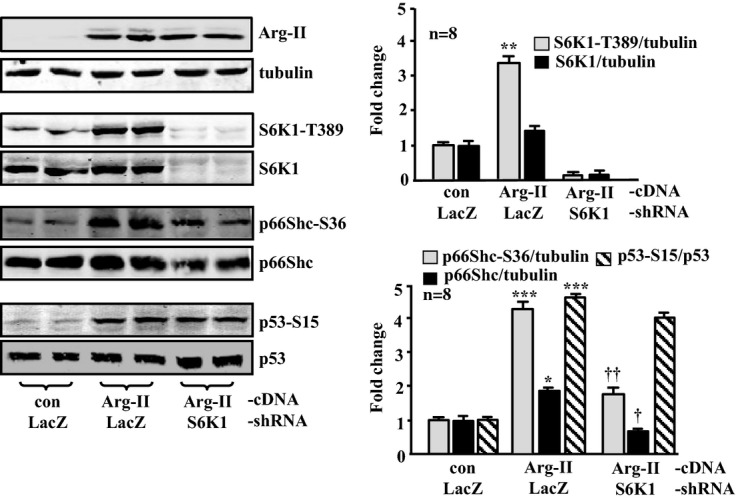
Silencing S6K1 in young VSMCs attenuates Arg‐II‐induced activation of p66Shc, but not that of p53. The young VSMCs were first transduced either with rAd/U6‐LacZshRNA as control or rAd/U6‐S6K1shRNA. Twenty‐four hours after the first transduction with rAd/U6‐shRNA, the cells were then transduced either with rAd/CMV as control (con) or with rAd/CMV‐Arg‐II to overexpress Arg‐II. Experiments were performed 72 hours after the second transduction (48 hours in 10% FCS‐DMEM plus overnight serum starvation in 0.2% BSA‐DMEM). Shown are immunoblotting analyses of Arg‐II levels and tubulin, S6K1‐T389 and total S6K1, p66Shc‐S36 and total p66Shc, and p53‐S15 and total p53. Tubulin served as a loading control. Quantification of the signals is shown in the bar graphs (n=8). *P<0.05, **P<0.01, ***P<0.001 vs control; †P<0.05, ††P<0.01 vs Arg‐II‐cDNA+LacZshRNA. Arg‐II indicates arginase‐II; VSMC, vascular smooth muscle cell; rAd, recombinant adenovirus.
Silencing p66Shc prevents the effects of Arg‐II overexpression in VSMCs
We further investigated whether p66Shc mediates Arg‐II‐induced mitochondrial dysfunction, cell senescence, and cell apoptosis. For this purpose, p66Shc was knocked down in the VSMCs overexpressing Arg‐II. The expression of Arg‐II and the silencing effect of p66Shc were validated by immunoblotting (Figure 6). The results showed that silencing p66Shc does not affect the effect of Arg‐II overexpression‐mediated activation of S6K1 or p53‐S15 (Figure 6), demonstrating that p66Shc functions downstream of S6K1 and that p53 activation by Arg‐II is independent of the S6K1‐p66Shc pathway (please also refer to the scheme in Figure 17A). Moreover, silencing p66Shc abolished mitochondrial H2O2 production (Figure 7A) and prevented the decrease in Δψm (Figure 7B) as well as the cell senescence (Figure 7C) and apoptosis (Figure 7D) induced by Arg‐II overexpression in the young VSMCs, demonstrating that p66Shc indeed mediates the effects of Arg‐II in VSMC.
Figure 6.
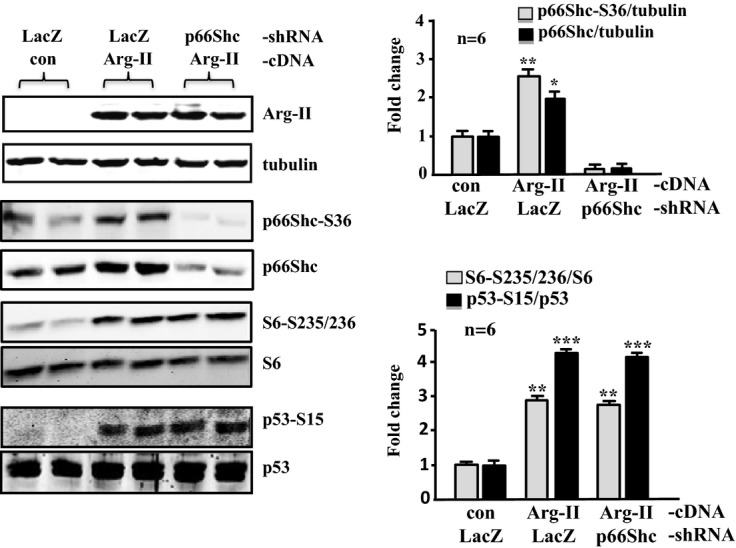
Silencing p66Shc in young VSMCs does not affect Arg‐II‐induced activation of S6K1 or p53. The young VSMCs were first transduced either with rAd/U6‐LacZshRNA as control or rAd/U6‐p66ShcshRNA. Twenty‐four hours after the first transduction with rAd/U6‐shRNA, cells were then transduced either with rAd/CMV as control (con) or rAd/CMV‐Arg‐II. Experiments were carried out 72 hours after the second transduction. Immunoblotting analysis (left) reveals the overexpression of Arg‐II, the silencing efficiency of p66Shc, and the effects on S6‐S235/236 and S6, p53‐S15, and total p53. Tubulin served as a loading control. Quantification of the signals is shown in the bar graphs on the right (n=6). *P<0.05, **P<0.01, ***P<0.001 vs control. Arg‐II indicates arginase‐II; VSMC, vascular smooth muscle cell; rAd, recombinant adenovirus; SA‐β‐gal, senescence‐associated β‐galactosidase; H2DCF, 2′,7′‐dichlorofluorescein.
Figure 7.
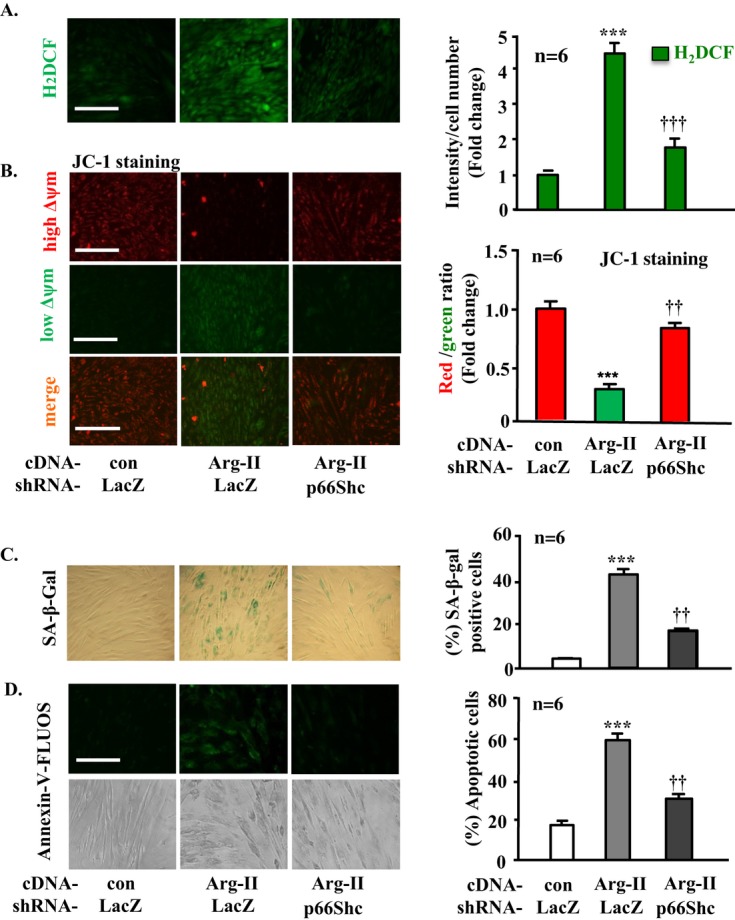
Silencing p66Shc in young VSMCs prevents Arg‐II‐induced mitochondrial dysfunction and cell senescence and apoptosis. The young VSMCs were transduced as described in Figure 6. A, H2DCF staining for detection of H2O2. B, Δψm Assessment by JC‐1 staining. C, SA‐β‐gal staining for senescent cells. D, Annexin‐V‐FLUOS staining for apoptotic cells. Quantification of the signals is shown in the corresponding bar graphs (n=6). ***P<0.001 vs control; ††P<0.01, †††P<0.001 vs Arg II. Scale bar=0.2 mm. Arg‐II indicates arginase‐II; VSMC, vascular smooth muscle cell; con, control; SA‐β‐gal, senescence‐associated β‐galactosidase; H2DCF, 2′,7′‐dichlorofluorescein; rAd, recombinant adenovirus.
Mechanisms of S6K1‐induced p66Shc activation in VSMCs
Because PKC‐β, ERK, and JNK have been reported to be involved in phosphorylation of p66Shc‐S36,16–18 we further investigated whether these signaling molecules are involved in S6K1‐mediated phosphorylation/activation of p66Shc in the young cells overexpressing Arg‐II. Treatment of the cells with the specific inhibitors of these signaling pathways overnight showed that the Arg‐II overexpression–induced activation of S6K1 (monitored by increased S6‐S235/236) was not affected by the inhibitors of these signaling pathways including the PKC‐α/β1 inhibitor Gö6976 (1 μmol/L; Figure 8A), PKC‐β2 inhibitor CGP53353 (1 μmol/L; Figure 8B), ERK inhibitor PD98059 (50 μmol/L; Figure 8A), and JNK inhibitor SP600125 (20 μmol/L; Figure 8A). However, activation of p66Shc (p66Shc‐S36) by Arg‐II overexpression was inhibited by PD98059 or SP600125, but not by Gö6976 (Figure 8A) or by CGP53353 (Figure 8B). These results demonstrate that ERK and JNK (not PKC‐α/β), in addition to S6K1 (see Figure 5), are required for Arg‐II‐induced activation of p66Shc.
Figure 8.
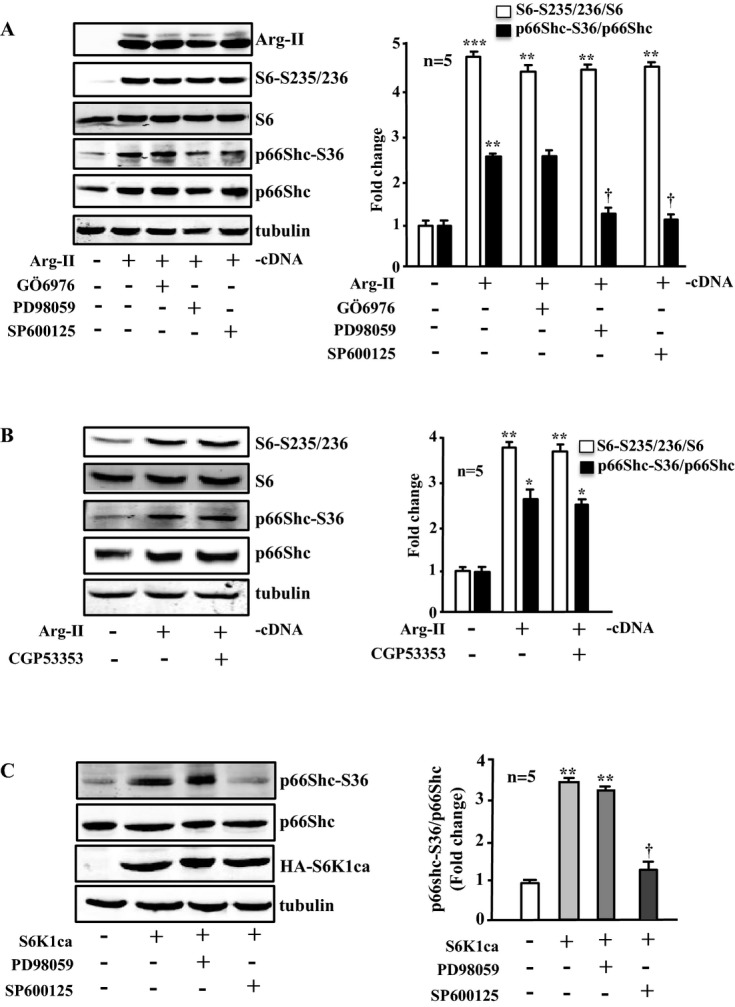
Effects of signaling inhibitors on Arg‐II‐ or S6K1‐induced phosphorylation of p66Shc‐S36. Immunoblotting analysis of the signaling and protein levels as indicated. A and B, Young VSMCs were transduced with empty‐vector rAd/CMV as control (Arg‐II: −) or rAd/CMV‐Arg‐II (Arg‐II: +). Cells were treated with or without the signaling inhibitor overnight during serum starvation before cell lysate preparation 72 hours posttransduction. Cells were treated with Gö6976 (1 μmol/L), PD98059 (50 μmol/L), or SP600125 (20 μmol/L) or with CGP53353 (1 μmol/L) as indicated. C, Young VSMCs were transduced with empty‐vector rAd/CMV as control (S6K1ca: −) or rAd/CMV‐HA‐S6K1ca (an active mutant of S6K1; S6K1ca: +). Cells were treated with PD98059 or SP600125 as described in A and B. Quantification of the signals is shown in the corresponding bar graphs (n=5). *P<0.05, **P<0.01, ***P<0.001 vs control; †P<0.05 vs Arg‐II (in A) or vs S6K1ca (in C). Arg‐II indicates arginase‐II; VSMC, vascular smooth muscle cell; SA‐β‐gal, senescence‐associated β‐galactosidase; H2DCF, 2′,7′‐dichlorofluorescein; rAd, recombinant adenovirus.
The relationship between S6K1, ERKs, and JNK in activation of p66Shc was further investigated in the young cells overexpressing an active S6K1 mutant (S6K1ca). As shown in Figure 8C, the increase in p66Shc‐S36 level on overexpression of S6K1ca was not affected by the ERK inhibitor PD98059, but was abolished by the JNK inhibitor SP600125. These results together with those presented in Figure 5 demonstrate that Arg‐II overexpression in young cells activates p66Shc, requiring 2 independent pathways, the S6K1‐JNK and ERK pathways (please refer to the scheme presented in Figure 17A).
Effects of Arg‐II on VSMCs are dependent on p66Shc‐S36 phosphorylation and p53
Next, we investigated whether the effects of p66Shc are dependent on its phosphorylation at S36. For this purpose, the dominant‐negative p66Shc mutant in which serine‐36 was mutated to alanine (S36A)30 was expressed together with Arg‐II in the young VSMCs (Figure 9A, left). The results showed that the stimulating effects of Arg‐II on H2O2 generation (Figure 9B), mitochondrial dysfunction (Figure 9C), cell senescence (Figure 9D), and cell apoptosis (Figure 9E) are further enhanced by coexpression of wild‐type p66Shc but are significantly inhibited by coexpression of the S36A mutant, demonstrating the functional importance of the phosphorylation of p66Shc at S36 in the cellular effects mediated by Arg‐II.
Figure 9.
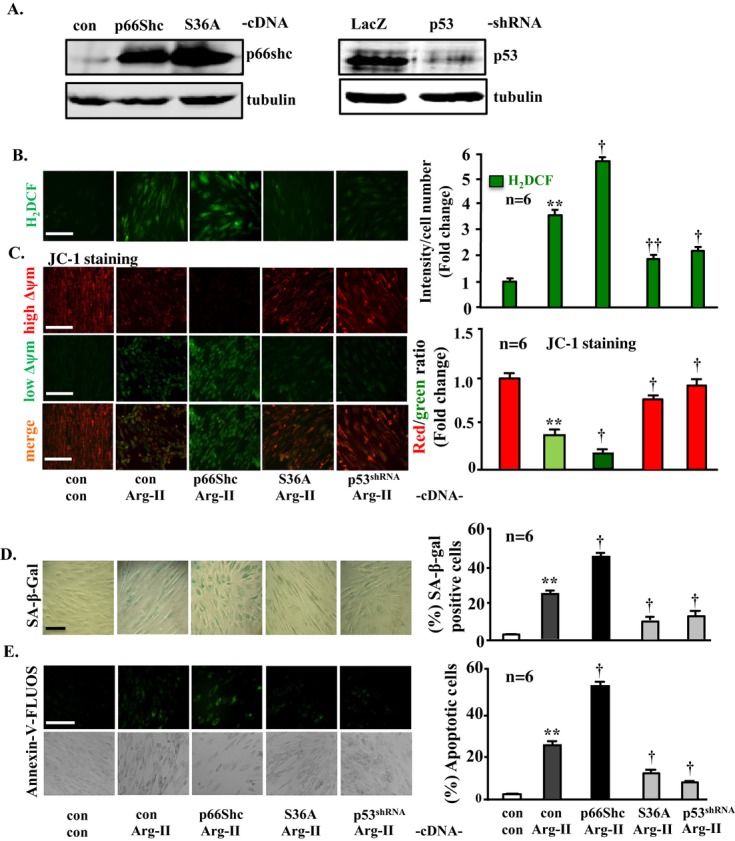
Phosphorylation of p66Shc at S36 as well as p53 signaling is required for the actions of Arg‐II in young VSMCs. The young VSMCs were first transduced with either rAd/CMV empty vector as control (con) or rAd/CMV‐p66Shc, ‐p66Shc‐S36A; rAd/U6‐LacZshRNA, or rAd/U6‐p53shRNA. Twenty‐four hours after the first transduction, the cells were then transduced with either rAd/CMV as control (con) or rAd/CMV‐Arg‐II to overexpress Arg‐II as indicated. Experiments were performed 72 hours after the second transduction. A, Immunoblotting analysis showing the overexpression of p66Shc and p66Shc‐S36A (left) and the efficient silencing of p53 (right). B, H2DCF staining for detection of H2O2. C, Δψm assessment by JC‐1 staining. D, SA‐β‐gal staining for senescent cells. E, Annexin‐V‐FLUOS staining for apoptotic cells. Quantification of the signals is shown in the corresponding bar graphs (n=6). **P<0.01 vs control; †P<0.05, ††P<0.01 vs Arg II. Scale bar=0.2 mm. Of note, in B through E, the images of the experimental group with LacZshRNA+Arg‐II are not shown because of space limitation and because the data are similar to those of the experimental group with con+Arg‐II. Arg‐II indicates arginase‐II; VSMC, vascular smooth muscle cell; SA‐β‐gal, senescence‐associated β‐galactosidase; H2DCF, 2′,7′‐dichlorofluorescein; rAd, recombinant adenovirus.
Furthermore, we examined whether p53, which is activated by Arg‐II independently from the S6K1‐JNK‐p66Shc pathway, is also involved in the actions of Arg‐II in VSMCs. Silencing p53 also inhibited Arg‐II‐mediated cellular effects including H2O2 production, mitochondrial dysfunction, senescence, and apoptosis (Figure 9A through 9E, the last lane or bar). These data demonstrate that both the S6K1‐JNK‐p66Shc and p53 pathways, which are independently activated by Arg‐II as shown by above experiments, are required for the actions of Arg‐II in VSMCs.
Effects of Silencing Arg‐II in Senescent VSMCs
Enhanced activation of Arg‐II‐S6K1‐p66Shc and p53 and decreased cell proliferation in senescent VSMCs
Next, we examined the effects of Arg‐II in senescent VSMCs. Compared with the young VSMCs, the senescent cells exhibited elevated arginase activity, which is associated with predominantly increased expression of Arg‐II (Arg‐I expression was very low, although it was also increased in senescent cells; Figure 10), suggesting that Arg‐II plays a dominant role, which is similar to that in human endothelial cells.36 Enhanced levels of S6K1‐T389, total S6K1, p66Shc‐S36, total p66Shc, p53‐S15, and total p53 were also observed, whereas the PCNA level was decreased in the senescent VSMCs (Figure 10), which is in line with the irreversible growth arrest characteristic of cell senescence.
Figure 10.
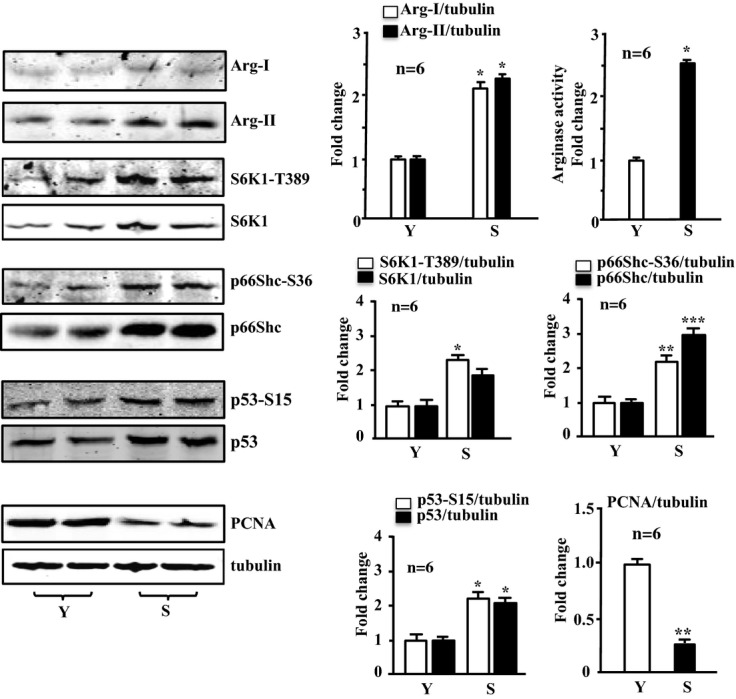
Enhanced expression and activity of arginases, S6K1, p66Shc, and p53 and decreased PCNA level in senescent VSMCs. Immunoblotting analysis of Arg‐I, Arg‐II, S6K1‐T389 and total S6K1, p66Shc‐S36 and total p66Shc, p53‐S15 and total p53, and PCNA expression, as well as arginase activity in the senescent VSMCs (S) compared with the young cells (Y). Quantification of the signals is shown in the corresponding bar graphs (n=6). *P<0.05, **P<0.01, ***P<0.001 vs young cells (Y). Arg‐II indicates arginase‐II; VSMC, vascular smooth muscle cell; PCNA, proliferating cell nuclear antigen; S6K1, ribosomal protein S6 kinase 1.
Effects of silencing Arg‐II or p53 and the importance of p66Shc‐S36 in senescent cell dysfunctions
Silencing Arg‐II in the senescent VSMCs suppressed almost all the senescence‐associated signaling, including S6K1‐T389 and total S6K1, S6‐S235/236 and total S6, p66Shc‐S36, and total p66Shc, as well as p53‐S15, but not total p53 (Figure 11A). The cell proliferation, as assessed by PCNA level with immunoblotting and immunostaining, which was decreased in the senescent VSMCs as shown in Figure 10, was further significantly reduced by Arg‐II silencing (Figure 11B), demonstrating that Arg‐II supports VSMC proliferation, which is in accordance with previously published studies28 and also in line with the finding in young cells (Figure 3C and 3D). Moreover, silencing Arg‐II in the senescent VSMCs reduced H2O2 generation, restored mitochondrial Δψm, and decreased SA‐β‐gal‐positive cell number and apoptosis (Figure 12).
Figure 11.
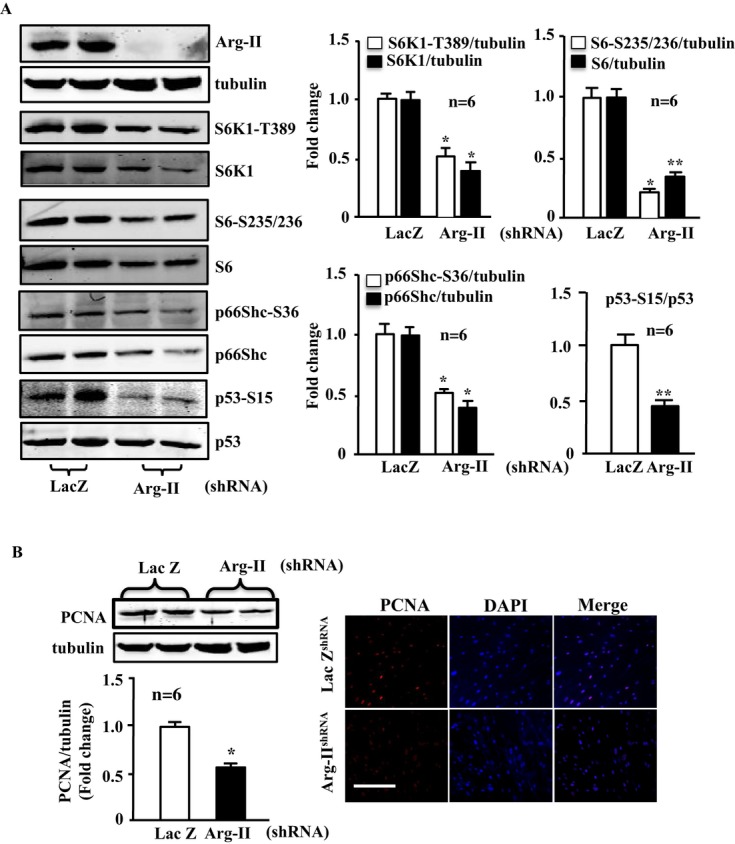
Effects of silencing Arg‐II in senescent VSMCs on activation of S6K1‐p66Shc and p53 and on PCNA level. Senescent VSMCs were transduced with either rAd/U6‐LacZshRNA or rAd/U6‐Arg‐IIshRNA. Seventy‐two hours after transduction cell lysates were prepared and subjected to (A) immunoblotting analysis of Arg‐II, tubulin, S6K1‐T389 and total S6K1, S6‐S235/236 and total S6, p66Shc‐S36 and total p66Shc, p53‐S15, and total p53. Quantification of the signals is shown in the right bar graphs (n=6). *P<0.05, **P<0.01 vs young cell (Y). B, Immunoblotting analysis and immunofluorescence staining of PCNA. Quantification of the immunoblotting signals of PCNA is shown in the bar graphs underneath the immunoblotting. *P<0.05 vs LacZ‐shRNA. Arg‐II indicates arginase‐II; VSMC, vascular smooth muscle cell; PCNA, proliferating cell nuclear antigen; DAPI, 4′6‐diamidino‐2‐phenyl‐indole, dihydrochloride.
Figure 12.
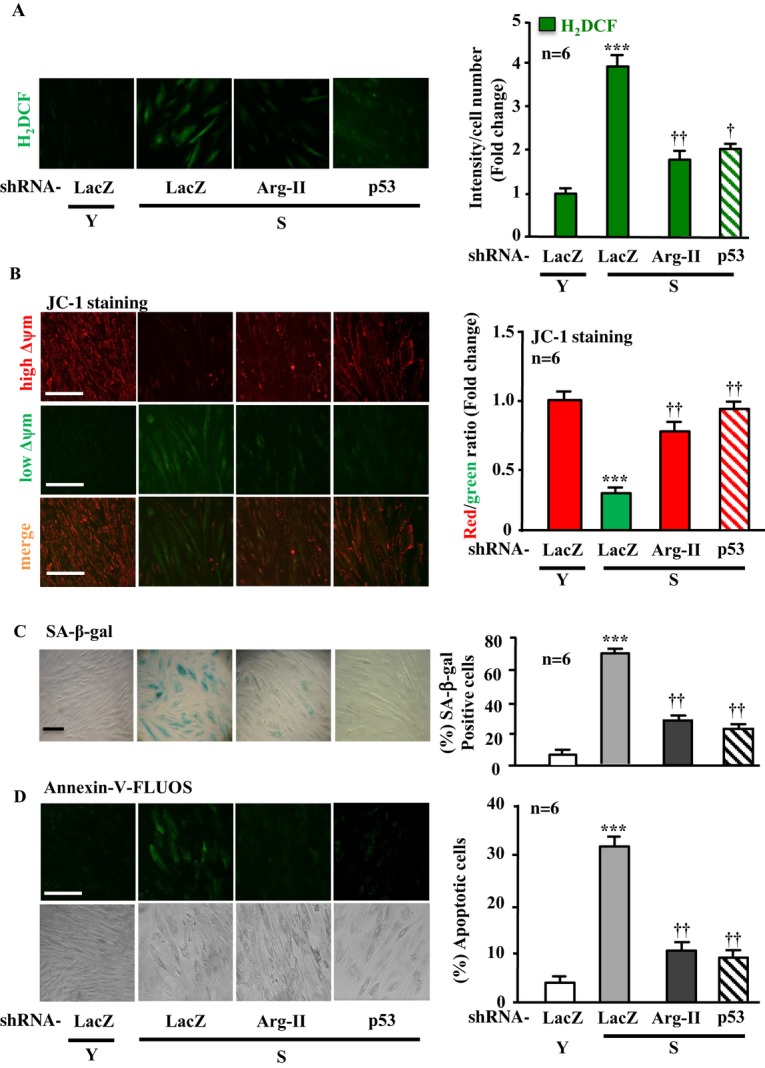
Silencing Arg‐II or p53 in senescent VSMCs prevents H2O2 production and decreased Δψm as well as senescence and apoptosis. Young (Y) and senescent (S) VSMCs were transduced with either rAd/U6‐LacZshRNA or rAd/U6‐Arg‐IIshRNA as indicated. Seventy‐two hours posttransduction, the cells were subjected to (A) H2DCF staining for detection of H2O2. B, JC‐1 staining for analysis of Δψm. C, SA‐β‐gal staining for senescent cells. D, Annexin‐V‐FLUOS staining for apoptotic cells. Quantification of the signals is shown in the corresponding bar graphs (n=6). ***P<0.001 vs Y+LacZshRNA; †P<0.05, ††P<0.01 vs S+LacZshRNA. Scale bar=0.2 mm. Arg‐II indicates arginase‐II; VSMC, vascular smooth muscle cell; H2DCF, 2′,7′‐dichlorofluorescein; SA‐β‐gal, senescence‐associated β‐galactosidase.
In agreement with the finding in young VSMCs that p53 is required for Arg‐II‐induced cellular senescence and apoptosis (Figure 9), silencing p53 in the senescent cells exerted effects similar to silencing Arg‐II (Figure 12, the last lane). Furthermore, over expression of wild‐type p66Shc in the senescent VSMCs, which showed high Arg‐II level and activity (Figure 10), further enhanced H2O2 generation, mitochondrial dysfunction, cell senescence, and cell apoptosis (Figure 13), whereas over expression of the p66Shc‐S36A in the cells significantly reduced these parameters (Figure 13A through 13D, last lane), further confirming the importance of S36 phosphorylation of p66Shc in the regulation of cellular functions. These results together with those shown in Figure 11 demonstrate that enhanced Arg‐II in senescent VSMCs accounts for deregulation of S6K1‐p66Shc and p53, leading to mitochondrial dysfunction, cell senescence, and cell apoptosis.
Figure 13.
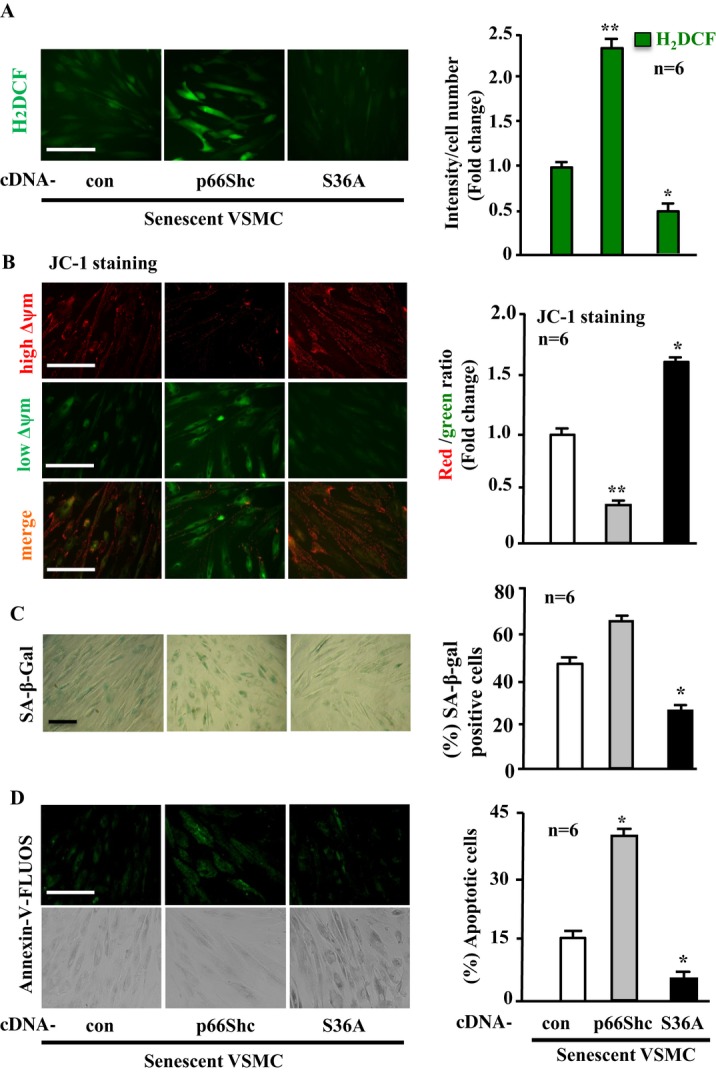
Enhanced phosphorylation of p66Shc‐S36 accounts for the phenotypic changes of senescent VSMCs. Senescent VSMCs were transduced with either rAd/CMV empty vector as control (con), rAd/CMV‐p66Shc or ‐p66Shc‐S36A (S36A) as indicated. Seventy‐two hours posttransduction, the cells were subjected to (A) H2DCF staining for detection of H2O2. B, JC‐1 staining for analysis of Δψm. C, SA‐β‐gal staining for senescent cells. D, Annexin‐V‐FLUOS staining for apoptotic cells. Quantification of the signals is shown in the corresponding bar graphs (n=6). *P<0.05, **P<0.01 vs con. Scale bar=0.2 mm. Arg‐II indicates arginase‐II; VSMC, vascular smooth muscle cell; H2DCF, 2′,7′‐dichlorofluorescein; SA‐β‐gal, senescence‐associated β‐galactosidase; rAd, recombinant adenovirus.
Role of JNK and ERK in S6K1 and p66Shc activation in senescent cells
The interaction between S6K1, JNK, and ERKs in the activation of p66Shc was also analyzed in the senescent VSMCs, which exhibited enhanced Arg‐II‐S6K1‐p66Shc signaling (Figure 10). In comparison with the young cells, in the senescent cells, the ERK inhibitor PD98059 or the JNK inhibitor SP600125 not only inhibited p66Shc‐S36, but also reduced S6K1 activity (Figure 14A), suggesting that ERK and JNK also act upstream of S6K1 in the senescent cells (please refer to Figure 17B). Moreover, silencing p66Shc in senescent VSMCs reduced activation of S6K1 and ERK and also reduced Arg‐II expression (Figure 14B). These results demonstrate that in senescent VSMCs, p66Shc also activates ERK, JNK, and S6K1, which upregulate expression of Arg‐II. These results suggest complex positive crosstalk among all the signaling components in senescent VSMCs (see Figure 17B).
Figure 14.
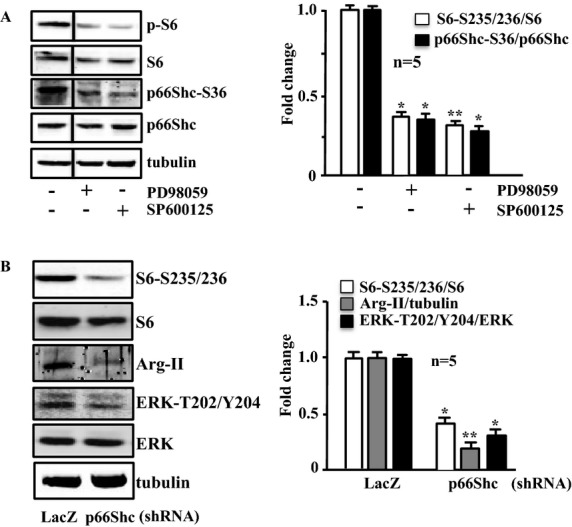
Interaction between S6K1, JNK, ERKs, and p66Shc in senescent VSMCs. Immunoblotting analysis of the parameters as indicated. A, Senescent VSMCs were either untreated (−) or treated with PD98059 (50 μmol/L) or SP600125 (20 μmol/L) overnight. The line between the first and second lanes indicates cutting of the same blots. B, Senescent VSMCs were transduced with either rAd/U6‐LacZshRNA or rAd/U6‐p66ShcshRNA as indicated. Experiments were performed 72 hours posttransduction. Quantification of the signals is shown in the corresponding bar graphs (n=5). *P<0.05, **P<0.01 vs untreated (in A) or LacZshRNA (in B). Arg‐II indicates arginase‐II; VSMC, vascular smooth muscle cell; JNK, c‐Jun N‐terminal kinases; ERK, extracellular signal‐regulated kinases; rAd, recombinant adenovirus.
Genetic ablation of Arg‐II inhibits activation of S6K1‐p66Shc as well as p53 and protects against cell apoptosis in atherosclerotic plaque
To further confirm the role of Arg‐II‐S6K1‐p66Shc in inducing VSMC apoptosis linked to cardiovascular diseases such as atherosclerosis and plaque vulnerability in vivo, we interbred Arg‐II−/− mice with atherosclerosis‐prone ApoE−/− mice and obtained ApoE−/−Arg‐II−/− double‐knockout mice.20 To accelerate the atherosclerotic lesion formation, ApoE−/−Arg‐II−/− mice (at age 10 weeks) were fed a HF diet for 10 weeks. Immunoblotting analysis with the aortas revealed reduced levels of S6K1, p66Shc‐S36, p66Shc, p53‐S15, p53, S6‐S235/236, and S6 in ApoE−/−Arg‐II−/− mice compared with the ApoE−/−Arg‐II+/+ control animals (Figure 15). Costaining of these proteins with α‐SMA revealed that these proteins are present in the VSMCs of aortic roots with atherosclerotic plaques, which was more pronounced in the ApoE−/−Arg‐II+/+ controls than in the ApoE−/−Arg‐II−/− mice (Figure 16A). A reduced number of apoptotic VSMCs was observed in the plaques of ApoE−/−Arg‐II−/− mice (Figure 16B), which is in agreement with the previous report of more stable plaque phenotypes in ApoE−/−Arg‐II−/− mice, that is, less macrophage accumulation, less production of proinflammatory cytokines, and smaller necrotic cores in ApoE−/−Arg‐II−/− mice compared with ApoE−/−Arg‐II+/+ mice.20 These data provide in vivo evidence of an important role for Arg‐II in inducing VSMC apoptosis through activation of S6K1‐p66Shc and p53, contributing to plaque vulnerability (Figure 17).
Figure 15.
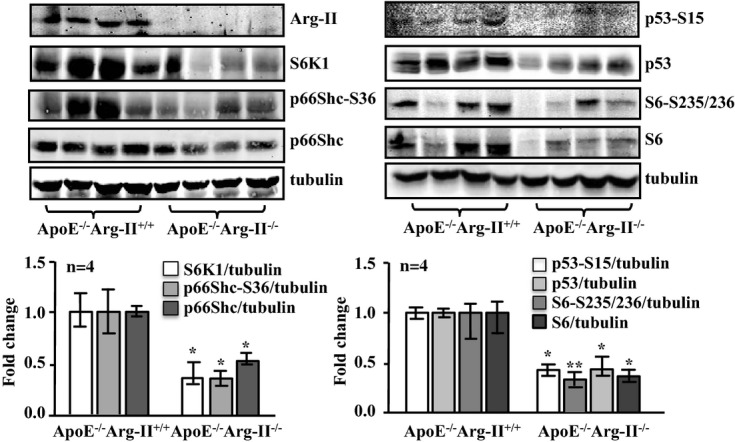
Ablation of Arg‐II in atherosclerosis‐prone ApoE−/− mice reduces signaling of S6K1, p66Shc, and p53 in the aortas. ApoE−/−Arg‐II+/+ and ApoE−/−Arg‐II−/− mice were fed a high‐fat diet for 10 weeks. Aortas from mice of both genotypes were cleaned of perivascular tissues and subjected to immunoblotting analysis of Arg‐II, S6K1, p66Shc‐S36 and total p66Shc, p53‐S15 and total p53, and S6‐S235/236 and total S6. Tubulin served as a loading control. Quantification of the signals is shown in the corresponding lower panels (n=4). *P<0.05, **P<0.001 vs ApoE−/−Arg‐II+/+. Arg‐II indicates arginase‐II; ApoE, apolipoprotein E; S6K1, ribosomal protein S6 kinase 1.
Figure 16.
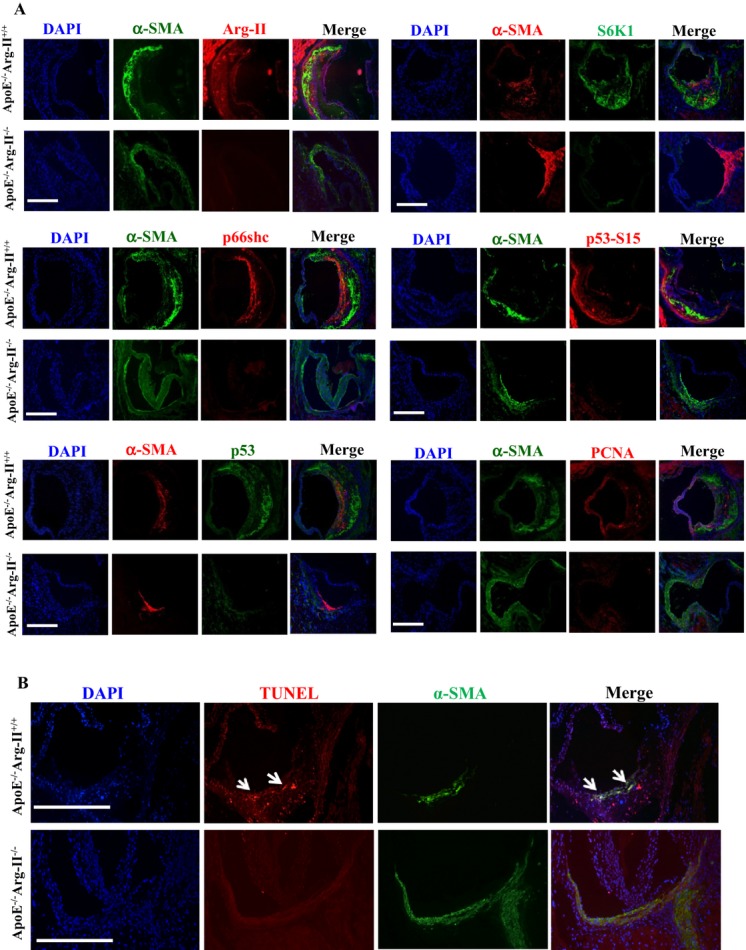
Ablation of Arg‐II in atherosclerosis‐prone ApoE−/− mice reduces levels of Arg‐II, S6K1, p66Shc, p53‐S15, and p53, as well as PCNA in VSMCs along with decreased apoptotic VSMCs in atherosclerotic plaque. ApoE−/−Arg‐II+/+ and ApoE−/−Arg‐II−/− mice were fed a high‐fat diet for 10 weeks. A, AR‐cryosections (7 μm) were subjected to immunofluorescence costaining of Arg‐II, S6K1, p66Shc, p53‐S15 and p53, and PCNA with anti‐α‐smooth muscle actin antibody (α‐SMA) for VSMCs. Mouse anti‐α‐SMA (green) was used for costaining with rabbit antibodies against Arg‐II, p66Shc, p53‐S15, and PCNA (red), whereas rabbit anti‐α‐SMA (red) was used for costaining with mouse anti‐p53 and ‐S6K1 (green). Alexa Fluor 488–conjugated goat anti‐mouse IgG (green) and Alexa Fluor 594–conjugated goat anti‐rabbit F(ab)2 (red) were used as secondary antibodies. All sections were counterstained with DAPI. Representative images of individual staining and merged images are shown (n=4). B, Representative images showing apoptotic cells detected by TUNEL staining (red) in the plaques in aortic roots of ApoE−/−Arg‐II+/+ and ApoE−/−Arg‐II−/− mice. All sections were stained for VSMCs with anti‐α‐SMA (green) followed by counterstaining with DAPI for nuclei (blue). The merged images are also shown (n=4). The white arrows indicate apoptotic VSMCs. Scale bars=0.2 mm. Arg‐II indicates arginase‐II; ApoE, apolipoprotein E; S6K1, ribosomal protein S6 kinase 1; VSMC, vascular smooth muscle cell; PCNA, proliferating cell nuclear antigen; DAPI, 4′6‐diamidino‐2‐phenyl‐indole, dihydrochloride; TUNEL, terminal dUTP nick end‐labeling.
Figure 17.
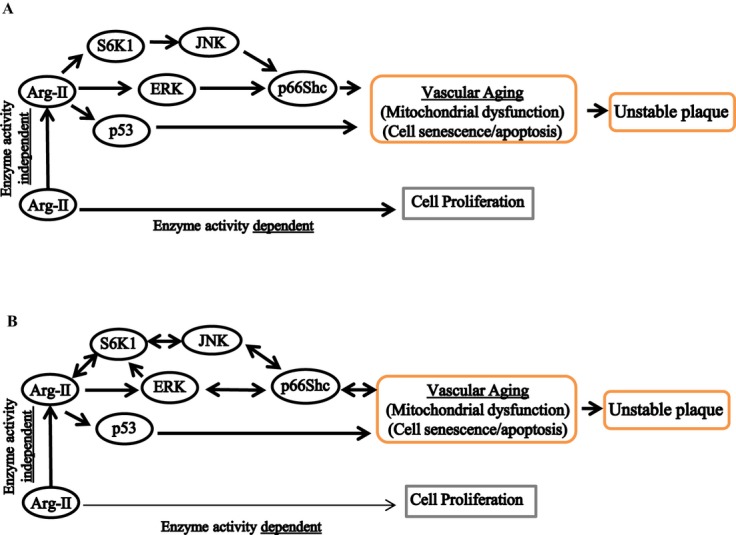
Schematic summary of the major findings of the study. A, Elevated Arg‐II leads to phosphorylation/activation of p66Shc through S6K1‐JNK and ERK in parallel. In addition, Arg‐II induces phosphorylation/activation of p53 independently of the S6K1‐JNK‐p66Shc pathway. Both S6K1‐JNK‐p66Shc and p53 are involved in Arg‐II‐induced H2O2 and mitochondrial dysfunction, which ultimately cause both VSMC senescence and apoptosis, contributing to plaque vulnerability. Arg‐II exerts all these effects independently of its l‐arginine ureahydrolase activity, whereas it is required for VSMC proliferation in an enzymatic activity–dependent manner. B, In senescent VSMCs, Arg‐II, S6K1, JNK, ERK, and p66Shc form a complex positive crosstalk network resulting in acceleration of VSMC aging. Arg‐II indicates arginase‐II; S6K1, ribosomal protein S6 kinase 1; VSMC, vascular smooth muscle cell; JNK, c‐Jun N‐terminal kinases; ERK, extracellular signal‐regulated kinases.
Discussion
Previous studies including our own demonstrated that vascular endothelial arginase is increased in numerous physiopathological conditions and plays an important role in endothelial dysfunction in aging, diabetes, and atherosclerosis.21,36,38–39 In endothelial cells, Arg‐II induces an increase in ROS and a decrease in NO production resulting from eNOS uncoupling, which promotes endothelial senescence and inflammation.21 Moreover, our recent study demonstrated that Arg‐II also promotes macrophage proinflammatory responses through inducing mitochondrial ROS and that genetic deficiency in Arg‐II in ApoE−/− mice reduces atherosclerotic lesion formation and confers features of stable plaques such as reduction in inflammatory macrophage accumulation, cytokine production, and necrotic cores.20 In the present study, we uncovered a novel mechanism by which Arg‐II, independently of its l‐arginine ureahydrolase activity, promotes mitochondrial dysfunction leading to VSMC senescence/apoptosis through complex positive crosstalk among S6K1‐JNK, ERK, p66Shc, and p53, contributing to atherosclerotic vulnerability phenotypes in mice.
Arginase Activity–Independent and –Dependent Effects in VSMCs
One of the major findings of the study is the l‐arginine ureahydrolase activity–independent functions of Arg‐II. In agreement with the role of Arg‐II in promoting cell proliferation through its enzymatic activity,28 we found that overexpression of wild‐type Arg‐II, but not of inactive Arg‐II, in young VSMCs enhances cell proliferation. In contrast to its effect on cell proliferation, Arg‐II activates S6K1‐p66Shc and p53 and promotes H2O2 production and mitochondrial dysfunction, leading to senescence and apoptosis independently of its enzymatic activity, as all these changes occur not only in VSMCs overexpressing wild‐type Arg‐II, but also in VSMCs overexpressing the inactive Arg‐II mutant. Moreover, the enzymatic activity–independent effects on S6K1‐p66Shc were also confirmed by the lack of the effects of arginase inhibitors. Thus, for the first time, we provide firm evidence that Arg‐II has dual opposing functions in promoting VSMC proliferation and senescence/apoptosis through different mechanisms. While it promotes VSMC proliferation and cytoplasmic and mitochondrial O2•− in an enzymatic activity–dependent manner, it induces H2O2 generation, mitochondrial dysfunction, and senescence/apoptosis through parallel activation of p66Shc and p53 independently of its enzymatic activity. Overall, the induction of senescence/apoptosis seems to be predominant over the induction of proliferation by Arg‐II, especially in senescent VSMCs, in which elevated Arg‐II results in a cellular outcome of senescence/apoptosis rather than proliferation, although Arg‐II is still involved in VSMC proliferation under this condition because silencing Arg‐II in senescent cells not only reduces senescence/apoptosis but also further decreases PCNA. Of note, enzymatic activity–independent effects have also been reported for another urea cycle enzyme, argininosuccinate lyase,40 which acts as a scaffold protein. Whether this is also true for Arg‐II requires further extensive investigation. We wish to point out that Arg‐I is expressed at a much lower level than Arg‐II and that the expression of Arg‐I is also increased in senescent VSMCs. However, silencing Arg‐II in senescent VSMCs or genetic ablation of Arg‐II in mice greatly inhibited the aging‐associated signaling pathways and senescence/apoptosis, demonstrating the predominant role of Arg‐II in VSMCs.
In contrast to endothelial cells, in which Arg‐II affects cell function mainly through interfering with eNOS function, in VSMCs the functional role of Arg‐II is independent of NOS because neither eNOS nor iNOS nor NO production is detectable in VSMCs under our experimental conditions. This is in agreement with our recent study showing that Arg‐II promotes proinflammatory responses in macrophages through enhancing mitochondrial ROS production independently of NOS.20 The effects of Arg‐II on mitochondrial dysfunction are demonstrated by its ability to decrease mitochondrial membrane potential, another hallmark of mitochondrial dysfunction.41 That the inactive mutant of Arg‐II is as potent as wild‐type Arg‐II in inducing H2O2 production and mitochondrial dysfunction demonstrates enzymatic activity–independent effects of Arg‐II and further supports our conclusion on NOS/NO‐independent effects of Arg‐II in VSMCs.
Key Roles of p66Shc and p53 in Arg‐II Enzymatic Activity–Independent Effects in VSMCs
In an attempt to elucidate the enzymatic activity–independent effects of Arg‐II, we have provided evidence that the mitochondrial redox protein p66Shc and p53, which are independently activated by Arg‐II, are the key molecules in mediating Arg‐II‐induced mitochondrial dysfunction independently of its enzymatic activity. This conclusion is supported by the following: (1) in young VSMCs, overexpression of either wild‐type or inactive Arg‐II increases levels of both p66Shc and phosphorylated p66Shc‐S36 as well as p53‐S15 with a concomitant increase in mitochondrial ROS production, a decrease in Δψm, and an increase in senescence/apoptosis; (2) in senescent VSMCs, p66Shc and p66Shc‐S36 as well as p53‐S15 levels are enhanced in parallel with the upregulation of Arg‐II expression and mitochondrial dysfunction and senescence/apoptosis, whereas silencing Arg‐II in senescent VSMCs decreases p66Shc‐S36 and p66Shc as well as p53‐S15 levels with simultaneous inhibition of mitochondrial dysfunction and senescence/apoptosis; (3) silencing p66Shc or p53, or overexpressing p66Shc‐S36A, prevents Arg‐II‐induced mitochondrial dysfunction and senescence/apoptosis; (4) targeted disruption of Arg‐II in atherosclerosis‐prone ApoE−/− mice (ApoE−/−Arg‐II−/− mice) decreases both p66Shc‐S36 and p66Shc as well as p53‐S15, and VSMC apoptosis in the plaque. That the dominant‐negative mutant of p66Shc‐S36A inhibits Arg‐II‐mediated cellular functions in young and senescent VSMCs demonstrates the functional significance of phosphorylation of p66Shc‐S36 in these processes. It is interesting to note that our findings are also supported by the observation that Arg‐II−/− and p66Shc−/− mice display similar protective phenotypes against the development of type 2 diabetes, aging, and atherosclerosis.11,19–21,42 Our results thus have uncovered a novel mechanism that Arg‐II induces mitochondrial dysfunction, leading to senescence/apoptosis in VSMCs through activation of p66Shc and p53. A noteworthy observation is that the activation of p53 by Arg‐II seems independent of the S6K1‐p66Shc pathway, because silencing either S6K1 or p66Shc does not affect Arg‐II‐induced p53 activation. However, its phosphorylation/activation by Arg‐II is also required for the actions of Arg‐II in an enzymatic activity–independent manner. How Arg‐II leads to p53 activation remains to be investigated.
Crosstalk Among Arg‐II, S6K1, JNK, ERK, and p66Shc in VSMC Senescence and Apoptosis
Another novel finding of this study is the role of S6K1 in Arg‐II‐induced p66Shc activation in VSMCs. Compelling evidence has been presented that both S6K1 and p66Shc are important life‐span regulators associated with oxidative stress and are involved in a number of age‐related diseases including type 2 diabetes and atherosclerosis.43 A linkage between p66Shc and S6K1 has been previously demonstrated in adipocytes, in which p66Shc is required for insulin and nutrient‐induced activation of S6K1.42 In our present study, we have provided evidence that S6K1 mediates Arg‐II‐induced activation of p66Shc in VSMCs. Overexpression of Arg‐II enhances S6K1‐T389 and S6‐S235/236 levels in young VSMCs, whereas silencing Arg‐II in senescent VSMCs mitigates S6K1‐T389 and S6‐S235/236 levels. These results are in line with the finding that Arg‐II positively regulates S6K1 in endothelial cells.21 Moreover, silencing S6K1 prevents Arg‐II‐induced elevation of p66Shc‐S36 in VSMCs. It is noteworthy that silencing p66Shc in VSMCs, in contrast to the adipocytes,42 does not significantly affect Arg‐II‐induced S6K1 activation in young VSMCs overexpressing Arg‐II. However, it reduces S6K1 activity in senescent VSMCs. These results demonstrate that p66Shc may operate both downstream and upstream of S6K1 in VSMCs depending on the environmental cues. Evidence has been presented that PKC‐β, ERK, and JNK are involved in phosphorylation of p66Shc‐S36.16–18 In the present study, we identified S6K1 as a protein kinase required for Arg‐II‐induced phosphorylation of p66Shc‐Ser36. In addition to mediating the phosphorylation/activation of p66Shc, S6K1 also upregulates the protein levels of p66Shc, which is in line with its well‐characterized function of promoting protein synthesis. Experiments exploring the interplay between S6K1 and PKC‐β, ERK, and JNK using specific inhibitors of these signaling pathways indicate a positive and complex crosstalk between Arg‐II, S6K1, JNK, ERK, and p66Shc in senescent cells. Although enhanced Arg‐II can activate S6K1‐JNK and ERK, leading to phosphorylation/activation of p66Shc that ultimately causes cell senescence, activated p66Shc in senescent cells can in turn, maybe in conjunction with other altered signaling, activate ERK, JNK, and S6K1, leading to increased expression of Arg‐II. The exact biochemical mechanisms, for example, whether and how S6K1, ERK, JNK, and p66Shc interact with each other leading to cellular senescence and apoptosis, are an interesting topic that requires further extensive investigation.
In Vivo Evidence of Arg‐II‐Induced Activation of S6K1, p66Shc, and p53 in Atherosclerotic Plaque‐Vulnerable Phenotypes in Mice
Persistent activation of S6K1 signaling, p66Shc, p53, and excessive ROS production have been shown to cause cellular senescence and apoptosis through oxidative damage.9–11,9–16,21,35,44–45 In aging and atherosclerotic plaques, cell senescence and apoptosis are found to be present in the vascular wall and are associated with plaque vulnerability.3,46–47 In addition to endothelial cells and macrophages, VSMC senescence and apoptosis are also implicated in unstable plaques.3–5,7,48–49 In support of this notion, we found that Arg‐II, which activates S6K1, p66Shc, and p53, leads to mitochondrial ROS production, VSMC senescence, and apoptosis. Furthermore, the role of Arg‐II in inducing VSMC apoptosis is reinforced by the finding that VSMC apoptosis in aortic plaques is decreased in ApoE−/−Arg‐II−/− mice compared with that in ApoE−/−Arg‐II+/+ control animals, in parallel with the striking decrease in S6K1, p66Shc, and p53 in aortic VSMCs. Noteworthy is that not only phosphorylated levels but also total levels of S6K1, S6, p66Shc, and p53 are strongly reduced in ApoE−/−Arg‐II−/− mice, which is consistent with the results obtained in senescent VSMCs with silencing Arg‐II, although total p53 is not affected in the cells. These may be explained by the finding that S6K1 is implicated in protein translation. The data on VSMC apoptosis and S6K1‐p66Shc and p53 signaling in ApoE−/−Arg‐II−/− mice are in agreement with the observations in our most recently published study showing that, in the atherosclerosis model, genetic ablation of Arg‐II reduces atherosclerotic plaque formation and confers features of stable plaques, that is, reduced macrophage accumulation, lower proinflammatory cytokine production, and decreased necrotic core in the atherosclerotic plaques.20 In addition to enhancing macrophage proinflammatory responses, as reported recently,20 our present study provides evidence that implicates the important role of enhanced Arg‐II in promoting VSMC senescence/apoptosis, contributing to unstable plaque. A definite answer for the specific role of Arg‐II in VSMCs in plaque vulnerability shall be further provided by generating VSMC‐specific Arg‐II‐knockout mouse.
In conclusion, (Figure 17), in line with a previous report,28 our current study has demonstrated the enzymatic activity‐dependent effect of Arg‐II on cell proliferation. Most importantly, the present study has uncovered a novel mechanism of Arg‐II, independently of its l‐arginine ureahydrolase activity, in promoting mitochondrial dysfunction, leading to VSMC senescence and apoptosis that ultimately contribute to atherosclerotic plaque instability through the parallel pathways consisting of Arg‐II–S6K1–JNK‐p66Shc, Arg‐II‐ERK‐p66Shc, and Arg‐II‐p53. Moreover, in senescent VSMCs, Arg‐II, S6K1, JNK, ERK, and p66Shc form a complex positive crosstalk network resulting in acceleration of VSMC aging. From a clinical point of view, along with protection against endothelial dysfunction and macrophage inflammation, targeting Arg‐II also exerts beneficial effects on atherosclerosis through stabilizing atherosclerotic plaque by preventing VSMC senescence and apoptosis.
Sources of Funding
This study was supported by the Swiss National Science Foundation (310030_141070/1), Swiss Heart Foundation. Yuyan Xiong and Yi Yu were supported by the Chinese Scholarship Council.
Disclosures
None.
References
- 1.Libby P, Aikawa M. Stabilization of atherosclerotic plaques: new mechanisms and clinical targets. Nat Med. 2002; 8:1257-1262 [DOI] [PubMed] [Google Scholar]
- 2.Finn AV, Nakano M, Narula J, Kolodgie FD, Virmani R. Concept of vulnerable/unstable plaque. Arterioscler Thromb Vasc Biol. 2010; 30:1282-1292 [DOI] [PubMed] [Google Scholar]
- 3.Wang JC, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ Res. 2012; 111:245-259 [DOI] [PubMed] [Google Scholar]
- 4.Clarke MC, Figg N, Maguire JJ, Davenport AP, Goddard M, Littlewood TD, Bennett MR. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med. 2006; 12:1075-1080 [DOI] [PubMed] [Google Scholar]
- 5.Matthews C, Gorenne I, Scott S, Figg N, Kirkpatrick P, Ritchie A, Goddard M, Bennett M. Vascular smooth muscle cells undergo telomere‐based senescence in human atherosclerosis: effects of telomerase and oxidative stress. Circ Res. 2006; 99:156-164 [DOI] [PubMed] [Google Scholar]
- 6.Gorenne I, Kavurma M, Scott S, Bennett M. Vascular smooth muscle cell senescence in atherosclerosis. Cardiovasc Res. 2006; 72:9-17 [DOI] [PubMed] [Google Scholar]
- 7.Kockx MM, De Meyer GR, Muhring J, Jacob W, Bult H, Herman AG. Apoptosis and related proteins in different stages of human atherosclerotic plaques. Circulation. 1998; 97:2307-2315 [DOI] [PubMed] [Google Scholar]
- 8.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007; 35:495-516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vicencio JM, Galluzzi L, Tajeddine N, Ortiz C, Criollo A, Tasdemir E, Morselli E, Ben Younes A, Maiuri MC, Lavandero S, Kroemer G. Senescence, apoptosis or autophagy? When a damaged cell must decide its path—a mini‐review. Gerontology. 2008; 54:92-99 [DOI] [PubMed] [Google Scholar]
- 10.Macip S, Igarashi M, Berggren P, Yu J, Lee SW, Aaronson SA. Influence of induced reactive oxygen species in p53‐mediated cell fate decisions. Mol Cell Biol. 2003; 23:8576-8585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Napoli C, Martin‐Padura I, de Nigris F, Giorgio M, Mansueto G, Somma P, Condorelli M, Sica G, De Rosa G, Pelicci P. Deletion of the p66Shc longevity gene reduces systemic and tissue oxidative stress, vascular cell apoptosis, and early atherogenesis in mice fed a high‐fat diet. Proc Natl Acad Sci USA. 2003; 100:2112-2116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moiseeva O, Bourdeau V, Roux A, Deschenes‐Simard X, Ferbeyre G. Mitochondrial dysfunction contributes to oncogene‐induced senescence. Mol Cell Biol. 2009; 29:4495-4507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Velarde MC, Flynn JM, Day NU, Melov S, Campisi J. Mitochondrial oxidative stress caused by Sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging. 2012; 4:3-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li N, Ragheb K, Lawler G, Sturgis J, Rajwa B, Melendez JA, Robinson JP. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J Biol Chem. 2003; 278:8516-8525 [DOI] [PubMed] [Google Scholar]
- 15.Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M, Pinton P, Rizzuto R, Bernardi P, Paolucci F, Pelicci PG. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005; 122:221-233 [DOI] [PubMed] [Google Scholar]
- 16.Pinton P, Rimessi A, Marchi S, Orsini F, Migliaccio E, Giorgio M, Contursi C, Minucci S, Mantovani F, Wieckowski MR, Del Sal G, Pelicci PG, Rizzuto R. Protein kinase C beta and prolyl isomerase 1 regulate mitochondrial effects of the life‐span determinant p66Shc. Science. 2007; 315:659-663 [DOI] [PubMed] [Google Scholar]
- 17.Hu Y, Wang X, Zeng L, Cai DY, Sabapathy K, Goff SP, Firpo EJ, Li B. ERK phosphorylates p66shcA on Ser36 and subsequently regulates p27kip1 expression via the Akt‐FOXO3a pathway: implication of p27kip1 in cell response to oxidative stress. Mol Biol Cell. 2005; 16:3705-3718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le S, Connors TJ, Maroney AC. c‐Jun N‐terminal kinase specifically phosphorylates p66ShcA at serine 36 in response to ultraviolet irradiation. J Biol Chem. 2001; 276:48332-48336 [DOI] [PubMed] [Google Scholar]
- 19.Francia P, delli GC, Bachschmid M, Martin‐Padura I, Savoia C, Migliaccio E, Pelicci PG, Schiavoni M, Luscher TF, Volpe M, Cosentino F. Deletion of p66shc gene protects against age‐related endothelial dysfunction. Circulation. 2004; 110:2889-2895 [DOI] [PubMed] [Google Scholar]
- 20.Ming XF, Rajapakse AG, Yepuri G, Xiong Y, Carvas JM, Ruffieux J, Scerri I, Wu Z, Popp K, Li J, Sartori C, Scherrer U, Kwak BR, Montani JP, Yang Z. Arginase II promotes macrophage inflammatory responses through mitochondrial reactive oxygen species, contributing to insulin resistance and atherogenesis. J Am Heart Assoc. 2012; 1:e000992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yepuri G, Velagapudi S, Xiong Y, Rajapakse AG, Montani JP, Ming XF, Yang Z. Positive crosstalk between arginase‐II and S6K1 in vascular endothelial inflammation and aging. Aging Cell. 2012; 11:1005-1016 [DOI] [PubMed] [Google Scholar]
- 22.Jastrzebski K, Hannan KM, Tchoubrieva EB, Hannan RD, Pearson RB. Coordinate regulation of ribosome biogenesis and function by the ribosomal protein S6 kinase, a key mediator of mTOR function. Growth Factors. 2007; 25:209-226 [DOI] [PubMed] [Google Scholar]
- 23.Burnett PE, Barrow RK, Cohen NA, Snyder SH, Sabatini DM. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E‐BP1. Proc Natl Acad Sci USA. 1998; 95:1432-1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pullen N, Dennis PB, Andjelkovic M, Dufner A, Kozma SC, Hemmings BA, Thomas G. Phosphorylation and activation of p70s6k by PDK1. Science. 1998; 279:707-710 [DOI] [PubMed] [Google Scholar]
- 25.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012; 149:274-293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Magnuson B, Ekim B, Fingar DC. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem J. 2012; 441:1-21 [DOI] [PubMed] [Google Scholar]
- 27.Yang Z, Ming XF. mTOR signalling: the molecular interface connecting metabolic stress, aging and cardiovascular diseases. Obes Rev. 2012; 13suppl 2:58-68 [DOI] [PubMed] [Google Scholar]
- 28.Chen B, Calvert AE, Cui H, Nelin LD. Hypoxia promotes human pulmonary artery smooth muscle cell proliferation through induction of arginase. Am J Physiol Lung Cell Mol Physiol. 2009; 297:L1151-L1159 [DOI] [PubMed] [Google Scholar]
- 29.Shi O, Morris SM, Jr, Zoghbi H, Porter CW, O'Brien WE. Generation of a mouse model for arginase II deficiency by targeted disruption of the arginase II gene. Mol Cell Biol. 2001; 21:811-813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith WW, Norton DD, Gorospe M, Jiang H, Nemoto S, Holbrook NJ, Finkel T, Kusiak JW. Phosphorylation of p66Shc and forkhead proteins mediates Abeta toxicity. J Cell Biol. 2005; 169:331-339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ming XF, Rajapakse AG, Carvas JM, Ruffieux J, Yang Z. Inhibition of S6K1 accounts partially for the anti‐inflammatory effects of the arginase inhibitor l‐norvaline. BMC Cardiovasc Disord. 2009; 9:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nemoto S, Finkel T. Redox regulation of forkhead proteins through a p66shc‐dependent signaling pathway. Science. 2002; 295:2450-2452 [DOI] [PubMed] [Google Scholar]
- 33.Xi G, Shen X, Clemmons DR. p66shc inhibits insulin‐like growth factor‐I signaling via direct binding to Src through its polyproline and Src homology 2 domains, resulting in impairment of Src kinase activation. J Biol Chem. 2010; 285:6937-6951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ming XF, Viswambharan H, Barandier C, Ruffieux J, Kaibuchi K, Rusconi S, Yang Z. Rho GTPase/Rho kinase negatively regulates endothelial nitric oxide synthase phosphorylation through the inhibition of protein kinase B/Akt in human endothelial cells. Mol Cell Biol. 2002; 22:8467-8477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rajapakse AG, Yepuri G, Carvas JM, Stein S, Matter CM, Scerri I, Ruffieux J, Montani JP, Ming XF, Yang Z. Hyperactive S6K1 mediates oxidative stress and endothelial dysfunction in aging: inhibition by resveratrol. PLoS ONE. 2011; 6:e19237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ming XF, Barandier C, Viswambharan H, Ruffieux J, Kaibuchi K, Rusconi S, Yang Z. Thrombin stimulates human endothelial arginase enzymatic activity via RhoA/ROCK pathway: implications for atherosclerotic endothelial dysfunction. Circulation. 2004; 110:3708-3714 [DOI] [PubMed] [Google Scholar]
- 37.Kubben FJ, Peeters‐Haesevoets A, Engels LG, Baeten CG, Schutte B, Arends JW, Stockbrügger RW, Blijham GH. Proliferating cell nuclear antigen (PCNA): a new marker to study human colonic cell proliferation. Gut. 1994; 35:530-535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ryoo S, Gupta G, Benjo A, Lim HK, Camara A, Sikka G, Lim HK, Sohi J, Santhanam L, Soucy K, Tuday E, Baraban E, Ilies M, Gerstenblith G, Nyhan D, Shoukas A, Christianson DW, Alp NJ, Champion HC, Huso D, Berkowitz DE. Endothelial arginase II: a novel target for the treatment of atherosclerosis. Circ Res. 2008; 102:923-932 [DOI] [PubMed] [Google Scholar]
- 39.Berkowitz DE, White R, Li D, Minhas KM, Cernetich A, Kim S, Burke S, Shoukas AA, Nyhan D, Champion HC, Hare JM. Arginase reciprocally regulates nitric oxide synthase activity and contributes to endothelial dysfunction in aging blood vessels. Circulation. 2003; 108:2000-2006 [DOI] [PubMed] [Google Scholar]
- 40.Erez A, Nagamani SC, Shchelochkov OA, Premkumar MH, Campeau PM, Chen Y, Garg HK, Li L, Mian A, Bertin TK, Black JO, Zeng H, Tang Y, Reddy AK, Summar M, O'Brien WE, Harrison DG, Mitch WE, Marini JC, Aschner JL, Bryan NS, Lee B. Requirement of argininosuccinate lyase for systemic nitric oxide production. Nat Med. 2011; 17:1619-1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kroemer G, Reed JC. Mitochondrial control of cell death. Nat Med. 2000; 6:513-519 [DOI] [PubMed] [Google Scholar]
- 42.Ranieri SC, Fusco S, Panieri E, Labate V, Mele M, Tesori V, Ferrara AM, Maulucci G, De Spirito M, Martorana GE, Galeotti T, Pani G. Mammalian life‐span determinant p66shcA mediates obesity‐induced insulin resistance. Proc Natl Acad Sci USA. 2010; 107:13420-13425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pani G. P66SHC and ageing: ROS and TOR? Aging. 2010; 2:514-518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gonzalez‐Rodriguez A, Alba J, Zimmerman V, Kozma SC, Valverde AM. S6K1 deficiency protects against apoptosis in hepatocytes. Hepatology. 2009; 50:216-229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vigneron A, Vousden KH. p53, ROS and senescence in the control of aging. Aging. 2010; 2:471-474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Minamino T, Komuro I. Vascular cell senescence: contribution to atherosclerosis. Circ Res. 2007; 100:15-26 [DOI] [PubMed] [Google Scholar]
- 47.Mallat Z, Tedgui A. Apoptosis in the vasculature: mechanisms and functional importance. Br J Pharmacol. 2000; 130:947-962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xu F, Sun Y, Chen Y, Sun Y, Li R, Liu C, Zhang C, Wang R, Zhang Y. Endothelial cell apoptosis is responsible for the formation of coronary thrombotic atherosclerotic plaques. Tohoku J Exp Med. 2009; 218:25-33 [DOI] [PubMed] [Google Scholar]
- 49.Tabas I, Seimon T, Timmins J, Li G, Lim W. Macrophage apoptosis in advanced atherosclerosis. Ann N Y Acad Sci. 2009; 1173suppl 1:E40-E45 [DOI] [PMC free article] [PubMed] [Google Scholar]