

Abstract
Common variable immune deficiency (CVID) is the most frequent symptomatic primary immune deficiency in adults. The standard of care is intravenous immunoglobulin (IVIG) or subcutaneous immunoglobulin (scIG) therapy. The cause of CVID is currently unknown, and there is no universally accepted definition of CVID. This creates problems in determining which patients will benefit from IVIG/scIG treatment. In this paper, we review the difficulties with the commonly used European Society of Immune Deficiencies (ESID) and the Pan American Group for Immune Deficiency (PAGID) definition of CVID. We propose new criteria for the diagnosis of CVID, which are based on recent scientific discoveries. Improved diagnostic precision will assist with treatment decisions including IVIG/scIG replacement. We suggest that asymptomatic patients with mild hypogammaglobulinaemia are termed hypogammaglobulinaemia of uncertain significance (HGUS). These patients require long-term follow-up, as some will evolve into CVID.
Keywords: common variable immunodeficiency (CVID), diagnostics, immunodeficiency–primary
Introduction
Common variable immune deficiency (CVID) is the most frequent symptomatic primary immune deficiency in adults. Current estimates suggest a prevalence of approximately one in 25 000 in the general population [1]. Significant ethnic variations exist and the frequency may be lower in some populations such as those in North East Asia [2,3].
Patients frequently become symptomatic later in life [4]. Careful clinical history, however, may reveal symptoms in some patients dating back to early childhood. The disorder is characterized by recurrent and/or severe infections, autoimmunity, malignancy and allergic disorders.
CVID probably represents a heterogeneous group of disorders culminating in late-onset antibody failure (LOAF). The genetic basis of these disorders has not been identified in the majority of individuals. It seems likely the majority of CVID patients have a polygenic disorder [5]. Rare patients with single gene defects have been identified. Causative mutations of ICOS, CD19, CD20, CD21 and CD81 lead to a severe cellular or humoral immune defect [6–9]. Identification of a single gene defect removes these patients from the umbrella diagnosis of CVID [10].
The significance of mutations and polymorphisms in other genes such as TACI, BAFF receptor and MSH5 is less certain. Mutations of these genes have been identified in healthy people, albeit at a lower frequency than symptomatic individuals [11,12]. We and others have shown that C104R mutations of the TACI gene do not segregate as expected with symptomatic family members [13]. Current thought is that these genes predispose to CVID rather than cause it. It is possible, of course, that some of these ‘healthy’ people with mutations of TACI and BAFF receptor may become symptomatic later in life. CVID can present in the seventh and eight decades of life [4].
Because the cause of the CVID is unknown, there is no universally accepted definition of the disorder. Various diagnostic criteria have been proposed. The definition published by the European Society of Immunodeficiencies (ESID) and the Pan American Group for Immune deficiency (PAGID) in 1999 is commonly used [14]. The ESID/PAGID diagnostic criteria comprise three parts: (1) hypogammaglobulinaemia with IgG levels two standard deviations below the mean; (2) impaired vaccine responses or absent isohemagglutinins; and (3) exclusion of other causes of hypogammaglobulinaemia.
CVID is thus a diagnosis of exclusion. It is important to note that the patient's symptomatic state and infective sequelae are not part of the ESID/PAGID definition of CVID. This can make it difficult to determine if asymptomatic patients with mild hypogammaglobulinaemia but abnormal vaccine responses have CVID and, more particularly, if they should be treated with intravenous immunoglobulin (IVIG) or subcutaneous immunoglobulin (scIG). The ESID/PAGID criteria were intended primarily for diagnosis, but are often used to make decisions about IVIG/scIG replacement. As discussed below, it is inappropriate to base treatment decisions mainly on vaccine responses, particularly if protective antibody levels are used.
The current standard of care for patients with CVID is lifelong replacement with IVIG/scIG. Studies have shown significant improvement in health outcomes when patients are treated with IVIG/scIG. There is both subjective improvement in quality of life as well as objective improvement in frequency and severity of infections [15]. Immunoglobulin treatment slows progression of complications, including suppurative lung disease.
IVIG use is increasing rapidly, given its efficacy in patients with autoimmune and inflammatory disorders [16,17]. It is an expensive treatment, and in some parts of the world supplies have been subject to shortages. Attempts have been made by various authorities to ensure that it is used effectively. IVIG/scIG use in single gene defects, such as Bruton's aggammaglobulinaemia, is facilitated by the availability of genetic testing to confirm the diagnosis [18]. In the case of CVID/hypogammaglobulinaemia the indications for IVIG/scIG use can be problematic given the lack of a precise diagnosis and spectrum of severity of the disorder [19]. A scoring system was suggested recently for IVIG/scIG treatment, although difficulties with diagnostic criteria for CVID was not addressed [20].
In this essay we explore the difficulties with the current definition of CVID and problems this causes in determining which patients should be treated with IVIG/scIG. In the future, studies such as the New Zealand asymptomatic hypogammaglobulinaemia/CVID cohort will assist in validating the proposed definition of CVID, which in turn may facilitate the appropriate use of IVIG/scIG.
Critical analysis of the ESID/PAGID criteria for CVID
The first criterion requires immunoglobulin levels to be two standard deviations below the mean. For most laboratories, the lower limit of normal for IgG (two standard deviations below the mean) is 7–8 g/l. This means that 2·5% of the general population meet this criterion [21]. It should be noted, however, that immunoglobulin levels are not distributed normally and percentiles should be used to determine reference ranges [22]. In clinical practice, patients with slightly reduced IgG levels would rarely be investigated, particularly if they are symptomatically well.
Impaired vaccine response is the second and most contentious criterion [13]. The ESID/PAGID diagnostic criteria do not specify which vaccines should be used [13]. Vaccine responses in CVID have been reviewed recently by an expert panel of the American Academy of Asthma Allergy and Immunology (AAAAI) [23]. A single booster dose of diphtheria–tetanus toxoid, conjugated Hemophilus influenzae type B (HIB) and Pneumovax® is often administered to assess the humoral immune response [24]. The Pneumovax® tests the response to carbohydrate antigens, while the other vaccines test responses to protein antigens. Serum is collected at baseline prior to vaccination and then a month later.
There are several problems with the use of each of these vaccines [13]. The tetanus toxoid is an excellent immunogen and many patients with CVID achieve a protective level of 0·1 IU/ml [20]. If they have lower levels, it has been suggested that patients should have a four fold increase in tetanus antibody titres. Many CVID patients are able to achieve a four fold increase in tetanus antibodies [25]. This may be a consequence of generation of tetanus-specific memory B cells prior to antibody failure. When there are higher baseline tetanus antibody levels, a four fold increase may not be achieved even in normal people. We have also questioned the validity of extrapolating responses from simple antigens to complex pathogens in vivo [13].
Therefore, using protective levels of tetanus antibodies is not appropriate. We suggest that patients' responses should be compared with age-matched controls undergoing immunization. A recent study of adults suggested that a level of 1 IU/ml is achieved by the majority of individuals receiving a single tetanus toxoid dose [26]. This threshold should therefore be applied to patients undergoing tetanus vaccination.
In contrast, diphtheria toxoid is a poor immunogen [21]. Poor responses to diphtheria toxoid are thus difficult to interpret. However, in the same study the majority of adults achieved a level of 1 IU/ml following diphtheria vaccination [26]. In our experience, many otherwise normal individuals do not reach this level following immunization.
Similarly, the response to immunization with conjugated H. influenzae vaccine should be compared with normal people. Recent studies suggest that the majority of children and adults given the conjugated HIB vaccine will reach an antibody level of at least 1 μg/ml [27,28]. Again, achievement of protective levels of H. influenzae antibodies of 0·15 μg/ml is less useful in assessing the adequacy of the response. The conjugated HIB vaccine has been available for the last two decades [29]. Recent data suggest a dramatic increase in the baseline HIB antibody level in cohorts of children who have received the conjugated HIB vaccine [30]. The response to a single dose of conjugated HIB vaccine may be very different in these vaccinated children compared with unvaccinated adults. This may complicate the assessment of the humoral immune response to this vaccine.
Pneumococcal immunization and assessment of individual serotype responses is the greatest source of difficulty. There are currently two types of pneumococcal vaccine, pneumococcal polysaccharide vaccine (PPV), Pneumovax® and pneumococcal conjugated vaccines (PCV), Prevnar 7® and Prevnar 13®. PCV tests responses to proteins, while PPV tests responses to carbohydrate antigens. Conjugation of polysaccharide vaccines to protein antigens such as toxoids converts them from T cell-independent to T cell-dependent antigens. Traditionally, PPV has not been administered to children aged less than 2 years, as the humoral immune response has not matured. Furthermore, there is concern that repeated doses of PPV may lead to a state of unresponsiveness [31]. In very young children, responses to carbohydrate antigens can be measured only by detection of isohaemagglutinins provided that the patient is not blood group AB [32]. Similarly, there may be differences in responses to the PPV between middle-aged and elderly adults [33].
Measuring pneumococcal antibody responses is also problematic. The commonly used World Health Organization's (WHO) ELIZA measures serotype-specific responses to the antigens contained in pneumococcal vaccine. Cross-reactive carbohydrate antigens (C group and 22F) can interfere with the assay and must be absorbed out [34,35]. There is currently no external quality assurance program for this technically challenging assay. Comparison of laboratories performing the WHO ELIZA show considerable variability in results [36]. The results from the WHO ELIZA cannot be compared with other commercially available assays, including the xMAP kit used on the Luminex platform [37]. Opsonophagocytic function has been measured in research laboratories, but is beyond the capability of most routine diagnostic laboratories [38,39]. The use of IgA and IgM responses to PPV may be useful as vaccine response markers in the future [40].
There is also no universal agreement as to what constitutes a ‘normal’ response to the PPV. There are at least five different criteria in the published literature [41]. Currently, the definition formulated by the AAAAI is commonly used, where adults should reach antibody levels above 1·3 μg/ml for at least 70% for each of the serotypes in the vaccine [42,43]. In children 50% is used. There is further variability, as some laboratories deem either 1 or 2 μg/ml as satisfactory responses [44,45]. When assessing the PPV responses in immune-deficient patients, protection against mucosal disease may also be important [23]. Higher levels of pneumococcal antibody may be needed to protect against mucosal disease in comparison with septicaemia [46].
To add to the difficulty is the observation that up to 18% of individuals diagnosed with CVID have adequate responses to the PPV [25]. Patients with high baseline pneumococcal titres are less likely to achieve a four fold increase in that serotype following vaccination [47]. Some serotypes are more immunogenic (serotype 3) than others (6B and 23F) [23]. It is also uncertain if patients with adequate but short-lived vaccine challenge responses should be considered to have an immune defect [13]. This may reflect an in-vivo defect in immunological memory. Prevnar 13® is now integrated into routine vaccine schedules. This will make it increasingly difficult to assess responses to carbohydrate antigens.
The use of other vaccines, such as the meningococcal vaccine or the typhoid vaccine [48], have yet to gain worldwide acceptance. In one study, 64% of CVID patients responded to the meningococcal vaccine [49]. Testing responses to other neoantigens such as the rabies virus may be logical, but there are currently few data in immune-deficient patients [50]. Experimental antigens such as ϕX174 are not licensed by the Food and Drug Administration (FDA) and cannot be used in routine clinical practice [51]. In our experience, the absence isohemagglutinins is not commonly used to make a diagnosis of CVID or to make decisions about IVIG/scIg treatment.
The last ESID/PAGID criterion requiring exclusion of secondary causes is the least contentious. Secondary hypogammaglobulinaemia can be caused by a variety of conditions, including gut or renal loss, adverse reactions to drugs. etc. [52,53].
This analysis underscores the difficulty in using the ESID/PAGID criteria to determine which patients have CVID and complicates decisions about IVIG/scIG treatment, which are often based inappropriately on vaccine responses alone.
Proposed new definition of CVID
Given that in the majority of patients the cause of CVID is unknown, the proposed new diagnostic criteria are primarily clinical with supportive laboratory evidence. Recurrent and severe infections are a common theme in the majority of CVID patients in large published series [4,54–57]. We believe that symptoms are an essential part of the diagnosis as it indicates in-vivo failure of the immune system. As discussed below, it is important to exclude other causes/predisposing factors for infections. There are small numbers of completely asymptomatic patients who have profound hypogammaglobulinaemia. As discussed below, some of these patients may need to be placed on long-term IVIG/scIG replacement [13] (Table 1).
Table 1.
Proposed definition of common variable immune deficiency (CVID)
| A Must meet all major criteria |
| • Hypogammaglobulinaemia: IgG below 5 g/l for adults [57] |
| • No other cause identified for immune defect [52] |
| • Age > 4 years [21] |
| B Clinical sequelae directly attributable to in-vivo failure of the immune system (one or more criteria) |
| • Recurrent, severe or unusual infections |
| • Poor response to antibiotics |
| • Breakthrough bacterial infections in spite of prophylactic antibiotics |
| • Infections in spite of immunization with the appropriate vaccine, e.g. HPV disease |
| • Bronchiectasis and/or chronic sinus disease |
| • Inflammatory disorders or autoimmunity [58] |
| C Supportive laboratory evidence (three or more criteria) |
| • Concomitant deficiency or reduction of IgA (<0·8 g/l) and/or IgM (<0·4 g/l) [4,56] |
| • Presence of B cells but reduced memory B cell subsets and/or increased CD21 low subsets by flow cytometry [59] |
| • IgG3 deficiency (<0·2 g/l) [60,61] |
| • Impaired vaccine responses compared to age-matched controls |
| • Transient responses to vaccines compared to age-matched controls [62] |
| • Absent isohaemagglutinins (if not blood group AB) [32] |
| • Serological support for autoimmunity in section B, e.g. positive Coombs' test |
| • Sequence variations of genes predisposing to CVID, e.g. TACI, BAFFR, MSH5, etc. [11,63] |
| D Presence of any one of relatively specific histological markers of CVID (not required for diagnosis but presence increases diagnostic certainty) |
| • Lymphoid interstitial pneumonitis [64] |
| • Granulomatous disorder [65,66] |
| • Nodular regenerative hyperplasia of the liver [67,68] |
| • Nodular lymphoid hyperplasia of the gut [69] |
| • Absence of plasma cells on gut biopsy [70,71] |
Meeting criteria in categories ABC or ABD indicates probable CVID. Patients meeting criteria ABC and ABD should be treated with intravenous immunoglobulin/subcutaneous immunoglobulin (IVIG/scIG). Patients meeting criteria A alone, AB or AC or AD but not B, are termed possible CVID. Some of these patients may need to be treated with IVIG/scIG. Patients with levels of immunoglobulin (Ig)G > 5 g/l, not meeting any other criteria are termed hypogammaglobulinaemia of uncertain significance (HGUS). A diagnostic algorithm is shown in Fig. 1. HPV: human papillomavirus.
Discussion
It is almost 15 years since the ESID/PAGID criteria were published [14]. There have been many advances in the field since that time, including several genetic discoveries. The proposed new definition incorporates these recent findings. A uniform definition of CVID has many advantages. It will facilitate treatment of individual patients and, in particular, will assist with decisions concerning whether or not to commence IVIG/scIG. A consistent definition may allow comparison of international cohorts of patients. There may be significant ethnic differences in presentation and complications, which may not be apparent if there is no consistent definition of CVID. The use of these criteria may have the added advantage of confirming the diagnosis in patients who have already commenced IVIG/scIG where there is doubt about the original diagnosis. It may obviate the need to stop IVIG/scIG and undertake vaccine responses in many such patients. This process can take several months, and the patient may be vulnerable to infections during the period they have discontinued their IVIG/scIG.
Meeting major criteria in category A is required for consideration of CVID. Patients must have significant hypogammaglobulinaemia compared to age-matched controls. We have suggested an IgG level below 5 g/l, which is the cut-off used by the French DEFI study [57]. This is similar to 4·5 g/l, as suggested previously [21]. It is, however, accepted that some patients with higher levels of IgG may be suitable candidates for IVIG/scIG if they have bronchiectasis for which no other cause has been identified [72]. As emphasized throughout this document, clinical judgement is paramount.
Secondly, other causes of hypogammaglobulinaemia must be excluded, including gut and renal loss as well as drug- and virus-induced immunoglobulin disorders [52]. The probable causes of secondary hypogammaglobulinaemia depend upon the age of the patient. Thorough clinical and laboratory evaluation is critical. Lastly, the diagnosis of CVID may not be secure in very young children who may have other monogenic immune defects [21]. We have chosen a cut-off of 4 years of age, as suggested by Chapel and Cunningham-Rundles [21]. Younger children with an immune deficiency will, of course, need to be treated pending a definitive diagnosis.
Group B criteria must be met. Patients must have at least one clinical sequel from their immune defect. This indicates in-vivo failure of their immune system. The majority of individuals in large series of CVID patients have had increased susceptibility to infections [4,54–57]. While there is debate about exactly what constitutes an increased number of infections, the number of infections will, be influenced by socio-demographic factors such as overcrowding, the number of young children attending daycare, etc. Patients with a host defect often need several courses of antibiotics to clear even uncomplicated bacterial infections. Similarly, infections in spite of vaccination may indicate in-vivo failure of the immune response. Patients who have recurrent proven bacterial infections in spite of prophylactic antibiotics also meet this criterion.
It is important to exclude other causes of recurrent infections, including anatomical and functional defects. For example, correction of gastroesophageal reflux may reduce the frequency and severity of chest infections. It is accepted that some patients may have multiple predisposing factors for infections. Clinical judgement is needed to determine the relative contribution of the immune defect to the patient's infections.
In most series of CVID patients there is a minority who present with significant autoimmunity but with minimal susceptibility to infections in spite of severe hypogammaglobulinaemia. These patients qualify as having CVID if they meet category A criteria as well as categories C or D criteria.
Meeting three or more criteria in category C offers laboratory support to the diagnosis. None of these criteria are specific for CVID, but in combination support the diagnosis. A significant proportion of CVID patients have a reduction or deficiency of IgG and IgA and/or IgM [56]. Memory B cell subsets are reduced in most patients with CVID [59]. However, caution must be exercised, as we have shown that these observations are not consistent when memory subsets are measured over time [73]. We suggest that only memory B cells, switched memory B cells and CD21low B cells be assessed on two occasions.
If there is an absence of B cells (<2% of total lymphocytes by flow cytometry), other disorders of B cell development such as Bruton's aggamaglobulinaemia and thymoma must be excluded. There is debate as to whether or not patients with absent B cells, but no other genetic defect, fall within the spectrum of CVID [59].
IgG subclass deficiency is not an indication per se for immunoglobulin replacement, but IgG3 deficiency may be considered a supportive marker for an immune defect [60,61]. In the context of a total IgG < 5 g/l, this will usually mean a reduction of IgG1 as well. IgG3 deficiency should be given similar status to impaired memory B cells. The significance of reduction or deficiency of IgG2 and IgG4 is less certain. Many normal individuals with absent IgG4 are in excellent health.
Given the difficulties measuring and interpreting vaccine responses, we suggest that these are placed in the category of supportive criteria. Again, if vaccine responses are being undertaken, it is important to compare the results with those of age-matched controls rather than protective levels. We suggest that adequate but transient vaccine responses should also be considered abnormal [62]. Serological support for patients with autoimmune manifestations in category B is included in this section.
Given the uncertainty about the relevance of mutations and polymorphisms of genes such as TACI, BAFF receptor and MSH5, we felt that these belong to this category. It is probable that more susceptibility genes/sequence variations will be identified with genome wide association studies and whole exome analysis [74]. Patients with well-characterized monogenic mutations of ICOS, CD19, CD20, CD21 and CD81 should not be considered to have CVID [10]. Mutation of any of these genes indicates permanent irreversible antibody failure, requiring lifelong immunoglobulin replacement. The indication to treat these rare patients with monogenic disorders with IVIG/scIg is clear, similar to those with Bruton's agammaglobulinaemia.
Category D criteria identify patients who have lesions which have been associated closely with CVID. Their presence requires histological confirmation. Only a minority of CVID patients will have the granulomatous variant, lymphoid interstitial pneumonitis, nodular hyperplasia of the gut or nodular regenerative hyperplasia of the liver, proved by biopsy [64–67,69]. Therefore, meeting this criterion is not required for the diagnosis, but their presence increases diagnostic certainty. Similarly, there may be histological support for CVID as many patients with CVID lack plasma cells on gut biopsy [70,71]. Other histological findings such as villus atrophy are less specific for CVID, and have not been included in these criteria [75].
Patients with CVID are predisposed to malignancy, particularly lymphoid malignancy [4]. We have not included malignancy in the proposed definition of CVID, as it may be difficult to determine if the hypogammaglobulinaemia is secondary to the cancer in these patients. Decisions to treat patients with IVIG/scIG who have malignancy and hypogammaglobulinaemia will depend upon clinical circumstances.
Patients classified as possible CVID include those meeting category A criteria alone as well as various other combinations, including AB, AC or AD, without meeting category B criteria. The combination AD may be less likely, as patients would usually be symptomatic before they underwent a biopsy. They would therefore fulfil category B criteria and be classified as probable CVID.
Some patients with possible CVID may not be symptomatic, i.e. they do not have category B symptoms. There are a small number of patients who have profound hypogammaglobulinaemia but are clinically well [13]. Many of these patients are identified during the course of other investigations, where their serum electrophoresis may reveal reduced staining in the gamma region. Some of these patients may have impaired vaccine responses within the limitations of the assays outlined above.
Such patients with possible CVID may also need to be treated with IVIG/scIG. For example, we recently described a patient with homozygous C104R deficiency of the TACI gene [13]. He has profound hypogammaglobulinaemia with an IgG of 1·6 g/l with excellent, albeit short-lived vaccine responses. He therefore does not meet the PAGID/ESID criteria for CVID. There is considerable uncertainty about his prognosis, given the unknown risk of bacterial sepsis and the risk of severe viral infections. Fatal septicaemia or meningitis may be the first clinical presentation in such patients. Clearly, these patients will require close long-term follow-up, as patients with symptomatic CVID can present in the seventh and eighth decades of life [4]. If such patients choose not to commence immunoglobulin treatment after in-depth discussion of risks, they should carry home antibiotics with a low threshold for seeking medical assistance. It is essential that sound clinical judgement is exercised in these challenging situations, which have serious medico-legal implications. In other cases of possible CVID there may be a role for a therapeutic trial of IVIG/scIG before a decision is made about long-term treatment.
There are patients with mild hypogammaglobulinaemia (IgG > 5 g/l) who do not meet any other criteria listed above. We suggest that these patients are termed hypogammaglobulinaemia of uncertain significance (HGUS) in analogy to patients with monoclonal gammopathies of undetermined significance (MGUS). Like MGUS patients, it is critical that HGUS patients are carefully evaluated periodically. A proportion of HGUS patients may develop symptoms or signs of an immune deficiency or autoimmunity over time. The New Zealand asymptomatic hypogammaglobulinaemia cohort was established in 2006 to identify risk factors for progression to CVID. Asymptomatic HGUS patients with slightly reduced IgG levels who do not meet criteria in categories A, B, C or D may not require immunoglobulin replacement therapy. These diagnostic criteria will also be helpful in distinguishing CVID from secondary causes of hypogammaglobulinemia [76].
These diagnostic and treatment criteria will require international consensus and can be modified over the course of time as large cohorts of patients with CVID and hypogammaglobulinaemia are analysed in the future. It will be important to confirm that these criteria identify subsets of patients with predominantly inflammatory and autoimmune manifestations of CVID. This review is based on the work of eminent investigators of learned societies, whose work has been cited. We hope that these criteria will either be endorsed or refined by groups such as the British Society for Immunology (BSI), Australasian Society for Clinical Immunology and Allergy (ASCIA), PAGID, ESID, AAAAI and the WHO/IUIS expert committee on immunodeficiencies. Improved precision in diagnosis will assist with difficult decisions about which patients should be placed on long-term IVIG.
In summary, these criteria will assist with diagnosis and treatment decisions for CVID patients. Patients with probable CVID must meet all major criteria in category A in addition to criteria in categories B and C or D. These patients should be treated with IVIG/scIG, as they have symptoms of immune system failure with supportive laboratory evidence. Patients classified as possible CVID must meet category A criteria as well as other combinations of criteria shown in Fig. 1. As shown here, some of these patients also need to be treated with IVIG/scIG. Clinical judgement is critical for this group of patients. Patients with mild hypogammaglobulinaemia (HGUS) may not need to be treated with IVIG/scIG.
Fig. 1.
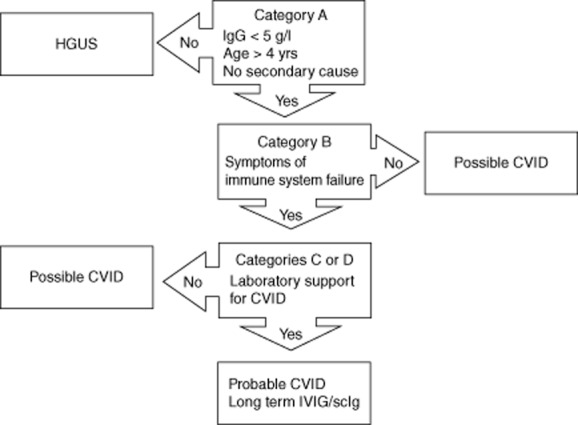
Diagnostic and treatment algorithm for patients with hypogammaglobulinaemia/common variable immune deficiency (CVID).
The majority of CVID patients have reductions in IgA and/ or IgM as well as reduced memory B cells and impaired vaccine responses. These patients will meet both the ESID/PAGID criteria as well as the proposed criteria in this paper. In the future, sensitivity analyses will indicate if the number of criteria within categories B and C needs to be adjusted to improve specificity but retain sensitivity.
This paper emphasizes the importance of clinical symptoms in the diagnosis of CVID. Secondly, we have shown why treatment decisions should not be based on vaccine responses alone, particularly if protective antibody levels are used. Lastly, while these criteria offer guidance for IVIG/scIG treatment, this is not a substitute for good clinical judgement. These patients should be under the long-term care of experienced clinical immunologists.
Acknowledgments
We thank our patients for participating in our studies for the benefit of others. We are grateful for the ongoing support of LabPlus and Auckland Hospital. We thank the A+ Trust, Paykel trust and Auckland Medical Research Foundation for grant support.
Disclosure
RA has received an unrestricted educational grant from Octapharma.
References
- 1.Kirkpatrick P, Riminton S. Primary immunodeficiency diseases in Australia and New Zealand. J Clin Immunol. 2007;27:517–524. doi: 10.1007/s10875-007-9105-z. [DOI] [PubMed] [Google Scholar]
- 2.Rhim JW, Kim KH, Kim DS, et al. Prevalence of primary immunodeficiency in Korea. J Korean Med Sci. 2012;27:788–793. doi: 10.3346/jkms.2012.27.7.788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ishimura M, Takada H, Doi T, et al. Nationwide survey of patients with primary immunodeficiency diseases in Japan. J Clin Immunol. 2011;31:968–976. doi: 10.1007/s10875-011-9594-7. [DOI] [PubMed] [Google Scholar]
- 4.Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol. 1999;92:34–48. doi: 10.1006/clim.1999.4725. [DOI] [PubMed] [Google Scholar]
- 5.Park JH, Resnick ES, Cunningham-Rundles C. Perspectives on common variable immune deficiency. Ann NY Acad Sci. 2011;1246:41–49. doi: 10.1111/j.1749-6632.2011.06338.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van Zelm MC, Reisli I, van der Burg M, et al. An antibody-deficiency syndrome due to mutations in the CD19 gene. N Engl J Med. 2006;354:1901–1912. doi: 10.1056/NEJMoa051568. [DOI] [PubMed] [Google Scholar]
- 7.Grimbacher B, Hutloff A, Schlesier M, et al. Homozygous loss of ICOS is associated with adult-onset common variable immunodeficiency. Nat Immunol. 2003;4:261–268. doi: 10.1038/ni902. [DOI] [PubMed] [Google Scholar]
- 8.Kuijpers TW, Bende RJ, Baars PA, et al. CD20 deficiency in humans results in impaired T cell-independent antibody responses. J Clin Invest. 2010;120:214–222. doi: 10.1172/JCI40231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Zelm MC, Smet J, Adams B, et al. CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J Clin Invest. 2010;120:1265–1274. doi: 10.1172/JCI39748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Al-Herz W, Bousfiha A, Casanova JL, et al. Primary immunodeficiency diseases: an update on the classification from the International Union of Immunological Societies Expert Committee for Primary Immunodeficiency. Front Immunol. 2011;2:54. doi: 10.3389/fimmu.2011.00054. doi: 10.3389/fimmu.2011.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pan-Hammarstrom Q, Salzer U, Du L, et al. Reexamining the role of TACI coding variants in common variable immunodeficiency and selective IgA deficiency. Nat Genet. 2007;39:429–430. doi: 10.1038/ng0407-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sekine H, Ferreira RC, Pan-Hammarstrom Q, et al. Role for Msh5 in the regulation of Ig class switch recombination. Proc Natl Acad Sci USA. 2007;104:7193–7198. doi: 10.1073/pnas.0700815104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koopmans W, Woon ST, Brooks AE, Dunbar PR, Browett P, Ameratunga R. Clinical variability of family members with the C104R mutation in transmembrane activator and calcium modulator and cyclophilin ligand interactor (TACI) J Clin Immunol. 2013;33:68–73. doi: 10.1007/s10875-012-9793-x. [DOI] [PubMed] [Google Scholar]
- 14.Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies) Clin Immunol. 1999;93:190–197. doi: 10.1006/clim.1999.4799. [DOI] [PubMed] [Google Scholar]
- 15.Quinti I, Soresina A, Guerra A, et al. Effectiveness of immunoglobulin replacement therapy on clinical outcome in patients with primary antibody deficiencies: results from a multicenter prospective cohort study. J Clin Immunol. 2011;31:315–322. doi: 10.1007/s10875-011-9511-0. [DOI] [PubMed] [Google Scholar]
- 16.Katz U, Shoenfeld Y, Zandman-Goddard G. Update on intravenous immunoglobulins (IVIg) mechanisms of action and off-label use in autoimmune diseases. Curr Pharm Des. 2011;17:3166–3175. doi: 10.2174/138161211798157540. [DOI] [PubMed] [Google Scholar]
- 17.Gelfand EW. Intravenous immune globulin in autoimmune and inflammatory diseases. N Engl J Med. 2012;367:2015–2025. doi: 10.1056/NEJMra1009433. [DOI] [PubMed] [Google Scholar]
- 18.Ameratunga R, Woon ST, Brewerton M, et al. Primary immune deficiency disorders in the South Pacific: the clinical utility of a customized genetic testing program in New Zealand. Ann NY Acad Sci. 2011;1238:53–64. doi: 10.1111/j.1749-6632.2011.06238.x. [DOI] [PubMed] [Google Scholar]
- 19.Gelfand EW, Ochs HD, Shearer WT. Controversies in IgG replacement therapy in patients with antibody deficiency diseases. J Allergy Clin Immunol. 2013;131:1001–1005. doi: 10.1016/j.jaci.2013.02.028. [DOI] [PubMed] [Google Scholar]
- 20.Agarwal S, Cunningham-Rundles C. Treatment of hypogammaglobulinemia in adults: a scoring system to guide decisions on immunoglobulin replacement. J Allergy Clin Immunol. 2013;131:1699–1701. doi: 10.1016/j.jaci.2013.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chapel H, Cunningham-Rundles C. Update in understanding common variable immunodeficiency disorders (CVIDs) and the management of patients with these conditions. Br J Haematol. 2009;145:709–727. doi: 10.1111/j.1365-2141.2009.07669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ritchie RF, Palomaki GE, Neveux LM, Navolotskaia O, Ledue TB, Craig WY. Reference distributions for immunoglobulins A, G, and M: a practical, simple, and clinically relevant approach in a large cohort. J Clin Lab Anal. 1998;12:363–370. doi: 10.1002/(SICI)1098-2825(1998)12:6<363::AID-JCLA6>3.0.CO;2-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Orange JS, Ballow M, Stiehm ER, et al. Use and interpretation of diagnostic vaccination in primary immunodeficiency: a working group report of the Basic and Clinical Immunology Interest Section of the American Academy of Allergy, Asthma and Immunology. J Allergy Clin Immunol. 2012;130:S1–24. doi: 10.1016/j.jaci.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 24.Empson M, Sinclair J, O'Donnell J, Ameratunga R, Fitzharris P, Steele R. The assessment and management of primary antibody deficiency. NZ Med J. 2004;117:U914. [PubMed] [Google Scholar]
- 25.Goldacker S, Draeger R, Warnatz K, et al. Active vaccination in patients with common variable immunodeficiency (CVID) Clin Immunol. 2007;124:294–303. doi: 10.1016/j.clim.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 26.Thierry-Carstensen B, Jordan K, Uhlving HH, et al. A randomised, double-blind, non-inferiority clinical trial on the safety and immunogenicity of a tetanus, diphtheria and monocomponent acellular pertussis (TdaP) vaccine in comparison to a tetanus and diphtheria (Td) vaccine when given as booster vaccinations to healthy adults. Vaccine. 2012;30:5464–5471. doi: 10.1016/j.vaccine.2012.06.073. [DOI] [PubMed] [Google Scholar]
- 27.Hawdon N, Nix EB, Tsang RS, Ferroni G, McCready WG, Ulanova M. Immune response to Haemophilus influenzae type B vaccination in patients with chronic renal failure. Clin Vaccine Immunol. 2012;19:967–969. doi: 10.1128/CVI.00101-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dentinger CM, Hennessy TW, Bulkow LR, et al. Immunogenicity and reactogenicity to Haemophilus influenzae type B (Hib) conjugate vaccine among rural Alaska adults. Hum Vaccin. 2006;2:24–28. doi: 10.4161/hv.2.1.2445. [DOI] [PubMed] [Google Scholar]
- 29.Ameratunga SN, Lennon DR, Entwistle B, Robinson E, Ameratunga RV. The immunogenicity of Haemophilus influenzae: meningococcal protein conjugate vaccine in Polynesian and non-Polynesian New Zealand infants. J Paediatr Child Health. 1997;33:138–141. doi: 10.1111/j.1440-1754.1997.tb01016.x. [DOI] [PubMed] [Google Scholar]
- 30.Ladhani S, Ramsay M, Flood J, et al. Haemophilus influenzae serotype B (Hib) seroprevalence in England and Wales in 2009. Euro Surveill. 2012;17:pii: 20313. doi: 10.2807/ese.17.46.20313-en. [DOI] [PubMed] [Google Scholar]
- 31.O'Brien KL, Hochman M, Goldblatt D. Combined schedules of pneumococcal conjugate and polysaccharide vaccines: is hyporesponsiveness an issue? Lancet Infect Dis. 2007;7:597–606. doi: 10.1016/S1473-3099(07)70210-4. [DOI] [PubMed] [Google Scholar]
- 32.Tiller TL, Jr, Buckley RH. Transient hypogammaglobulinemia of infancy: review of the literature, clinical and immunologic features of 11 new cases, and long-term follow-up. J Pediatr. 1978;92:347–353. doi: 10.1016/s0022-3476(78)80417-x. [DOI] [PubMed] [Google Scholar]
- 33.Lee H, Nahm MH, Kim KH. The effect of age on the response to the pneumococcal polysaccharide vaccine. BMC Infect Dis. 2010;10:60. doi: 10.1186/1471-2334-10-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Henckaerts I, Goldblatt D, Ashton L, Poolman J. Critical differences between pneumococcal polysaccharide enzyme-linked immunosorbent assays with and without 22F inhibition at low antibody concentrations in pediatric sera. Clin Vaccine Immunol. 2006;13:356–360. doi: 10.1128/CVI.13.3.356-360.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Concepcion NF, Frasch CE. Pneumococcal type 22f polysaccharide absorption improves the specificity of a pneumococcal–polysaccharide enzyme-linked immunosorbent assay. Clin Diagn Lab Immunol. 2001;8:266–272. doi: 10.1128/CDLI.8.2.266-272.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ballow M. Vaccines in the assessment of patients for immune deficiency. J Allergy Clin Immunol. 2012;130:283–284. doi: 10.1016/j.jaci.2012.04.028. e5. [DOI] [PubMed] [Google Scholar]
- 37.Balloch A, Licciardi PV, Tang ML. Serotype-specific anti-pneumococcal IgG and immune competence: critical differences in interpretation criteria when different methods are used. J Clin Immunol. 2013;33:335–341. doi: 10.1007/s10875-012-9806-9. [DOI] [PubMed] [Google Scholar]
- 38.Russell FM, Carapetis JR, Burton RL, et al. Opsonophagocytic activity following a reduced dose 7-valent pneumococcal conjugate vaccine infant primary series and 23-valent pneumococcal polysaccharide vaccine at 12 months of age. Vaccine. 2011;29:535–544. doi: 10.1016/j.vaccine.2010.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Romero-Steiner S, Frasch CE, Carlone G, Fleck RA, Goldblatt D, Nahm MH. Use of opsonophagocytosis for serological evaluation of pneumococcal vaccines. Clin Vaccine Immunol. 2006;13:165–169. doi: 10.1128/CVI.13.2.165-169.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cavaliere FM, Milito C, Martini H, et al. Quantification of IgM and IgA anti-pneumococcal capsular polysaccharides by a new ELISA assay: a valuable diagnostic and prognostic tool for common variable immunodeficiency. J Clin Immunol. 2013;33:838–846. doi: 10.1007/s10875-012-9856-z. [DOI] [PubMed] [Google Scholar]
- 41.Sorensen RU, Leiva LE, Javier FC, III, et al. Influence of age on the response to Streptococcus pneumoniae vaccine in patients with recurrent infections and normal immunoglobulin concentrations. J Allergy Clin Immunol. 1998;102:215–221. doi: 10.1016/s0091-6749(98)70089-2. [DOI] [PubMed] [Google Scholar]
- 42.Bonilla FA, Bernstein IL, Khan DA, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. Ann Allergy Asthma Immunol. 2005;94:S1–63. doi: 10.1016/s1081-1206(10)61142-8. [DOI] [PubMed] [Google Scholar]
- 43.Paris K, Sorensen RU. Assessment and clinical interpretation of polysaccharide antibody responses. Ann Allergy Asthma Immunol. 2007;99:462–464. doi: 10.1016/S1081-1206(10)60572-8. [DOI] [PubMed] [Google Scholar]
- 44.Jodar L, Butler J, Carlone G, et al. Serological criteria for evaluation and licensure of new pneumococcal conjugate vaccine formulations for use in infants. Vaccine. 2003;21:3265–3272. doi: 10.1016/s0264-410x(03)00230-5. [DOI] [PubMed] [Google Scholar]
- 45.Lee LH, Frasch CE, Falk LA, Klein DL, Deal CD. Correlates of immunity for pneumococcal conjugate vaccines. Vaccine. 2003;21:2190–2196. doi: 10.1016/s0264-410x(03)00025-2. [DOI] [PubMed] [Google Scholar]
- 46.Jokinen JT, Ahman H, Kilpi TM, Makela PH, Kayhty MH. Concentration of antipneumococcal antibodies as a serological correlate of protection: an application to acute otitis media. J Infect Dis. 2004;190:545–550. doi: 10.1086/422531. [DOI] [PubMed] [Google Scholar]
- 47.Hare ND, Smith BJ, Ballas ZK. Antibody response to pneumococcal vaccination as a function of preimmunization titer. J Allergy Clin Immunol. 2009;123:195–200. doi: 10.1016/j.jaci.2008.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ferry BL, Misbah SA, Stephens P, et al. Development of an anti-Salmonella typhi Vi ELISA: assessment of immunocompetence in healthy donors. Clin Exp Immunol. 2004;136:297–303. doi: 10.1111/j.1365-2249.2004.02439.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rezaei N, Siadat SD, Aghamohammadi A, et al. Serum bactericidal antibody response 1 year after meningococcal polysaccharide vaccination of patients with common variable immunodeficiency. Clin Vaccine Immunol. 2010;17:524–528. doi: 10.1128/CVI.00389-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Brinkman DM, Jol-van der Zijde CM, ten Dam MM, et al. Vaccination with rabies to study the humoral and cellular immune response to a T-cell dependent neoantigen in man. J Clin Immunol. 2003;23:528–538. doi: 10.1023/b:joci.0000010429.36461.6b. [DOI] [PubMed] [Google Scholar]
- 51.Ochs HD, Davis SD, Wedgwood RJ. Immunologic responses to bacteriophage phi-X 174 in immunodeficiency diseases. J Clin Invest. 1971;50:2559–2568. doi: 10.1172/JCI106756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Agarwal S, Cunningham-Rundles C. Assessment and clinical interpretation of reduced IgG values. Ann Allergy Asthma Immunol. 2007;99:281–283. doi: 10.1016/S1081-1206(10)60665-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Smith J, Fernando T, McGrath N, Ameratunga R. Lamotrigine-induced common variable immune deficiency. Neurology. 2004;62:833–834. doi: 10.1212/01.wnl.0000113754.29225.5d. [DOI] [PubMed] [Google Scholar]
- 54.Hermaszewski RA, Webster AD. Primary hypogammaglobulinaemia: a survey of clinical manifestations and complications. Q J Med. 1993;86:31–42. [PubMed] [Google Scholar]
- 55.Quinti I, Soresina A, Spadaro G, et al. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol. 2007;27:308–316. doi: 10.1007/s10875-007-9075-1. [DOI] [PubMed] [Google Scholar]
- 56.Chapel H, Lucas M, Lee M, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. 2008;112:277–286. doi: 10.1182/blood-2007-11-124545. [DOI] [PubMed] [Google Scholar]
- 57.Oksenhendler E, Gerard L, Fieschi C, et al. Infections in 252 patients with common variable immunodeficiency. Clin Infect Dis. 2008;46:1547–1554. doi: 10.1086/587669. [DOI] [PubMed] [Google Scholar]
- 58.Knight AK, Cunningham-Rundles C. Inflammatory and autoimmune complications of common variable immune deficiency. Autoimmun Rev. 2006;5:156–159. doi: 10.1016/j.autrev.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 59.Wehr C, Kivioja T, Schmitt C, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. 2008;111:77–85. doi: 10.1182/blood-2007-06-091744. [DOI] [PubMed] [Google Scholar]
- 60.Abrahamian F, Agrawal S, Gupta S. Immunological and clinical profile of adult patients with selective immunoglobulin subclass deficiency: response to intravenous immunoglobulin therapy. Clin Exp Immunol. 2010;159:344–350. doi: 10.1111/j.1365-2249.2009.04062.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Olinder-Nielsen AM, Granert C, Forsberg P, Friman V, Vietorisz A, Bjorkander J. Immunoglobulin prophylaxis in 350 adults with IgG subclass deficiency and recurrent respiratory tract infections: a long-term follow-up. Scand J Infect Dis. 2007;39:44–50. doi: 10.1080/00365540600951192. [DOI] [PubMed] [Google Scholar]
- 62.Musher DM, Manof SB, Liss C, et al. Safety and antibody response, including antibody persistence for 5 years, after primary vaccination or revaccination with pneumococcal polysaccharide vaccine in middle-aged and older adults. J Infect Dis. 2010;201:516–524. doi: 10.1086/649839. [DOI] [PubMed] [Google Scholar]
- 63.Salzer U, Bacchelli C, Buckridge S, et al. Relevance of biallelic versus monoallelic TNFRSF13B mutations in distinguishing disease-causing from risk-increasing TNFRSF13B variants in antibody deficiency syndromes. Blood. 2009;113:1967–1976. doi: 10.1182/blood-2008-02-141937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Popa V. Lymphocytic interstitial pneumonia of common variable immunodeficiency. Ann Allergy. 1988;60:203–206. [PubMed] [Google Scholar]
- 65.Ameratunga R, Becroft DM, Hunter W. The simultaneous presentation of sarcoidosis and common variable immune deficiency. Pathology. 2000;32:280–282. [PubMed] [Google Scholar]
- 66.Fasano MB, Sullivan KE, Sarpong SB, et al. Sarcoidosis and common variable immunodeficiency. Report of 8 cases and review of the literature. Medicine (Balt) 1996;75:251–261. doi: 10.1097/00005792-199609000-00002. [DOI] [PubMed] [Google Scholar]
- 67.Fuss IJ, Friend J, Yang Z, et al. Nodular regenerative hyperplasia in common variable immunodeficiency. J Clin Immunol. 2013;33:748–758. doi: 10.1007/s10875-013-9873-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Malamut G, Ziol M, Suarez F, et al. Nodular regenerative hyperplasia: the main liver disease in patients with primary hypogammaglobulinemia and hepatic abnormalities. J Hepatol. 2008;48:74–82. doi: 10.1016/j.jhep.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 69.Luzi G, Zullo A, Iebba F, et al. Duodenal pathology and clinical–immunological implications in common variable immunodeficiency patients. Am J Gastroenterol. 2003;98:118–121. doi: 10.1111/j.1572-0241.2003.07159.x. [DOI] [PubMed] [Google Scholar]
- 70.Malamut G, Verkarre V, Suarez F, et al. The enteropathy associated with common variable immunodeficiency: the delineated frontiers with celiac disease. Am J Gastroenterol. 2010;105:2262–2275. doi: 10.1038/ajg.2010.214. [DOI] [PubMed] [Google Scholar]
- 71.Agarwal S, Smereka P, Harpaz N, Cunningham-Rundles C, Mayer L. Characterization of immunologic defects in patients with common variable immunodeficiency (CVID) with intestinal disease. Inflamm Bowel Dis. 2011;17:251–259. doi: 10.1002/ibd.21376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hill AT, Pasteur M, Cornford C, Welham S, Bilton D. Primary care summary of the British Thoracic Society Guideline on the management of non-cystic fibrosis bronchiectasis. Prim Care Respir J. 2011;20:135–140. doi: 10.4104/pcrj.2011.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Koopmans W, Woon ST, Zeng IS, et al. Variability of memory B cell markers in a cohort of common variable immune deficiency patients over six months. Scand J Immunol. 2013;77:470–475. doi: 10.1111/sji.12028. [DOI] [PubMed] [Google Scholar]
- 74.Orange JS, Glessner JT, Resnick E, et al. Genome-wide association identifies diverse causes of common variable immunodeficiency. J Allergy Clin Immunol. 2011;127:1360–1376. doi: 10.1016/j.jaci.2011.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Biagi F, Bianchi PI, Zilli A, et al. The significance of duodenal mucosal atrophy in patients with common variable immunodeficiency: a clinical and histopathologic study. Am J Clin Pathol. 2012;138:185–189. doi: 10.1309/AJCPEIILH2C0WFYE. [DOI] [PubMed] [Google Scholar]
- 76.Ameratunga R, Casey P, Parry S, et al. Allergy Asthma Clin Immunol. 2013. Hypogammaglobulinemia factitia: Munchausen syndrome masquerading as common variable immune deficiency. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]