

Abstract
Mannan binding lectin (MBL)-associated serine protease type 1 (MASP-1) has a central role in the lectin pathway of complement activation and is required for the formation of C3 convertase. The activity of MASP-1 in the peripheral blood has been identified previously as a highly significant predictor of the severity of liver fibrosis in hepatitis C virus (HCV) infection, but not in liver disease of other aetiologies. In this study we tested the hypotheses that expression of MASP-1 may promote disease progression in HCV disease by direct activation of hepatic stellate cells (HSCs) and may additionally be up-regulated by HCV. In order to do so, we utilized a model for the maintenance of primary human HSC in the quiescent state by culture on basement membrane substrate prior to stimulation. In comparison to controls, recombinant MASP-1 stimulated quiescent human HSCs to differentiate to the activated state as assessed by both morphology and up-regulation of HSC activation markers α-smooth muscle actin and tissue inhibitor of metalloproteinase 1. Further, the expression of MASP-1 was up-regulated significantly by HCV infection in hepatocyte cell lines. These observations suggest a new role for MASP-1 and provide a possible mechanistic link between high levels of MASP-1 and the severity of disease in HCV infection. Taken together with previous clinical observations, our new findings suggest that the balance of MASP-1 activity may be proinflammatory and act to accelerate fibrosis progression in HCV liver disease.
Keywords: fibrosis, hepatitis C, innate immune response, MASP-1
Introduction
Hepatic stellate cells (HSCs) play a central role in the initiation and modulation of hepatic fibrogenesis. Following liver injury, HSCs differentiate to a myofibroblast phenotype and acquire a range of proliferative and proinflammatory functions which result in deposition of excess fibrogenic extracellular matrix proteins [1–3]. Elucidation of the mechanisms by which HSCs become activated is essential for an understanding of the pathogenesis of chronic liver disease. Hepatitis C virus (HCV) is a major cause of chronic liver disease. Up to 200 million people worldwide are estimated to be infected with the virus and at least 30% develop liver fibrosis, with the consequent life-threatening complications of end-stage liver disease and hepatocellular carcinoma [4]. We have previously shown a highly significant correlation between the severity of liver fibrosis in a cohort of HCV-infected patients and the activity of mannan binding lectin (MBL)-associated serine protease (MASP)-1, a central component of the complement lectin activation pathway [5].
Three types of MASPs have been identified, designated MASP-1, MASP-2 and MASP-3. MASPs circulate in association with pattern recognition molecules MBL and ficolins L, H and M. The lectin pathway of complement activation is initiated by binding of MBL or ficolins to their targets, followed by activation of the associated proteases [6]. It is established that MASP-2 can autoactivate and cleave complement components C2 and C4, leading to the generation of C3 convertase [7], but the role of MASP-1 has been unclear. Recent evidence, however, has established an essential role for MASP-1 in the initiation of the complement lectin pathway. The use of monospecific inhibitors has confirmed that both MASP-1 and MASP-2 are necessary for lectin pathway activation under physiological conditions [8–10]. Further, MASP-1 is both absolutely required for autoactivation of MASP-2 and cleaves 60% of C2 required to generate C3 convertase [8]. Consonant with these findings, autoactivation of MASP-1 has been found to be highly efficient, and is crucial for activation of complexes with MBL/ficolin [11]. There is therefore compelling evidence that MASP-1 has a regulatory role in the complement lectin activation pathway. We also note that MASP-1 is related structurally and functionally to thrombin [12,13], and several lines of evidence suggest that thrombin has an important role in the generation of fibrosis in chronic liver disease [14,15]. Against this background, we set out to test the hypothesis that MASP-1 directly activates HSC, and may therefore promote liver fibrosis in HCV infection. In order to do so, we have developed a model in which HSCs are maintained in the deactivated state on basement membrane substrate prior to stimulation [16].
Materials and methods
Isolation and culture of human HSCs
For the isolation of HSCs, liver resections were perfused ex-vivo with Hanks's HEPES buffer containing ethylene glycol tetraacetic acid (EGTA) (Gibco-BRL, Life Technologies, Paisley, UK), followed by perfusion with Hanks's HEPES buffer without EGTA. Tissue was then digested for 20 min with Hanks's HEPES buffer containing calcium, collagenase (0·13 U/ml NB4G grade collagenase; Serva Electrophoresis GmbH, Heidelberg, Germany) and trypsin inhibitor (Sigma, Poole, UK). Digested tissue was minced and agitated gently in Hanks's HEPES buffer without EGTA and filtered through 250 μm and 100 μm nylon membranes. The cell suspension was spun down at 10 g for 2 min and washed in Williams' medium E (Gibco-BRL) containing 10% heat-inactivated fetal calf serum (FCS) (Sigma) and 2 mM L-glutamine (Sigma). Supernatants from centrifugal spins of the hepatocyte isolation process were pooled and retained for HSC isolation: supernatants were centrifuged three times at 50 g for 5 min until no pellet was formed, then centrifuged at 4000 g for 20 min to form a pellet containing HSC. The supernatant was discarded and the pellet resuspended in 20 ml of Dulbecco's modified Eagles medium (DMEM) (Gibco-BRL) containing 10% FCS and seeded in a 75 cm2 tissue culture flask. Medium was changed 24 h after seeding and every 48 h thereafter. HSC became confluent after 7–10 days. Confluent HSC were detached by trypsinization and seeded at a density of 3 × 105 cells per well on six-well plates. Liver tissue was obtained from patients undergoing partial liver resection for metastatic cancer and was obtained following written informed consent and with full ethical approval.
Activated HSCs were then either left untreated or cultured in basement membrane substrate as Matrigel™ (Becton Dickinson Labware, Bedford, MA, USA) using the thin gel method (50 μl/cm2 of growth surface area), according to the manufacturer's instructions. Morphological observation was performed using a phase contrast inverted Leica microscope and images captured on a QICAM digital camera.
Activation of MASP-1 with enterokinase
MASP-1 protein was obtained as a zymogen with an enterokinase cleavage site replacing the natural cleavage sequence. The protein was produced with an N-terminal His-tag in Chinese hamster ovary (CHO) cells and purified by affinity chromatography on Ni-Sepharose followed by gel filtration on a Superdex 200 Column (GE Healthcare Life Sciences, Chalfont, UK) [17]. Following optimization of the cleavage reaction, MASP-1 was released by incubation with enterokinase (New England Biolabs, Hitchin, UK) at concentration of 0·02% at 37°C for 1 h. In order to standardize the quantity of MASP-1 used in subsequent experiments, the activities of both MASP-1 and thrombin were assayed by comparing the cleavage of VPR-AMC (Boc-Val-Pro-Arg-aminomethylcoumarin), a fluorescent substrate common to both proteases (Bachem, St Helens, UK). The activity of each batch of MASP-1 was highly consistent (data not shown).
Treatment of quiescent HSC with MASP-1, thrombin and TGF-β
After 24 h on Matrigel™, quiescent HSCs were treated with purified MASP-1 at a concentration of 2·5–3·5 μg/ml. Experimental controls were as follows: (i) HSC exposed to 5 ng/ml of recombinant transforming growth factor (TGF)-β (R&D Systems, Abingdon, UK); (ii) HSC treated with thrombin (Sigma, UK) at 30 mU/ml; (iii) 3·5 μg/ml MASP-1 which had not been cleaved with enterokinase; and (iv) cells cultured on Matrigel™ with the addition of enterokinase alone to a final concentration of 0·02%. All experiments were performed in at least three biological replicates.
RNA extraction and cDNA production
Total RNA was isolated using the RNeasy Mini Kit (Qiagen Ltd, Manchester, UK), according to the manufacturer's protocol, in a final volume of 50 μl and measured using the Nanodrop ND-1000 UV-Vis spectrophotometer. Consecutive samples were obtained from the following time-points: (i) freshly isolated HSCs; (ii) activated HSCs following 7 days' growth on tissue culture plastic; (iii) HSCs fully deactivated following 48 h culture on basement membrane substrate; and (iv) deactivated HSC subsequently stimulated by MASP-1 and controls for 24 h. In order to ensure comparability of results, only 50% of cells at each time-point were harvested for RNA extraction and the remaining cells allowed to progress through the protocol. HSCs cultured on tissue culture plastic were released by trypsinization prior to processing for RNA extraction. HSCs cultured in Matrigel™ substrate were recovered using dispase (Becton Dickinson Biosciences, Bedford, MA, USA), according to the manufacturer's instructions. A two-step cDNA synthesis was then performed. An initial oligo : template annealing mix was made up using 1 μg RNA template, 200 ng oligo (dT) and ×1 concentration reverse transcription (RT) buffer in a final volume of 10 μl, heated to 70°C for 5 min and snap-chilled on ice. Synthesis of cDNA was then performed via addition of 200 U Moloney murine leukaemia virus reverse transcriptase (MMLV) reverse transcriptase (Promega, Madison, WI, USA) and 1·2 mM deoxyribonucleoside triphosphates (dNTPs) and made up to a final volume of 25 μl with diethylpyrocarbonate (DEPC)-treated water. Reactions were incubated at 42°C for 1 h and stored at 4°C.
Reverse transcription polymerase chain reaction (RT–qPCR)
In order to interrogate HSC activation status further, primers were designed to amplify sequences from genes encoding alpha smooth muscle actin (α-SMA) and tissue inhibitor of metalloproteinase-1 (TIMP-1) using the Primer 3 program (http://frodo.wi.mit.edu/primer3) and Molecular Biology Work Bench version 3·2 (http://workbench.sdsc.edu). Primers were also designed to detect MASP-1 and MBL in HCV-infected cell lines. The sequences of all primers and probes are shown in Table 1. Primer sets were validated in standard PCR using a dilution series of recombinant plasmids containing amplicons as template. For quantitative PCR, standard curves were prepared for logarithmic serial dilution of recombinant plasmids containing cloned genes with a copy range of 107–10−1 with hypoxanthine–guanine phosphoribosyltransferase (HPRT) for normalization. Quantitative PCR was performed using BRILLIANT® SYBR® Green Master Mix (Stratagene, La Jolla, CA, USA). Twenty-five-μl reactions were made up of 12·5 μl SYBR® Green Master Mix, 5 pmol forward and reverse primer; 0·375 μl of 1:500 reference dye, before making up to 24 μl with nuclease-free water and addition of 1 μl of cDNA template and cycled on a Stratagene MX4000 real-time PCR machine using an initial denaturation step of 95°C for 15 min, followed by 50 cycles of 94°C for 30 s, 55°C for 30 s and extension at 72°C for 1 min. MASP-1 and MBL-2 gene expression levels were quantified using a standard curve based on consecutive 1:2 dilution and PCR conditions were as follows: denaturation at 95°C for 10 min followed by 40 cycles of 30 s denaturation at 95°C; 1 min annealing at 55°C and 30 s extension at 72°C, after which product melting temperature was determined in 30 s segments at 1°C intervals between 55 and 95°C. Results were analysed by Maxpro software (Stratagene). All experiments were performed at least in triplicate.
Table 1.
Primer sequences for reverse transcription–quantitative polymerase chain reaction (RT–qPCR)
| Primer sequence (5′–3′) | |
|---|---|
| HPRT | Forward: AAATTCTTTGCTGACCTGCTG |
| Reverse: TCCCCTGTTGACTGGTCATT | |
| α-SMA | Forward: AGCCAACAGGGAGAAGATGA |
| Reverse: CAGCTGTGGTCACAAAGGAA | |
| TIMP-1 | Forward: TCCCCAGAAATCATCGAGAC |
| Reverse: TCAGATTATGCCAGGGAACC | |
| MASP-1 | Forward: GGAATCCTCCTACCTTTGTGAA |
| Reverse: ATCTGACCGGAAAGTGATGG | |
| MBL2 | Forward: CAGAAACTGTGACCTGTGAGGA |
| Reverse: TGTAAGCCTCTGAGCCCTTG |
HPRT: hypoxanthine–guanine phosphoribosyltransferase; α-SMA: α-smooth muscle actin; TIMP: tissue inhibitor of metalloproteinase; MASP: mannan binding lectin-associated serine protease; MBL: mannan binding lectin.
HCV infections
In order to test the hypothesis that the expression of MASP-1 is up-regulated by HCV infection, we established in-vitro replication of infectious molecular clones JFH-1 and replication defective mutant JFH-1GND[18,19] in Huh 7·5 hepatocyte cell lines. HCV replication was assessed by monoclonal antibody (mAb) 9E10 anti-NS5A staining and infectious virus production measured using the TCID50 protocol.
Statistics
All statistical values shown were calculated using one-way analysis of variance (anova) with Bonferroni's multiple comparison test.
Results
Cell culture and reversal of the activated HSC phenotype in Matrigel™
HSCs grown on tissue plastic became fully confluent and required passaging after 7–10 days. Passaged cells recultured on plastic surfaces maintained a typical myofibroblast phenotype as expected. In contrast, HSCs recultured on basement membrane substrate showed a rapid reversion to the quiescent state (Fig. 1). In order to confirm phenotypic evidence of activation and reversion to quiescence, we tested molecular markers of HSC activation. α-SMA and TIMP-1 were detectable in quiescent cells but showed an unequivocal increase in expression in activated HSCs, and were therefore informative of HSC activation. In addition to up-regulation following activation on tissue culture plastic, α-SMA and TIMP-1 were both down-regulated subsequently 24 h after exposure of HSCs to basement membrane substrate (Fig. 2).
Fig. 1.

Human hepatic stellate cells (HSCs) are deactivated by culture on basement membrane substrate (Matrigel™). Human HSCs: in the quiescent state immediately following retrieval and plating on non-treated tissue culture plastic (a) and following 24 h of culture on the same surface (b). Cells fully activated by culture on tissue culture plastic for a further 7 days (c), followed by splitting and reculture on Matrigel™ for 48 h with full loss of activated phenotype and reversion to an appearance characteristic of the quiescent state (d). Images were visualized using a phase contrast Leica microscope. Magnification was selected to best illustrate the key features: (a,d) ×100 magnification and (b,c) ×200 magnification.
Fig. 2.
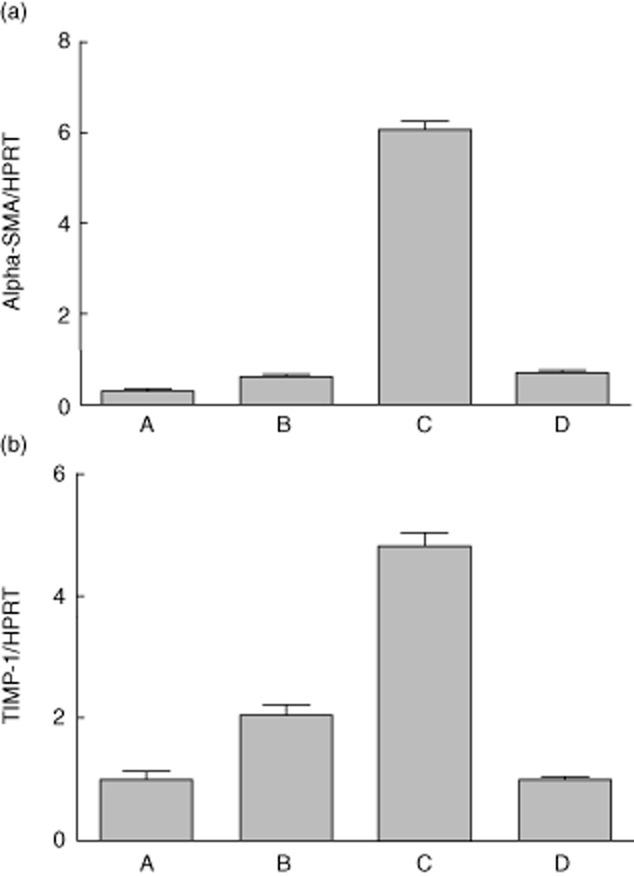
Expression of core activation genes in hepatic stellate cells (HSCs) following deactivation by basement membrane substrate (Matrigel™). Reverse transcription–quantitative polymerase chain reaction (RT–qPCR) for α-smooth muscle actin (SMA) (a) and tissue inhibitor of metalloproteinase (TIMP)-1 (b) was performed as described using RNA extracted from human HSC at the following time-points: quiescent HSCs immediately following isolation and prior to plating on tissue culture plastic (column A); HSCs cultured on tissue culture plastic for 24 h (column B); HSCs activated following reculture on tissue culture plastic for 7 days (column C) and activated HSCs recultured in Matrigel™ for 48 h with loss of activated phenotype (column D). Expression of α-SMA and TIMP-1 is shown normalized relative to housekeeping gene hypoxanthine–guanine phosphoribosyltransferase (HPRT). For (a), one-way analysis of variance (anova) with Bonferroni's multiple comparison test of A versus C, B versus C and C versus D found significant difference with a P-value of <0·0001. Other comparisons were not significant. For (b), all comparisons found significant difference with a P-value of <0·0001 except A versus D.
Activation of HSCs by MASP-1
In order to assess the capacity of MASP-1 to activate HSC from the quiescent state, HSC which had been recultured on Matrigel™ were exposed to MASP-1 and appropriate controls. HSCs stimulated with positive controls thrombin (30 mU/ml) and recombinant TGF-β (5 ng/ml) showed clear evidence of activation after 24 h. HSCs stimulated with MASP-1 also showed unequivocal evidence of differentiation to the activated phenotype, as assessed by typical changes in morphology (Fig. 3). In contrast, HSCs that had either been exposed to control MASP-1 which had not been cleaved with enterokinase, or cultured in the presence of enterokinase alone, showed no evidence of activation (Fig. 3). Consistent with phenotypic evidence, HSCs stimulated with MASP-1 and positive controls up-regulated α-SMA and TIMP-1 expression significantly as assessed by RT–quantitative (q)PCR, whereas cells which had been exposed to uncleaved MASP-1 as a control showed no up-regulation of these activation markers (Fig. 4). For α-SMA, levels of expression in cells stimulated by MASP-1 increased more than 10-fold at 24 h in comparison to both control HSCs cultured on Matrigel™ alone for the same time-period (P < 0·001) and HSCs which had been exposed MASP-1 zymogen which had not been cleaved with enterokinase (P < 0·001). For TIMP-1, increases at 24 h were also significant (P < 0·001 for comparisons with quiescent cells and P < 0·001 for comparisons with cells cultured for 24 h in the presence of uncleaved zymogen).
Fig. 3.

Mannan binding lectin-associated serine protease (MASP-1) activates human hepatic stellate cells (HSCs) cultured on Matrigel™-coated plates. Human HSCs were cultured in Matrigel™ thin gel as described treated with: thrombin at a dose of 30 mU/ml (a); purified MASP-1 cleaved with enterokinase at a dose of 2·5–3·5 μg/ml [with an equivalent capacity to cleave Boc-Val-Pro-Arg-aminomethylcoumarin (VPR-AMC) as 30 mU/ml of thrombin] (b); transforming growth factor (TGF)-β at a dose of 5 ng/ml (c). HSC treated with an equivalent dose of MASP-1 zymogen which had not been cleaved with 0·2% enterokinase (d) and HSCs cultured on Matrigel™ exposed to 0·2% enterokinase alone (e) were used as controls. (a–c) The ‘myofibroblast’ morphology typical of fully activated HSCs, whereas HSC shown in (d,e) remain in the quiescent state. Magnification was selected to best illustrate the key features: (a,b,c) ×200 magnification and (d,e) ×100 magnification.
Fig. 4.
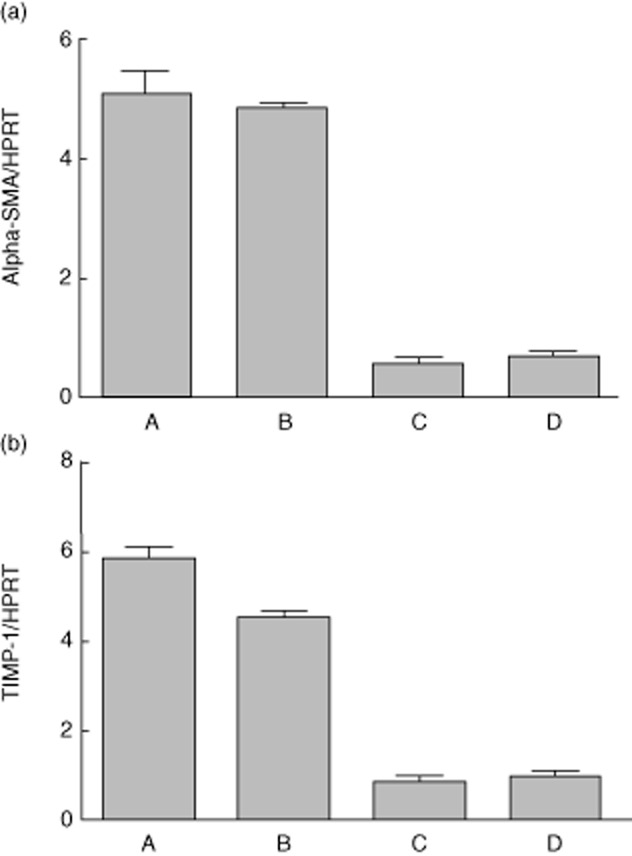
Expression of core activation genes in hepatic stellate cells (HSCs) cultured in Matrigel™ following stimulation by mannan binding lectin-associated serine protease (MASP-1) and controls. Reverse transcription–quantitative polymerase chain reaction (RT–qPCR) for α-smooth muscle actin (SMA) (a) and tissue inhibitor of metalloproteinase (TIMP)-1 (b) was performed as described using RNA extracted from HSCs cultured in Matrigel™ thin gel and treated with: thrombin at a dose of 30 mU/ml (column A); purified MASP-1 cleaved with enterokinase at a dose of 2·5–3·5 μg/ml (column B); HSC treated with an equivalent dose of MASP-1 zymogen which had not been cleaved with enterokinase (column C) and control HSCs cultured on Matrigel™ without further stimulus (column D). Expression of α-SMA and TIMP-1 is shown normalized relative to housekeeping gene hypoxanthine–guanine phosphoribosyltransferase (HPRT). For (a), one-way analysis of variance (anova) with Bonferroni's multiple comparison test found that all comparisons found a significant difference (P < 0·0001) except for two (A versus B and C versus D). For (b), all comparisons except one (C versus D) found a significant difference with a P-value of <0·001).
Expression of MBL and MASP-1 in HCV infected cells
Anti-NS5A staining confirmed high levels of JFH-1 replication in Huh 7·5 cells (Fig. 5). Expression of MASP-1 and MBL was assessed by RT–qPCR at 24 and 72 h following transfection of infectious clone JFH-1, replication defective mutant JFH-1GND and in mock-transfected control cultures. MASP-1 was not up-regulated at 24 h but became up-regulated significantly in productively infected Huh 7·5 cells in comparison to controls at 72 h (P = 0·0015) (Fig. 6), corresponding to the onset of peak viral replication in our system. We did not find evidence of up-regulation of MBL-2 in our system (data not shown).
Fig. 5.
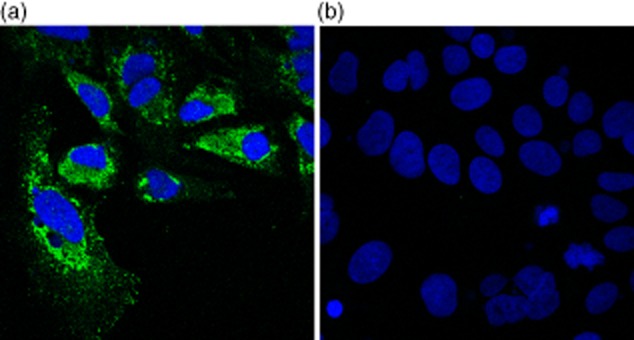
Immunofluorescence of Huh 7·5 cells 48 h after transfection of infectious molecular clone JFH-1. Blue = 4′,6-diamidino-2-phenylindole (DAPI) nuclear stain; green = 9E10 anti-NS5A. Anti-NS5A and DAPI nuclear staining are shown together in (a) and DAPI staining alone in (b).
Fig. 6.
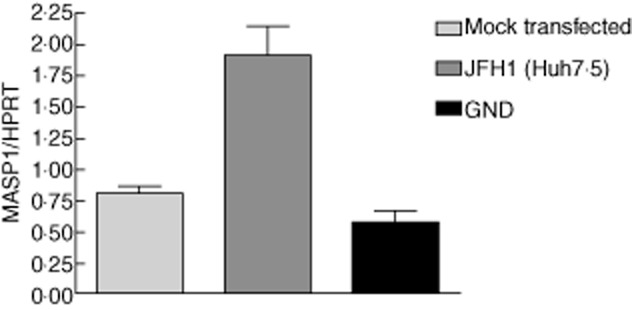
Hepatitis C virus (HCV) infection increases expression of mannan binding lectin-associated serine protease (MASP-1) in the Huh 7·5 hepatocyte cell line. Reverse transcription–quantitative polymerase chain reaction (RT–qPCR) was conducted in the Huh 7·5 hepatocyte cell line 72 h after transfection with either infectious molecular clone JFH-1 or replication defective mutant GND. Mock transfected cells were used as controls. Using the one-way analysis of variance (anova) test with Bonferroni's multiple comparison test to compare MASP-1 expression in mock transfected, Huh 7·5 and GND found that MASP-1 was expressed significantly more highly in Huh7·5 (P = 0·0015).
Discussion
This study has, for the first time, provided unequivocal evidence that MASP-1 can induce differentiation of human HSCs to the activated state under experimental conditions. Furthermore, these effects occur at a concentration equivalent to that found in vivo [20]. These observations suggest a new role for MASP-1 and provide a possible mechanistic link between the high levels of MASP-1 activity and fibrosis in HCV infection which we have demonstrated previously.
Two lines of evidence are consistent with our finding of a direct role for MASP-1 in HSC activation. First, MASP-1 has a broad structural relationship with thrombin and can also promote the formation of a fibrin clot [12,13], suggesting functional relatedness. Thrombin exerts multiple actions on HSCs, including stimulation of proliferation, calcium influx and contractility, and induces secretion of key mediators of the process of activation [21,22]. Secondly, MASP-1 has been shown recently to activate human vascular endothelial cells (HUVECs) in a manner dependent upon calcium signalling, nuclear factor (NF)-kB and p38 mitogen-activated protein kinase (MAPK) pathways [23]. MASP-1 appears to exert its actions on HUVEC in a protease-activated receptor 4 (PAR4)-dependent manner [23], and we note that the PAR family is expressed at the protein level on quiescent stellate cells (K.B. and B.J.T., unpublished observations).
There is good evidence that MBL and ficolin deficiency predisposes to opportunistic infections in both adults and children [24–26], and accumulating evidence that innate immunity is central to the host response to HCV infection [27–29]. HCV surface glycoproteins E1 and E2 possess up to 15 glycosylation sites which present mannose targets to MBL [30]. Two recent studies have found evidence of functional interaction between MBL/ficolins and surface determinants of HCV. L-ficolin has been shown to recognize and bind HCV envelope glycoproteins E1 and E2, with consequent lectin complement pathway-mediated cytolytic activity against HCV-infected hepatocytes [31]. A second study has found unequivocal evidence that MBL binds to HCV E1 and E2 glycoproteins [32]. Further, recognition of E1/E2 by MBL-activated complement in a MASP-dependent manner [32]. These studies suggest that HCV glycoproteins are naturally recognized by MBL/ficolins, and this pattern recognition event may be important in the innate immune response to HCV infection. In this context, our observations that levels of MASP-1 are elevated in severe HCV disease [5], and that MASP-1 may be profibrogenic, are important and suggest the novel notion that the balance between host defence and inflammatory response central to innate immune responses may act to amplify HCV-related disease in certain individuals by a direct action of elevated levels of MASP-1 on HSCs. The role of MASP-1 in HCV pathogenesis may be amplified further by increased expression of MASP-1 in hepatocytes. These findings, obtained by quantitative PCR, confirm a recent expression profile analysis that found increased expression of MASP-1 in hepatocyte cell lines infected by JFH-1 [33].
There are two broad caveats associated with our observations. First, we have demonstrated MASP-1 activation of HSCs that have been isolated recently from a complex cellular architecture in human liver, and studied in isolation in a two-dimensional experimental system. Secondly, we have studied the effect of purified MASP-1, whereas its physiological role is a function of complexes between proteases and pattern recognitions molecules [6]. Our study was, however, prompted by the clinical observation of association between elevated MASP-1 activity and the severity on fibrosis in HCV-related liver disease [5]. Further, recent studies have found evidence of an interaction between HCV envelope glycoproteins and MBL/ficolins with consequent activation of complement and such interactions would almost certainly occur within the productively infected liver [31,32]. There is therefore a plausible clinical and disease pathogenesis context for our findings. Finally, in support of the notion that lectin pathway complement activation may have a role in liver fibrosis, both MASP-2 [34] and MBL-2 [35] single nucleotide polymorphisms have been shown to correlate with the severity of liver fibrosis and outcomes in HCV-infected individuals.
Taken together, the clinical and experimental evidence supports the notion that MASP-1 could be a driver of disease progression in HCV infection, and that innate immune responses may promote progression of HCV-related disease in certain individuals. In this context, it is interesting to note that the structure of the substrate binding groove in MASP-1 has been resolved [36] and that recent work suggests the possibility of monospecific inhibition of MASP-1 activity which may, in due course, have therapeutic potential [8,9].
Acknowledgments
The authors would like to acknowledge the invaluable contribution of Dr Alexander Tarr, Professor Jonathan K. Ball and Professor William L. Irving of the School of Molecular Medical Sciences, University of Nottingham. We would also like to thank Professor Charles Rice of Rockefeller University for the kind gift of anti-NS5a monoclonal antibody 9E10 and the Huh7·5 hepatocyte cell line. This work was supported by international studentships to: Amanj Saeed (Government of Iraq); Kanwal Baloch (University of Liaquat, Pakistan); and Liqiong Chen (University of Nottingham, UK).
Disclosures
No competing interests are declared.
References
- 1.Henderson NC, Iredale JP. Liver fibrosis: cellular mechanisms of progression and resolution. Clin Sci (Lond) 2007;112:265–280. doi: 10.1042/CS20060242. [DOI] [PubMed] [Google Scholar]
- 2.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kisseleva T, Brenner DA. Role of hepatic stellate cells in fibrogenesis and the reversal of fibrosis. J Gastroenterol Hepatol. 2007;22(Suppl. 1):P73–78. doi: 10.1111/j.1440-1746.2006.04658.x. [DOI] [PubMed] [Google Scholar]
- 4.Thomson BJ. Hepatitis C virus: the growing challenge. Br Med Bull. 2009;89:153–167. doi: 10.1093/bmb/ldp003. [DOI] [PubMed] [Google Scholar]
- 5.Brown KS, Keogh MJ, Tagiuri N, et al. Severe fibrosis in hepatitis C virus-infected patients is associated with increased activity of the mannan-binding lectin (MBL)/MBL-associated serine protease 1 (MASP-1) complex. Clin Exp Immunol. 2007;147:90–98. doi: 10.1111/j.1365-2249.2006.03264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thiel S. Complement activating soluble pattern recognition molecules with collagen-like regions, mannan-binding lectins, ficolins, and associated proteins. Mol Immunol. 2007;44:3875–3888. doi: 10.1016/j.molimm.2007.06.005. [DOI] [PubMed] [Google Scholar]
- 7.Gal P, Harmat V, Kocsis A, et al. A true autoactivating enzyme. Structural insight into mannose-binding lectin-associated serine protease-2 activations. J Biol Chem. 2005;280:33435–33444. doi: 10.1074/jbc.M506051200. [DOI] [PubMed] [Google Scholar]
- 8.Heja D, Kocsis A, Dobo J, et al. Revised mechanism of complement lectin-pathway activation revealing the role of serine protease MASP-1 as the exclusive activator of MASP-2. Proc Natl Acad Sci USA. 2012;109:10498–10503. doi: 10.1073/pnas.1202588109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heja D, Harmat V, Fodor K, et al. Monospecific inhibitors show that both mannan-binding lectin-associated serine protease-1 (MASP-1) and -2 are essential for lectin pathway activation and reveal structural plasticity of MASP-2. J Biol Chem. 2012;287:20290–20300. doi: 10.1074/jbc.M112.354332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Degn SE, Jensen L, Hansen AG, et al. Mannan-binding lectin-associated serine protease (MASP)-1 is crucial for lectin pathway activation in human serum, whereas neither MASP-1 nor MASP-3 is required for alternative pathway function. J Immunol. 2012;189:3957–3969. doi: 10.4049/jimmunol.1201736. [DOI] [PubMed] [Google Scholar]
- 11.Megyeri M, Harmat V, Major B, et al. Quantitative characterisation of the activation steps of mannan-binding lectin (MBL)-associated serine proteases (MASPs) points to the central role of MASP-1 in the initiation of the complement lectin pathway. J Biol Chem. 2013;288:8922–8934. doi: 10.1074/jbc.M112.446500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gulla KC, Gupta K, Krarup A, et al. Activation of mannan-binding lectin-associated serine proteases leads to generation of a fibrin clot. Immunology. 2010;129:482–495. doi: 10.1111/j.1365-2567.2009.03200.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hess K, Ajjan R, Phoenix F, et al. Effects of MASP-1 of the complement system on activation of coagulation factors and plasma clot formation. PLoS. 2012;7:e35690. doi: 10.1371/journal.pone.0035690. doi: 10.1371/journal.pone.0035690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marra F, DeFranco R, Grappone C, et al. Expression of the thrombin receptor in human liver: up-regulation during acute and chronic injury. Hepatology. 1998;27:462–471. doi: 10.1002/hep.510270221. [DOI] [PubMed] [Google Scholar]
- 15.Calvaruso V, Maimone S, Gatt A, et al. Coagulation and fibrosis in chronic liver disease. Gut. 2008;57:1722–1727. doi: 10.1136/gut.2008.150748. [DOI] [PubMed] [Google Scholar]
- 16.Gaca MD, Zhou X, Issa R, et al. Basement membrane-like matrix inhibits proliferation and collagen synthesis by activated rat hepatic stellate cells: evidence for matrix-dependent deactivation of stellate cells. Matrix Biol. 2003;22:229–239. doi: 10.1016/s0945-053x(03)00017-9. [DOI] [PubMed] [Google Scholar]
- 17.Chen CB, Wallis R. Stoichiometry of complexes between mannose-binding protein and its associated serine proteases. Defining functional units of complement activation. J Biol Chem. 2001;276:25894–25902. doi: 10.1074/jbc.M103539200. [DOI] [PubMed] [Google Scholar]
- 18.Wakita T, Pietschmann T, Kato T, et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med. 2005;11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lindenbach BD, Evans MJ, Syder AJ, et al. Complete replication of hepatitis C virus in cell culture. Science. 2005;309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 20.Terai I, Kobayashi K, Matsushita M, Fujita T. Human serum mannose-binding lectin (MBL)-associated serine protease-1 (MASP-1): determination of levels in body fluids and identification of two forms in serum. Clin Exp Immunol. 1997;110:317–323. doi: 10.1111/j.1365-2249.1997.tb08334.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mallat A, Gallois C, Tao J, et al. Platelet-derived growth factor-BB and thrombin generate positive and negative signals from human hepatic stellate cell proliferation. Role of a prostaglandin/cyclic AMP pathway and cross-talk with endothelin receptors. J Biol Chem. 1998;273:27300–27305. doi: 10.1074/jbc.273.42.27300. [DOI] [PubMed] [Google Scholar]
- 22.Marra F, Grandaliano G, Valente AJ, et al. Thrombin stimulates proliferation of liver fat storing cells and expression of monocyte-chemotactic protein-1: potential role in liver injury. Hepatology. 1995;22:780–787. [PubMed] [Google Scholar]
- 23.Megyeri M, Mako V, Beinrohr L, et al. Complement protease MASP-1 activates human endothelial cells: PAR4 activation is a link between complement and endothelial function. J Immunol. 2009;183:3409–3416. doi: 10.4049/jimmunol.0900879. [DOI] [PubMed] [Google Scholar]
- 24.Schlapbach LJ, Mattmann M, Thiel S, et al. Differential role of the lectin pathway of complement activation in susceptibility to neonatal sepsis. Clin Infect Dis. 2010;15:153–162. doi: 10.1086/653531. [DOI] [PubMed] [Google Scholar]
- 25.Azad AK, Sadee W, Schlesinger LS. Innate immune gene polymorphisms in tuberculosis. Infect Immun. 2012;80:3343–3359. doi: 10.1128/IAI.00443-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ali YM, Lynch NJ, Haleem KS, et al. The lectin pathway of complement activation is a critical component of the innate immune response to pneumococcal infection. PLoS Pathog. 2012;8:e1002793. doi: 10.1371/journal.ppat.1002793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang S, Kodys K, Babcock GJ, Szabo G. CD81/CD9 tetraspanins aid plasmacytoid dendritic cells in recognition of HCV-infected cells and induction of IFNα. Hepatology. 2012 doi: 10.1002/hep.25827. doi: 10.1002/hep.25827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang S, Kodys K, Li K, Szabo G. Human type 2 myeloid dendritic cells produce interferon-λ and amplify interferon-α in response to hepatitis C virus infection. Gastroenterology. 2013;144:414–425. doi: 10.1053/j.gastro.2012.10.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tarr AW, Urbanowicz RA, Ball JK. The role of humoral innate immunity in hepatitis C virus infection. Viruses. 2012;4:1–27. doi: 10.3390/v4010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iacob RE, Perdivara I, Przybylski M, et al. Mass spectrometric characterization of glycosylation of hepatitis C virus E2 envelope glycoprotein reveals extended microheterogeneity of N-glycans. J Am Soc Mass Spectrom. 2008;19:428–444. doi: 10.1016/j.jasms.2007.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu J, Ali MA, Shi Y, et al. Specifically binding of L-ficolin to N-glycans of HCV envelope glycoproteins E1 and E2 leads to complement activation. Cell Mol Immunol. 2009;6:235–244. doi: 10.1038/cmi.2009.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brown K, Keogh MJ, Owsianka AM, et al. Specific interaction of hepatitis C virus glycoproteins with mannan binding lectin inhibits virus entry. Protein Cell. 2010;1:664–674. doi: 10.1007/s13238-010-0088-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Blackham S, Baillie A, Al-Hababi F, et al. Gene expression profiling indicates the roles of host oxidative stress, apoptosis, lipid metabolism, and intracellular transport genes in the replication of hepatitis C virus. J Virol. 2010;84:5404–5414. doi: 10.1128/JVI.02529-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tulio S, Faucz FR, Werneck RI, et al. MASP2 gene polymorphism is associated with susceptibility to hepatitis C virus infection. Hum Immunol. 2011;72:912–915. doi: 10.1016/j.humimm.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eurich D, Boas-Knoop S, Morawietz L, et al. Association of mannose-binding lectin-2 gene polymorphism with the development of hepatitis C-induced hepatocellular carcinoma. Liver Int. 2011;31:1006–1012. doi: 10.1111/j.1478-3231.2011.02522.x. [DOI] [PubMed] [Google Scholar]
- 36.Dobo J, Harmat V, Beinrohr L, Sebestyen E, Zavodsky P, Gal P. MASP-1, a promiscuous complement protease: structure of its catalytic region reveals the basis of its broad specificity. J Immunol. 2009;183:1207–1214. doi: 10.4049/jimmunol.0901141. [DOI] [PubMed] [Google Scholar]