

Abstract
Atherosclerosis is a multifactorial disease with a severe burden on western society. Recent insights into the pathogenesis of atherosclerosis underscore the importance of chronic inflammation in both the initiation and progression of vascular remodelling. Expression of immunoregulatory molecules by vascular wall components within the atherosclerotic lesions is accordingly thought to contribute to the ongoing inflammatory process. Besides gene regulatory proteins (transcription factors), epigenetic mechanisms also play an essential and fundamental role in the transcriptional control of gene expression. These epigenetic mechanisms change the accessibility of chromatin by DNA methylation and histone modifications. Epigenetic modulators are thus critically involved in the regulation of vascular, immune and tissue-specific gene expression within the atherosclerotic lesion. Importantly, epigenetic processes are reversible and may provide an excellent therapeutic target. The concept of epigenetic regulation is gradually being recognized as an important factor in the pathogenesis of atherosclerosis. Recent research provides an essential link between inflammation and reprogramming of the epigenome. In this review we therefore discuss the basis of epigenetic regulation – and the contribution thereof in the regulation of inflammatory processes in general and during atherosclerosis in particular. Moreover we highlight potential therapeutic interventions based on epigenetic mechanisms.
Keywords: atherosclerosis, epigenetics, inflammation, chromatin, histone modifications, CAD, KDAC inhibitors
Introduction
Cardiovascular diseases (CVD) are the number one cause of death in western societies [1, 2]. Atherosclerosis is the primary cause of coronary artery disease (CAD) and stroke and is regarded as a (chronic) inflammatory disease [3, 4]. Atherosclerosis is characterized by asymmetrical focal thickenings of the arterial intima that are often referred to as atherosclerotic lesions or atheromas [5]. These thickenings are caused by the accumulation of lipids and inflammatory cells in the vessel wall [5]. Especially T cells and macrophages play an important role in pathogenesis of atherosclerosis [6]. Besides lipids and inflammatory cells, the lesion – depending on the stage of the disease – further consists of vascular endothelial cells (vECs) and vascular smooth muscle cells (vSMCs), and extracellular matrix (ECM) [7]. Extreme thickening of the intima or even plaque rupture with subsequent thrombus formation may seriously hamper coronary blood flow, eventually leading to myocardial infarction [8]. Thus, atherosclerosis is a potential life-threatening condition and understanding the cause of atherosclerosis may contribute to the treatment and possibly prevention of this disease.
Atherosclerotic plaque formation is a dynamic multi-cellular process in which the activity of the different cell types involved is essentially determined by the regulation of different genes [3–8]. Nowadays we are fully aware of the important involvement of epigenetic processes in the regulation of gene expression. Understanding these processes is critical for our understanding of inflammatory responses and disease. For instance, not only is cytokine expression under epigenetic control, cytokines themselves induce (indirect) changes to the chromatin, providing an essential link between inflammation and epigenetic programming [9]. Here, we review the role of epigenetic regulation in vascular remodelling and in the activity of immune cells involved in atherosclerotic plaque formation.
Epigenetics explained
Although all cells in our body contain the same genetic material, each cell acts in a cell-type specific manner, as determined by its gene expression profile. Epigenetic mechanisms control gene expression without modifying the actual genetic code, while the altered gene expression patterns can be passed on to the daughter cells upon cell division or even transgenerationally [10–12].
In its natural state DNA is packaged into chromatin, a highly organized and dynamic protein-DNA complex which consists of DNA, histones and non-histone proteins [13]. Epigenetic mechanisms alter the accessibility of chromatin by modifying DNA and by modification or rearrangement of nucleosomes, which include post-translational modifications of histones [14–16]. Accessible chromatin allows gene-regulatory proteins (transcription factors) to interact with their cognate binding sites within the regulatory regions of genes, such as proximal promoters and enhancers/ silencers. In this way, global gene activation and local control of gene-specific transcription is exerted by components of the epigenetic machinery. Moreover, environmental factors have an important role in the establishment of the epigenome [17–21]. Since epigenetic alterations can accumulate in time, environmental factors can have profound effects on the cellular repertoire of expressed genes [10].
Already in 1975 DNA methylation was proposed to play a role in regulating gene transcription [22, 23]. Generally speaking, DNA methylation is associated with low gene activity. Actively transcribed genes are usually maintained in an unmethylated state in their promoter regions [24–26]. DNA methylation involves methylation at the C5 position of cytosine residues in a CpG dinucleotide (i.e. cytosine followed by a guanine) context, exerted by DNA methyltransferases (DNMTs). DNMTs are capable of both methylation and demethylation – as has been postulated in two recent reports – thus making the modification reversible [27, 28]. Notably, CpG dinucleotides are underrepresented in the genome of eukaryotes, but can be found in clusters – so-called ‘CpG islands’– which in turn are mainly found in promoter regions. The term ‘CpG island’ is defined as a region of at least 500 base pairs with a CG content greater then 55%[29].
For many years it was thought that functionally relevant DNA methylation occurs at the CpG islands within the promoter region. However, in a recent study by Irizarry et al. it was shown that most tissue-specific DNA methylation occurs in regions up to 2 kb distant of the promoter region [30], which were dubbed ‘CpG island shores’ by the authors. Furthermore they showed that cancer-specific methylation also occurs at conserved tissue-specific CpG island shores.
Besides methylation of DNA, post-translational modifications of N-terminal tails of histone proteins are key components in the epigenetic regulation of genes. Over 60 distinct modifications are currently known – mostly in the histone tails – although some have been observed in the globular domain [31, 32]. Modifications of histone tails include (amongst others) acetylation and methylation of lysine residues. Whereas acetylation of histone tails is correlated with gene activation [33–35], the influence of histone methylation depends on the exact residue methylated and the number of added methyl groups [36–38]. Histone modifications and DNA methylation provide a close interplay with respect to gene regulation as both activities are functionally linked [39].
Whereas lysine methylation and acetylation are the most studied modifications, there are many more histone modifications known. Arginine residues can also be methylated and acetylated, just as lysine residues. As is the case with lysine methylation, whether arginine methylation results in repression or activation of transcription depends on which arginine residue is methylated [40]. In addition, SUMOylation and ubiquitination of histones has also been observed [32]. SUMOylation appears to be associated with transcriptional repression [41], whereas ubiquitination has been suggested to play a role in transcriptional activation and elongation [42]. However, the exact function of these modifications remains to be elucidated.
Epigenetic alterations are reversible
Interestingly, epigenetic modifications are reversible, which is illustrated by the counterbalancing actions of the various enzymes that are responsible for maintaining the epigenome. Lysine acetyltransferases (KATs, formerly known as histone acetyltransferases) are counteracted by lysine deacetylases (KDACs, formerly known as histone deacetylases) and sirtuins in establishing histone acetylation modifications at lysine residues in the N-terminal tails. Lysine methyltransferases KMTs, (formerly known as histone methyltransferases), finally, are counteracted by the recently discovered lysine demethylases (KDMs, formerly known as histone demethylases) in establishing histone methylation modifications (Fig. 1). In this way these enzymes promote a return to, respectively, repressive or active chromatin structure [43–45].
Fig 1.
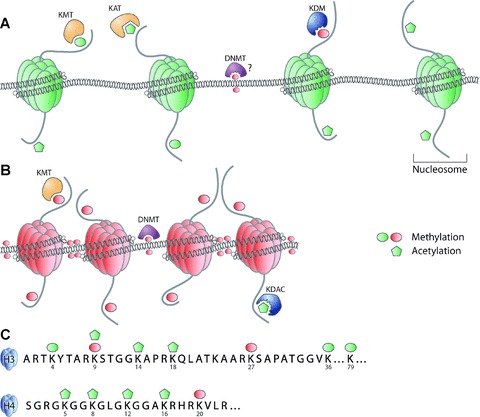
Schematic representation of chromatin. DNA is wrapped around the octamer of core histones to form a structure called the nucleosome. Transcriptionally active chromatin (euchromatin) is hallmarked by low frequency of DNA methylation. High levels of histone acetylation and histone methylation correlated with activation (MeK4H3, MeK36H3, MeK79H3) are detected in euchromatin. KATs are responsible for acetylation of histone tails, KMTs for methylation, whereas KDMs remove methylation marks of histones. DNMTs probably remove methylation marks from DNA, although this has not yet been proven definitively (A). Transcriptionally silent chromatin (heterochromatin), is hallmarked by high frequencies of repressive methylation markers (i.e. DNA methylation and MeK9H3, MeK27H3 and MeK20H4). Methylation of histone tails is catalysed by KMTs whereas DNA methylation is catalysed by DNMT. Acetylation markers, associated with activation, are removed by KDACs (B). Post-translational modifications of histone tails include (but are not limited to) acetylation (Ac) and methylation (Me), which can be associated with transcriptionally active (green) or silent (red) chromatin (C).
Importantly, the reversible nature of these epigenetic modifications makes the chromatin-modifying enzymes interesting therapeutic targets [46–48] and a myriad of small molecule inhibitors that can influence the enzymatic activity of these chromatin-modifying enzymes are currently being tested for their efficacy [49 and references therein]. These inhibitors are mostly evaluated in the field of cancer research, in cell lines, in animal models as well as in clinical trials [49]. Notably suberoylanilide hydroxamic acid (SAHA, or Vorinostat) and valproic acid (KDAC inhibitors [KDACi]) are already FDA approved for cancer treatment and epilepsy, respectively [50–52]. With respect to atherosclerosis, administration of curcumin (a KAT inhibitor) resulted in significantly lowered low-density lipoprotein levels and raised high-density lipoprotein levels in healthy volunteers [53–55]. It has also been reported that curcumin has an anti-proliferative effect on peripheral blood mononuclear cells (PBMCs) and vSMCs [56].
Atherosclerosis
The first step in atherosclerosis is thought to be endothelial dysfunction [57], possibly triggered by oxidized low-density lipoprotein (OxLDL) [5]. Endothelial activation results in the expression of cytokines and chemokines, enhancement in the permeability of the endothelial cell (EC) layer and an increment in the expression of adhesion molecules. Immune and inflammatory cells, such as monocytes and T lymphocytes, are then attracted by chemokines (chemotaxis), followed by firm adhesion and transendothelial migration. These cells subsequently infiltrate the subendothelium of the vascular wall and form the main players in the formation of the ‘fatty streak’– the first identifiable lesion – (Fig. 2) [57].
Fig 2.
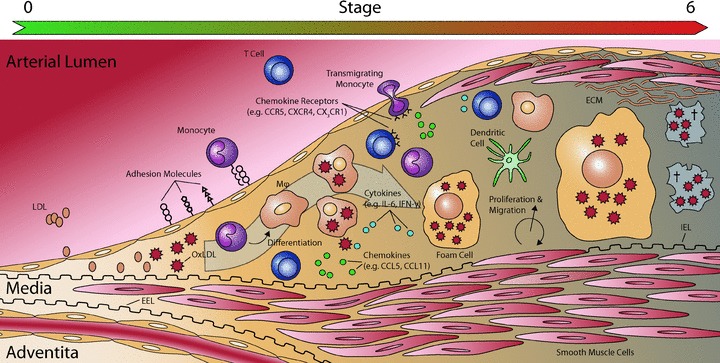
Schematic representation of plaque development. OxLDL is thought to trigger endothelial dysfunction. OxLDL is often associated with proteoglycans in the subendothelium. Activated ECs start to express various adhesion molecules. Immune cells, mainly monocytes and T cells, are attracted by chemokines and migrate across the endothelium, using adhesion molecules. Monocytes then differentiate into macrophages (Mϕ) and DCs. These macrophages take op OxLDL and transform into ‘foam cells’. This process of foam cell formation is indicated with the large grey arrow. T cells recruited to the plaque express several cytokines and chemokines. In response to the stimuli, vSMCs start to proliferate and migrate, and start to produce ECM. Abbreviations: ECM: extra cellular matrix; EEL: external elastic lamina; IEL: internal elastic lamina; LDL: low-density lipoprotein; OxLDL: oxidized low-density lipoprotein. Illustration inspired on [192].
Foam cells – cholesterol-engorged monocyte-derived macrophages – are the dominant type of immune cells found within the lesions. Macrophages take up the OxLDL present in the subendothelium, but are unable to digest it sufficiently, resulting in the formation of foam cells [5]. Although fatty streaks are not clinically significant, they are thought to be the precursors to more advanced lesions, although there is some dispute whether fatty streaks are truly precursors of advanced lesions [7, 58]. If the causative agents for this endothelial dysfunction are not effectively removed, the inflammatory response can continue indefinitely. The atherosclerosis-associated immune response is driven by monocyte-derived macrophages and specific subsets of T cells [6, 59]. Monocytes and T lymphocytes migrate from the blood and proliferate within the lesion, resulting in the accumulation of inflammatory cells. If these cells become activated they release cytokines, chemokines and growth factors [57], contributing to the ongoing inflammatory process. Moreover, immune activation eventually induces focal necrosis leading to the necrotic core found in advanced lesions.
In later stages of the disease other cell types (e.g. vSMCs) become involved. vSMCs start to proliferate as a response to the various signalling molecules present in the lesion, thereby thickening the arterial wall. Eventually the lesion will evolve into a ‘fibrolipid plaque’, consisting of a lipid-rich necrotic core covered by a fibrous cap of vSMCs and a collagen-rich matrix [3, 60]. The resulting thickening of the arterial wall caused by the plaque formation will partially be compensated by gradual dilation (remodelling). If the plaque volume becomes too large, this process cannot compensate enough, leading to decreased lumen size and hampered blood flow [61].
As stated above, the development of the atherosclerotic lesion is for the larger part determined by the release of various cytokines and chemokines. Pro-inflammatory cytokines such as tumour necrosis factor-α (TNF-α) and interferon-γ (IFN-γ) released by activated T cells – recruited to the vascular cell wall – play an essential role in disease development. This critical role for pro-inflammatory cytokines in atherosclerosis development is illustrated in atherosclerotic mouse models. It has been shown that deletion of the genes coding for pro-inflammatory cytokines (e.g. IL-12, IFN-γ, IFN-γ–receptor and TNF-α) resulted in reduced atherosclerosis development in these knockout mice [62–67]. Conversely, deletion of anti-inflammatory cytokine genes (e.g. IL-10 and TGF-β) resulted in increased atherosclerosis [68, 69]. Different genetic variants of the TNF-α gene also showed to be important for the outcome of percutaneous coronary intervention (PCI, one of the treatments used for opening of occluded vessels) with respect to restenosis – one of the major limitations of the PCI technique [66]. Genetic variants of the IL-10 gene have also proven to be important for the development of restenosis [70].
The cross-talk between immune system and vascular wall results in up-regulation of major histocompatibility complex (MHC) class II (MHC-II) molecules and of the MHC class Ib molecule, HLA-E [3, 71]. Furthermore, it results in up-regulation of chemokines such as CX3CL1 (fractalkine), CCL2 (MCP-1), CCL5 (RANTES) and their receptors [3, 72–75]. All of these factors influence the gene expression profiles of cells present within the lesion. These observations not only feed the current theory that atherosclerosis is an inflammatory disease – and not just a disease of lipid metabolism. For many of these inflammatory and immune modulating factors epigenetic components have been identified that are responsible for the transcriptional regulation of the genes encoding these factors. Thus epigenetic processes might prove to be important contributing factors to disease pathogenesis that should be taken into consideration.
Epigenetics and association with atherosclerosis
Epigenetics provides an attractive explanation how diet, environment and lifestyle may contribute to disease. In principle, epigenetics explains how such external factors can impose aberrant gene expression patterns in an individual lifetime and even trans-generationally. One of the earliest studies linking DNA methylation to atherosclerosis showed that the extracellular superoxide dismutase (ec-SOD) gene was hypomethylated in atherosclerotic lesions in rabbits [76]. The importance of DNA methylation as a contributing factor to the pathogenesis of atherosclerosis is underscored by a study linking global DNA hypermethylation with predisposition to, and natural history of atherosclerosis [77]. In this study the correlation was made by comparing methylation sensitive restriction of peripheral blood leucocyte DNA. However, this approach evaluates only the inflammatory component of the disease process in PBMCs and not other components such as vECs and vSMCs. In particular, DNA hypermethylation was found to be significantly associated with both all-cause and cardiovascular mortality, even following the adjustment for age, CVD and diabetes mellitus. Hypermethylation was found in patients suffering from inflammation (high C-reactive protein levels) and also in 13 patients who died from CVD. This paper suggests that global DNA hypermethylation is associated with inflammation and increased mortality in chronic kidney disease. Importantly, hypermethylation was found to be the strongest independent risk factor for CVD mortality.
In the case of CAD an epigenetic component associated with disease was also identified [78]. Sharma et al., found that angiographically confirmed CAD patients showed a higher level of genomic DNA methylation – determined in peripheral blood lymphocytes – when compared to healthy controls [78]. Furthermore, a positive correlation was found between global DNA methylation and homocysteine levels. Homocysteine is known to be an independent risk factor for CAD [79, 80]. Sharma and coworkers also tested the specific methylation status of the ApoE gene by bisulphite-sequencing. The ApoE promoter was previously shown to have a degree of methylation that varies with homocysteine levels [81]. However, no significant difference could be detected between patients and controls [78]. Both these studies underline the potential effect DNA methylation can have with respect to disease development and outcome. However, the precise genes targeted by DNA methylation and thus the precise mechanisms that induce DNMT activity remain to be elucidated.
Although DNA methylation is one of the most studied epigenetic phenomena, more and more research is performed on the specific contribution of histone modifications. Recently, a study was published directly linking epigenetic histone modifications and atherosclerosis. Hastings et al. showed that atherosclerosis-prone shear-stress profiles globally decreased histone H4 acetylation and increased H4 acetylation at the c-fos promoter in smooth muscle cells (SMCs) [82]. Perhaps more importantly, they showed that atheroprone haemodynamics resulted in differentially regulated phenotypes for ECs and SMCs. Shear stress (flow) was applied to ECs that were layered on top of SMCs in forces that correlate to atheroprone or atheroprotective conditions. Atheroprone flow reduced the expression of endothelial nitric oxide synthetase (eNOS), Tie-2 and kruppel-like factor in ECs, and smooth muscle actin and myocardin in SMCs, whereas vascular cell adhesion molecule 1 (VCAM-1), IL-8 and MCP-I were increased in both cell types. This correlated to a decrease in total H4 acetylation and serum response factor (SRF) binding to the SMC actin CArG-box in the promoter region. Furthermore atheroprotective conditions induced a polygonal shape in EC whereas atheroprone conditions induced an elongated EC phenotype. In vSMCs atheroprotective conditions induced an elongated shape whereas atheroprone conditions showed a random SMC organization and cells aligned toward a perpendicular orientation relative to the direction of flow [82].
Another linkage between atherosclerosis and histone modifications was found when the administration of the KDACi trichostatin A (TSA) in ldr−/− mice led to exacerbated neointimal lesions [83]. TSA increased CD36 mRNA, protein and cell surface expression levels, which were related to increased acetylated histone binding at the promoter region. CD36 recognizes OxLDL, thus the obtained results might be due to increased OxLDL uptake by macrophages. Also levels of TNF-α and VCAM-1 were increased in aortic plaques. It has also been noted that application of TSA resulted in inhibition of SMC migration [84]. Unfortunately, the exact mechanisms are not yet fully understood, but these studies clearly indicate a role for histone acetylation in disease pathogenesis.
Interestingly, some components relevant for the development of atherosclerosis, such as OxLDL, are able to modulate the activity of lysine deacetylases [85]. In cultured ECs it was shown that OxLDL reduced expression of KDAC1 and KDAC2. Global KDAC activities were partly restored by statins: pretreatment of ECs for 24 hrs with simvastatin or fluvastatin blocked the OxLDL-related modifications in H3 and H4. Addition of mevalonate could revert the effects observed after statin treatment. The researchers postulate that this, at least in part, may be due to inhibition of small GTP-binding Rho proteins. Furthermore decreased expression of KDAC2 in ECs was shown in atherosclerotic plaques of human coronary arteries [85]. Taken together, these data show the importance of histone modifications in atherosclerosis.
A study performed by Barker et al. in 1989 sheds light on how long-term cellular memory can have an effect on the development of atherosclerosis. Nowadays it is generally accepted that long-term cellular memory is mediated via epigenetic phenomena. Barker et al. demonstrated that low birth weight is associated with a higher risk of cardiovascular events in adult live [86]. This led to the hypothesis that the environment the foetus is exposed to during pregnancy might partially determine cardiovascular risk. This so-called ‘foetal origins hypothesis’ states that adaptation to an unfavourable maternal environment is beneficial to the developing embryo in utero[86]. However, when the adult environment differs from the in utero environment, this may lead to an increased risk for CVD. Studies in pregnant rats show that a protein-restricted diet results in reduced expression of Dnmt1, leading to hypomethylation of specific promoters [87]. These observations provide a strong indication that the in utero environment leads to epigenetic (re)programming, which later in life might contribute to disease development.
Epigenetic regulation of cell activity
Cell differentiation (e.g. monocyte differentiation into macrophages) and cell-specific gene expression in most cases is controlled by epigenetic mechanisms. Below we will discuss the most important cellular factors that contribute to atherosclerotic plaque formation and how their phenotype is regulated by epigenetic processes.
T cells
Several lines of investigation illustrate that T cells are important in the formation of (early) lesions [88–91]. Within the plaque, several subsets of T cells can be found. Mainly CD4+ T cells are dominantly found in plaques, but there are some CD8+ T cells found as well [5]. The T cells present in human plaques are predominantly of the Th1 phenotype [91]. Th1 cells produce IFN-γ– a macrophage activating cytokine – and are generally considered pro-atherogenic. Th2 cells are rarely detected in lesions [92]. They are generally considered anti-atherogenic, but may also contribute to aneurysm formation [93]. Regulatory T cells (Treg cells) control the balance between Th1 and Th2. Tregs are considered to be atheroprotective [94].
All T cells derive from the same precursor: the naïve T-cell. Not surprisingly, differentiation of e.g. CD4+ T cells into a Th1, Th2 or Treg phenotype is regulated by epigenetic processes [95, 96]. The differentiation of naïve T cells into Th1 or Th2 is determined by IL-12 and IL-4 cytokines, respectively. In response to these signals, transcription is initiated of lineage specific cytokine genes including IFN-γ and IL-4[97]. The IFN-γ and IL-4 loci are maintained in a poised state in naïve T cells, meaning they show both repressive and activating epigenetic marks. In 1998 it was first proposed that epigenetic, chromatin-based structural changes, underlie the Th1/Th2 differentiation [98]. These epigenetic mechanisms – that are necessary to stably maintain gene expression patterns and eliminate the need for feedback loops – will be discussed below.
In Th1 cells expression of IFN-γ is preceded by remodelling of the IFN-γ locus [99]. Upon initial stimulation of naïve T cells, the lineage determining factors GATA3 and T-bet mediate many of the structural changes to the chromatin [95]. These factors will, respectively, render the IFN-γ or IL-4 genes accessible to regulatory enzymes and other transcription factors [100–102]. On the level of DNA methylation, there are numerous findings supporting the epigenetic regulation of IFN-γ expression. Increased expression of IFN-γ was found in T cells from Dnmt knockout mice [103] and in T cells treated with DNMT inhibitors (DNMTi) [104, 105]. In addition, expression of IFN-γ by NK and NKT cells is also controlled by epigenetic processes [99]. The ability of NK and NKT cells to rapidly produce substantial amounts of IFN-γ implies that the IFN-γ locus is accessible to transcription factors, meaning that the chromatin at the IFN-γ locus is maintained in an ‘open’ state. Indeed the IFN-γ locus in NK cells was found to be hyperacetylated and in a poised state [106, 107].
In the differentiation to Th2 cells, IL-4 expression is preceded by remodelling of the IL-4 locus, similarly to the IFN-γ locus remodelling in Th1 cells. In naïve T cells, the IL-4 locus is ‘poised’, allowing rapid, early transcription. The IL-4 promoter region exhibits a low basal level of H3 acetylation and DNA hypomethylation, but also shows 3MeK27H3 (i.e. triple methylation of lysine 27 on histone H3) modifications. When naïve T cells are stimulated under Th1 conditions, transcription activating chromatin marks at the IL-4 locus are replaced with repressive marks, whereas the contrary happens under Th2 stimulating conditions (e.g. at the IFN-γlocus). These processes have been excellently reviewed by Ansel et al.[108 and references therein].
The Foxp3 transcription factor is considered the master switch for Treg. The promoter of this transcription factor showed a difference in methylation levels between Tregs and non-Treg CD4+ cells [109]. Furthermore, this study also showed a difference in activating histone marks (AcH3, AcH4 and 3MeK4H3) in Foxp3 promoter chromatin. Epigenetic regulation of T cell subtypes has also been shown in vivo. Mice which were treated with the KDACi TSA showed an increase in Foxp3+ CD4+ Treg cells – both proportionally as well as in absolute numbers – in the lymphoid tissues [96]. Besides influencing T cell populations, the differentiation of monocytes to dendritic cells (DCs) can also be modulated by KDACi [110].
Monocytes
Monocytes are critical players in the formation of atherosclerotic plaques. Monocytes migrate from the vessel lumen into the arterial wall, where they differentiate into either DCs or macrophages (Fig. 2). These macrophages later transform into foam cells, one of the major components of the atherosclerotic plaque. It has been shown by two independent researches that interfering with KDAC function during differentiation resulted in the formation of function-impaired DCs. Notably, treatment with the KDACi’s butyrate and MS-275 resulted in lowered expression of CD1. CD1 expression is a marker for mature DCs, but notably it is also important in the presentation of lipid antigens and thus may play an important role in the development of the atherosclerotic plaque [110, 111].
Endothelial cells
Epigenetic mechanisms are also directly involved in the transcriptional regulation of immune genes in the vascular wall. For instance, in cultured human ECs it was found that administration of the KDACi TSA inhibited TNF-α induced monocyte adhesion. This was the result of down-regulation of the VCAM-1 gene; however, ICAM-1 and E-selectin – two other cytokines inducible genes – were not affected [112]. This suggests that the KDACi modulated TNF-α-mediated signalling specifically targets the VCAM-1 promoter, instead of general inhibition of the NF-κB pathway. This notion is interesting, since NF-κB is a target for chromatin modifying enzymes as well (see ‘Non-histone targets’ below). Similar results have been presented for the induction of tissue factor (TF) in human ECs. The human TF promoter contains a NF-κB related binding site: TF-κB. KDACi administration inhibited agonist induced (TNF-α, IL-1β or LPS) TF activity in ECs by blocking activation of TF-κB (TF activity was reduced 90% in HUVECs; 50% reduced in PBMCs, in vitro). TSA nearly abolishes TF-κB binding, without affecting NF-κB binding (as determined by electrophoretic mobility shift assays (EMSAs), chromatin immunoprecipitation (ChIP) and luciferase promoter reporter assays). These results were achieved using a variety of structurally distinct KDACi’s [113].
Work performed in the group of Stefanie Dimmeler showed an important role for KDACs in regulating several endothelial-specific genes. Using KDACi’s, it was shown that eNOS mRNA and protein levels decreased after KDACi administration [114]. Furthermore the ‘master-switch’ for some typical endothelial expressed genes (HoxA9, which regulates eNOS, VEGF receptor 2 and VE-cadherin expression) was also down-regulated in its expression by KDACi administration [115].
Smooth muscle cells
The data reported in literature regarding epigenetic regulation in SMCs provides an explanation how epigenetics may control lineage specificity. Nearly all SMC-restricted protein genes contain the CArG box DNA sequence within their promoter and this sequence is also required for SMC gene transcription in vivo. Genes important for SMC phenotypic switching (e.g. genes important for migration, proliferation and ECM production) also contain CArG boxes within their promoters [116]. CArG boxes serve as a binding site for the transcription factor SRF [117]. However, SRF expression is not limited to SMCs and serves many functions in different cell types including cell migration and cell–cell adhesion [118].
Multiple pieces of evidence suggest that SRF binding to CArG boxes in SMCs is regulated at the level of the chromatin structure. First of all, the x-ray crystal structure of SRF showed that SRF requires several contacts within the minor groove of DNA. The minor groove would be obscured if DNA was wrapped around the nucleosome structure. Also from thermodynamic considerations, binding of SRF to the CArG box would be greatly inhibited if the CArG box was bound to nucleosomes [119 and references therein].
Secondly, using EMSAs it was shown that SRF can bind to synthetic DNA with the same affinity as to DNA obtained from lysates of both SMC and non-SMC cell cultures. Conversely, by using ChIP assays, it was shown that SRF binds to promoter regions in SMCs much more effectively then to promoter regions in non-SMCs. This suggests that in SMCs the chromatin is in such a state that it is accessible to SRF [120–123]. This observation is supported by micrococcal nuclease digestion experiments, which showed that the CArG-box of gene promoters in SMCs is more accessible to digestion then in non-SMCs [124].
Finally, it was shown that histone H3 and H4 acetylation was increased in SMCs when compared to numerous different cell-types, including cultured endothelial stem cells. In addition, non-SMCs were enriched for the repressive chromatin marks 3MeK27H3 and 2MeK20H4 at SMC-specific promoter regions [119 and references therein].
Evidence from diabetic mouse models suggests an important role for epigenetic regulation in the inflammatory phenotype of vSMCs. The recently discovered KDM, lysine-specific demethylase (LSD-1 or KDM1, a 2MeH3K27 demethylase), was shown to play a role in the pro-inflammatory phenotype of vSMCs in diabetic mice [125]. In this mouse model levels of 2MeH3K4 (a histone modification correlated with expression) were increased at the MCP-1 and IL-6 promoters. Upon stimulation with TNF-α there was recruitment of RNA polymerase II to the promoter and increased expression of MCP-1 and IL-6. Expression of LSD-1 was shown to be decreased in vSMCs. Knockdown of LSD-1 with siRNA increased inflammatory gene expression whereas over expression decreased inflammatory gene expression in vSMCs of these mice.
It may very well be possible that similar mechanisms apply in the case of atherosclerosis. As discussed in a previous section, vSMCs isolated from rat thoracic aorta, which were treated with the KDACi TSA, displayed an inhibition of vSMC proliferation [84]. It was subsequently shown that expression of p21WAF1 was induced by this treatment. However, KDACi treatment had no effect on SMCs isolated from p21WAF1 knockout mice.
Together, these findings illustrate that epigenetic mechanisms play an important role in the processes that control differentiation and activation of haematopoietic cells, but also of vECs and vSMCs [119, 126].
Chemokines, their receptors and other genes involved in inflammation
Expression of immune response genes in vECs and vSMCs is a major determinant in atherosclerosis onset and development because it contributes to the vascular-immune cross talk. The vascular wall modulates inflammation by the expression of numerous cytokines and chemokines. For many of these genes epigenetic components, influencing expression, have been identified. Below, we will discuss some of the best-studied examples in detail.
eNOS
As stated in the previous sections, many inflammatory processes are recognized by an epigenetic component. One of the best-studied cases of epigenetic regulation in the vascular wall involves the transcription of eNOS, an enzyme which is regarded as endothelial specific. Its catalytic product, nitric oxide, was first identified as a vasodilator [127], although recent evidence also suggest a role in inflammation. Nitric oxide has been attributed roles in leucocyte adhesion and vSMC proliferation [128]. Furthermore, dysfunctional eNOS is believed to be implicated in atherosclerosis development [129].
The human eNOS promoter does not contain a canonical TATA box, nor does it contain a proximal CpG island. Expression of eNOS is restricted to ECs in vivo. Various assays in both expressing and non-expressing cell types showed that a majority of the non-expressing cell types displayed transgene promoter activity [130–132]. In eNOS expressing ECs, eNOS promoter DNA was only slightly methylated or unmethylated, whereas non-expressing cells showed a high amount of DNA methylation. Treatment of non-expressing cell types with 5-azacytidine (a DNMTi) led to an increase in eNOS mRNA levels [132]. Later experiments showed the involvement of histone modifications in the expression of eNOS as well. ChIP showed enrichment of AcH3, AcH4 and (2/3)MeK4H3 in chromatin of the eNOS promoter in eNOS expressing cells. Treatment of non-expressing cells with the KDACi TSA – effectively increasing histone acetylation – also led to an increase in eNOS mRNA [128 and references therein].
iNOS
Not only is eNOS regulated by epigenetic mechanisms, of interest is the observation that also inducible nitric oxide synthase (iNOS) is actively regulated by the epigenetic mechanisms [133]. This gene is expressed in macrophages, but also in ECs and vSMCs, under inflammatory conditions. It has been suggested that nitric oxide – derived from macrophages expressing iNOS – can result in apoptosis of vSMCs, promoting plaque instability [134]. A study performed by Mellott et al. showed that cytokine induction of iNOS resulted in a change of chromatin structure at the iNOS promoter [9]. Later work showed that the NOS2A gene, encoding iNOS, had high levels of DNA methylation and of the 2MeK9H3 and 3MeK9H3 repressive marks in non-expressing cells. Cell lines capable of iNOS induction had lower levels of CpG-methylation and histone 3 lysine 9 methylation at the NOS2A promoter. Treatment with a DNMTi resulted in an increase of iNOS mRNA [133].
CCL11 (eotaxin)
In human atherosclerotic plaques, the chemokine CCL11 (also know as eotaxin) was shown by immunohistochemistry to be expressed [135]. In healthy tissue only negligible amounts of CCL11 were found. Polymorphisms within the CCL11 gene are also associated with the development of restenosis after PCI [136]. CCL11, an eosinophil chemoattractant, has been suggested to play a role in atherosclerosis development, although the precise relation remains to be elucidated [135, 137, 138]. At the same time CCL11 is expressed by SMCs, while macrophages in the human atherosclerotic plaque express the CCL11 receptor, CCR3 [135]. CCL11 transcription is induced by inflammatory TNF-α signalling and is mediated through NF-κB. TNF-α induces acetylation of histone H4 in the CCL11 promoter. This results in an open chromatin structure, promoting subsequent binding of the NF-κB subunit p65 to the CCL11 promoter (as shown by ChIP) [139]. Notably, glucocorticoids repress CCL11 transcription through selective inhibition of histone H4 acetylation [139]. Although only histone H4 acetylation and no other epigenetic markers were investigated, this research provides a very strong indication for epigenetic regulation of the CCL11 gene [139].
CCR5
CCR5 is important for plaque formation as it attracts T cells and mononuclear cells. Ccr5 knockout mice show less neointima formation and an increase in production of the anti-inflammatory IL-10 molecule by SMCs [140, 141]. One of the ligands for the CCR5 receptor, CCL5 or RANTES, has also been shown to be involved in unstable angina pectoris (UAP) [142]. UAP is generally the result of erosion or rupture of an atherosclerotic plaque. CCR5 is epigenetically regulated in T cells. In naïve T cells, lacking CCR5 expression, DNA of the CCR5 receptor is hypermethylated and the acetylation level of histone tails is strongly reduced in CCR5 promoter chromatin. When T cells are activated and express CCR5, DNA-methylation is significantly lowered combined with an increase in histone acetylation modifications (our own unpublished observations).
The observations above indicate that epigenetic mechanisms play a key role also in regulation of (immune) genes in cells that participate in atherosclerosis and vascular remodelling, and in the cross-talk of immune cells and vascular wall components.
Epigenetics in (vascular) Inflammation
KDM6B
De Santa et al. recently showed that inflammation and Polycomb-mediated gene silencing are linked [143]. Polycomb group (PcG) proteins are important for the maintenance of a repressive chromatin state that is stable over many cell divisions. As such, PcG proteins play a critical role in differentiation processes and maintenance of cellular identity. Gene silencing is mediated by the Polycomb repressive complex 2 (PRC2) through the 3MeK27H3 chromatin mark, which is subsequently read by the PRC1 maintenance complex [144 and references therein].
The JmjC-domain protein Jmjd3 (KDM6B, a H3K27 demethylase) is expressed in macrophages upon stimulation with bacterial products and pro-inflammatory cytokines [143]. Jmjd3 binds to PcG target genes, regulates 3MeK27H3 levels and therewith their transcriptional activity [145]. For the first time, an inducible enzyme was shown to erase epigenetic modifications. Later work showed that 70% of lipopolysaccharide-inducible genes in macrophages are Jmjd3 targets [146]. This provides an essential link between inflammation and reprogramming of the epigenome and might prove to be of vital importance in chronic inflammation and autoimmune diseases including atherosclerosis in the near future.
Oestrogen receptor
Oestrogen receptors (ERs) are present in the coronary arterial wall on both SMCs and ECs [147–149]. ERs may play an important role in protection against atherosclerosis [148]. Deficiencies in ER-α lead to accelerated atherosclerosis in men, furthermore ER-α mediates the loss of expression of some cytokine induced cell-adhesion molecules [150 and references therein]. ER-α was shown to have a varying degree of methylation throughout the cardiovascular system [151]. In the same study it was shown that an age related increase of methylation occurred in the right atrium. Furthermore, higher degrees of methylation were found in coronary atheromas when compared to macroscopically normal tissues.
Similarly, the ER-β also displays a correlation in methylation of its promoter and atherosclerosis [152]. Contrary to ER-α expression, expression of ER-β correlates with atherosclerosis independent of age [153]. By comparing plaque versus non-plaque tissue from the same vessels, it was recently shown that the ER-β promoter has higher methylation levels in atherosclerotic lesions [152]. In this study, increased expression of ER-β was also observed –in vitro– in ECs and SMCs after administration of a DNMTi (5-aza-2′-deoxycytidine) and a KDACi (TSA). These experiments provide supporting evidence for epigenetic regulation of the ER-β receptor.
COX2
Another example for the linkage between epigenetics and CVD-related inflammation is transcription of the cyclooxygenase-2 (Cox-2) gene. TNF-α– as well as several other cytokines – can induce expression of Cox-2, a protein associated with atherosclerosis development [154]. Cytokines (such as TNF-α and IFN-γ) can also induce (indirect) changes to the chromatin, allowing effector genes to be expressed upon stimulation [9]. For Cox-2 it was demonstrated that mRNA expression and protein synthesis can be repressed in vitro, in cell lines by the KDACi’s sodium butyrate and TSA revealing a significant role for epigenetic components in Cox-2 expression [155]. These experiments were performed in a colon cancer cell line, and thus need to be confirmed in cells relevant for vascular biology and atherosclerosis (e.g. vECs, vSMCs or monocytes) to extrapolate these results to atherosclerosis.
Transcriptional regulation of MHC molecules – the role of CIITA
Constitutive expression of MHC-II surface molecules is restricted to professional antigen presenting cells (APC) of the immune system. On all other cell types their expressions can be induced in an environment rich in inflammatory cytokines – such as the atherosclerotic plaque – or upon activation such as in human T cells [156]. Therefore under inflammatory conditions MHC-II molecule expression can be induced on vECs and vSMCs, which normally are not expressing MHC-II [157, 158].
The ‘master regulator’ of MHC-II expression is the co-activator CIITA (class II transactivator) [159]. Within the context of atherosclerosis, CIITA is of importance, since it has been shown to be involved in transcriptional regulation of collagen type 1 [160, 161]. Collagen 1 is one of the main components of ECM and is essential in cap formation and plaque stabilization [57].
Transcriptional regulation of MHC2TA, the gene encoding CIITA, is mediated through the activity of four independent promoter units (CIITA-PI through CIITA-PIV, Fig. 3) [162]. These promoter units are employed in a cell type- and activation-specific manner. In aortic SMCs CIITA-PIII and -PIV are expressed after IFN-γ stimulation and CIITA is needed for IFN-γ–mediated repression of collagen type 1 genes (COL1A1 and COL1A2) [160, 161]. Furthermore, collagen transcription is also repressed by the RFX family proteins. RFX1 only weakly interacts with the unmethylated collagen promoter, but binds with higher affinity when the promoter is in a methylated state [161].
Fig 3.
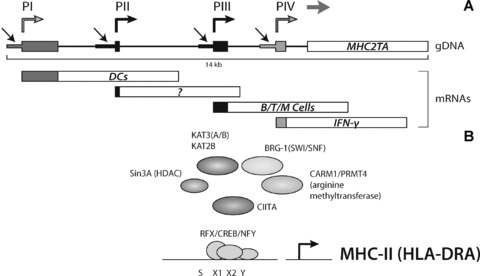
Organization of the MHC2TA multipromoter region. Each promoter (indicated with black arrows) transcribes a unique first exon (coloured boxes). Transcription start sites are indicated with coloured, angles arrows. Promoter PI is mainly used for constitutive CIITA expression in DCs, whereas PIII is mainly used in B Cells, activated T cells and macrophages. Promoter PIV is mainly used for IFN-γ induced CIITA expression in non-bone-marrow-derived cells. The function of PII is still poorly understood (A). Factor assembly on the MHC-II promoter. Shown is the regulatory SXY module, to which the MHC enhanceosome binds (B).
In MHC-II transcription, CIITA exerts its transactivating function through protein–protein interactions with a multi-protein complex, which comprises RFX, CREB/ATF and NFY (Fig. 3B) [163–165]. Together with CIITA, this complex acts as an enhanceosome driving transactivation of these genes [163–165]. In the MHC-enhanceosome, CIITA acts as a platform for recruitment of chromatin-modifying activities which include KATs, KDACs and an arginine methyltransferase [166–168]. Interactions of these chromatin-modifying enzymes and CIITA result in higher-order structural changes within the chromatin, effectively activating or silencing MHC-II gene transcription [167, 169]. A number of studies now have shown that epigenetic processes not only control MHC-II transcription, but also contribute to the activating/silencing of MHC2TA transcription. This is illustrated by the observation that in DC maturation deacetylation occurs at the MHC2TA locus coinciding with transcriptional inactivation [170]. During DC differentiation histone acetylation of the type I promoter is increased. This increase was blocked by IL-10, inhibiting MHC2TA transcription [171].
Non-histone targets
Acetyltransferases/deacetylases and methyltransferases/demethylases discussed in the previous sections also target non-histone proteins which could lead to alterations in function of the targeted proteins or the generation of novel epitopes which now can evoke an adaptive (auto-) immune response [172]. Especially since NF-κB – playing crucial roles in inflammation – is showing up as one of the non-histone substrates. Both NF-κB and p53 are acetylated by KAT3A (CBP) and KAT3B (p300) [173–176], whereas p53 can also be acetylated by KAT2B (PCAF) [177, 178].
As is the case with most genes, transcription of pro-inflammatory cytokine genes such as IFN-γ and TNF-α is positively regulated by transcription factors. However, these cytokines can also induce changes to the epigenome. Thus not only are cytokines under epigenetic control, cytokines themselves induce (indirect) changes to the chromatin, allowing effector genes to be expressed upon stimulation. Activation of the IFN-γ pathway results in activation of e.g. NF-κB. NF-κB is found in the cytoplasm of unstimulated cells, together with its inhibitor, IκB, that prevents NF-κB from entering the nucleus [179]. Upon stimulation with inducers –e.g. reactive oxygen species (ROS) and lipid peroxidase products – IκB is rapidly degraded via the ubiquitin–proteasome pathway [180, 181]. Hereupon, NF-κB is transported to the nucleus, where it can activate the transcription of various targets with κB-elements in their promoter [182, 183]. This process is known to be activated by KATs and repressed by KDACs [172 and references therein]. The p65 subunit of NF-κB is acetylated by KAT3A/B (P300/CBP) at lysines 218, 221 and 310 [184]. The acetylated form of p65 shows weak affinity for IκB. KDAC3 in turn deacetylates p65, promoting association with IκBα and thereby nuclear export of NF-κB [175]. In addition to KDAC3, KDAC1 has been shown in vitro and in vivo to deacetylate p53 [185].
Besides to acetylation, methylation of non-histone proteins has also been observed. For example, methylation occurs at lysine 831 of the vascular endothelial growth factor receptor 1, which is di-methylated by Smyd3 (a H3K4 methylase) [186]. The biological function of methylation at lysine 831 is yet to be discovered.
These examples perfectly illustrate how epigenetic and inflammatory processes are interwoven.
MicroRNAs
In recent years it became apparent that microRNAs (miRNAs) are critically involved in the regulation of gene expression. Synthesized as a larger precursor in the nucleus, miRNAs are processed in the cytoplasm into mature miRNAs, where they target specific mRNAs for degradation or translational inhibition [187]. A large number of miRNAs have already been identified at the moment and, not surprisingly, miRNAs have also been found to be involved in inflammation and atherosclerosis [188]. Unfortunately for most miRNAs the target they act upon is yet to be identified.
For some miRNAs, which might play a role in atherosclerosis development or inflammation the miRNA target gene has been identified. For instance the EC specific miR-126 inhibits the (TNF-α mediated) expression of VCAM-1 [189, 190]. This miRNA might thus be involved in transmigration of mononuclear cells into the subendothelium. Several other miRNAs have also been identified to be involved in inflammation [191 and references therein].
Conclusions
Epigenetic control is one of the most fundamental regulatory systems within the cell. Not surprisingly it fulfils essential roles in the regulation of inflammatory and immune responses (summarized in Table 1). The link between inflammation and reprogramming of the epigenome, shown by the induction of jmjd3 after LPS treatment, is an important discovery. Furthermore, the involvement of KDM1 in the inflammatory phenotype of vSMCs is of importance, as it does provide evidence for a link between inflammation and epigenetics, relevant for atherosclerosis. Correlations between global DNA methylation and atherosclerosis have provided valuable clues that epigenetics plays a role in atherosclerosis development; however, it remains to be further elucidated which genes are directly affected by DNA methylation.
Table 1.
Overview of genes regulated by epigenetic processes involved in atherosclerosis development. Genes for which evidence of epigenetic regulation is solely based on administration of inhibitors of histone modifying enzymes are not shown.
| Gene | Type of regulation | References |
|---|---|---|
| ec-sod | DNA methylation | [76] |
| c-fos | Histone code | [82] |
| CD36 | Histone code | [83] |
| IFN-γ | DNA methylation and histone code | [99–101] |
| IL-4 | DNA methylation and histone code | [108] |
| Foxp3 | DNA methylation and histone code | [109] |
| eNOS | DNA methylation and histone code | [128, 132] |
| iNOS | DNA methylation and histone code | [9, 133] |
| CCL11 | Histone code | [139] |
| CCR5 | DNA methylation and histone code | Own observations |
| ER-α | DNA methylation | [151] |
| ER-β | DNA methylation and histone code | [152] |
| CIITA | DNA methylation and histone code | [166–171] |
Similarly, precise determination of histone modifications at specific genes might prove its value in understanding gene expression profiles in vascular disease. Especially since small molecules can inhibit the function of histone modifying enzymes, altering the expression of genes. Therefore intervention in epigenetic gene regulation might prove to be the therapeutic intervention of the future, especially in complex multi-factorial diseases such as atherosclerosis, multiple sclerosis and rheumatoid arthritis.
Acknowledgments
The authors would like to acknowledge financial support from the Macropa Foundation, the TeRM Smart Mix Program of the Netherlands Ministry of Economic Affairs and the Netherlands Ministry of Education, Culture and Science, and the Department of Immunohematology and Blood Transfusion. We thank Alice Muggen for critically reading the manuscript.
References
- 1.Braunwald E. Cardiovascular medicine at the turn of the millennium: triumphs, concerns, and opportunities. N Engl J Med. 1997;337:1360–9. doi: 10.1056/NEJM199711063371906. [DOI] [PubMed] [Google Scholar]
- 2.Breslow JL. Cardiovascular disease burden increases, NIH funding decreases. Nat Med. 1997;3:600–1. doi: 10.1038/nm0697-600. [DOI] [PubMed] [Google Scholar]
- 3.Hansson GK, Robertson AK, Soderberg-Naucler C. Inflammation and atherosclerosis. Annu Rev Pathol. 2006;1:297–329. doi: 10.1146/annurev.pathol.1.110304.100100. [DOI] [PubMed] [Google Scholar]
- 4.Libby P. Inflammation in atherosclerosis. Nature. 2002;420:868–74. doi: 10.1038/nature01323. [DOI] [PubMed] [Google Scholar]
- 5.Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–95. doi: 10.1056/NEJMra043430. [DOI] [PubMed] [Google Scholar]
- 6.Jonasson L, Holm J, Skalli O, et al. Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis. 1986;6:131–8. doi: 10.1161/01.atv.6.2.131. [DOI] [PubMed] [Google Scholar]
- 7.Stary HC, Chandler AB, Dinsmore RE, et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis: a report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. 1995;92:1355–74. doi: 10.1161/01.cir.92.5.1355. [DOI] [PubMed] [Google Scholar]
- 8.Schroeder AP, Falk E. Vulnerable and dangerous coronary plaques. Atherosclerosis. 1995;118:S141–9. [PubMed] [Google Scholar]
- 9.Mellott JK, Nick HS, Waters MF, et al. Cytokine-induced changes in chromatin structure and in vivo footprints in the inducible NOS promoter. Am J Physiol Lung Cell Mol Physiol. 2001;280:390–9. doi: 10.1152/ajplung.2001.280.3.L390. [DOI] [PubMed] [Google Scholar]
- 10.Fraga MF, Ballestar E, Paz MF, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci USA. 2005;102:10604–9. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaminsky ZA, Tang T, Wang S-C, et al. DNA methylation profiles in monozygotic and dizygotic twins. Nat Genet. 2009;41:240–5. doi: 10.1038/ng.286. [DOI] [PubMed] [Google Scholar]
- 12.Heijmans BT, Tobi EW, Stein AD, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci USA. 2008;105:17046–9. doi: 10.1073/pnas.0806560105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luger K, Mader AW, Richmond RK, et al. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997;389:251–60. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 14.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 15.Wu C. Chromatin remodeling and the control of gene expression. J Biol Chem. 1997;272:28171–4. doi: 10.1074/jbc.272.45.28171. [DOI] [PubMed] [Google Scholar]
- 16.Kuo MH, Allis CD. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays. 1998;20:615–26. doi: 10.1002/(SICI)1521-1878(199808)20:8<615::AID-BIES4>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 17.Lahiri DK, Maloney B, Basha MR, et al. How and when environmental agents and dietary factors affect the course of Alzheimer’s disease: the “LEARn” model (latent early-life associated regulation) may explain the triggering of AD. Curr Alzheimer Res. 2007;4:219–28. doi: 10.2174/156720507780362164. [DOI] [PubMed] [Google Scholar]
- 18.Muskiet FAJ. The importance of (early) folate status to primary and secondary coronary artery disease prevention. Reprod Toxicol. 2005;20:403–10. doi: 10.1016/j.reprotox.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 19.Davis CD, Uthus EO. DNA methylation, cancer susceptibility, and nutrient interactions. Exp Biol Med. 2004;229:988–95. doi: 10.1177/153537020422901002. [DOI] [PubMed] [Google Scholar]
- 20.Foley DL, Craig JM, Morley R, et al. Prospects for epigenetic epidemiology. Am J Epidemiol. 2009;169:389–400. doi: 10.1093/aje/kwn380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cobiac L. Epigenomics and nutrition. Forum Nutr. 2007;60:31–41. doi: 10.1159/000107065. [DOI] [PubMed] [Google Scholar]
- 22.Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science. 1975;187:226–32. [PubMed] [Google Scholar]
- 23.Riggs AD. X inactivation, differentiation, and DNA methylation. Cytogenet Cell Genet. 1975;14:9–25. doi: 10.1159/000130315. [DOI] [PubMed] [Google Scholar]
- 24.Razin A. CpG methylation, chromatin structure and gene silencing – a three-way connection. EMBO J. 1998;17:4905–8. doi: 10.1093/emboj/17.17.4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Razin A, Levine A, Kafri T, et al. Relationship between transient DNA hypomethylation and erythroid differentiation of murine erythroleukemia cells. Proc Natl Acad Sci USA. 1988;85:9003–6. doi: 10.1073/pnas.85.23.9003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guccione E, Martinato F, Finocchiaro G, et al. Myc-binding-site recognition in the human genome is determined by chromatin context. Nat Cell Biol. 2006;8:764–70. doi: 10.1038/ncb1434. [DOI] [PubMed] [Google Scholar]
- 27.Kangaspeska S, Stride B, Metivier R, et al. Transient cyclical methylation of promoter DNA. Nature. 2008;452:112–5. doi: 10.1038/nature06640. [DOI] [PubMed] [Google Scholar]
- 28.Metivier R, Gallais R, Tiffoche C, et al. Cyclical DNA methylation of a transcriptionally active promoter. Nature. 2008;452:45–50. doi: 10.1038/nature06544. [DOI] [PubMed] [Google Scholar]
- 29.Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci USA. 2002;99:3740–5. doi: 10.1073/pnas.052410099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Irizarry RA, Ladd-Acosta C, Wen B, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet. 2009;41:178–86. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669–81. doi: 10.1016/j.cell.2007.01.033. [DOI] [PubMed] [Google Scholar]
- 32.Wang GG, Allis CD, Chi P. Chromatin remodeling and cancer, part I: covalent histone modifications. Trends Mol Med. 2007;13:363–72. doi: 10.1016/j.molmed.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 33.Grunstein M. Histone acetylation in chromatin structure and transcription. Nature. 1997;389:349–52. doi: 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- 34.Struhl K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 1998;12:599–606. doi: 10.1101/gad.12.5.599. [DOI] [PubMed] [Google Scholar]
- 35.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of Rna synthesis. Proc Natl Acad Sci USA. 1964;51:786–94. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cao R, Wang L, Wang H, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298:1039–43. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 37.Ng HH, Ciccone DN, Morshead KB, et al. Lysine-79 of histone H3 is hypomethylated at silenced loci in yeast and mammalian cells: a potential mechanism for position-effect variegation. Proc Natl Acad Sci USA. 2003;100:1820–5. doi: 10.1073/pnas.0437846100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stewart MD, Li J, Wong J. Relationship between histone H3 lysine 9 methylation, transcription repression, and heterochromatin protein 1 recruitment. Mol Cell Biol. 2005;25:2525–38. doi: 10.1128/MCB.25.7.2525-2538.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vaissiere T, Sawan C, Herceg Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat Res. 2008;659:40–8. doi: 10.1016/j.mrrev.2008.02.004. [DOI] [PubMed] [Google Scholar]
- 40.Kim JK, Samaranayake M, Pradhan S. Epigenetic mechanisms in mammals. Cell Mol Life Sci. 2009;66:596–612. doi: 10.1007/s00018-008-8432-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garcia-Dominguez M, Reyes JC. SUMO association with repressor complexes, emerging routes for transcriptional control. Biochim Biophys Acta. 2009;1789:451–9. doi: 10.1016/j.bbagrm.2009.07.001. [DOI] [PubMed] [Google Scholar]
- 42.Shukla A, Chaurasia P, Bhaumik SR. Histone methylation and ubiquitination with their cross-talk and roles in gene expression and stability. Cell Mol Life Sci. 2009;66:1419–33. doi: 10.1007/s00018-008-8605-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.de Ruijter AJ, van Gennip AH, Caron HN, et al. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–49. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grozinger CM, Schreiber SL. Deacetylase enzymes: biological functions and the use of small-molecule inhibitors. Chem Biol. 2002;9:3–16. doi: 10.1016/s1074-5521(02)00092-3. [DOI] [PubMed] [Google Scholar]
- 45.Holbert MA, Marmorstein R. Structure and activity of enzymes that remove histone modifications. Curr Opin Struct Biol. 2005;15:673–80. doi: 10.1016/j.sbi.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 46.Adcock IM. Histone deacetylase inhibitors as novel anti-inflammatory agents. Curr Opin Investig Drugs. 2006;7:966–73. [PubMed] [Google Scholar]
- 47.Cole PA. Chemical probes for histone-modifying enzymes. Nat Chem Biol. 2008;4:590–7. doi: 10.1038/nchembio.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pons D, de Vries FR, van den Elsen PJ, et al. Epigenetic histone acetylation modifiers in vascular remodelling: new targets for therapy in cardiovascular disease. Eur Heart J. 2009;30:266–77. doi: 10.1093/eurheartj/ehn603. [DOI] [PubMed] [Google Scholar]
- 49.Mai A. The therapeutic uses of chromatin-modifying agents. Expert Opin Ther Targets. 2007;11:835–51. doi: 10.1517/14728222.11.6.835. [DOI] [PubMed] [Google Scholar]
- 50.Duenas-Gonzalez A, Candelaria M, Perez-Plascencia C, et al. Valproic acid as epigenetic cancer drug: preclinical, clinical and transcriptional effects on solid tumors. Cancer Treat Rev. 2008;34:206–22. doi: 10.1016/j.ctrv.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 51.Mann BS, Johnson JR, He K, et al. Vorinostat for treatment of cutaneous manifestations of advanced primary cutaneous T-cell lymphoma. Clin Cancer Res. 2007;13:2318–22. doi: 10.1158/1078-0432.CCR-06-2672. [DOI] [PubMed] [Google Scholar]
- 52.Gerstner T, Bell N, Konig S. Oral valproic acid for epilepsy–long-term experience in therapy and side effects. Expert Opinion on Pharmacotherapy. 2008;9:285–92. doi: 10.1517/14656566.9.2.285. [DOI] [PubMed] [Google Scholar]
- 53.Soni KB, Kuttan R. Effect of oral curcumin administration on serum peroxides and cholesterol levels in human volunteers. Indian J Physiol Pharmacol. 1992;36:273–5. [PubMed] [Google Scholar]
- 54.Ramirez-Boscá A, Soler A, Carrión Gutierrez M, et al. Antioxidant Curcuma extracts decrease the blood lipid peroxide levels of human subjects. AGE. 1995;18:167–9. [Google Scholar]
- 55.Ramirez Boscá A, Carrión Gutierrez M, Soler A, et al. Effects of the antioxidant turmeric on lipoprotein peroxides: implications for the prevention of atherosclerosis. AGE. 1997;20:165–8. doi: 10.1007/s11357-997-0015-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huang HC, Jan TR, Yeh SF. Inhibitory effect of curcumin, an anti-inflammatory agent, on vascular smooth muscle cell proliferation. Eur J Pharmacol. 1992;221:381–4. doi: 10.1016/0014-2999(92)90727-l. [DOI] [PubMed] [Google Scholar]
- 57.Ross R. Atherosclerosis – an inflammatory disease. N Engl J Med. 1999;340:115–26. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 58.Virmani R, Kolodgie FD, Burke AP, et al. Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler Thromb Vasc Biol. 2000;20:1262–75. doi: 10.1161/01.atv.20.5.1262. [DOI] [PubMed] [Google Scholar]
- 59.van der Wal AC, Das PK, Bentz van de Berg D, et al. Atherosclerotic lesions in humans. In situ immunophenotypic analysis suggesting an immune mediated response. Lab Invest. 1989;61:166–70. [PubMed] [Google Scholar]
- 60.Lusis AJ. Atherosclerosis. Nature. 2000;407:233–41. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Glagov S, Weisenberg E, Zarins CK, et al. Compensatory enlargement of human atherosclerotic coronary arteries. N Engl J Med. 1987;316:1371–5. doi: 10.1056/NEJM198705283162204. [DOI] [PubMed] [Google Scholar]
- 62.Davenport P, Tipping PG. The role of interleukin-4 and interleukin-12 in the progression of atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol. 2003;163:1117–25. doi: 10.1016/S0002-9440(10)63471-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gupta S, Pablo AM, Jiang X, et al. IFN-gamma potentiates atherosclerosis in ApoE knock-out mice. J Clin Invest. 1997;99:2752–61. doi: 10.1172/JCI119465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Whitman SC, Ravisankar P, Daugherty A. IFN-gamma deficiency exerts gender-specific effects on atherogenesis in apolipoprotein E−/− mice. J Interferon Cytokine Res. 2002;22:661–70. doi: 10.1089/10799900260100141. [DOI] [PubMed] [Google Scholar]
- 65.Buono C, Come CE, Stavrakis G, et al. Influence of interferon-gamma on the extent and phenotype of diet-induced atherosclerosis in the LDLR-deficient mouse. Arterioscler Thromb Vasc Biol. 2003;23:454–60. doi: 10.1161/01.ATV.0000059419.11002.6E. [DOI] [PubMed] [Google Scholar]
- 66.Monraats PS, Pires NM, Schepers A, et al. Tumor necrosis factor-alpha plays an important role in restenosis development. FASEB J. 2005;19:1998–2004. doi: 10.1096/fj.05-4634com. [DOI] [PubMed] [Google Scholar]
- 67.Boesten LS, Zadelaar AS, van Nieuwkoop A, et al. Tumor necrosis factor-alpha promotes atherosclerotic lesion progression in APOE*3-Leiden transgenic mice. Cardiovasc Res. 2005;66:179–85. doi: 10.1016/j.cardiores.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 68.Robertson AK, Rudling M, Zhou X, et al. Disruption of TGF-beta signaling in T cells accelerates atherosclerosis. J Clin Invest. 2003;112:1342–50. doi: 10.1172/JCI18607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Caligiuri G, Rudling M, Ollivier V, et al. Interleukin-10 deficiency increases atherosclerosis, thrombosis, and low-density lipoproteins in apolipoprotein E knockout mice. Mol Med. 2003;9:10–7. [PMC free article] [PubMed] [Google Scholar]
- 70.Monraats PS, Kurreeman FA, Pons D, et al. Interleukin 10: a new risk marker for the development of restenosis after percutaneous coronary intervention. Genes Immun. 2007;8:44–50. doi: 10.1038/sj.gene.6364343. [DOI] [PubMed] [Google Scholar]
- 71.Coupel S, Moreau A, Hamidou M, et al. Expression and release of soluble HLA-E is an immunoregulatory feature of endothelial cell activation. Blood. 2007;109:2806–14. doi: 10.1182/blood-2006-06-030213. [DOI] [PubMed] [Google Scholar]
- 72.Boring L, Gosling J, Cleary M, et al. Decreased lesion formation in CCR2−/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. 1998;394:894–7. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- 73.Gu L, Okada Y, Clinton SK, et al. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol Cell. 1998;2:275–81. doi: 10.1016/s1097-2765(00)80139-2. [DOI] [PubMed] [Google Scholar]
- 74.Mach F, Sauty A, Iarossi AS, et al. Differential expression of three T lymphocyte-activating CXC chemokines by human atheroma-associated cells. J Clin Invest. 1999;104:1041–50. doi: 10.1172/JCI6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Veillard NR, Kwak B, Pelli G, et al. Antagonism of RANTES receptors reduces atherosclerotic plaque formation in mice. Circ Res. 2004;94:253–61. doi: 10.1161/01.RES.0000109793.17591.4E. [DOI] [PubMed] [Google Scholar]
- 76.Laukkanen MO, Mannermaa S, Hiltunen MO, et al. Local hypomethylation in atherosclerosis found in rabbit ec-sod gene. Arterioscler Thromb Vasc Biol. 1999;19:2171–8. doi: 10.1161/01.atv.19.9.2171. [DOI] [PubMed] [Google Scholar]
- 77.Stenvinkel P, Karimi M, Johansson S, et al. Impact of inflammation on epigenetic DNA methylation – a novel risk factor for cardiovascular disease. J Intern Med. 2007;261:488–99. doi: 10.1111/j.1365-2796.2007.01777.x. [DOI] [PubMed] [Google Scholar]
- 78.Sharma P, Kumar J, Garg G, et al. Detection of altered global DNA methylation in coronary artery disease patients. DNA Cell Biol. 2008;27:357–65. doi: 10.1089/dna.2007.0694. [DOI] [PubMed] [Google Scholar]
- 79.Chao CL, Tsai HH, Lee CM, et al. The graded effect of hyperhomocysteinemia on the severity and extent of coronary atherosclerosis. Atherosclerosis. 1999;147:379–86. doi: 10.1016/s0021-9150(99)00208-7. [DOI] [PubMed] [Google Scholar]
- 80.Wald DS, Law M, Morris J. Serum homocysteine and the severity of coronary artery disease. Thromb Res. 2003;111:55–7. doi: 10.1016/j.thromres.2003.08.015. [DOI] [PubMed] [Google Scholar]
- 81.Yi-Deng J, Tao S, Hui-Ping Z, et al. Folate and ApoE DNA methylation induced by homocysteine in human monocytes. DNA Cell Biol. 2007;26:737–44. doi: 10.1089/dna.2007.0619. [DOI] [PubMed] [Google Scholar]
- 82.Hastings NE, Simmers MB, McDonald OG, et al. Atherosclerosis-prone hemodynamics differentially regulates endothelial and smooth muscle cell phenotypes and promotes pro-inflammatory priming. Am J Physiol Cell Physiol. 2007;293:C1824–33. doi: 10.1152/ajpcell.00385.2007. [DOI] [PubMed] [Google Scholar]
- 83.Choi JH, Nam KH, Kim J, et al. Trichostatin A exacerbates atherosclerosis in low density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25:2404–9. doi: 10.1161/01.ATV.0000184758.07257.88. [DOI] [PubMed] [Google Scholar]
- 84.Okamoto H, Fujioka Y, Takahashi A, et al. Trichostatin A, an inhibitor of histone deacetylase, inhibits smooth muscle cell proliferation via induction of p21(WAF1) J Atheroscler Thromb. 2006;13:183–91. doi: 10.5551/jat.13.183. [DOI] [PubMed] [Google Scholar]
- 85.Dje N’Guessan P, Riediger F, Vardarova K, et al. Statins control oxidized LDL-mediated histone modifications and gene expression in cultured human endothelial cells. Arterioscler Thromb Vasc Biol. 2009;29:380–6. doi: 10.1161/ATVBAHA.108.178319. [DOI] [PubMed] [Google Scholar]
- 86.Barker DJ, Winter PD, Osmond C, et al. Weight in infancy and death from ischaemic heart disease. Lancet. 1989;2:577–80. doi: 10.1016/s0140-6736(89)90710-1. [DOI] [PubMed] [Google Scholar]
- 87.Lillycrop KA, Slater-Jefferies JL, Hanson MA, et al. Induction of altered epigenetic regulation of the hepatic glucocorticoid receptor in the offspring of rats fed a protein-restricted diet during pregnancy suggests that reduced DNA methyltransferase-1 expression is involved in impaired DNA methylation and changes in histone modifications. Br J Nutr. 2007;97:1064–73. doi: 10.1017/S000711450769196X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dansky HM, Charlton SA, Harper MM, et al. T and B lymphocytes play a minor role in atherosclerotic plaque formation in the apolipoprotein E-deficient mouse. Proc Natl Acad Sci USA. 1997;94:4642–6. doi: 10.1073/pnas.94.9.4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Buono C, Binder CJ, Stavrakis G, et al. T-bet deficiency reduces atherosclerosis and alters plaque antigen-specific immune responses. Proc Natl Acad Sci USA. 2005;102:1596–601. doi: 10.1073/pnas.0409015102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Daugherty A, Pure E, Delfel-Butteiger D, et al. The effects of total lymphocyte deficiency on the extent of atherosclerosis in apolipoprotein E−/− mice. J Clin Invest. 1997;100:1575–80. doi: 10.1172/JCI119681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ait-Oufella H, Taleb S, Mallat Z, et al. Cytokine network and T cell immunity in atherosclerosis. Semin Immunopathol. 2009;31:23–33. doi: 10.1007/s00281-009-0143-x. [DOI] [PubMed] [Google Scholar]
- 92.Mallat Z, Taleb S, Ait-Oufella H, et al. The role of adaptive T cell immunity in atherosclerosis. Journal of Lipid Research. 2009;50:S364–9. doi: 10.1194/jlr.R800092-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shimizu K, Shichiri M, Libby P, et al. Th2-predominant inflammation and blockade of IFN-gamma signaling induce aneurysms in allografted aortas. J Clin Invest. 2004;114:300–8. doi: 10.1172/JCI19855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis. Annu Rev Immunol. 2009;27:165–97. doi: 10.1146/annurev.immunol.021908.132620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sanders VM. Epigenetic regulation of Th1 and Th2 cell development. Brain Behav Immun. 2006;20:317–24. doi: 10.1016/j.bbi.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 96.Tao R, de Zoeten EF, Ozkaynak E, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med. 2007;13:1299–307. doi: 10.1038/nm1652. [DOI] [PubMed] [Google Scholar]
- 97.Amsen D, Spilianakis CG, Flavell RA. How are TH1 and TH2 effector cells made. Curr Opin Immunol. 2009;21:153–60. doi: 10.1016/j.coi.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Agarwal S, Rao A. Long-range transcriptional regulation of cytokine gene expression. Curr Opin Immunol. 1998;10:345–52. doi: 10.1016/s0952-7915(98)80174-x. [DOI] [PubMed] [Google Scholar]
- 99.Schoenborn JR, Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol. 2007;96:41–101. doi: 10.1016/S0065-2776(07)96002-2. [DOI] [PubMed] [Google Scholar]
- 100.Ansel KM, Lee DU, Rao A. An epigenetic view of helper T cell differentiation. Nat Immunol. 2003;4:616–23. doi: 10.1038/ni0703-616. [DOI] [PubMed] [Google Scholar]
- 101.Murphy KM, Reiner SL. The lineage decisions of helper T cells. Nat Rev Immunol. 2002;2:933–44. doi: 10.1038/nri954. [DOI] [PubMed] [Google Scholar]
- 102.Rao A, Avni O. Molecular aspects of T-cell differentiation. Br Med Bull. 2000;56:969–84. doi: 10.1258/0007142001903634. [DOI] [PubMed] [Google Scholar]
- 103.Makar KW, Wilson CB. DNA methylation is a nonredundant repressor of the Th2 effector program. J Immunol. 2004;173:4402–6. doi: 10.4049/jimmunol.173.7.4402. [DOI] [PubMed] [Google Scholar]
- 104.Young HA, Dray JF, Farrar WL. Expression of transfected human interferon-gamma DNA: evidence for cell-specific regulation. J Immunol. 1986;136:4700–3. [PubMed] [Google Scholar]
- 105.Young HA, Ghosh P, Ye J, et al. Differentiation of the T helper phenotypes by analysis of the methylation state of the IFN-gamma gene. J Immunol. 1994;153:3603–10. [PubMed] [Google Scholar]
- 106.Chang S, Aune TM. Histone hyperacetylated domains across the Ifng gene region in natural killer cells and T cells. Proc Natl Acad Sci USA. 2005;102:17095–100. doi: 10.1073/pnas.0502129102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Stetson DB, Mohrs M, Reinhardt RL, et al. Constitutive cytokine mRNAs mark natural killer (NK) and NK T cells poised for rapid effector function. J Clin Invest. 2003;198:1069–76. doi: 10.1084/jem.20030630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ansel KM, Djuretic I, Tanasa B, Rao A. Regulation of Th2 differentiation and Il4 locus accessibility. Annu Rev Immunol. 2006;24:607–56. doi: 10.1146/annurev.immunol.23.021704.115821. [DOI] [PubMed] [Google Scholar]
- 109.Floess S, Freyer J, Siewert C, et al. Epigenetic control of the foxp3 locus in regulatory T cells. PLoS Biol. 2007;5:e38. doi: 10.1371/journal.pbio.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nencioni A, Beck J, Werth D, et al. Histone deacetylase inhibitors affect dendritic cell differentiation and immunogenicity. Clin Cancer Res. 2007;13:3933–41. doi: 10.1158/1078-0432.CCR-06-2903. [DOI] [PubMed] [Google Scholar]
- 111.Wang B, Morinobu A, Horiuchi M, et al. Butyrate inhibits functional differentiation of human monocyte-derived dendritic cells. Cell Immunol. 2008;253:54–8. doi: 10.1016/j.cellimm.2008.04.016. [DOI] [PubMed] [Google Scholar]
- 112.Inoue K, Kobayashi M, Yano K, et al. Histone deacetylase inhibitor reduces monocyte adhesion to endothelium through the suppression of vascular cell adhesion molecule-1 expression. Arterioscler Thromb Vasc Biol. 2006;26:2652–9. doi: 10.1161/01.ATV.0000247247.89787.e7. [DOI] [PubMed] [Google Scholar]
- 113.Wang J, Mahmud SA, Bitterman PB, et al. Histone deacetylase inhibitors suppress TF-kappaB-dependent agonist-driven tissue factor expression in endothelial cells and monocytes. J Biol Chem. 2007;282:28408–18. doi: 10.1074/jbc.M703586200. [DOI] [PubMed] [Google Scholar]
- 114.Rossig L, Li H, Fisslthaler B, et al. Inhibitors of histone deacetylation downregulate the expression of endothelial nitric oxide synthase and compromise endothelial cell function in vasorelaxation and angiogenesis. Circ Res. 2002;91:837–44. doi: 10.1161/01.res.0000037983.07158.b1. [DOI] [PubMed] [Google Scholar]
- 115.Rossig L, Urbich C, Bruhl T, et al. Histone deacetylase activity is essential for the expression of HoxA9 and for endothelial commitment of progenitor cells. J Exp Med. 2005;201:1825–35. doi: 10.1084/jem.20042097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Miano JM. Serum response factor: toggling between disparate programs of gene expression. J Mol Cell Cardiol. 2003;35:577–93. doi: 10.1016/s0022-2828(03)00110-x. [DOI] [PubMed] [Google Scholar]
- 117.Chang VK, Donato JJ, Chan CS, et al. Mcm1 promotes replication initiation by binding specific elements at replication origins. Mol Cell Biol. 2004;24:6514–24. doi: 10.1128/MCB.24.14.6514-6524.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sun Q, Chen G, Streb JW, et al. Defining the mammalian CArGome. Genome Res. 2006;16:197–207. doi: 10.1101/gr.4108706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.McDonald OG, Owens GK. Programming smooth muscle plasticity with chromatin dynamics. Circ Res. 2007;100:1428–41. doi: 10.1161/01.RES.0000266448.30370.a0. [DOI] [PubMed] [Google Scholar]
- 120.Mack CP, Thompson MM, Lawrenz-Smith S, et al. Smooth muscle alpha-actin CArG elements coordinate formation of a smooth muscle cell-selective, serum response factor-containing activation complex. Circ Res. 2000;86:221–32. doi: 10.1161/01.res.86.2.221. [DOI] [PubMed] [Google Scholar]
- 121.Manabe I, Owens GK. The smooth muscle myosin heavy chain gene exhibits smooth muscle subtype-selective modular regulation in vivo. J Biol Chem. 2001;276:39076–87. doi: 10.1074/jbc.M105402200. [DOI] [PubMed] [Google Scholar]
- 122.Manabe I, Owens GK. CArG elements control smooth muscle subtype-specific expression of smooth muscle myosin in vivo. J Clin Invest. 2001;107:823–34. doi: 10.1172/JCI11385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Qiu P, Li L. Histone acetylation and recruitment of serum responsive factor and CREB-binding protein onto SM22 promoter during SM22 gene expression. Circ Res. 2002;90:858–65. doi: 10.1161/01.res.0000016504.08608.b9. [DOI] [PubMed] [Google Scholar]
- 124.McDonald OG, Wamhoff BR, Hoofnagle MH, et al. Control of SRF binding to CArG box chromatin regulates smooth muscle gene expression in vivo. J Clin Invest. 2006;116:36–48. doi: 10.1172/JCI26505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Reddy MA, Villeneuve LM, Wang M, et al. Role of the lysine-specific demethylase 1 in the proinflammatory phenotype of vascular smooth muscle cells of diabetic mice. Circ Res. 2008;103:615–23. doi: 10.1161/CIRCRESAHA.108.175190. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 126.Matouk CC, Marsden PA. Epigenetic regulation of vascular endothelial gene expression. Circ Res. 2008;102:873–87. doi: 10.1161/CIRCRESAHA.107.171025. [DOI] [PubMed] [Google Scholar]
- 127.Ignarro LJ. Nitric oxide as a unique signaling molecule in the vascular system: a historical overview. J Physiol Pharmacol. 2002;53:503–14. [PubMed] [Google Scholar]
- 128.Fish JE, Marsden PA. Endothelial nitric oxide synthase: insight into cell-specific gene regulation in the vascular endothelium. Cell Mol Life Sci. 2006;63:144–62. doi: 10.1007/s00018-005-5421-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kawashima S, Yokoyama M. Dysfunction of endothelial nitric oxide synthase and atherosclerosis. Arterioscler Thromb Vasc Biol. 2004;24:998–1005. doi: 10.1161/01.ATV.0000125114.88079.96. [DOI] [PubMed] [Google Scholar]
- 130.Guillot PV, Liu L, Kuivenhoven JA, et al. Targeting of human eNOS promoter to the Hprt locus of mice leads to tissue-restricted transgene expression. Physiol Genomics. 2000;2:77–83. doi: 10.1152/physiolgenomics.2000.2.2.77. [DOI] [PubMed] [Google Scholar]
- 131.Teichert AM, Miller TL, Tai SC, et al. In vivo expression profile of an endothelial nitric oxide synthase promoter-reporter transgene. Am J Physiol Heart Circ Physiol. 2000;278:H1352–61. doi: 10.1152/ajpheart.2000.278.4.H1352. [DOI] [PubMed] [Google Scholar]
- 132.Chan Y, Fish JE, D’Abreo C, et al. The cell-specific expression of endothelial nitric-oxide synthase: a role for DNA methylation. J Biol Chem. 2004;279:35087–100. doi: 10.1074/jbc.M405063200. [DOI] [PubMed] [Google Scholar]
- 133.Chan GC, Fish JE, Mawji IA, et al. Epigenetic basis for the transcriptional hyporesponsiveness of the human inducible nitric oxide synthase gene in vascular endothelial cells. J Immunol. 2005;175:3846–61. doi: 10.4049/jimmunol.175.6.3846. [DOI] [PubMed] [Google Scholar]
- 134.Boyle JJ. Macrophage activation in atherosclerosis: pathogenesis and pharmacology of plaque rupture. Curr Vasc Pharmacol. 2005;3:63–8. doi: 10.2174/1570161052773861. [DOI] [PubMed] [Google Scholar]
- 135.Haley KJ, Lilly CM, Yang JH, et al. Overexpression of eotaxin and the CCR3 receptor in human atherosclerosis: using genomic technology to identify a potential novel pathway of vascular inflammation. Circulation. 2000;102:2185–9. doi: 10.1161/01.cir.102.18.2185. [DOI] [PubMed] [Google Scholar]
- 136.Monraats PS, Pires NM, Agema WR, et al. Genetic inflammatory factors predict restenosis after percutaneous coronary interventions. Circulation. 2005;112:2417–25. doi: 10.1161/CIRCULATIONAHA.105.536268. [DOI] [PubMed] [Google Scholar]
- 137.Sheikine Y, Olsen B, Gharizadeh B, et al. Influence of eotaxin 67G>A polymorphism on plasma eotaxin concentrations in myocardial infarction survivors and healthy controls. Atherosclerosis. 2006;189:458–63. doi: 10.1016/j.atherosclerosis.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 138.Emanuele E, Falcone C, D’Angelo A, et al. Association of plasma eotaxin levels with the presence and extent of angiographic coronary artery disease. Atherosclerosis. 2006;186:140–5. doi: 10.1016/j.atherosclerosis.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 139.Nie M, Knox AJ, Pang L. beta2-Adrenoceptor agonists, like glucocorticoids, repress eotaxin gene transcription by selective inhibition of histone H4 acetylation. J Immunol. 2005;175:478–86. doi: 10.4049/jimmunol.175.1.478. [DOI] [PubMed] [Google Scholar]
- 140.Zernecke A, Shagdarsuren E, Weber C. Chemokines in atherosclerosis: an update. Arterioscler Thromb Vasc Biol. 2008;28:1897–908. doi: 10.1161/ATVBAHA.107.161174. [DOI] [PubMed] [Google Scholar]
- 141.Zernecke A, Liehn EA, Gao JL, et al. Deficiency in CCR5 but not CCR1 protects against neointima formation in atherosclerosis-prone mice: involvement of IL-10. Blood. 2006;107:4240–3. doi: 10.1182/blood-2005-09-3922. [DOI] [PubMed] [Google Scholar]
- 142.Kraaijeveld AO, de Jager SC, de Jager WJ, et al. CC chemokine ligand-5 (CCL5/RANTES) and CC chemokine ligand-18 (CCL18/PARC) are specific markers of refractory unstable angina pectoris and are transiently raised during severe ischemic symptoms. Circulation. 2007;116:1931–41. doi: 10.1161/CIRCULATIONAHA.107.706986. [DOI] [PubMed] [Google Scholar]
- 143.De Santa F, Totaro MG, Prosperini E, et al. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell. 2007;130:1083–94. doi: 10.1016/j.cell.2007.08.019. [DOI] [PubMed] [Google Scholar]
- 144.Kohler C, Villar CB. Programming of gene expression by Polycomb group proteins. Trends Cell Biol. 2008;18:236–43. doi: 10.1016/j.tcb.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 145.Hong S, Cho YW, Yu LR, et al. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc Natl Acad Sci USA. 2007;104:18439–44. doi: 10.1073/pnas.0707292104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.De Santa F, Narang V, Yap ZH, et al. Jmjd3 contributes to the control of gene expression in LPS-activated macrophages. EMBO J. 2009;28:3341–52. doi: 10.1038/emboj.2009.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Venkov CD, Rankin AB, Vaughan DE. Identification of authentic estrogen receptor in cultured endothelial cells. A potential mechanism for steroid hormone regulation of endothelial function. Circulation. 1996;94:727–33. doi: 10.1161/01.cir.94.4.727. [DOI] [PubMed] [Google Scholar]
- 148.Losordo DW, Rosenfield K, Kaufman J, et al. Focal compensatory enlargement of human arteries in response to progressive atherosclerosis. In vivo documentation using intravascular ultrasound. Circulation. 1994;89:2570–7. doi: 10.1161/01.cir.89.6.2570. [DOI] [PubMed] [Google Scholar]
- 149.Karas RH, Patterson BL, Mendelsohn ME. Human vascular smooth muscle cells contain functional estrogen receptor. Circulation. 1994;89:1943–50. doi: 10.1161/01.cir.89.5.1943. [DOI] [PubMed] [Google Scholar]
- 150.Miller VM, Duckles SP. Vascular actions of estrogens: functional implications. Pharmacol Rev. 2008;60:210–41. doi: 10.1124/pr.107.08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Post WS, Goldschmidt-Clermont PJ, Wilhide CC, et al. Methylation of the estrogen receptor gene is associated with aging and atherosclerosis in the cardiovascular system. Cardiovasc Res. 1999;43:985–91. doi: 10.1016/s0008-6363(99)00153-4. [DOI] [PubMed] [Google Scholar]
- 152.Kim J, Kim JY, Song KS, et al. Epigenetic changes in estrogen receptor beta gene in atherosclerotic cardiovascular tissues and in-vitro vascular senescence. Biochim Biophys Acta. 2007;1772:72–80. doi: 10.1016/j.bbadis.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 153.Christian RC, Liu PY, Harrington S, et al. Intimal estrogen receptor (ER)beta, but not ERalpha expression, is correlated with coronary calcification and atherosclerosis in pre- and postmenopausal women. J Clin Endocrinol Metab. 2006;91:2713–20. doi: 10.1210/jc.2005-2672. [DOI] [PubMed] [Google Scholar]
- 154.Cipollone F, Prontera C, Pini B, et al. Overexpression of functionally coupled cyclooxygenase-2 and prostaglandin E synthase in symptomatic atherosclerotic plaques as a basis of prostaglandin E(2)-dependent plaque instability. Circulation. 2001;104:921–7. doi: 10.1161/hc3401.093152. [DOI] [PubMed] [Google Scholar]
- 155.Tong X, Yin L, Giardina C. Butyrate suppresses Cox-2 activation in colon cancer cells through HDAC inhibition. Biochem Biophys Res Commun. 2004;317:463–71. doi: 10.1016/j.bbrc.2004.03.066. [DOI] [PubMed] [Google Scholar]
- 156.Holling TM, van der Stoep N, Quinten E, et al. Activated human T cells accomplish MHC class II expression through T cell-specific occupation of class II transactivator promoter III. J Immunol. 2002;168:763–70. doi: 10.4049/jimmunol.168.2.763. [DOI] [PubMed] [Google Scholar]
- 157.Collins T, Korman AJ, Wake CT, et al. Immune interferon activates multiple class II major histocompatibility complex genes and the associated invariant chain gene in human endothelial cells and dermal fibroblasts. Proc Natl Acad Sci USA. 1984;81:4917–21. doi: 10.1073/pnas.81.15.4917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Leeuwenberg JF, Van Damme J, Meager T, et al. Effects of tumor necrosis factor on the interferon-gamma-induced major histocompatibility complex class II antigen expression by human endothelial cells. Eur J Immunol. 1988;18:1469–72. doi: 10.1002/eji.1830180925. [DOI] [PubMed] [Google Scholar]
- 159.Steimle V, Otten LA, Zufferey M, et al. Complementation cloning of an MHC class II transactivator mutated in hereditary MHC class II deficiency (or bare lymphocyte syndrome) Cell. 1993;75:135–46. [PubMed] [Google Scholar]
- 160.Buttice G, Miller J, Wang L, et al. Interferon-gamma induces major histocompatibility class II transactivator (CIITA), which mediates collagen repression and major histocompatibility class II activation by human aortic smooth muscle cells. Circ Res. 2006;98:472–9. doi: 10.1161/01.RES.0000204725.46332.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sengupta P, Xu Y, Wang L, et al. Collagen alpha1(I) gene (COL1A1) is repressed by RFX family. J Biol Chem. 2005;280:21004–14. doi: 10.1074/jbc.M413191200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Muhlethaler-Mottet A, Otten LA, Steimle V, et al. Expression of MHC class II molecules in different cellular and functional compartments is controlled by differential usage of multiple promoters of the transactivator CIITA. EMBO J. 1997;16:2851–60. doi: 10.1093/emboj/16.10.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Zhu XS, Linhoff MW, Li G, et al. Transcriptional scaffold: CIITA interacts with NF-Y, RFX, and CREB to cause stereospecific regulation of the class II major histocompatibility complex promoter. Mol Cell Biol. 2000;20:6051–61. doi: 10.1128/mcb.20.16.6051-6061.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Masternak K, Muhlethaler-Mottet A, Villard J, et al. CIITA is a transcriptional coactivator that is recruited to MHC class II promoters by multiple synergistic interactions with an enhanceosome complex. Genes Dev. 2000;14:1156–66. [PMC free article] [PubMed] [Google Scholar]
- 165.Jabrane-Ferrat N, Nekrep N, Tosi G, et al. MHC class II enhanceosome: how is the class II transactivator recruited to DNA-bound activators. Int Immunol. 2003;15:467–75. doi: 10.1093/intimm/dxg048. [DOI] [PubMed] [Google Scholar]
- 166.van den Elsen PJ, Holling TM, Kuipers HF, et al. Transcriptional regulation of antigen presentation. Curr Opin Immunol. 2004;16:67–75. doi: 10.1016/j.coi.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 167.Wright KL, Ting JP. Epigenetic regulation of MHC-II and CIITA genes. Trends Immunol. 2006;27:405–12. doi: 10.1016/j.it.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 168.Zika E, Ting JP. Epigenetic control of MHC-II: interplay between CIITA and histone-modifying enzymes. Curr Opin Immunol. 2005;17:58–64. doi: 10.1016/j.coi.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 169.Ni Z, Abou El Hassan M, Xu Z, et al. The chromatin-remodeling enzyme BRG1 coordinates CIITA induction through many interdependent distal enhancers. Nat Immunol. 2008;9:785–93. doi: 10.1038/ni.1619. [DOI] [PubMed] [Google Scholar]
- 170.Landmann S, Muhlethaler-Mottet A, Bernasconi L, et al. Maturation of dendritic cells is accompanied by rapid transcriptional silencing of class II transactivator (CIITA) expression. J Exp Med. 2001;194:379–91. doi: 10.1084/jem.194.4.379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Choi YE, Yu HN, Yoon CH, et al. Tumor-mediated down-regulation of MHC class II in DC development is attributable to the epigenetic control of the CIITA type I promoter. Eur J Immunol. 2009;39:858–68. doi: 10.1002/eji.200838674. [DOI] [PubMed] [Google Scholar]
- 172.Glozak MA, Sengupta N, Zhang X, et al. Acetylation and deacetylation of non-histone proteins. Gene. 2005;363:15–23. doi: 10.1016/j.gene.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 173.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 174.Luo J, Li M, Tang Y, et al. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc Natl Acad Sci USA. 2004;101:2259–64. doi: 10.1073/pnas.0308762101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Chen LF, Greene WC. Regulation of distinct biological activities of the NF-kappaB transcription factor complex by acetylation. J Mol Med. 2003;81:549–57. doi: 10.1007/s00109-003-0469-0. [DOI] [PubMed] [Google Scholar]
- 176.Liu Y, Denlinger CE, Rundall BK, et al. Suberoylanilide hydroxamic acid induces Akt-mediated phosphorylation of p300, which promotes acetylation and transcriptional activation of RelA/p65. J Biol Chem. 2006;281:31359–68. doi: 10.1074/jbc.M604478200. [DOI] [PubMed] [Google Scholar]
- 177.Liu L, Scolnick DM, Trievel RC, et al. p53 sites acetylated in vitro by PCAF and p300 are acetylated in vivo in response to DNA damage. Mol Cell Biol. 1999;19:1202–9. doi: 10.1128/mcb.19.2.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Di Stefano V, Soddu S, Sacchi A, et al. HIPK2 contributes to PCAF-mediated p53 acetylation and selective transactivation of p21Waf1 after nonapoptotic DNA damage. Oncogene. 2005;24:5431–42. doi: 10.1038/sj.onc.1208717. [DOI] [PubMed] [Google Scholar]
- 179.Rahman I, Marwick J, Kirkham P. Redox modulation of chromatin remodeling: impact on histone acetylation and deacetylation, NF-kappaB and pro-inflammatory gene expression. Biochem Pharmacol. 2004;68:1255–67. doi: 10.1016/j.bcp.2004.05.042. [DOI] [PubMed] [Google Scholar]
- 180.Bowie AG, Moynagh PN, O’Neill LA. Lipid peroxidation is involved in the activation of NF-kappaB by tumor necrosis factor but not interleukin-1 in the human endothelial cell line ECV304. Lack of involvement of H2O2 in NF-kappaB activation by either cytokine in both primary and transformed endothelial cells. J Biol Chem. 1997;272:25941–50. doi: 10.1074/jbc.272.41.25941. [DOI] [PubMed] [Google Scholar]
- 181.Ginn-Pease ME, Whisler RL. Optimal NF kappa B mediated transcriptional responses in Jurkat T cells exposed to oxidative stress are dependent on intracellular glutathione and costimulatory signals. Biochem Biophys Res Commun. 1996;226:695–702. doi: 10.1006/bbrc.1996.1416. [DOI] [PubMed] [Google Scholar]
- 182.Rahman I, MacNee W. Regulation of redox glutathione levels and gene transcription in lung inflammation: therapeutic approaches. Free Radic Biol Med. 2000;28:1405–20. doi: 10.1016/s0891-5849(00)00215-x. [DOI] [PubMed] [Google Scholar]
- 183.Rahman I, MacNee W. Role of transcription factors in inflammatory lung diseases. Thorax. 1998;53:601–12. doi: 10.1136/thx.53.7.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Chen L, Fischle W, Verdin E, et al. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science. 2001;293:1653–7. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- 185.Luo J, Su F, Chen D, et al. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature. 2000;408:377–81. doi: 10.1038/35042612. [DOI] [PubMed] [Google Scholar]
- 186.Kunizaki M, Hamamoto R, Silva FP, et al. The lysine 831 of vascular endothelial growth factor receptor 1 is a novel target of methylation by SMYD3. Cancer Res. 2007;67:10759–65. doi: 10.1158/0008-5472.CAN-07-1132. [DOI] [PubMed] [Google Scholar]
- 187.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Urbich C, Kuehbacher A, Dimmeler S. Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc Res. 2008;79:581–8. doi: 10.1093/cvr/cvn156. [DOI] [PubMed] [Google Scholar]
- 189.Harris TA, Yamakuchi M, Ferlito M, et al. MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc Natl Acad Sci USA. 2008;105:1516–21. doi: 10.1073/pnas.0707493105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.van Solingen C, Seghers L, Bijkerk R, et al. Antagomir-mediated silencing of endothelial cell specific microRNA-126 impairs ischemia-induced angiogenesis. J Cell Mol Med. 2009;13:1577–85. doi: 10.1111/j.1582-4934.2008.00613.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Pedersen I, David M. MicroRNAs in the immune response. Cytokine. 2008;43:391–4. doi: 10.1016/j.cyto.2008.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Sherer Y, Shoenfeld Y. Mechanisms of disease: atherosclerosis in autoimmune diseases. Nat Clin Pract Rheumatol. 2006;2:99–106. doi: 10.1038/ncprheum0092. [DOI] [PubMed] [Google Scholar]