

Abstract
Systemic sclerosis (SSc, scleroderma) is a chronic, multisystem connective tissue disorder affecting the skin and various internal organs. Although the disease is characterized by a triad of widespread microangiopathy, fibrosis and autoimmunity, increasing evidence indicates that vascular damage is a primary event in the pathogenesis of SSc. The progressive vascular injury includes persistent endothelial cell activation/damage and apoptosis, intimal thickening, delamination, vessel narrowing and obliteration. These profound vascular changes lead to vascular tone dysfunction and reduced capillary blood flow, with consequent tissue ischemia and severe clinical manifestations, such as digital ulceration or amputation, pulmonary arterial hypertension and scleroderma renal crisis. The resulting tissue hypoxia induces complex cellular and molecular mechanisms in the attempt to recover endothelial cell function and tissue perfusion. Nevertheless, in SSc patients there is no evidence of significant angiogenesis and the disease evolves towards chronic tissue ischemia, with progressive and irreversible structural changes in multiple vascular beds culminating in the loss of capillaries. A severe imbalance between pro-angiogenic and angiostatic factors may also lead to impaired angiogenic response during SSc. Besides insufficient angiogenesis, defective vasculogenesis with altered numbers and functional defects of bone marrow-derived endothelial progenitor cells may contribute to the vascular pathogenesis of SSc. The purpose of this article is to review the contribution of recent studies to the understanding of the complex mechanisms of impaired vascular repair in SSc. Indeed, understanding the pathophysiology of SSc-associated vascular disease may be the key in dissecting the disease pathogenesis and developing novel therapies. Either angiogenic or vasculogenic mechanisms may potentially become in the future the target of therapeutic strategies to promote capillary regeneration in SSc.
Keywords: systemic sclerosis, scleroderma, endothelial cell, endothelial progenitor cell, angiogenesis, vasculogenesis, vascular repair
Introduction
Evidence for endothelial disease and loss of capillaries in SSc
Systemic sclerosis (SSc, scleroderma) is characterized by widespread microangiopathy, activation of humoural and cellular immune responses and progressive tissue fibrosis, which affect the skin and a variety of internal organs, including the lung, heart, kidney and gastrointestinal tract [1, 2]. The aetiology of SSc remains unknown, and currently no therapy has been able to modify disease evolution and progression.
Extensive clinical and pathological findings of endothelial cell (EC) activation and damage and structural vascular changes strongly support the hypothesis of a vascular disease as an important and primary process in SSc pathogenesis [3–5]. In fact, the dysregulation of vascular tone control, which manifests clinically as Raynaud’s phenomenon, and microcirculatory abnormalities are the earliest clinical manifestations of SSc, and may precede the onset of fibrosis by months or years [3–5]. The vascular damage in SSc affects primarily the microvasculature (small and medium-sized arteries, arterioles and capillaries) and can be observed in all affected organs [3, 4]. Increasing evidence suggests that microvascular endothelial cell (MVEC) injury and apoptosis occur early in SSc, and that progressive proliferation of the intima leads to vessel narrowing and obliteration [4, 6–10]. In the very early phase, increased vascular permeability favours plasma and mononuclear cell extravasation, with formation of perivascular inflammatory infiltrates, clinically represented by oedema of the fingers (puffy hands) [5]. The involvement of microvessels causes recurrent episodes of ischemia-reperfusion injury and reduces capillary blood flow leading to a state of chronic tissue ischemia, lack of nutrients and severe tissue hypoxia, thus contributing to organ dysfunction and significant morbidity and mortality [3–5]. With disease progression, vessels lose their elasticity, the vessel media and adventitia become fibrotic, and occlusion of the small arteries, together with platelet activation and aggregation, facilitates thrombotic events with severe organ complications, such as digital ulceration or amputation, pulmonary arterial hypertension, and scleroderma renal crisis [3, 10–14]. The disarray of the microcirculation is testified by the extensive morphological modifications detected by nailfold capillaroscopy. The capillaroscopic features are enlarged, giant and bushy capillaries, microhaemorrhages and, in particular, a variable loss of capillaries with formation of avascular areas (vascular desertification) [5, 15–17] (see Fig. 1). These specific capillaroscopic changes are also referred as ‘scleroderma pattern’ and differentiate SSc from primary Raynaud’s phenomenon, in which usually capillaries are normal in number and size [15].
Fig 1.
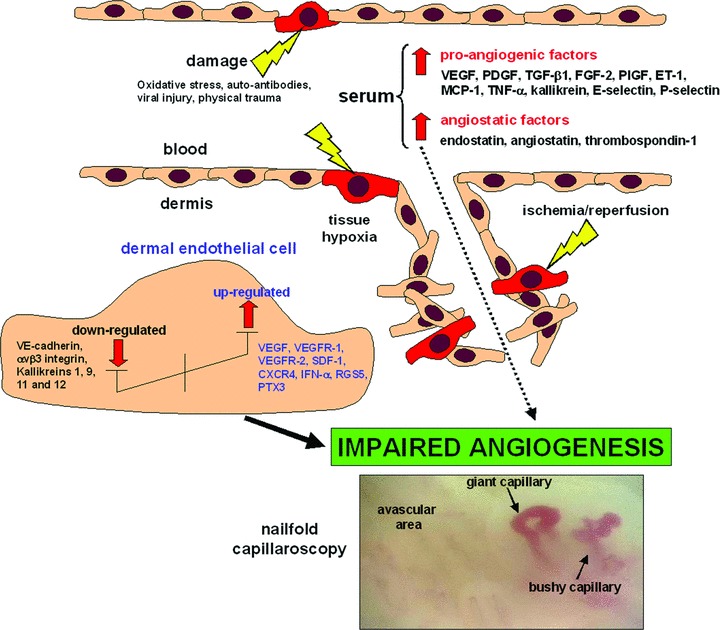
Mechanisms of impaired angiogenesis in SSc. A complex imbalance between pro-angiogenic and anti-angiogenic (angiostatic) mediators results in an impaired and decreased ability to form new microvessels. This leads to the formation of enlarged, giant and bushy capillaries, microhaemorrhages and avascular areas (vascular desertification) as shown by nailfold capillaroscopy. See text for abbreviations.
The abnormal capillary morphology followed by the loss of capillaries is the clear evidence of the disturbance of the angiogenic process that does not allow the regeneration of functional microvessels. This may be seen as a paradox because usually tissue hypoxia stimulates angiogenesis with the formation of new capillaries from pre-existing vessels. However, despite the increase of vascular endothelial growth factor (VEGF) and others pro-angiogenic molecules in SSc skin and serum, the disease evolves towards a progressive loss of capillaries [4, 6, 18, 19]. In the past, it had been thought that impaired angiogenesis and increased apoptosis of mature ECs were exclusively responsible for the microvascular abnormalities in SSc. However, several studies published over the last few years suggest that an impairment of vasculogenesis and vascular repair may be also involved in SSc capillary loss, with altered numbers and functional defects of circulating endothelial progenitor cells (EPCs), as well as abnormalities in the commitment of bone marrow-derived mesenchymal stem cells (MSCs) towards the EC lineage [20, 21].
This review presents current knowledge of mechanisms of defective vascular repair in SSc and discusses the role that abnormalities in the angiogenic and vasculogenic processes may play in the pathophysiology of SSc. We also discuss how the angiogenic and vasculogenic processes might be manipulated to develop novel vascular treatment strategies for this complex disease.
Angiogenesis and vasculogenesis
Angiogenesis is a complex and finely balanced process that consists in the formation of new vessels from the pre-existing ones such as capillaries and post-capillary venules, and plays a pivotal role during embryonal development and later, in adult life, in several physiological and pathological conditions, such as corpus luteum formation, tumours and chronic inflammation [22, 23]. Sprouting angiogenesis encompasses an increase in vasopermeability, leading to the extravasation of plasma proteins that function as a temporary scaffold for migrating ECs. Matrix metalloproteinases (MMPs), secreted by the endothelium, break down the vascular basement membrane and allow the invasion of the surrounding stroma by ECs, in the direction of the pro-angiogenic stimulus. EC migration and invasion are accompanied by EC proliferation and the organization of newly formed ECs into three-dimensional tubular structures. Lumen formation and vessel wall stabilization by pericytes are the final processes of sprouting angiogenesis and lead to the formation of a functional network of new capillary vessels [24].
Under physiological conditions, angiogenesis is dependent on the tight balance of positive and negative angiogenic regulators (pro-angiogenic and angiostatic factors, respectively) within the perivascular and vascular microenvironment and requires the functional activities of a number of molecules, including growth factors and their receptors, extracellular matrix proteins, adhesion molecules and proteolytic enzymes [24, 25]. During angiogenesis, ECs have a distinct gene expression profile characterized by a switch of the cell proteolytic balance towards an invasive phenotype as well as the expression of specific adhesion molecules and growth factor receptors. In normal tissues, vascular quiescence is maintained by the dominant influence of endogenous angiogenic inhibitors over pro-angiogenic stimuli [24, 25]. In contrast, pathological angiogenesis is mainly linked to an imbalance between pro-angiogenic and anti-angiogenic mediators [23]. The major pro-angiogenic and anti-angiogenic factors are listed in Table 1.
Table 1.
Major pro-angiogenic and anti-angiogenic mediators
| Type of mediator | Molecule |
|---|---|
| Pro-angiogenic | VEGF |
| FGF-2 | |
| PlGF | |
| SDF-/XCL12 | |
| PDGF | |
| TGF-α and -β | |
| ET-1 | |
| Hepatocyte growth factor (HGF) | |
| Platelet activating factor (PAF) | |
| TNF-α | |
| Insulin-like growth factor (IGF) | |
| Angiopoietin-1 | |
| Granulocyte colony-stimulating factor (G-CSF) | |
| Granulocyte-macrophage colony-stimulating factor (GM-CSF) | |
| Erythropoietin | |
| MCP-1 | |
| Tissue kallikrein | |
| Fractalkine/CX3CL1 | |
| IL-6 | |
| IL-8 | |
| Anti-angiogenic | Angiostatin |
| Endostatin | |
| Thrombospondin-1 | |
| IFN-α and -γ | |
| PTX3 | |
| IL-12 | |
| Angiopoietin-2 | |
| Tissue inhibitors of metalloproteinases | |
Vasculogenesis, the generation of new blood vessels by stem or progenitor cells, was long regarded to be confined to embryogenesis. However, the discovery of EPCs in adult bone marrow and peripheral blood has challenged this theory [26, 27]. In their landmark study, Asahara et al. demonstrated that new blood vessels can be formed in the adult not only by the sprouting of fully differentiated ECs, but also by circulating progenitor cells, independently of the pre-existing vasculature [26]. They found that bone marrow-derived CD34+ progenitor cells could acquire the characteristics of mature ECs, express EC markers, and incorporate into new capillary vessels at sites of ischemia [26]. Subsequent analyses revealed that postnatal vasculogenesis contributes to vascular healing in response to vascular injury through the processes of rapid re-endothelialization of denuded vessels and collateral vessel formation in ischemic tissues [28]. In this process, EPCs are mobilized from their bone marrow niches into the circulation in response to stress- and/or damage-related signals, migrate through the bloodstream and home to the sites of vascular injury, where they extravasate through the endothelium and contribute to the formation of neovessels and the repair of damaged vessels working in concert with pre-existing mature ECs [28]. Because EPCs may critically contribute to the homeostasis of the physiological vascular network, these progenitor cells might be considered interesting candidates for novel cell therapies for the treatment of various ischemic diseases [28]. EPCs have now become clinically relevant, as trials of therapeutic neovascularization based on autologous bone marrow mononuclear cell transplantation in patients affected by severe limb or myocardial ischemia have been recently carried out [29, 30]. In this regard, recent studies indicate that autologous bone marrow stem cell transplantation may improve peripheral microcirculation in SSc, including nailfold capillary changes and ischemic digital ulcers [31–33]. Alternatively, EPCs may be isolated from the peripheral blood, in vitro expanded and implanted into the ischemic tissue, where they are involved in the restoration of vascular perfusion. However, controversy exists regarding the mechanisms by which human EPCs may induce neovascularization in such a therapeutic setting [34].
In fact, two different types of EPCs appear to exist: the ‘so-called’ early- and late-outgrowth EPCs, according to their time-dependent appearance when isolated with cell culture-based methods [35, 36]. These two types of EPCs have different cell surface markers (CD14+ early-outgrowth EPCs, CD14− late-outgrowth EPCs), morphologies, gene expression profiles and survival behaviours. However, both types of EPCs appear to contribute to neovasculogenesis in vivo. In particular, while early-outgrowth EPCs are mainly involved in the secretion of pro-angiogenic cytokines and vascular growth factors, late-outgrowth EPCs may proliferate and differentiate at sites of vascular injury after integration into the vessel wall, thus supplying a sufficient number of mature ECs for vascular repair [36].
Mechanisms and biomarkers of endothelial damage in SSc
In SSc pathogenesis, EC injury and apoptosis are early events observed in the affected skin before any evidence of tissue fibrosis [37, 38].
The identity of the initial trigger leading to EC damage remains unknown [5, 39]. Numerous environmental and infectious agents have been suggested as possible initiating factors of SSc [1, 40]. Auto-antibodies showing cross-reactivity between cytomegalovirus epitopes and specific surface molecules of ECs, inducing EC apoptosis, have been implicated in endothelial injury [41, 42]. Anti-EC antibodies are commonly found in SSc serum and can activate ECs, stimulating the production of adhesion molecules and cytokines, and also induce EC apoptosis [43–45]. Oxidative stress with consequent elevated levels of reactive oxygen species has also been suggested to be involved in EC damage, despite the fact that the cellular source and mechanisms of action of reactive oxygen species in SSc remain unknown [46–48].
The endothelium is a metabolically active tissue that, under normal circumstances, regulates regional blood flow, transportation of nutrients, transendothelial migration of blood cells, as well as coagulation and fibrinolysis with the maintenance of an antithrombotic lining along the vascular tree. These biological functions are regulated by several molecules synthetized and released by ECs, including vasodilators (e.g. nitric oxide and prostacyclin), vasoconstrictors (e.g. endothelin-1 [ET-1] and platelet-activating factor) and cell adhesion molecules (e.g. selectins, integrins).
In SSc, the production of vasoactive molecules is dysregulated following EC perturbation [49]. In the early stage of SSc, activated ECs highly express vascular adhesion molecules, which enable the transmigration of inflammatory and immune cells through the endothelium leading to the formation of perivascular inflammatory infiltrates [50–54]. This results in a complex perivascular cellular reaction involving injured ECs, immune and inflammatory cells, and fibroblasts that become constitutively activated and transdifferentiate into myofibroblasts [1, 55]. Altered secretion of vasodilators and vasoconstrictors, and chronic activation of platelets and fibrinolytic pathways secondary to EC activation are also found [49, 56, 57]. Activated ECs release ET-1, a potent vasoconstrictor that also promotes leucocyte adhesion to the endothelium as well as vascular smooth muscle cell proliferation and fibroblast activation [58]. Studies on the peripheral blood of SSc patients demonstrate abnormalities in several biomarkers of endothelial perturbation, including factor VIII/von Willebrand factor, soluble adhesion molecules, thrombospondin, thrombomodulin, nitric oxide, prostacyclin, N-terminal pro-brain natriuretic peptide, plasminogen activators and thromboxane metabolites [59–62]. Furthermore, increased numbers of EC-derived circulating microparticles in SSc plasma suggest their possible role as marker of EC damage [63]. There is also evidence that circulating mature ECs are increased in the blood of SSc patients as a marker of EC activation in comparison with healthy controls, and that this increase correlates with disease activity scores [64].
In SSc, persistent EC activation results in EC damage, and once vascular perturbation has occurred there is evidence that ischemia-reperfusion injury is an ongoing pathological process which inevitably evolves towards chronic underperfusion. In fact, ischemia-reperfusion injury is a complex inflammatory process that results from interaction between humoural and cellular components including the complement, vasoactive cytokines and the contact activation cascade within ischemic vascular beds. Soon after the start of reperfusion, EC dysfunction is perpetuated by superoxide radicals mainly produced by damaged ECs and neutrophils. Indeed, superoxide radicals inhibit the release of nitric oxide, prostacyclin and tissue plasminogen activator from ECs leading to the impairment of the vascular tone control and favouring thrombotic events with consequent chronic tissue hypoxia [39, 49].
Hence, a crucial question in SSc is why the damaged microvessels are not replaced by new capillaries via angiogenesis or vasculogenesis.
Mechanisms of defective angiogenesis in SSc
Clinical and in vitro evidence indicate an impaired angiogenic response in SSc [6]. Comparative immunohistological studies of SSc and normal skin biopsies have shown that SSc involved skin has significantly fewer dermal capillaries [31, 65]. In vitro studies demonstrated that serum from SSc patients is toxic for ECs and may inhibit EC migration and vascular tube formation [66].
During the pro-angiogenic switch, both the innate and adaptive immune cells are involved in the mechanisms of EC activation, proliferation and migration through the production and release of a large spectrum of pro-angiogenic mediators [67]. In this regard, numerous in vitro studies on peripheral blood mononuclear cells suggest a defective contribution of SSc immune cells to angiogenesis. Impaired angiogenic activity has been reported in peripheral blood lymphocytes and monocytes from SSc patients [68, 69]. Furthermore, supernatants from SSc peripheral blood mononuclear cells decreased EC chemotaxis [70], and SSc serum failed to enhance normal mononuclear cell angiogenic capacity [71]. In contrast, another study reported that a subset of mononuclear cells from SSc patients displays enhanced angiogenic activity [72].
Increasing evidence also suggests a severe imbalance between pro-angiogenic and anti-angiogenic factors [6]. In fact, the capillaroscopic changes in the course of SSc may be explained by the action of different factors on angiogenesis. In the early stages of the disease, a pro-inflammatory state and an increased production of pro-angiogenic factors may stimulate angiogenesis. As a result, capillaroscopic analysis of the nailfold bed demonstrates the presence of microhaemorrages and tortuous, giant capillary loops, which are immature and instable microvessels formed during an uncontrolled angiogenic response. This short pro-angiogenic response is followed by a dramatic impairment of the angiogenic process which might in part be explained by the action of several angiostatic factors, ultimately resulting in reduced capillary density and extensive avascular areas.
Despite the overall decrease in the angiogenic response, several pro-angiogenic mediators are up-regulated in the skin and serum of SSc patients [6] (Fig. 1). Consistent with increased pro-angiogenic factors in SSc skin, Ribatti et al. have shown that angiogenesis is stimulated in the chick embryo chorioallantoic membrane by co-culture with skin biopsy tissue from SSc patients [73]. Up-regulated pro-angiogenic factors in SSc include: VEGF, platelet-derived growth factor (PDGF), transforming growth factor-β1 (TGF-β1), fibroblast growth factor-2 (FGF-2), placental growth factor (PlGF), ET-1, monocyte chemoattractant protein-1 (MCP-1), stromal cell-derived factor-1 (SDF-1/CXCL12), tumour necrosis factor-α (TNF-α), interleukin (IL)-8, fractalkine/CX3CL1, and vascular adhesion molecules E-selectin, P-selectin and their soluble forms [6, 19, 50, 62, 74–77].
Interestingly, several of these pro-angiogenic cytokines (e.g. PDGF, TGF-β1, FGF-2, ET-1 and MCP-1) are also potent activators of vascular smooth muscle cells, pericytes and stromal fibroblasts and may be involved in the complex relationship among angiogenesis, proliferative vasculopathy and fibrosis in SSc [1, 58, 75]. In fact, vascular smooth muscle cells and fibroblasts proliferate and synthetize extracellular matrix in response to EC-released growth factors leading to perivascular fibrosis, intimal proliferation, matrix deposition in the vessel wall, vessel obliteration and irreversible loss of vascular integrity.
VEGF is one of the major regulators of angiogenesis [78]. Several studies have shown that VEGF expression is markedly increased in different cell types both in the epidermis and dermis of patients with SSc [19, 79, 80]. VEGF exerts its biological functions by binding to the tyrosine kinase receptors VEGFR-1 (flt-1) and VEGFR-2 (flk-1/KDR), which both are up-regulated on dermal ECs in SSc affected skin [19, 79]. In agreement with these findings, a number of studies demonstrated that serum levels of VEGF are significantly increased in SSc patients throughout different disease stages and correlate with organ manifestations [52, 74, 77, 81, 82]. Another recent study showed an altered expression of VEGF, PlGF and VEGF receptors which was associated with decidual and villous vasculopathy in placental tissue from pregnancies in women with SSc [83].
Sufficient tissue vascularization depends on the tight regulation of the expression of VEGF and its receptors [78, 84]. In fact, time-dependent regulation of VEGF expression seems of critical importance for an adequate vascularization and tissue perfusion, as well as for blood flow restoration in ischemic tissues. Using a transgenic mouse model in which VEGF expression can be conditionally switched off in an organ-dependent manner, Dor et al.[84] demonstrated that, while short-term overexpression of VEGF induced the formation of new mature and functional capillaries, prolonged exposure to VEGF resulted in the formation of a chaotic capillary network with irregularly shaped, enlarged capillaries. These capillary modifications are similar to the altered capillary morphology commonly seen in SSc by nailfold capillaroscopy [3, 17]. Therefore, the uncontrolled and chronic overexpression of VEGF found in SSc might lead to the formation of a chaotic capillary network rather than promote the formation of new functional and stable capillaries [19]. Moreover, in SSc skin Konttinen and coworkers [65] found a low tissue expression of αvβ3 integrin, an endothelial receptor complex facilitating the pro-angiogenic action of VEGF, which therefore could further contribute to disordered angiogenesis. In addition, MVECs isolated from the dermis of SSc patients exhibited an impaired response to VEGF in the in vitro capillary morphogenesis assay on Matrigel substrate, suggesting that VEGF receptor signalling might be impaired in these ECs [85]. Because the expression of VEGF is stimulated by hypoxia, in SSc one would expect that chronic hypoxia could even contribute to persistent overexpression of VEGF, that is one of the main transcriptional targets of hypoxia-inducible factor (HIF)-1α[86]. However, HIF-1α protein expression was not increased and did not correlate with up-regulated VEGF in the skin of SSc patients [19, 86]. Alternatively, the persistent overexpression of VEGF in SSc might also be driven by fibrogenic cytokines, such as IL-1β, PDGF and TGF-β, which are up-regulated in SSc and can induce VEGF expression [86].
An alternative explanation for the loss of angiogenesis in the course of SSc is that the up-regulated pro-angiogenic factors could be exceeded by an even greater up-regulation of angiostatic factors. In fact, although conflicting results have been reported, several studies found elevated angiostatic factors in SSc patients, including endostatin, angiostatin and thrombospondin-1 [66, 74, 77, 87–90] (Fig. 1). Endostatin, an endogenous angiogenesis inhibitor derived from the breakdown of type XVIII collagen, is increased in SSc serum and associates with the presence of more severe clinical involvement [74, 77, 87, 89]. A recent study showed that plasmin activity is reduced while the amount of circulating angiostatin, a cleavage product of plasminogen, is increased in SSc plasma [66]. Furthermore, normal human MVECs exhibited reduced migration and proliferation when exposed to SSc plasma, as well as a significant impaired ability to form vascular structures in collagen after exposure to angiostatin in amounts similar to those detected in SSc plasma [66].
There is also evidence that platelets may be key players in the profound imbalance between pro-angiogenic and angiostatic factors in SSc. In fact, human platelets carry in their alpha granules a set of angiogenesis stimulators, such as VEGF, FGF-2, PDGF and TGF-β1, and inhibitors, such as endostatin, platelet factor-4 and thrombospondin-1 [91]. Moreover, these angiogenesis-regulatory molecules are sequestered in platelets in higher concentration than in plasma, and are packed into separate and distinct alpha granules and differentially released after platelet activation [91, 92]. Many reports document evidence in SSc for ongoing platelet activation, aggregation and release of bioactive molecules into the circulation and in injured endothelium [56, 57]. A recent study has shown that SSc platelets store and transport high levels of VEGF [93]. Therefore, platelets may be a source of circulating VEGF in the course of SSc because of their activation at the contact of injured endothelium, possibly contributing to impaired angiogenesis [93]. Besides angiogenesis, the contribution of platelets to SSc pathogenesis is thought to involve multiple and different pathways. Indeed, aggregating platelets also release several molecules that affect vascular tone, such as nitric oxide, serotonin, thromboxane A2 and prostaglandins, as well as a large array of factors that regulate inflammation, growth of vascular smooth muscle cells and fibroblasts, and synthesis of extracellular matrix components and matrix-degrading enzymes [56].
Gene expression levels of pro- and anti-angiogenic factors have been analysed in microarray studies which compared the transcriptosome profiling of dermal MVECs isolated from normal individuals and SSc patients [94, 95]. One microarray gene expression study detected differences between normal and SSc MVECs in the kallikrein gene family [94]. Pro-angiogenic kallikreins 9, 11 and 12 were down-regulated in SSc MVECs, whereas anti-angiogenic kallikrein 3 was up-regulated. The microarray data were further validated in experiments using normal MVECs treated with antibodies against kallikreins 9, 11 and 12 and subsequently analysed in migration, proliferation, and capillary morphogenesis functional assays. All three antibodies were able to block angiogenesis in healthy MVECs [94]. Tissue kallikrein (also known as kallikrein 1, or ‘true’ tissue kallikrein) is a serine protease that cleaves kininogen and thereby regulates the kininogen-kinin pathway. Tissue kallikrein synthetized at vessel level acts through kinins which modulate a broad spectrum of vascular functions, playing an important role in the regulation of vascular homeostasis and angiogenesis [96, 97]. Del Rosso et al. found that tissue kallikrein is increased in the serum of SSc patients, particularly in those with signs of early and active vascular disease, suggesting a role in the development of SSc microvascular abnormalities [98]. Moreover, another study documented a progressive decrease of tissue kallikrein expression in dermal MVECs of SSc skin from the early to the advanced disease stage [99]. A second microarray gene expression study has shown that SSc MVECs overexpress a number of pro-angiogenic transcripts but also a variety of genes that have a negative effect on angiogenesis [95]. Therefore, the authors proposed dermal SSc MVECs as ‘a model of anti-angiogenesis’. In particular, the angiogenesis inhibitor pentraxin-3 (PTX3), which is known to inhibit the pro-angiogenic effects of FGF-2, was strongly up-regulated in SSc MVECs compared with control MVECs [95]. Instead, several genes that promote cell migration and adhesion to the extracellular matrix were down-regulated in SSc MVECs, suggesting an anti-invasive phenotype of these cells [95].
In fact, MVECs can perform angiogenesis only when provided with a proper enzymatic machinery, enabling them to lyse the extracellular matrix and invade the surrounding tissue. In this regard, the cell-associated plasminogen activator system is known to play a crucial role in angiogenesis by modulating the adhesive properties of ECs in their interactions with the extracellular matrix and in the degradation of matrix components [100, 101]. The urokinase-type plasminogen activator (uPA) promotes growth, chemotaxis and matrix invasion of ECs by interaction with its receptor, uPAR, which is constitutively expressed on ECs [102]. uPAR not only functions as uPA receptor, but also plays an important role in cell motility and adhesion through its binding interactions with vitronectin and intracellular signalling mediators, such as the integrin receptors [103]. A recent in vitro study has shown that in dermal MVECs isolated from SSc skin, uPAR undergoes truncation between domains 1 and 2, a cleavage that is known to impair uPAR functions [85]. The uPAR cleavage occurring in SSc MVECs was associated with the overexpression of MMP-12 [85]. Indeed, overproduction of MMP-12 by SSc dermal MVECs and fibroblasts accounted for endothelial uPAR cleavage leading to impaired uPA-induced EC migration, invasion, proliferation, and capillary morphogenesis on Matrigel [85, 104] (Fig. 2). Interestingly, treatment with an anti-MMP-12 monoclonal antibody was able to restore the angiogenic activity of MVECs, including cell migration, chemoinvasion and tube formation [85, 105]. Furthermore, the same authors have shown that in SSc MVECs uPAR cleavage results in loss of an integrin-mediated uPAR connection with the actin cytoskeleton [105]. The uncoupling of cleaved uPAR from β2 integrins impaired the activation of the small Rho GTPases Rac and Cdc42, thus inhibiting their mediation of uPAR-dependent cytoskeletal rearrangement and cell motility, and ultimately blocked the integrin-engagement-delivered signals to the actin cytoskeleton resulting in loss of angiogenesis [105] (Fig. 2).
Fig 2.

MMP-12-dependent cleavage of uPAR in SSc microvascular endothelial cells (MVECs) results in impaired angiogenesis. uPAR is a glycosylphosphatidylinositol-anchored 3-extracellular domain (D1-D2-D3) cell surface receptor that concentrates the serine protease activity of the uPA in the pericellular region and promotes extracellular matrix remodelling. The main binding site for uPA is located in D1, and interaction of uPA and uPAR activates the proteolytic cascade necessary to open a path within tissues to migrating cells. Moreover, uPAR not only functions as uPA receptor but also plays a role in growth factor activation, cell adhesion, differentiation, proliferation and migration by interacting with extracellular matrix molecules, including vitronectin (VN), and intracellular signalling mediators, such as the integrin receptors. VN/uPAR interaction occurs directly with D1, but requires the integrity of the full-length receptor (D1-D2-D3). The removal of D1 also abolishes the interaction of uPAR with integrins and its ability to regulate the integrin adhesive functions. The constitutive overproduction and secretion of MMP-12 by SSc dermal MVECs and fibroblasts accounts for endothelial cell uPAR cleavage between D1 and D2, leaving a truncated receptor (D2-D3) on MVEC surface which results in loss of an integrin-mediated uPAR connection with the actin cytoskeleton. The uncoupling of cleaved uPAR from integrins impairs the activation of Rac and Cdc42, thus inhibiting their ability to bind the p21-activated protein kinase 1 (PAK-1), which regulates the downstream signalling cascades of small Rho GTPases and the uPAR-dependent cytoskeletal rearrangement. The net result is the impairment of cell proliferation, migration, invasion, and tube formation, thus preventing SSc MVECs from entering a suitable angiogenic programme in vitro. See text for abbreviations.
In another recent study, the expression of the CXC chemokine SDF-1 and its receptor CXCR4 was investigated in the skin and dermal MVECs isolated from SSc patients [76]. The SDF-1/CXCR4 axis regulates specific steps in new vessel formation. SDF-1 released by or expressed on ECs creates a local chemokine gradient, dictating directional responses of ECs expressing CXCR4 [106]. Moreover, SDF-1–CXCR4 interaction further amplifies angiogenesis by increasing VEGF release by ECs [107]. SDF-1 is also a strong chemoattractant for CXCR4-expressing EPCs which are mobilized from the bone marrow in response to tissue ischemia [108]. The expression of SDF-1 and CXCR4 is elevated in capillaries of lesional skin and MVECs from early stage SSc, suggesting a role in the pathogenesis of SSc microvascular abnormalities [76]. Interestingly, it has also been shown that a functional gene polymorphism which affects SDF-1 gene expression is associated with major SSc vascular manifestations, such as pulmonary arterial hypertension and ischemic digital ulcers [109]. Besides the SDF-1 gene, polymorphisms of other genes which regulate EC plasticity and functions have been shown to be associated with the vascular phenotype of SSc [110]. Furthermore, epigenetic mechanisms may also modulate EC dysfunction in SSc. It has recently been shown that DNA methylation and histone deacethylation cause an epigenetic repression of the bone morphogenetic protein type 2 receptor in dermal SSc MVECs, and that this modification results in an enhanced vulnerability of these ECs to apoptosis [111].
Finally, Fleming and coworkers [31] have recently demonstrated that in SSc skin, along with the loss of capillaries, there is a dramatic change in the endothelial phenotype of residual microvessels. The SSc vascular phenotype was characterized by loss of vascular endothelial cadherin (VE-cadherin), a supposedly universal endothelial marker required for tube formation, as well as the overexpression of the anti-angiogenic interferon-α (IFN-α) and overexpression of RGS5, a signalling molecule whose expression coincides with the end of branching morphogenesis during embryo development and tumour angiogenesis [31].
Altogether, the current laboratory and clinical data clearly indicate that, despite the increased levels of pro-angiogenic factors, there are also a number of ongoing anti-angiogenic mechanisms that ultimately result in defective angiogenesis preventing the formation of new functional capillaries in SSc.
Mechanisms of defective vasculogenesis in SSc
Besides impaired angiogenesis, there is evidence that defective vasculogenesis may contribute to the vascular pathophysiology of SSc [20]. However, conflicting results have been published concerning the presence and role of circulating EPCs in SSc. Currently, different protocols for isolation, enrichment, in vitro culture and quantification of EPCs, as well as differences in the prevalence of cardiovascular risk factors, medications, mean disease duration and severity of the study populations, might account for the discrepancy between the findings of different studies [20].
Many studies have analysed the levels of circulating EPCs isolated from the peripheral blood of SSc patients in comparison with healthy controls and/or other rheumatic conditions. The first study by Kuwana et al. demonstrated reduced absolute numbers of circulating CD34+/CD133+/VEGFR-2+ EPCs with impaired functions in SSc [112]. The lowest numbers of EPCs were observed in SSc patients with pitting scars and active fingertip ulcers, and early-outgrowth EPCs showed faint or no expression of markers for mature endothelium, such as VE-cadherin, CD146 and von Willebrand factor, thus suggesting their defective capacity to differentiate into mature ECs [112]. Similar results were also found in the recent study by Zhu et al. which demonstrated substantial depletion of circulating CD34+/CD133+/VEGFR-2+ EPCs in SSc, regardless of disease subtypes, disease stage and different methodologies used to quantify early-outgrowth EPCs [113]. In contrast, Del Papa et al. reported significantly increased numbers of circulating CD34+/CD133+/VEGFR-2+ EPCs, as well as a negative correlation between EPC counts and disease duration [64]. Nevskaya and coworkers also found that early stage SSc and high disease activity were accompanied by a rise in circulating EPC levels that correlated positively with the severity of peripheral vascular manifestations [114]. Furthermore, EPC reduction with disease progression was linked to endothelial dysfunction and capillary loss, as well as the development of severe cardiac involvement and pulmonary arterial hypertension [114]. Allanore et al. showed higher numbers of CD34+/CD133+ EPCs in SSc than in patients with osteoarthritis, but lower than in rheumatoid arthritis patients [115]. These authors also observed a positive correlation between the EPC counts and the European Scleroderma Study Group disease activity index, and, in agreement with other studies [64, 114], the numbers of EPCs tended to be higher in the early SSc stages [115]. Very recently, the same group of investigators demonstrated that the levels of circulating CD34+/CD133+/VEGFR-2+ EPCs were significantly increased in SSc patients compared to healthy individuals, supporting their active mobilization from the bone marrow [116]. The subset of patients with digital vascular lesions and higher Medsger severity score displayed lower circulating EPC counts, suggesting increased homing to peripheral ischemic tissues and consequent EPC depletion in the blood at this disease stage [116]. In addition, the levels of late-outgrowth EPCs were not different between SSc patients and healthy individuals, as determined by the in vitro colony-forming assay [116].
Altogether, the conflicting data on EPCs suggest that different scenarios might be responsible for the vascular repair defect in SSc (Fig. 3). In fact, the findings of decreased EPC levels regardless of SSc stage [113] could suggest that EPC depletion might occur within the bone marrow and/or early after their mobilization into the circulation (Fig. 3). Alternatively, the findings of increased EPC levels in early/active stage and subsequent reduction with disease progression and severity [64, 114, 116] could suggest that EPCs might be mobilized from the bone marrow and home to ischemic tissues, where they might fail to repair vascular damage due to intrinsic functional defects. Moreover, persistent peripheral endothelial injury and ischemia-reperfusion episodes might eventually lead to depletion of EPCs in the peripheral circulation (Fig. 3).
Fig 3.
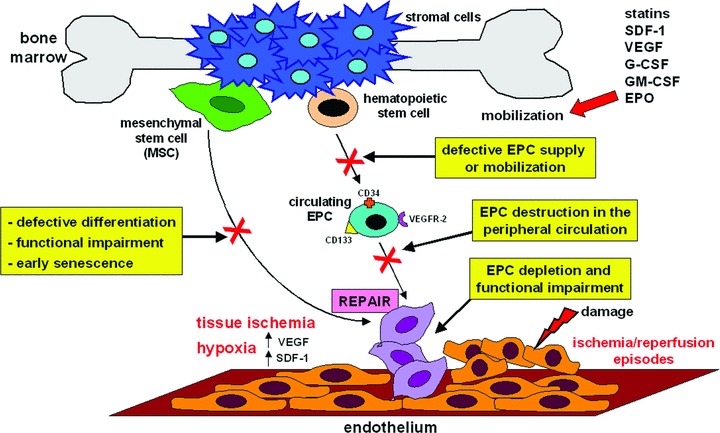
Mechanisms of impaired vasculogenesis in SSc. Different scenarios may be responsible for the altered numbers and defective vascular repair ability of bone marrow-derived CD34+/CD133+/VEGFR-2+ EPCs and MSCs in SSc. Aside from haemangioblast-derived CD34+ EPCs, there is evidence that MSCs may also differentiate into EPCs. VEGF and SDF-1 are oxygen-sensitive cytokines that are induced by hypoxia, and act as molecular mediators to rapidly mobilize EPCs from the bone marrow and to guide them into ischemic tissues. Besides VEGF and SDF-1, other cytokines and growth factors, such as granulocyte colony-stimulating factor (G-CSF), granulocyte-macrophage colony-stimulating factor (GM-CSF) and erythropoietin (EPO), are also important for the maturation and mobilization of bone marrow resident CD34+ EPCs. See text for abbreviations.
In this regard, two recent studies have suggested that the bone marrow microenvironment for stem cells is impaired in SSc [21, 117]. Del Papa et al. reported that SSc bone marrow had reduced numbers of CD133+ EPCs and stromal cells, both of which were also functionally impaired [117]. In particular, the ability of bone marrow-derived CD133+ cells to differentiate into ECs in vitro was impaired, while the number and size of colonies formed by stromal cells were reduced. The second study by Cipriani et al. examined the in vitro capacity of bone marrow-derived MSCs to differentiate towards the EC lineage [21]. In SSc patients, the percentage of VEGFR-2+ and CXCR4+ MSCs and endothelial-like MSCs was significantly lower than in controls. Accordingly, both SSc MSCs and endothelial-like MSCs displayed impaired responses to VEGF- and SDF-1-induced migration, invasion and capillary-like structure formation on Matrigel, as well as an early senescence [21]. These data collectively suggest that endothelial repair may be affected in SSc starting from bone marrow stem niches. In agreement with these findings, other authors have shown that SSc EPCs mobilized during atorvastatin treatment are functionally defective [118]. Moreover, the atorvastatin-induced increase of circulating EPCs was transient and even with treatment, however, the numbers of circulating EPCs in SSc patients did not reach those in healthy individuals [118, 119]. Del Papa et al. also reported that simvastatin treatment failed to increase the number of circulating EPCs in SSc patients [120].
A very recent study suggested that SSc serum can induce EPC apoptosis, and that this might account, at least in part, for the decreased circulating EPC numbers in SSc patients [113]. In fact, freshly isolated SSc EPCs displayed an increased rate of apoptosis, and depletion of the IgG fraction from SSc serum was able to abolish apoptosis in EPCs from healthy individuals, thus suggesting the involvement of auto-antibodies in this process [113]. In particular, SSc serum induced EPC apoptosis through the inhibition of Akt phosphorylation, which prevented FOXO3a degradation with consequent up-regulation of the pro-apoptotic protein Bim [113]. Consistent with these findings, another study demonstrated an increased membrane expression of the pro-apoptotic Fas (CD95) receptor on SSc EPCs, suggesting a pre-determined commitment to programmed cell death [114]. Finally, a recent report analysed the in vitro angiogenic potential of late-outgrowth EPCs cultured after isolation from the peripheral blood of SSc patients using different functional assays [121]. These authors found that SSc late-outgrowth EPCs had the phenotype of genuine ECs and displayed in vitro angiogenic properties similar to those of control EPCs, including migratory and tube formation ability [121]. However, SSc late-outgrowth EPC exhibited defective up-regulation of VEGFR-1 under hypoxic conditions, and the authors suggested that this might promote the VEGF/VEGFR-2 pathway, favouring chronic and uncontrolled stimulation of VEGF and ultimately resulting in altered vessel morphology and functionality [121].
Conclusions and perspectives on vascular treatment strategies
The extensive vascular damage and lack of compensatory vascular repair mechanisms make SSc an ideal model to study novel therapeutic strategies for vascular regeneration.
It is most important to realize that in SSc there are parallel vascular pathologies that may require different and specific therapeutic approaches. In fact, the SSc vascular pathophysiology consists of an uncompensated loss of capillaries following peripheral ischemia on one hand, and a vascular remodelling process with intimal proliferation and vessel obliteration in multiple vascular beds on the other. Therefore, future investigations in the field of SSc peripheral vasculopathy should mainly focus on the understanding of the complex biology of ECs, pericytes, vascular smooth muscle cells and perivascular fibroblasts/ myofibroblasts, as well as the molecular mechanisms of cross-talk among these different cell types.
Still, the mechanisms of the impaired response to vascular injury and lack of angiogenesis remain poorly understood. Currently, several studies have shown that angiogenesis is impaired in SSc despite the up-regulation of a large array of pro-angiogenic factors. Conversely, there are controversial data on the up-regulation of angiostatic factors, which might serve as targets for therapeutic approaches. In addition, functional in vitro and in vivo (i.e. using preclinical animal models) studies targeting pro-angiogenic or angiostatic factors are lacking. There is also evidence that the uncontrolled and chronic expression of pro-angiogenic mediators (e.g. VEGF) may have deleterious effects rather than promote the formation of new functional capillaries. Moreover, many pro-angiogenic factors can stimulate not only EC functions, but also induce vascular smooth muscle cell and pericyte proliferation and hyperthrophy. Therefore, an important issue to address in SSc is whether it would be therapeutically useful to promote angiogenesis and whether it would be possible to reverse the complex imbalance between pro-angiogenic and angiostatic factors. In this regard, strategies for dosing and timing of pro-angiogenic factors might be of major importance.
For therapeutic neovascularization, it was shown that only the proper and timely delivery of EC-inducing factors (i.e. VEGF and FGF-2) and vessel maturating factors (i.e. PDGF and TGF-β) was able to induce the formation of functionally mature vessels composed of ECs and smooth muscle cells in vivo. For example, Hao et al. reported that the sequential release of VEGF and PDGF-BB in a mouse model of myocardial infarction could promote vascularization and improve vessel maturation compared with the release of either factor alone [122]. Obviously, mimicking such complicated series of events is difficult to achieve in human beings using conventional drug-delivery systems, which generally function via a burst-release mechanism. However, several release systems are being successfully established [122–124]. A highly interesting development in this field is the generation of ‘on-demand release’ platforms that use the principles of enzyme-mediated growth factor release. In fact, physiologically growth factors are stored in the extracellular matrix, from which they are released by degrading enzymes secreted by invading or growing cells. The development of such ‘on-demand release’ depots, containing multiple pro-angiogenic factors that are secreted in temporally distinct or cell-dependent patterns, could mimic paracrine signalling events during neovascularization more closely [123]. However, the development of such release mechanisms is still in its early stages, and the efficacy of such protein-based therapies needs to be elucidated in disease-specific animal models and subsequently in clinical trials. In SSc, a potential difficulty in the clinical success of this therapeutic approach is the status of the affected endothelium in the ischemic regions. Compromised endothelium will not migrate nor proliferate as well as healthy endothelium, therefore potentially limiting the extent of neovascularization induced by angiogenic factor delivery system-based therapies. This impairment represents an important challenge for vascular regenerative medicine, and strategies to overcome EC defects need to be addressed in future research.
Besides angiogenesis, the role of vasculogenesis in SSc appears even less clear. Whether EPC counts are decreased or increased in the peripheral blood of SSc patients is still a matter of controversy. In fact, it is still to be determined whether poor vasculogenesis is due to any defect in the EPC supply within and mobilization from the bone marrow, an excessive immune-mediated EPC destruction in the peripheral circulation, a progressive depletion of EPCs following homing to ischemic tissues under persistent peripheral vascular injury, or an intrinsic functional impairment of EPCs through genetic and/or epigenetic mechanisms. Moreover, the biology of EPCs is complex and currently deserves further dissection. It appears that the incorporation of bone marrow-derived EPCs in new vessels is strictly determined by the local microenvironment, and that the main contribution of EPCs to neovascularization may be through the paracrine secretion of pro-angiogenic factors acting on pre-existing mature ECs rather than via their incorporation into the neovasculature [34, 125]. Therefore, further investigations are needed to evaluate the potential therapeutic use of EPCs in SSc. In this regard, recent pilot studies reported that autologous haematopoietic stem cell transplantation or local implantation of CD34+ cells in ischemic tissues may improve microcirculation in SSc patients, as shown by morphological changes on nailfold capillaroscopy, increased dermal capillary density and healing of ischemic digital ulcers [31–33]. Drugs that enhance the mobilization of EPCs or injection of in vitro amplified autologous EPCs might offer new therapeutic options for the treatment of severe vascular disease in SSc. Alternatively, since EPCs home to sites of ischemic injury and tissue hypoxia, ‘engineered EPCs’ might be used as ‘on-demand release’ depots of pro-angiogenic factors or as vehicles for the selective transport of drugs to the injured endothelium. However, intrinsic functional defects, early senescence and increased susceptibility to apoptosis of SSc EPCs might limit the efficacy of these potential approaches.
Therefore, new insights into the disease pathogenesis are needed to allow the translation of basic research into more effective treatment of SSc patients. In this regard, suitable animal models for the peripheral vasculopathy of SSc allowing studies on pathophysiological mechanisms and preclinical testing of potential drugs are now becoming available and promise to be useful tools [126]. Either angiogenic or vasculogenic mechanisms may potentially become in the future the target of novel therapeutic strategies to promote vascular regeneration in SSc.
References
- 1.Varga J, Abraham D. Systemic sclerosis: a prototypic multisystem fibrotic disorder. J Clin Invest. 2007;117:557–67. doi: 10.1172/JCI31139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jimenez SA, Derk CT. Following the molecular pathways towards an understanding of the pathogenesis of systemic sclerosis. Ann Intern Med. 2004;140:37–50. [PubMed] [Google Scholar]
- 3.Guiducci S, Giacomelli R, Matucci-Cerinic M. Vascular complications of scleroderma. Autoimmun Rev. 2007;6:520–3. doi: 10.1016/j.autrev.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 4.Kahaleh MB. Vascular involvement in systemic sclerosis. Clin Exp Rheumatol. 2004;22:S19–23. [PubMed] [Google Scholar]
- 5.LeRoy EC. Systemic sclerosis: a vascular perspective. Rheum Dis Clin North Am. 1996;22:675–94. doi: 10.1016/s0889-857x(05)70295-7. [DOI] [PubMed] [Google Scholar]
- 6.Koch AE, Distler O. Vasculopathy and disordered angiogenesis in selected rheumatic diseases: rheumatoid arthritis and systemic sclerosis. Arthritis Res Ther. 2007;9:S3. doi: 10.1186/ar2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campbell PM, LeRoy EC. Pathogenesis of systemic sclerosis: a vascular hypothesis. Semin Arthritis Rheum. 1975;4:351–68. doi: 10.1016/0049-0172(75)90017-7. [DOI] [PubMed] [Google Scholar]
- 8.Trotta F, Biagini G, Cenacchi G, et al. Microvascular changes in progressive systemic sclerosis: immunohistochemical and ultrastructural study. Clin Exp Rheumatol. 1984;2:209–15. [PubMed] [Google Scholar]
- 9.Ibba-Manneschi L, Del Rosso A, Milia AF, et al. Damage of cutaneous peripheral nervous system evolves differently according to the disease phase and subset of systemic sclerosis. Rheumatology. 2005;44:607–13. doi: 10.1093/rheumatology/keh559. [DOI] [PubMed] [Google Scholar]
- 10.Rodnan GP, Myerowitz RL, Justh GO. Morphologic changes in the digital arteries of patients with systemic sclerosis (scleroderma) and Raynaud phenomenon. Medicine. 1980;59:393–408. doi: 10.1097/00005792-198011000-00001. [DOI] [PubMed] [Google Scholar]
- 11.Al-Sabbagh MR, Steen VD, Zee BC, et al. Pulmonary arterial histology and morphometry in systemic sclerosis: a case-control autopsy study. J Rheumatol. 1989;16:1038–42. [PubMed] [Google Scholar]
- 12.Cool CD, Kennedy D, Voelkel NF, et al. Pathogenesis and evolution of plexiform lesions in pulmonary hypertension associated with scleroderma and human immunodeficiency virus infection. Hum Pathol. 1997;28:434–42. doi: 10.1016/s0046-8177(97)90032-0. [DOI] [PubMed] [Google Scholar]
- 13.Penn H, Howie AJ, Kingdon EJ, et al. Scleroderma renal crisis: patient characteristics and long-term outcomes. Q J Med. 2007;100:485–94. doi: 10.1093/qjmed/hcm052. [DOI] [PubMed] [Google Scholar]
- 14.Rhew EY, Barr WG. Scleroderma renal crises: new insights and developments. Curr Rheumatol Rep. 2004;6:129–36. doi: 10.1007/s11926-004-0057-5. [DOI] [PubMed] [Google Scholar]
- 15.Cutolo M, Grassi W, Matucci-Cerinic M. Raynaud’s phenomenon and the role of capillaroscopy. Arthritis Rheum. 2003;48:3023–30. doi: 10.1002/art.11310. [DOI] [PubMed] [Google Scholar]
- 16.Maricq HR. Wide-field capillary microscopy: technique and rating scale for abnormalities seen in scleroderma and related disorders. Arthritis Rheum. 1981;24:1159–65. doi: 10.1002/art.1780240907. [DOI] [PubMed] [Google Scholar]
- 17.Sulli A, Secchi ME, Pizzorni C, et al. Scoring the nailfold microvascular changes during the capillaroscopic analysis in systemic sclerosis patients. Ann Rheum Dis. 2008;67:885–7. doi: 10.1136/ard.2007.079756. [DOI] [PubMed] [Google Scholar]
- 18.Kahaleh B. Progress in research into systemic sclerosis. Lancet. 2004;364:561–2. doi: 10.1016/S0140-6736(04)16864-5. [DOI] [PubMed] [Google Scholar]
- 19.Distler O, Distler JH, Scheid A, et al. Uncontrolled expression of vascular endothelial growth factor and its receptors leads to insufficient skin angiogenesis in patients with systemic sclerosis. Circ Res. 2004;95:109–16. doi: 10.1161/01.RES.0000134644.89917.96. [DOI] [PubMed] [Google Scholar]
- 20.Distler JH, Beyer C, Schett G, et al. Endothelial progenitor cells: novel players in the pathogenesis of rheumatic diseases. Arthritis Rheum. 2009;60:3168–79. doi: 10.1002/art.24921. [DOI] [PubMed] [Google Scholar]
- 21.Cipriani P, Guiducci S, Miniati I, et al. Impairment of endothelial cell differentiation from bone marrow-derived mesenchymal stem cells: new insight into the pathogenesis of systemic sclerosis. Arthritis Rheum. 2007;56:1994–2004. doi: 10.1002/art.22698. [DOI] [PubMed] [Google Scholar]
- 22.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–6. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 23.Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 24.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6:389–95. doi: 10.1038/74651. [DOI] [PubMed] [Google Scholar]
- 25.Distler J, Hirth A, Kurowska-Stolarska M, et al. Angiogenic and angiostatic factors in the molecular control of angiogenesis. Q J Nucl Med. 2003;47:149–61. [PubMed] [Google Scholar]
- 26.Asahara T, Murohara T, Sullivan A, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–7. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 27.Shi Q, Rafii S, Wu MH, et al. Evidence for circulating bone marrow-derived endothelial cells. Blood. 1998;92:362–7. [PubMed] [Google Scholar]
- 28.Urbich C, Dimmeler S. Endothelial progenitor cells: characterization and role in vascular biology. Circ Res. 2004;95:343–53. doi: 10.1161/01.RES.0000137877.89448.78. [DOI] [PubMed] [Google Scholar]
- 29.Tateishi-Yuyama E, Matsubara H, Murohara T, et al. Therapeutic angiogenesis for patients with limb ischaemia by autologous transplantation of bone-marrow cells: a pilot study and a randomised controlled trial. Lancet. 2002;360:427–35. doi: 10.1016/S0140-6736(02)09670-8. [DOI] [PubMed] [Google Scholar]
- 30.Stamm C, Westphal B, Kleine HD, et al. Autologous bone-marrow stem cell transplantation for myocardial regeneration. Lancet. 2003;361:45–6. doi: 10.1016/S0140-6736(03)12110-1. [DOI] [PubMed] [Google Scholar]
- 31.Fleming JN, Nash RA, McLeod DO, et al. Capillary regeneration in scleroderma: stem cell therapy reverses phenotype. PLoS ONE. 2008;3:e1452. doi: 10.1371/journal.pone.0001452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miniati I, Guiducci S, Conforti ML, et al. Autologous stem cell transplantation improves microcirculation in systemic sclerosis. Ann Rheum Dis. 2009;68:94–8. doi: 10.1136/ard.2007.082495. [DOI] [PubMed] [Google Scholar]
- 33.Nevskaya T, Ananieva L, Bykovskaia S, et al. Autologous progenitor cell implantation as a novel therapeutic intervention for ischemic digits in systemic sclerosis. Rheumatology. 2009;48:61–4. doi: 10.1093/rheumatology/ken407. [DOI] [PubMed] [Google Scholar]
- 34.Krenning G, van Luyn MJ, Harmsen MC. Endothelial progenitor cell-based neovascularization: implications for therapy. Trends Mol Med. 2009;15:180–9. doi: 10.1016/j.molmed.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 35.Deschaseaux F, Selmani Z, Falcoz PE, et al. Two types of circulating endothelial progenitor cells in patients receiving long term therapy by HMG-CoA reductase inhibitors. Eur J Pharmacol. 2007;562:111–8. doi: 10.1016/j.ejphar.2007.01.045. [DOI] [PubMed] [Google Scholar]
- 36.Hur J, Yoon CH, Kim HS, et al. Characterization of two types of endothelial progenitor cells and their different contributions to neovasculogenesis. Arterioscler Thromb Vasc Biol. 2004;24:288–93. doi: 10.1161/01.ATV.0000114236.77009.06. [DOI] [PubMed] [Google Scholar]
- 37.Prescott RJ, Freemont AJ, Jones CJ, et al. Sequential dermal microvascular and perivascular changes in the development of scleroderma. J Pathol. 1992;166:255–63. doi: 10.1002/path.1711660307. [DOI] [PubMed] [Google Scholar]
- 38.Sgonc R, Gruschwitz MS, Dietrich H, et al. Endothelial cell apoptosis is a primary pathogenetic event underlying skin lesions in avian and human scleroderma. J Clin Invest. 1996;98:785–92. doi: 10.1172/JCI118851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kahaleh B. Vascular disease in scleroderma: mechanisms of vascular injury. Rheum Dis Clin North Am. 2008;34:57–71. doi: 10.1016/j.rdc.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 40.Nietert PJ, Silver RM. Systemic sclerosis: environmental and occupational risk factors. Curr Opin Rheumatol. 2000;12:520–6. doi: 10.1097/00002281-200011000-00008. [DOI] [PubMed] [Google Scholar]
- 41.Pandey JP, LeRoy EC. Human cytomegalovirus and the vasculopathies of autoimmune diseases (especially scleroderma), allograft rejection, and coronary restenosis. Arthritis Rheum. 1998;41:10–5. doi: 10.1002/1529-0131(199801)41:1<10::AID-ART2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 42.Lunardi C, Bason C, Navone R, et al. Systemic sclerosis immunoglobulin G autoantibodies bind the human cytomegalovirus late protein UL94 and induce apoptosis in human endothelial cells. Nat Med. 2000;6:1183–6. doi: 10.1038/80533. [DOI] [PubMed] [Google Scholar]
- 43.Renaudineau Y, Revelen R, Levy Y, et al. Anti-endothelial cell antibodies in systemic sclerosis. Clin Diagn Lab Immunol. 1999;6:156–60. doi: 10.1128/cdli.6.2.156-160.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pignone A, Scaletti C, Matucci-Cerinic M, et al. Anti-endothelial cell antibodies in systemic sclerosis: significant association with vascular involvement and alveolo-capillary impairment. Clin Exp Rheumatol. 1998;16:527–32. [PubMed] [Google Scholar]
- 45.Sgonc R, Gruschwitz MS, Boeck G, et al. Endothelial cell apoptosis in systemic sclerosis is induced by antibody-dependent cell-mediated cytotoxicity via CD95. Arthritis Rheum. 2000;43:2550–62. doi: 10.1002/1529-0131(200011)43:11<2550::AID-ANR24>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 46.Gabrielli A, Svegliati S, Moroncini G, et al. Oxidative stress and the pathogenesis of scleroderma: the Murrell’s hypothesis revisited. Semin Immunopathol. 2008;30:329–37. doi: 10.1007/s00281-008-0125-4. [DOI] [PubMed] [Google Scholar]
- 47.Casciola-Rosen L, Wigley F, Rosen A. Scleroderma autoantigens are uniquely fragmented by metal-catalyzed oxidation reactions: implications for pathogenesis. J Exp Med. 1997;185:71–9. doi: 10.1084/jem.185.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blake DR, Winyard P, Scott DG, et al. Endothelial cell cytotoxicity in inflammatory vascular diseases: the possible role of oxidised lipoproteins. Ann Rheum Dis. 1985;44:176–82. doi: 10.1136/ard.44.3.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Matucci-Cerinic M, Valentini G, Sorano GG, et al. Blood coagulation, fibrinolysis, and markers of endothelial dysfunction in systemic sclerosis. Semin Arthritis Rheum. 2003;32:285–95. doi: 10.1053/sarh.2002.50011. [DOI] [PubMed] [Google Scholar]
- 50.Koch AE, Kronfeld-Harrington LB, Szekanecz Z, et al. In situ expression of cytokines and cellular adhesion molecules in the skin of patients with systemic sclerosis. Their role in early and late disease. Pathobiology. 1993;61:239–46. doi: 10.1159/000163802. [DOI] [PubMed] [Google Scholar]
- 51.Sollberg S, Peltonen J, Uitto J, et al. Elevated expression of beta 1 and beta 2 integrins, intercellular adhesion molecule 1, and endothelial leukocyte adhesion molecule 1 in the skin of patients with systemic sclerosis of recent onset. Arthritis Rheum. 1992;35:290–8. doi: 10.1002/art.1780350307. [DOI] [PubMed] [Google Scholar]
- 52.Kuryliszyn-Moskal A, Klimiuk PA, Sierakowski S. Soluble adhesion molecules (sVCAM-1, sE-selectin), vascular endothelial growth factor (VEGF) and endothelin-1 in patients with systemic sclerosis: relationship to organ systemic involvement. Clin Rheumatol. 2005;24:111–6. doi: 10.1007/s10067-004-0987-3. [DOI] [PubMed] [Google Scholar]
- 53.Sato S. Abnormalities of adhesion molecules and chemokines in scleroderma. Curr Opin Rheumatol. 1999;11:503–7. [PubMed] [Google Scholar]
- 54.Manetti M, Neumann E, Müller A, et al. Endothelial/lymphocyte activation leads to prominent CD4+ T cell infiltration in the gastric mucosa of patients with systemic sclerosis. Arthritis Rheum. 2008;58:2866–73. doi: 10.1002/art.23806. [DOI] [PubMed] [Google Scholar]
- 55.Sakkas LI, Chikanza IC, Platsoucas CD. Mechanisms of disease: the role of immune cells in the pathogenesis of systemic sclerosis. Nat Clin Pract Rheumatol. 2006;2:679–85. doi: 10.1038/ncprheum0346. [DOI] [PubMed] [Google Scholar]
- 56.Postlethwaite AE, Chiang TM. Platelet contributions to the pathogenesis of systemic sclerosis. Curr Opin Rheumatol. 2007;19:574–9. doi: 10.1097/BOR.0b013e3282eeb3a4. [DOI] [PubMed] [Google Scholar]
- 57.Kahaleh MB, Osborn I, LeRoy EC. Elevated levels of circulating platelet aggregates and beta-thromboglobulin in scleroderma. Ann Intern Med. 1982;96:610–3. doi: 10.7326/0003-4819-96-5-610. [DOI] [PubMed] [Google Scholar]
- 58.Abraham D, Distler O. How does endothelial cell injury start? The role of endothelin in systemic sclerosis. Arthritis Res Ther. 2007;9:S2. doi: 10.1186/ar2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hummers LK. Biomarkers of vascular disease in scleroderma. Rheumatology. 2008;47:v21–2. doi: 10.1093/rheumatology/ken281. [DOI] [PubMed] [Google Scholar]
- 60.Denton CP, Bickerstaff MC, Shiwen X, et al. Serial circulating adhesion molecule levels reflect disease severity in systemic sclerosis. Br J Rheumatol. 1995;34:1048–54. doi: 10.1093/rheumatology/34.11.1048. [DOI] [PubMed] [Google Scholar]
- 61.Kahaleh MB, Osborn I, LeRoy EC. Increased factor VIII/von Willebrand factor antigen and von Willebrand factor activity in scleroderma and in Raynaud’s phenomenon. Ann Intern Med. 1981;94:482–4. doi: 10.7326/0003-4819-94-4-482. [DOI] [PubMed] [Google Scholar]
- 62.Wigley FM. Vascular disease in scleroderma. Clin Rev Allerg Immunol. 2009;36:150–75. doi: 10.1007/s12016-008-8106-x. [DOI] [PubMed] [Google Scholar]
- 63.Guiducci S, Distler JH, Jungel A, et al. The relationship between plasma microparticles and disease manifestations in patients with systemic sclerosis. Arthritis Rheum. 2008;58:2845–53. doi: 10.1002/art.23735. [DOI] [PubMed] [Google Scholar]
- 64.Del Papa N, Colombo G, Fracchiolla N, et al. Circulating endothelial cells as a marker of ongoing vascular disease in systemic sclerosis. Arthritis Rheum. 2004;50:1296–304. doi: 10.1002/art.20116. [DOI] [PubMed] [Google Scholar]
- 65.Konttinen YT, Mackiewicz Z, Ruuttila P, et al. Vascular damage and lack of angiogenesis in systemic sclerosis skin. Clin Rheumatol. 2003;22:196–202. doi: 10.1007/s10067-003-0698-1. [DOI] [PubMed] [Google Scholar]
- 66.Mulligan-Kehoe MJ, Drinane MC, Mollmark J, et al. Antiangiogenic plasma activity in patients with systemic sclerosis. Arthritis Rheum. 2007;56:3448–58. doi: 10.1002/art.22861. [DOI] [PubMed] [Google Scholar]
- 67.Ribatti D, Crivellato E. Immune cells and angiogenesis. J Cell Mol Med. 2009;13:2822–33. doi: 10.1111/j.1582-4934.2009.00810.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kaminski MJ, Majewski S, Jablonska S, et al. Lowered angiogenic capability of peripheral blood lymphocytes in progressive systemic sclerosis (scleroderma) J Invest Dermatol. 1984;82:239–43. doi: 10.1111/1523-1747.ep12260139. [DOI] [PubMed] [Google Scholar]
- 69.Koch AE, Litvak MA, Burrows JC, et al. Decreased monocyte-mediated angiogenesis in scleroderma. Clin Immunol Immunopathol. 1992;64:153–60. doi: 10.1016/0090-1229(92)90193-r. [DOI] [PubMed] [Google Scholar]
- 70.Kahaleh MB, DeLustro F, Bock W, et al. Human monocyte modulation of endothelial cells and fibroblast growth: possible mechanism for fibrosis. Clin Immunol Immunopathol. 1986;39:242–55. doi: 10.1016/0090-1229(86)90088-7. [DOI] [PubMed] [Google Scholar]
- 71.Majewski S, Skopinska-Rozewska E, Jablonska S, et al. Modulatory effect of sera from scleroderma patients on lymphocyte-induced angiogenesis. Arthritis Rheum. 1985;28:1133–9. doi: 10.1002/art.1780281009. [DOI] [PubMed] [Google Scholar]
- 72.Marczak M, Majewski S, Skopinska-Rozewska E, et al. Enhanced angiogenic capability of monocyte-enriched mononuclear cell suspension from patients with systemic scleroderma. J Invest Dermatol. 1986;86:355–8. doi: 10.1111/1523-1747.ep12285572. [DOI] [PubMed] [Google Scholar]
- 73.Ribatti D, Cantatore FP, Vacca A, et al. Systemic sclerosis stimulates angiogenesis in the chick embryo chorioallantoic membrane. Clin Rheumatol. 1998;17:115–20. doi: 10.1007/BF01452256. [DOI] [PubMed] [Google Scholar]
- 74.Distler O, Del Rosso A, Giacomelli R, et al. Angiogenic and angiostatic factors in systemic sclerosis: increased levels of vascular endothelial growth factor are a feature of the earliest disease stages and are associated with the absence of fingertip ulcers. Arthritis Res. 2002;4:R11. doi: 10.1186/ar596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Distler O, Pap T, Kowal-Bielecka O, et al. Overexpression of monocyte chemoattractant protein 1 in systemic sclerosis: role of platelet-derived growth factor and effects on monocyte chemotaxis and collagen synthesis. Arthritis Rheum. 2001;44:2665–78. doi: 10.1002/1529-0131(200111)44:11<2665::aid-art446>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 76.Cipriani P, Milia AF, Liakouli V, et al. Differential expression of stromal cell-derived factor 1 and its receptor CXCR4 in the skin and endothelial cells of systemic sclerosis patients: pathogenetic implications. Arthritis Rheum. 2006;54:3022–33. doi: 10.1002/art.22047. [DOI] [PubMed] [Google Scholar]
- 77.Hummers LK, Hall A, Wigley FM, et al. Abnormalities in the regulators of angiogenesis in patients with scleroderma. J Rheumatol. 2009;36:576–82. doi: 10.3899/jrheum.080516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Byrne AM, Bouchier-Hayes DJ, Harmey JH. Angiogenic and cell survival functions of vascular endothelial growth factor (VEGF) J Cell Mol Med. 2005;9:777–94. doi: 10.1111/j.1582-4934.2005.tb00379.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mackiewicz Z, Sukura A, Povilenaite D, et al. Increased but imbalanced expression of VEGF and its receptors has no positive effect on angiogenesis in systemic sclerosis skin. Clin Exp Rheumatol. 2002;20:641–6. [PubMed] [Google Scholar]
- 80.Davies CA, Jeziorska M, Freemont AJ, et al. The differential expression of VEGF, VEGFR-2, and GLUT-1 proteins in disease subtypes of systemic sclerosis. Hum Pathol. 2006;37:190–7. doi: 10.1016/j.humpath.2005.10.007. [DOI] [PubMed] [Google Scholar]
- 81.Allanore Y, Borderie D, Lemaréchal H, et al. Nifedipine decreases sVCAM-1 concentrations and oxidative stress in systemic sclerosis but does not affect the concentrations of vascular endothelial growth factor or its soluble receptor 1. Arthritis Res Ther. 2004;6:R309–14. doi: 10.1186/ar1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Choi JJ, Min DJ, Cho ML, et al. Elevated vascular endothelial growth factor in systemic sclerosis. J Rheumatol. 2003;30:1529–33. [PubMed] [Google Scholar]
- 83.Ibba-Manneschi L, Manetti M, Milia AF, et al. Severe fibrotic changes and altered expression of angiogenic factors in maternal scleroderma: placental findings. Ann Rheum Dis. 2010;69:458–61. doi: 10.1136/ard.2009.107623. [DOI] [PubMed] [Google Scholar]
- 84.Dor Y, Djonov V, Abramovitch R, et al. Conditional switching of VEGF provides new insights into adult neovascularization and pro-angiogenic therapy. EMBO J. 2002;21:1939–47. doi: 10.1093/emboj/21.8.1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.D’Alessio S, Fibbi G, Cinelli M, et al. Matrix metalloproteinase 12-dependent cleavage of urokinase receptor in systemic sclerosis microvascular endothelial cells results in impaired angiogenesis. Arthritis Rheum. 2004;50:3275–85. doi: 10.1002/art.20562. [DOI] [PubMed] [Google Scholar]
- 86.Beyer C, Schett G, Gay S, et al. Hypoxia in the pathogenesis of systemic sclerosis. Arthritis Res Ther. 2009;11:220. doi: 10.1186/ar2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hebbar M, Peyrat JP, Hornez L, et al. Increased concentrations of the circulating angiogenesis inhibitor endostatin in patients with systemic sclerosis. Arthritis Rheum. 2000;43:889–93. doi: 10.1002/1529-0131(200004)43:4<889::AID-ANR21>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 88.Macko RF, Gelber AC, Young BA, et al. Increased circulating concentrations of the counteradhesive proteins SPARC and thrombospondin-1 in systemic sclerosis (scleroderma). Relationship to platelet and endothelial cell activation. J Rheumatol. 2002;29:2565–70. [PubMed] [Google Scholar]
- 89.Dziankowska-Bartkowiak B, Waszczykowska E, Dziankowska-Zaboroszczyk E, et al. Decreased ratio of circulatory vascular endothelial growth factor to endostatin in patients with systemic sclerosis – association with pulmonary involvement. Clin Exp Rheumatol. 2006;24:508–13. [PubMed] [Google Scholar]
- 90.Dziankowska-Bartkowiak B, Waszczykowska E, Zalewska A, Sysa-Jedrzejowska A. Correlation of endostatin and tissue inhibitor of metalloproteinases 2 (TIMP2) serum levels with cardiovascular involvement in systemic sclerosis patients. Mediators Inflamm. 2005;3:144–9. doi: 10.1155/MI.2005.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Italiano JE, Jr, Richardson JL, Paltel-Hett S, et al. Angiogenesis is regulated by a novel mechanism: pro- and anti-angiogenic proteins are organized into separate platelet alpha granules and differentially released. Blood. 2008;111:1227–33. doi: 10.1182/blood-2007-09-113837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Klement GL, Yip TT, Cassiola F, et al. Platelets actively sequester angiogenesis regulators. Blood. 2009;113:2835–42. doi: 10.1182/blood-2008-06-159541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Solanilla A, Villeneuve J, Auguste P, et al. The transport of high amounts of vascular endothelial growth factor by blood platelets underlines their potential contribution in systemic sclerosis angiogenesis. Rheumatology. 2009;48:1036–44. doi: 10.1093/rheumatology/kep154. [DOI] [PubMed] [Google Scholar]
- 94.Giusti B, Serratì S, Margheri F, et al. The antiangiogenic tissue kallikrein pattern of endothelial cells in systemic sclerosis. Arthritis Rheum. 2005;52:3618–28. doi: 10.1002/art.21383. [DOI] [PubMed] [Google Scholar]
- 95.Giusti B, Fibbi G, Margheri F, et al. A model of anti-angiogenesis: differential transcriptosome profiling of microvascular endothelial cells from diffuse systemic sclerosis patients. Arthritis Res Ther. 2006;8:R115. doi: 10.1186/ar2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Emanueli C, Minasi A, Zacheo A, et al. Local delivery of Human Tissue Kallikrein Gene accelerates spontaneous angiogenesis in mouse model of hindlimb ischemia. Circulation. 2001;103:125–32. doi: 10.1161/01.cir.103.1.125. [DOI] [PubMed] [Google Scholar]
- 97.Mahabeer R, Bhoola KD. Kallikrein and kinin receptor genes. Pharmacol Ther. 2000;88:77–89. doi: 10.1016/s0163-7258(00)00080-2. [DOI] [PubMed] [Google Scholar]
- 98.Del Rosso A, Distler O, Milia AF, et al. Increased circulating levels of tissue kallikrein in systemic sclerosis correlate with microvascular involvement. Ann Rheum Dis. 2005;64:382–7. doi: 10.1136/ard.2004.023382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Milia AF, Del Rosso A, Pacini A, et al. Differential expression of tissue kallikrein in the skin of systemic sclerosis. Histol Histopathol. 2005;20:415–22. doi: 10.14670/HH-20.415. [DOI] [PubMed] [Google Scholar]
- 100.Mazar AP, Henkin J, Goldfarb RH. The urokinase plasminogen activator system in cancer: implications for tumor angiogenesis and metastasis. Angiogenesis. 1999;3:15–32. doi: 10.1023/a:1009095825561. [DOI] [PubMed] [Google Scholar]
- 101.Kroon ME, Koolwijk P, van Goor H, et al. Role and localization of urokinase receptor in the formation of new microvascular structures in fibrin matrices. Am J Pathol. 1999;154:1731–42. doi: 10.1016/S0002-9440(10)65429-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fibbi G, Caldini R, Chevanne M, et al. Urokinase-dependent angiogenesis in vitro and diacylglycerol production are blocked by antisense oligonucleotides against the urokinase receptor. Lab Invest. 1998;78:1109–19. [PubMed] [Google Scholar]
- 103.Ragno P. The urokinase receptor: a ligand or a receptor? Story of a sociable molecule. Cell Mol Life Sci. 2006;63:1028–37. doi: 10.1007/s00018-005-5428-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Serratì S, Cinelli M, Margheri F, et al. Systemic sclerosis fibroblasts inhibit in vitro angiogenesis by MMP-12-dependent cleavage of the endothelial cell urokinase receptor. J Pathol. 2006;210:240–8. doi: 10.1002/path.2048. [DOI] [PubMed] [Google Scholar]
- 105.Margheri F, Manetti M, Serratì S, et al. Domain 1 of the urokinase-type plasminogen activator receptor is required for its morphologic and functional, β2 integrin-mediated connection with actin cytoskeleton in human microvascular endothelial cells: failure of association in systemic sclerosis endothelial cells. Arthritis Rheum. 2006;54:3926–38. doi: 10.1002/art.22263. [DOI] [PubMed] [Google Scholar]
- 106.Salvucci O, Yao L, Villalba S, et al. Regulation of endothelial cell branching morphogenesis by endogenous chemokine stromal-derived factor-1. Blood. 2002;99:2703–11. doi: 10.1182/blood.v99.8.2703. [DOI] [PubMed] [Google Scholar]
- 107.Salcedo R, Wasserman K, Young HA, et al. Vascular endothelial growth factor and basic fibroblast growth factor induce expression of CXCR4 on human endothelial cells: in vivo neovascularization induced by stromal-derived factor-1alpha. Am J Pathol. 1999;154:1125–35. doi: 10.1016/s0002-9440(10)65365-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mohle R, Bautz F, Rafii S, et al. The chemokine receptor CXCR4 is expressed on CD34+ hematopoietic progenitors and leukemic cells and mediates transendothelial migration induced by stromal cell-derived factor-1. Blood. 1998;91:4523–30. [PubMed] [Google Scholar]
- 109.Manetti M, Liakouli V, Fatini C, et al. Association between a stromal cell-derived factor 1 (SDF-/XCL12) gene polymorphism and microvascular disease in systemic sclerosis. Ann Rheum Dis. 2009;68:408–11. doi: 10.1136/ard.2008.098277. [DOI] [PubMed] [Google Scholar]
- 110.Allanore Y, Wipff J, Kahan A, et al. Genetic basis for systemic sclerosis. Joint Bone Spine. 2007;74:577–83. doi: 10.1016/j.jbspin.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 111.Wang Y, Kahaleh B. Epigenetic repression of the bone morphogenetic protein type II receptor expression in scleroderma endothelial cells: a possible basis for enhanced SSc endothelial vulnerability to apoptosis. Arthritis Rheum. 2008;58:S928. [Google Scholar]
- 112.Kuwana M, Okazaki Y, Yasuoka H, et al. Defective vasculogenesis in systemic sclerosis. Lancet. 2004;364:603–10. doi: 10.1016/S0140-6736(04)16853-0. [DOI] [PubMed] [Google Scholar]
- 113.Zhu S, Evans S, Yan B, et al. Transcriptional regulation of Bim by FOXO3a and Akt mediates scleroderma serum-induced apoptosis in endothelial progenitor cells. Circulation. 2008;118:2156–65. doi: 10.1161/CIRCULATIONAHA.108.787200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nevskaya T, Bykovskaia S, Lyssuk E, et al. Circulating endothelial progenitor cells in systemic sclerosis: relation to impaired angiogenesis and cardiovascular manifestations. Clin Exp Rheumatol. 2008;26:421–9. [PubMed] [Google Scholar]
- 115.Allanore Y, Batteux F, Avouac J, et al. Levels of circulating endothelial progenitor cells in systemic sclerosis. Clin Exp Rheumatol. 2007;25:60–6. [PubMed] [Google Scholar]
- 116.Avouac J, Juin F, Wipff J, et al. Circulating endothelial progenitor cells in systemic sclerosis: association with disease severity. Ann Rheum Dis. 2008;67:1455–60. doi: 10.1136/ard.2007.082131. [DOI] [PubMed] [Google Scholar]
- 117.Del Papa N, Quirici N, Soligo D, et al. Bone marrow endothelial progenitors are defective in systemic sclerosis. Arthritis Rheum. 2006;54:2605–15. doi: 10.1002/art.22035. [DOI] [PubMed] [Google Scholar]
- 118.Kuwana M, Kaburaki J, Okazaki Y, et al. Increase in circulating endothelial precursors by atorvastatin in patients with systemic sclerosis. Arthritis Rheum. 2006;54:1946–51. doi: 10.1002/art.21899. [DOI] [PubMed] [Google Scholar]
- 119.Kuwana M, Okazaki Y, Kaburaki J. Long-term beneficial effects of statins on vascular manifestations in patients with systemic sclerosis. Mod Rheumatol. 2009;19:530–5. doi: 10.1007/s10165-009-0199-4. [DOI] [PubMed] [Google Scholar]
- 120.Del Papa N, Cortiana M, Vitali C, et al. Simvastatin reduces endothelial activation and damage but is partially ineffective in inducing endothelial repair in systemic sclerosis. J Rheumatol. 2008;35:1323–8. [PubMed] [Google Scholar]
- 121.Avouac J, Wipff J, Goldman O, et al. Angiogenesis in systemic sclerosis: impaired expression of vascular endothelial growth factor receptor 1 in endothelial progenitor-derived cells under hypoxic conditions. Arthritis Rheum. 2008;58:3550–61. doi: 10.1002/art.23968. [DOI] [PubMed] [Google Scholar]
- 122.Hao X, Silva EA, Mansson-Broberg A, et al. Angiogenic effects of sequential release of VEGF-A165 and PDGF-BB with alginate hydrogels after myocardial infarction. Cardiovasc Res. 2007;75:178–85. doi: 10.1016/j.cardiores.2007.03.028. [DOI] [PubMed] [Google Scholar]
- 123.Chen RR, Silva EA, Yuen WW, et al. Integrated approach to designing growth factor delivery systems. FASEB J. 2007;21:3896–903. doi: 10.1096/fj.06-7873com. [DOI] [PubMed] [Google Scholar]
- 124.Zisch AH, Lutolf MP, Ehrbar M, et al. Cell-demanded release of VEGF from synthetic, biointeractive cell-ingrowth matrices for vascularized tissue growth. FASEB J. 2003;17:2260–2. doi: 10.1096/fj.02-1041fje. [DOI] [PubMed] [Google Scholar]
- 125.Popa ER, van der Strate BW, Brouwer LA, et al. Dependence of neovascularization mechanisms on the molecular microenvironment. Tissue Eng. 2007;13:2913–21. doi: 10.1089/ten.2007.0031. [DOI] [PubMed] [Google Scholar]
- 126.Maurer B, Busch N, Jüngel A, et al. Transcription factor Fos-related antigen-2 induces progressive peripheral vasculopathy in mice closely resembling human systemic sclerosis. Circulation. 2009;120:2367–76. doi: 10.1161/CIRCULATIONAHA.109.855114. [DOI] [PubMed] [Google Scholar]