

Abstract
Pancreatic ductal adenocarcinoma (PDAC) is a dismal disease with a median survival below 6 months and a 5-year survival rate below 1%. Effective therapies for locally advanced or metastatic tumours are missing and curatively resected patients relapse in over 80% of the cases. Although histone deacetylases (HDACs) are involved in the control of proliferation, apoptosis, differentiation, migration and angiogenesis of cancer cells, knowledge about the expression patterns and functions of individual HDAC isoenzymes in pancreatic cancer is sparse. This review summarizes the roles of HDACs as novel therapeutic targets and the molecular mode of action of HDAC-inhibitors (HDACI) in PDACs. Success of HDACI in clinical settings will depend on an increased knowledge of HDAC functions as well as on a better understanding of the mode of action of HDACI. Pre-clinical experimental data that constitute the basis for rational therapeutic strategies to treat PDAC are described here. Translating these rational-based therapies into the clinic will finally increase our chance to establish an effective HDACI-containing combination therapy effective against PDAC.
Keywords: pancreatic cancer, therapy, apoptosis, cell cycle, HDAC, HDAC inhibitors
Introduction
In the last decades, conventional chemotherapy has become one of the major medical intervention strategies for certain malignancies. The introduction of targeted molecular therapies directed against cancer-specific molecules and signalling pathways has further increased therapy responses and survival rates of patients with solid and haematological malignancies. However, one prominent exception is pancreatic ductal adenocarcinoma (PDAC), where 5-year survival rates are below 1% and effective conservative therapies are missing [1, 2]. ‘Biologicals’, e.g. the epidermal growth factor receptor inhibitor erlotinib, are only effective in subsets of PDAC patients [3]. Therefore, there is the need to develop new concepts for the treatment of PDAC. Targeting histone deacetylases (HDACs) could be a promising approach. However, functions of HDAC isoenzymes in PDAC and rationally based combination therapies still have to be identified for successful applications of HDAC inhibitors (HDACI) in the clinic. Since a recent phase II study revealed no advantage of combining gemcitabine with the HDACI CI-994 in patients with advanced PDAC, alternative HDACI-based combinations should be considered [4]. This review recapitulates the current knowledge on molecular functions and actions of HDACs and HDACI in PDACs.
The HDAC family
According to phylogenetic analyses and sequence homology, deacetylases can be grouped in class I to IV enzymes (Fig. 1). The yeast Rpd3 homologues HDACs 1, 2, 3 and 8 represent class I and the yeast Hda1 homologous enzymes HDACs 4, 5, 6, 7, 9 and 10 represent class II HDACs. Class II HDACs are subdivided according to the presence of one or two catalytical domains. HDACs 4, 5, 7 and 9 harbour one catalytically active site and are grouped into class IIa in contrast to class IIb, comprising HDACs 6 and 10, containing two catalytic domains (Fig. 1). HDAC11 shares homology with class I as well as class II HDACs and is grouped in class IV. Apart from HDAC3, class I HDACs primarily localize to the nucleus, whereas class II enzymes shuttle into the nucleus upon specific stimulation. In contrast to the zinc-dependent catalysis of class I, II and IV enzymes, the class III deacetylases (SIRT1–7), homologues of the yeast SIR2 enzyme, use NAD+ as co-factor [5, 6]. Since class III enzymes are not inhibited by HDACI currently used in clinical trials and SIRT deacetylases are poorly investigated in PDAC, we will focus on class I, II and IV HDACs. Nevertheless, since (I) the contribution of SIRT to other solid tumours is documented, (II) SIRT1 negatively regulates important molecules like the tumour-suppressor p53 [7] and (III) SIRT inhibitors reduce the viability of PDAC cells [8], SIRT enzymes as well as SIRT inhibitors should be analysed in PDAC in molecular detail in the future.
Fig 1.
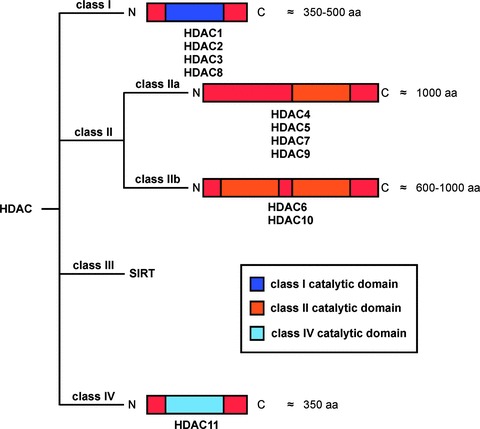
The HDAC family. HDACs can be classified according to their homology in the catalytic domain into class I (HDAC1, 2, 3 and 8), class II (HDAC4, 5, 6, 7, 9 and 10) and class IV (HDAC11) enzymes. Class II is subdivided depending onto the presence of one (class IIa) or two (class IIb) catalytic domains. The NAD±-dependent sirtuin protein deacetylases, SIRT1–7, represent class III. aa: amino acids.
The first identified substrates of HDACs were the histones. HDACs deacetylate the ɛ-amino group of lysines located at the N-terminal tail of histones, which leads to a repressive chromatin formation (heterochromatin) and the suppression of gene expression [5, 6]. In contrast, histone acetyl transferases (HATs) counteract histone deacetylation, which generates an open chromatin structure (euchromatin), enabling transcription factors to activate their target genes. Among other post-translational modifications, reversible acetylation of histones contributes to a ‘histone code’. For example, acetylation of lysine 9 of histone H3 is a mark of active transcription [9].
Considering phylogenetic analyses, which demonstrate that classical HDACs precede the evolution of histone proteins, it is not surprising that a continuously growing number of non-histone substrates of HDACs and HATs are described [9, 10]. Many of these proteins are transcription factors, such as p53, NF-κB and STATs, and therefore changes in the transcriptome upon HDACI treatment can be due to a direct modulation of the ‘histone code’ or the consequence of an indirect modulation of transcription factor activities [9–11] (Fig. 2). HDACs function in multiprotein complexes containing co-repressors and co-activators. Since HDACs are involved in the control of proliferation, apoptosis, differentiation, migration and angiogenesis in cancer [12] (Fig. 2), they represent attractive therapeutic targets. In 2006, the HDACI suberoylanilide hydroxamic acid (SAHA) was approved by the FDA for the treatment of cutaneous T-cell lymphoma [13, 14].
Fig 2.

HDAC functions and responses in cancer cells. In addition to the deacetylation of histones, HDACs can deacetylate various other proteins. These proteins are often transcription factors. Therefore, HDACs can regulate gene expression by the modulation of chromatin condensation (histone code) and the regulation of transcription factor activity. HDAC-dependent changes in the transcriptome mediate several biological HDAC effects, which are described in the figure.
HDAC inhibitors
Several natural and synthetic compounds are currently known to inhibit HDACs [15, 16]. Since HDACI do not inhibit all HDAC isoforms to the same extent, these agents can be grouped into pan- and class I-specific inhibitors [16]. Hydroxamic acids, for example TSA, SAHA, NVP-LAQ824, NVP-LBH589, CBHA, pyroxamic acid, PXD-101 and CRA-026440, are pan-HDACI targeting class I, II and IV HDACs in the nanomolar range [13, 14]. In contrast, the carboxylic acids valproic acid (VPA) and sodium butyrate, and the benzamides MS-27–275, CI-994 and MGCD0103, or the cyclic tetrapeptides trapoxin, depsipeptide (FK228) and spiruchostatin A are rather class I-specific HDACI [17]. The main side effects of HDACI are fatigue, nausea, dehydration, diarrhoea, thrombocytopenia as well as QT time prolongations and other ECG abnormalities. Currently, at least 15 different HDACI are used in clinical trials as part of mono- or combination therapies.
HDAC expression in PDAC
Although overexpression of class I HDACs is a common feature of solid tumours, systematic analysis of HDAC expression in larger PDAC cohorts have not been reported yet [11, 18]. 56% of PDACs show positive immunohistochemical staining for HDAC1, and the coexpression of HDAC1 and HIF-1α remarkably correlates with poor prognosis [19]. Using tissue microarrays, we recently detected overexpression of HDAC2, especially in moderately differentiated (G2) and undifferentiated (G3) PDACs [20]. In addition, increased expression of HDAC7 has recently been demonstrated in 11 well to moderately differentiated PDACs [21]. These scanty data hint to a role for HDAC isoform overexpression in PDAC. However, the investigation of HDAC expression in PDACs in more detail is necessary, especially in larger cohorts and in correlation with clinical and prognostic parameters.
HDACs control proliferation of PDAC cells
Many studies concerning HDAC functions in PDAC used HDACI targeting several HDAC isoenzymes. Therefore, little is known about non-redundant isoenzyme-specific molecular functions of HDACs in this disease (see Table 1). In 2003, the first systematic analysis describing the response of PDAC cell lines towards the pan-HDACI TSA was published [22]. Donadelli et al. observed reduced growth of PDAC cells and an IC50 in a range between 30 to 170 nM TSA was observed. Furthermore, it was demonstrated that TSA treatment results in impaired proliferation due to an arrest in the G2 phase of the cell cycle and the induction of the caspase-dependent programmed cell death pathway [22]. At the molecular level an increase of the cyclin-dependent kinase inhibitor (CDKI) p21Cip1/Waf1 at the mRNA and protein level was observed. The retinoblastoma (pRB) protein tumour suppressor can determine whether p21Cip1/Waf1 affects G1/S or G2/M phase progression [23]. Since pRB protein is functionally inactivated in PDACs [2], up-regulation of p21Cip1/Waf1 could causally determine the G2 arrest induced with TSA in such cells (Fig. 3). The ability of HDACI to inhibit the cell cycle in G2 and to up-regulate p21Cip1/Waf1 in PDAC cells was equally found for the HDACI NVP-LAQ824 or NVP-LBH589 [24], SAHA [25] and FR901228 [26]. These data argue for general mechanisms by which HDACI induce cell cycle arrest in PDACs. In addition, reduced expression of cyclin B1 [25, 27] and accumulation of the CDKI p27Kip1[28] were observed after the treatment of PDAC cells with HDACI. Nonetheless, SAHA-dependent induction of G1 arrest was observed in BxPc3 and Colo357 PDAC cells. Hence, cell type-specific regulatory circuits determine biological responses induced by HDACI [29].
Table 1.
Function and expression of individual HDACs in PDAC
| HDAC | Function/expression | References |
|---|---|---|
| HDAC1 | • Co-expression of HIF-1α and HDAC1 correlates with poor prognosis | [19] |
| • Included in a SNAIL recruited repressor complex that controls E-cadherin expression, EMT and metastasis | [54] | |
| HDAC2 | • Overexpressed, especially in G2 (moderately-) and G3 (un-) differentiated PDAC | [20] |
| • Mediates resistance towards DNA-damage induced apoptosis by controlling expression of the pro-apoptotic BH3-only protein NOXA | [20] | |
| • Included in a SNAIL recruited repressor complex that controls E-cadherin expression, EMT and metastasis | [54] | |
| HDAC6 | • Contributes to aggresome formation and reduces efficiency of proteasome inhibitors | [49] |
| HDAC7 | • Overexpressed in PDAC | [21] |
Fig 3.
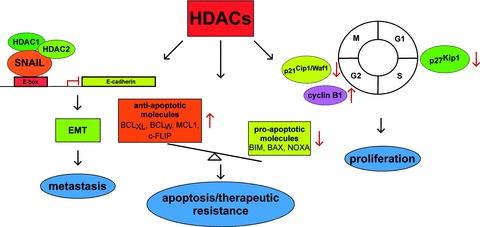
Characterized pathways engaged by HDACs in PDAC. Three molecular well characterized HDAC controlled processes in PDAC are illustrated. Left part: A HDAC1, 2 containing repressor complex is recruited to the E-box of the E-cadherin promoter by the transcription factor SNAIL, contributing to EMT and metastasis. Middle part: HDACs contribute to the imbalanced expression of anti-apoptotic (BCLXL, BCLw, MCL1, c-Flip) and pro-apoptotic (BIM, BAX, NOXA) genes, contributing to apop totic and therapeutic resistance of PDAC cells. Right part: HDACs control expression of the CDKI p21Cip1/Waf1 and cyclin B1 to control G2/M-phase or the CDKI p27Kip1 to control G1/S-phase of the cell cycle.
Class I selective inhibitors show different biological effects. Although Capan1 cells responded to MS-27–275 [30] and BxPc3 cells to MGCD0103 [31], which both are class I HDAC-specific inhibitors [9], the IC50s for both compounds were relatively high and the exact impact towards cell cycle progression of PDAC cells is unclear. Consistently, we observed no reduced proliferative capacity of PDAC cells treated with the HDACI VPA, which when applied in clinically relevant doses, selectively inhibits class I HDACs [9, 20, 32]. Furthermore, the influence of the rather class I selective HDACI butyrate towards cell cycle distribution and proliferation was rather marginal, when used in concentrations up to 2 mM [33, 34]. These results indicate that class II HDACs contribute to cell cycle progression of PDAC cells. Recently, HDAC4 was shown to suppress p21Cip1/Waf1 in ovarian carcinoma cells, cervical cancer cells, glioblastoma cells and breast cancer cells in a non-redundant fashion [35]. Since some rather class I-selective HDACI, like SK-7041 [36] and FR901228 [37] also target HDAC4 at higher concentrations and both HDACI induce up-regulation of p21Cip1/Waf1 expression in PDAC cells [26, 27], HDAC4 might contribute to cell cycle regulation in PDAC. This speculation surely awaits further experimental validation in PDAC models and redundant contribution of several HDACs towards cell cycle progression has obviously to be considered.
Molecular effects observed after the treatment of PDAC cells with various HDACI are summarized in Table 2.
Table 2.
Molecular action of HDACI in PDAC cells
| HDACI | Molecular action in PDAC cells | References |
|---|---|---|
| TSA | • G2/M-phase arrest with up-regulation of p21Cip1/Waf1 and down-regulation of cyclin B1 | [22, 27] |
| (Hydroxamic acid; pan-HDACI) | • Caspase-dependent apoptosis with up-regulation of BIM and down-regulation of BCLXL, BCLW and MCL1 | [22, 27, 40, 41] |
| • Increased p27Kip1 expression | [28] | |
| • Synergizes with gemcitabine, oxaliplatin, CPT11, gefitinib and bortezomib in vitro | [39, 41, 46, 48] | |
| • Synergizes with gemcitabine in a s.c. nude mouse T3M4 cell xenotransplant model | [41] | |
| • Restores E-cadherin expression in mesenchymal pancreatic cancer cells | [54] | |
| SAHA | • G2/M-phase arrest with up-regulation of p21Cip1/Waf1 and down-regulation of cyclin B1 | [25] |
| (Hydroxamic acid; pan-HDACI) | • Synergizes with bortezomib in vitro and in an orthotopic xenotransplant model using L3.6pl cells in vivo | [49] |
| • induction of apoptosis | [25, 49] | |
| • Synergizes with gemcitabine and the smoothend antagonist SANT-1 in vitro | [29, 52] | |
| NVP-LBH589 | • G2/M-phase arrest with up-regulation of p21Cip1/Waf1 and down-regulation of cyclin B1 | [24] |
| (Hydroxamic acid; pan-HDACI) | • induction of apoptosis | [24] |
| • combination with gemcitabine more efficient than each alone in a s.c. nude mouse model using HPAF-2 and L3.6pl cells | [24] | |
| FR901228 | • G2/M-phase arrest with up-regulation of p21Cip1/Waf1 | [26] |
| Depsipeptide (tetrapeptide; class I-selective) | • induction of caspase-dependent apoptosis and down-regulation of survivin | [26] |
| SK-7041 | • G2/M-phase arrest with up-regulation of p21Cip1/Waf1 and down-regulation of cyclin B1 | [27] |
| (hybrid from hydroxamic acid and pyridyl ring of MS-275; class I-selective) | • induction of apoptosis | [27] |
| • down-regulation of MCL1 and BCLXL | [27] | |
| Butyrate | • Synergizes with gemcitabine in vitro | [34] |
| (carboxylic acid; low doses rather class I-selective) | • Sensitizes towards FAS-mediated apoptosis | [33] |
| • down-regulation of BCLXL | [33] | |
| VPA | • Synergizes with etoposide to induce caspase-dependent apoptosis in vitro | [20] |
| (carboxylic acid; low doses rather class I-selective) | • Restores E-cadherin expression in mesenchymal pancreatic cancer cells | [54] |
HDACs control apoptosis and mediate therapeutic resistance
Although PDAC cells are characterized by profound apoptosis resistance [38], pan-HDACI efficiently induce caspase-dependent apoptosis [22, 27, 39]. In nine PDAC cell lines apoptosis induced by TSA correlates with increased mRNA expression of the pro-apoptotic BH3-only protein BIM, an initiator of the mitochondrial cell death pathway, together with attenuation of the anti-apoptotic BCL2 family members BCLXL and BCLW[40] (Fig. 3). Using siRNA approaches targeting BIM, the contribution of BIM towards TSA-induced apoptosis was validated [41]. In addition to BCLXL and BCLW, the expression of the anti-apoptotic protein MCL1 was decreased upon treatment with the HDACI SK-7041 [27].
In contrast to the activation of the cell death pathway by pan-HDACI, low doses of VPA (<1.5 mM) or butyrate (<2 mM) negligibly induce apoptosis [20, 33, 34]. In addition, caspase-independent apoptosis is induced by treatment with the pan-HDACI TSA in the PDAC cell lines IMIM-PC1, IMIM-PC2 and RWP-1 [42]. This process correlates with an initially increased expression of the multidomain pro-apoptotic BCL2 family member BAX and subsequent release of apoptosis-inducing factor (AIF) and OMI/HTR-A2 from mitochondria. Although translocation of AIF into the nucleus and dependency of TSA-induced apoptosis on the serine-protease activity of OMI/HTR-A2 could be demonstrated, it is presently unclear whether caspase-independent apoptosis is a specific feature of the cell lines investigated. Furthermore, considering that the IC50 of most PDAC cell lines for TSA ranges below 200 nM [22], a high dose of 1 μM TSA used in the mentioned study might have contributed to cell death different from caspase-dependent apoptosis. In addition, functional genetics using RNA interference was not used to prove contribution of AIF and OMI/HTR-A2. Whether other forms of cell deaths, like autophagy, are induced upon HDACI treatment of PDAC cells is presently unclear but possible.
Although HDACI demonstrate certain single agent activity against haematological malignancies, efficient combination therapies are clearly required for the treatment of solid tumours [13, 43–45]. In a systematic study, the pan-HDACI TSA was applied in combination with several currently used chemotherapeutics to 10 PDAC cell lines [46]. With the exception of 5-flourouracil (5-FU), TSA cooperated positively with current standard therapeutics, especially with CPT11 [46]. Consistently, a combination of TSA [39, 41], SAHA [29], NVP-LBH589 [24] or sodium butyrate [34] with gemcitabine-induced apoptosis in PDAC cell lines more efficiently in vitro than either agent on its own. In vivo, a TSA/gemcitabine combination also proved efficacy in a subcutaneous (s.c.) xenotransplantation model using the T3M4 PDAC cell line [41]. Furthermore, an NVP-LBH589/gemcitabine combination demonstrated some efficiency in s.c. xenotransplant models using HPAF-2 PDAC cells (reduction in tumour volume versus controls: gemcitabine 52%, NVP-LBH589 73%, combination 79%) and L3.6pl PDAC cells (reduction in tumour volume versus controls: gemcitabine 31%, NVP-LBH589 78%, combination 85%) [24]. Considering the limited predictive potential of s.c. xenotransplants [47] and the negative results of the recent HDACI (CI-994)/gemcitabine phase II study in PDAC patients [4], there are some doubts whether gemcitabine is the right compound for combinatorial approaches with HDACI in PDAC. Keeping in mind that CI-994 is a weak HDACI, one cannot exclude that a combination of a potent HDACI, like SAHA or NVP-LBH589, with gemcitabine is effective in patients with PDAC. Here, the definite clarification awaits further clinical trials. Nevertheless, alternative rationally based treatment strategies using HDACI must be considered.
We could recently reveal that inhibition of class I HDACs with VPA distinctly synergizes with the topoisomerase II inhibitor etoposide, but not with gemcitabine, oxaliplatin or 5-FU, to induce apoptosis in PDAC cells. We further characterized this effect as a non-redundant, HDAC2-dependent function. At the molecular level, HDAC2 inhibition opens the locus of the epigenetically silenced NOXA gene, a BH3-only protein and apical initiator of apoptosis [20]. Consistent with this, HDACI efficiently combine with other classical NOXA activating agents like UV-light or proteasome inhibition in PDAC cells [48, 49 and unpublished data]. The observation that a class I HDACI (VPA)/topoisomerase II inhibitor combination efficiently induces apoptosis of PDAC cells, appears important, since a phase I VPA/topoisomerase II inhibitor (epirubicin) trial was already conducted in patients with solid tumours [50]. In this study, a remarkable degree of antitumour activity was observed in a pre-treated patient population, which had already undergone a median of three prior treatment regimes. Interestingly, one partial response was also demonstrated in a patient with PDAC in this trial [50].
In addition, recent work revealed a contribution of HDAC6 towards therapeutic resistance of PDAC cells, especially against proteasome inhibitors, like bortezomib [49]. The cytoplasmatic and cytoskeleton-associated HDAC6 plays an important role in the proteolysis pathway of misfolded proteins and deacetylates proteins like α-tubulin or HSP90 [44]. Bortezomib induces ER stress signalling to activate the mitochondrial apoptosis pathway. Electron-dense structures, the so-called aggresomes, occur upon bortezomib treatment of PDAC cells. Aggregated, ubiquitylated proteins are sequestered in aggresomes for lysosomal degradation, which attenuate ER stress. It was demonstrated that HDAC6 contributes to aggresome formation and HDACI profoundly synergize with bortezomib in PDAC cells to induce apoptosis in vitro[49]. Furthermore, distinct efficacy of a bortezomib/SAHA combination was demonstrated in an orthotopic xenotransplantation model of L3.6pl PDAC cells [49]. This rationally based strategy is now translated into a clinical phase I trial in patients with PDAC using the proteasome Inhibitor NPI-0052 and SAHA (http://www.clinicaltrials.gov).
Beyond targeting the intrinsic apoptotic machinery, HDACs can modulate the extrinsic death receptor pathways of apoptosis [44, 51]. In line with this, sodium butyrate sensitizes PDAC cells to FAS-induced apoptosis correlating with a decrease of the anti-apoptotic proteins c-FLIP and BCLXL[33]. Considering the tumour-selective activity of TRAIL and the availability of agonistic TRAIL receptor antibodies, the modulation of TRAIL sensitivity by HDACs in PDACs should be investigated in more detail.
Other potential combination partners for HDACI to treat PDACs are Smoothened (Smo) antagonists, which interfere with the Hedgehog pathway [52]. Hedgehog signalling is involved in the initiation and progression of PDAC. Furthermore, NF-κB mediated Sonic hedgehog transcription contributes to apoptotic resistance of PDAC cells [53]. Indeed, a combination of SAHA with the Smo antagonist SANT-1 was demonstrated to evoke caspase-dependent apoptosis of PDAC cells [52]. Therapeutic resistance in PDACs is a combination of cell intrinsic and extrinsic resistance. Cell intrinsic therapeutic resistance is due to a fundamental change in gene expression in PDAC cells, resulting in a transcriptome favouring survival of PDAC upon therapeutic stress. For example, there is a distinct change in the expression of pro- and anti-apoptotic molecules in PDAC leading to an increased threshold for conventional chemo- and radiotherapy-induced cell death [38]. In addition to cell intrinsic resistance, recent work by the Tuveson lab demonstrated that cell extrinsic resistance contributes to therapeutic failure in PDAC [47]. In a genetically engineered KrasG12D-dependent mouse model of PDAC, which recapitulates many aspects of the human disease, it was demonstrated that PDAC is characterized (I) by a very low blood vessel density and (II) by an exceptional stromal matrix (desmoplasia) embedding the blood vessels [47]. The authors nicely demonstrated that both factors contribute to impaired drug delivery and therapeutic failure. Furthermore, it was shown demonstrated that the Smo antagonists IPI-926 reduces desmoplasia, increases vessel density and therefore therapeutic efficiency of conventional chemotherapy [47]. Since Smo antagonists target PDAC cell intrinsic [52] as well as extrinsic therapeutic resistance [47] it would be important to demonstrate whether a HDACI/Smo antagonist combination is effective in relevant genetically engineered murine endogenous PDAC models.
HDACs, EMT and metastasis of PDAC
In addition to the clear contribution of HDACs towards the proliferation, apoptosis and therapeutic resistance, HDAC activity equally contributes to PDAC metastasis in vivo[54]. By serial in vivo passaging of parental pancreatic cancer cells with low metastatic potential we selected for cells with high metastatic potential. Molecular analysis of such cell lines revealed the induction of an epithelial–mesenchymal transition (EMT) constituting an early step of metastasis [55]. EMT correlates with a loss of E-cadherin expression due to epigenetic silencing by a transcriptional repressor complex containing the transcription factor SNAIL acting in concert with HDAC1 and HDAC2 (Fig. 3) [54]. Since signs of EMT with loss of E-cadherin expression were also observed in a genetically engineered KrasG12D-dependent murine PDAC model and HDAC activity was necessary for the silencing of E-cadherin in murine and human models of EMT, HDACI might be a tailored approach for interference with PDAC metastasis [54].
Concluding remarks
Well characterized molecular pathways that HDACs engage in PDACs are summarized in Fig. 3. Inhibiting these enzymes is a promising approach for the treatment of cancers, especially in rationally and molecularly defined combination schedules. Considering the inefficiency of current therapies for PDAC and the negative outcome of many large gemcitabine-based phase III studies [1], HDACI should not be refused as a therapeutic approach for PDAC, solely because gemcitabine/HDACI doublets fail [4]. Instead, we should characterize HDAC functions in PDAC at the molecular level, define biomarkers for HDACI-responsiveness, discover efficient combinatorial therapies and decipher their molecular mode of action. Translating such knowledge in genetically defined animal models of PDACs and ultimately into the clinic would be great steps ahead.
Acknowledgments
We are supported by Deutsche Forschungsgemeinschaft (Grant SCHN 959/1–2), SFB456, SFB824, Else Kröner-Fresenius-Stiftung, Fritz-Thyssen Stiftung, Bayerische Forschungsstiftung and Deutsche Krebshilfe. We apologize for not citing any relevant reports due to lack of space, the need to selectively choose examples or an oversight on our part.
Conflict of interest
Nothing to declare.
References
- 1.Eckel F, Schneider G, Schmid RM. Pancreatic cancer: a review of recent advances. Expert opinion on investigational drugs. 2006;15:1395–410. doi: 10.1517/13543784.15.11.1395. [DOI] [PubMed] [Google Scholar]
- 2.Schneider G, Siveke JT, Eckel F, et al. Pancreatic cancer: basic and clinical aspects. Gastroenterology. 2005;128:1606–25. doi: 10.1053/j.gastro.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 3.Moore MJ, Goldstein D, Hamm J, et al. Erlotinib plus gemcitabine compared with gemcitabine alone in patients with advanced pancreatic cancer: a phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J Clin Oncol. 2007;25:1960–6. doi: 10.1200/JCO.2006.07.9525. [DOI] [PubMed] [Google Scholar]
- 4.Richards DA, Boehm KA, Waterhouse DM, et al. Gemcitabine plus CI-994 offers no advantage over gemcitabine alone in the treatment of patients with advanced pancreatic cancer: results of a phase II randomized, double-blind, placebo-controlled, multicenter study. Ann Oncol. 2006;17:1096–102. doi: 10.1093/annonc/mdl081. [DOI] [PubMed] [Google Scholar]
- 5.Yang XJ, Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat Rev Mol Cell Biol. 2008;9:206–18. doi: 10.1038/nrm2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet. 2009;10:32–42. doi: 10.1038/nrg2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luo J, Nikolaev AY, Imai S, et al. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–48. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 8.Ouaissi M, Cabral S, Tavares J, et al. Histone deacetylase (HDAC) encoding gene expression in pancreatic cancer cell lines and cell sensitivity to HDAC inhibitors. Cancer Biol Ther. 2008;7:523–31. doi: 10.4161/cbt.7.4.5480. [DOI] [PubMed] [Google Scholar]
- 9.Spange S, Wagner T, Heinzel T, et al. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int J Biochem Cell Biol. 2009;41:185–98. doi: 10.1016/j.biocel.2008.08.027. [DOI] [PubMed] [Google Scholar]
- 10.Buchwald M, Krämer OH, Heinzel T. HDACi–targets beyond chromatin. Cancer Lett. 2009;280:160–7. doi: 10.1016/j.canlet.2009.02.028. [DOI] [PubMed] [Google Scholar]
- 11.Witt O, Deubzer HE, Milde T, et al. HDAC family: what are the cancer relevant targets. Cancer Lett. 2009;277:8–21. doi: 10.1016/j.canlet.2008.08.016. [DOI] [PubMed] [Google Scholar]
- 12.Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene. 2007;26:5420–32. doi: 10.1038/sj.onc.1210610. [DOI] [PubMed] [Google Scholar]
- 13.Marks PA. Discovery and development of SAHA as an anticancer agent. Oncogene. 2007;26:1351–6. doi: 10.1038/sj.onc.1210204. [DOI] [PubMed] [Google Scholar]
- 14.Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26:5541–52. doi: 10.1038/sj.onc.1210620. [DOI] [PubMed] [Google Scholar]
- 15.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–84. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 16.Balasubramanian S, Verner E, Buggy JJ. Isoform-specific histone deacetylase inhibitors: the next step. Cancer Lett. 2009;280:211–21. doi: 10.1016/j.canlet.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 17.Marks PA, Xu WS. Histone deacetylase inhibitors: potential in cancer therapy. J Cell Biochem. 2009;107:600–8. doi: 10.1002/jcb.22185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weichert W. HDAC expression and clinical prognosis in human malignancies. Cancer Lett. 2009;280:168–76. doi: 10.1016/j.canlet.2008.10.047. [DOI] [PubMed] [Google Scholar]
- 19.Miyake K, Yoshizumi T, Imura S, et al. Expression of hypoxia-inducible factor-1alpha, histone deacetylase 1, and metastasis-associated protein 1 in pancreatic carcinoma: correlation with poor prognosis with possible regulation. Pancreas. 2008;36:e1–9. doi: 10.1097/MPA.0b013e31815f2c2a. [DOI] [PubMed] [Google Scholar]
- 20.Fritsche P, Seidler B, Schüler S, et al. HDAC2 mediates therapeutic resistance of pancreatic cancer cells via the BH3-only protein NOXA. Gut. 2009;58:1399–409. doi: 10.1136/gut.2009.180711. [DOI] [PubMed] [Google Scholar]
- 21.Ouaissi M, Sielezneff I, Silvestre R, et al. High histone deacetylase 7 (HDAC7) expression is significantly associated with adenocarcinomas of the pancreas. Ann Surg Oncol. 2008;15:2318–28. doi: 10.1245/s10434-008-9940-z. [DOI] [PubMed] [Google Scholar]
- 22.Donadelli M, Costanzo C, Faggioli L, et al. Trichostatin A, an inhibitor of histone deacetylases, strongly suppresses growth of pancreatic adenocarcinoma cells. Mol Carcinog. 2003;38:59–69. doi: 10.1002/mc.10145. [DOI] [PubMed] [Google Scholar]
- 23.Niculescu AB, 3rd, Chen X, Smeets M, et al. Effects of p21(Cip1/Waf1) at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol Cell Biol. 1998;18:629–43. doi: 10.1128/mcb.18.1.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haefner M, Bluethner T, Niederhagen M, et al. Experimental treatment of pancreatic cancer with two novel histone deacetylase inhibitors. World J Gastroenterol. 2008;14:3681–92. doi: 10.3748/wjg.14.3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kumagai T, Wakimoto N, Yin D, et al. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (Vorinostat, SAHA) profoundly inhibits the growth of human pancreatic cancer cells. Int J Cancer. 2007;121:656–65. doi: 10.1002/ijc.22558. [DOI] [PubMed] [Google Scholar]
- 26.Sato N, Ohta T, Kitagawa H, et al. FR901228, a novel histone deacetylase inhibitor, induces cell cycle arrest and subsequent apoptosis in refractory human pancreatic cancer cells. Int J Oncol. 2004;24:679–85. [PubMed] [Google Scholar]
- 27.Ryu JK, Lee WJ, Lee KH, et al. SK-7041, a new histone deacetylase inhibitor, induces G2-M cell cycle arrest and apoptosis in pancreatic cancer cell lines. Cancer Lett. 2006;237:143–54. doi: 10.1016/j.canlet.2005.05.040. [DOI] [PubMed] [Google Scholar]
- 28.Schneider G, Reichert M, Saur D, et al. HDAC3 is linked to cell cycle machinery in MiaPaCa2 cells by regulating transcription of skp2. Cell Prolif. 2007;40:522–31. doi: 10.1111/j.1365-2184.2007.00454.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arnold NB, Arkus N, Gunn J, et al. The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces growth inhibition and enhances gemcitabine-induced cell death in pancreatic cancer. Clin Cancer Res. 2007;13:18–26. doi: 10.1158/1078-0432.CCR-06-0914. [DOI] [PubMed] [Google Scholar]
- 30.Saito A, Yamashita T, Mariko Y, et al. A synthetic inhibitor of histone deacetylase, MS-27–275, with marked in vivo antitumor activity against human tumors. Proc Natl Acad Sci USA. 1999;96:4592–7. doi: 10.1073/pnas.96.8.4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fournel M, Bonfils C, Hou Y, et al. MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol Cancer Ther. 2008;7:759–68. doi: 10.1158/1535-7163.MCT-07-2026. [DOI] [PubMed] [Google Scholar]
- 32.Krämer OH, Knauer SK, Zimmermann D, et al. Histone deacetylase inhibitors and hydroxyurea modulate the cell cycle and cooperatively induce apoptosis. Oncogene. 2008;27:732–40. doi: 10.1038/sj.onc.1210677. [DOI] [PubMed] [Google Scholar]
- 33.Natoni F, Diolordi L, Santoni C, et al. Sodium butyrate sensitises human pancreatic cancer cells to both the intrinsic and the extrinsic apoptotic pathways. Biochim Biophys Acta. 2005;1745:318–29. doi: 10.1016/j.bbamcr.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 34.Ammerpohl O, Trauzold A, Schniewind B, et al. Complementary effects of HDAC inhibitor 4-PB on gap junction communication and cellular export mechanisms support restoration of chemosensitivity of PDAC cells. Br J Cancer. 2007;96:73–81. doi: 10.1038/sj.bjc.6603511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mottet D, Pirotte S, Lamour V, et al. HDAC4 represses p21(WAF1/Cip1) expression in human cancer cells through a Sp1-dependent, p53-independent mechanism. Oncogene. 2009;28:243–56. doi: 10.1038/onc.2008.371. [DOI] [PubMed] [Google Scholar]
- 36.Park JH, Jung Y, Kim TY, et al. Class I histone deacetylase-selective novel synthetic inhibitors potently inhibit human tumor proliferation. Clin Cancer Res. 2004;10:5271–81. doi: 10.1158/1078-0432.CCR-03-0709. [DOI] [PubMed] [Google Scholar]
- 37.Furumai R, Matsuyama A, Kobashi N, et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Cancer Res. 2002;62:4916–21. [PubMed] [Google Scholar]
- 38.Hamacher R, Schmid RM, Saur D, et al. Apoptotic pathways in pancreatic ductal adenocarcinoma. Mol Cancer. 2008;7:64. doi: 10.1186/1476-4598-7-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gahr S, Ocker M, Ganslmayer M, et al. The combination of the histone-deacetylase inhibitor trichostatin A and gemcitabine induces inhibition of proliferation and increased apoptosis in pancreatic carcinoma cells. Int J Oncol. 2007;31:567–76. [PubMed] [Google Scholar]
- 40.Moore PS, Barbi S, Donadelli M, et al. Gene expression profiling after treatment with the histone deacetylase inhibitor trichostatin A reveals altered expression of both pro- and anti-apoptotic genes in pancreatic adenocarcinoma cells. Biochim Biophys Acta. 2004;1693:167–76. doi: 10.1016/j.bbamcr.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 41.Donadelli M, Costanzo C, Beghelli S, et al. Synergistic inhibition of pancreatic adenocarcinoma cell growth by trichostatin A and gemcitabine. Biochim Biophys Acta. 2007;1773:1095–106. doi: 10.1016/j.bbamcr.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 42.Garcia-Morales P, Gomez-Martinez A, Carrato A, et al. Histone deacetylase inhibitors induced caspase-independent apoptosis in human pancreatic adenocarcinoma cell lines. Mol Cancer Ther. 2005;4:1222–30. doi: 10.1158/1535-7163.MCT-04-0186. [DOI] [PubMed] [Google Scholar]
- 43.Bug G, Ritter M, Wassmann B, et al. Clinical trial of valproic acid and all-trans retinoic acid in patients with poor-risk acute myeloid leukemia. Cancer. 2005;104:2717–25. doi: 10.1002/cncr.21589. [DOI] [PubMed] [Google Scholar]
- 44.Bots M, Johnstone RW. Rational combinations using HDAC inhibitors. Clin Cancer Res. 2009;15:3970–7. doi: 10.1158/1078-0432.CCR-08-2786. [DOI] [PubMed] [Google Scholar]
- 45.Frew AJ, Johnstone RW, Bolden JE. Enhancing the apoptotic and therapeutic effects of HDAC inhibitors. Cancer Lett. 2009;280:125–33. doi: 10.1016/j.canlet.2009.02.042. [DOI] [PubMed] [Google Scholar]
- 46.Piacentini P, Donadelli M, Costanzo C, et al. Trichostatin A enhances the response of chemotherapeutic agents in inhibiting pancreatic cancer cell proliferation. Virchows Arch. 2006;448:797–804. doi: 10.1007/s00428-006-0173-x. [DOI] [PubMed] [Google Scholar]
- 47.Olive KP, Jacobetz MA, Davidson CJ, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324:1457–61. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bai J, Demirjian A, Sui J, et al. Histone deacetylase inhibitor trichostatin A and proteasome inhibitor PS-341 synergistically induce apoptosis in pancreatic cancer cells. Biochem Biophys Res Commun. 2006;348:1245–53. doi: 10.1016/j.bbrc.2006.07.185. [DOI] [PubMed] [Google Scholar]
- 49.Nawrocki ST, Carew JS, Pino MS, et al. Aggresome disruption: a novel strategy to enhance bortezomib-induced apoptosis in pancreatic cancer cells. Cancer Res. 2006;66:3773–81. doi: 10.1158/0008-5472.CAN-05-2961. [DOI] [PubMed] [Google Scholar]
- 50.Munster P, Marchion D, Bicaku E, et al. Phase I trial of histone deacetylase inhibition by valproic acid followed by the topoisomerase II inhibitor epirubicin in advanced solid tumors: a clinical and translational study. J Clin Oncol. 2007;25:1979–85. doi: 10.1200/JCO.2006.08.6165. [DOI] [PubMed] [Google Scholar]
- 51.Fulda S. Modulation of TRAIL-induced apoptosis by HDAC inhibitors. Curr Cancer Drug Targets. 2008;8:132–40. doi: 10.2174/156800908783769355. [DOI] [PubMed] [Google Scholar]
- 52.Chun SG, Zhou W, Yee NS. Combined targeting of histone deacetylases and hedgehog signaling enhances cytoxicity in pancreatic cancer. Cancer Biol Ther. 2009;8:1328–39. doi: 10.4161/cbt.8.14.8633. [DOI] [PubMed] [Google Scholar]
- 53.Kasperczyk H, Baumann B, Debatin KM, et al. Characterization of sonic hedgehog as a novel NF-kappaB target gene that promotes NF-kappaB-mediated apoptosis resistance and tumor growth in vivo. FASEB J. 2009;23:21–33. doi: 10.1096/fj.08-111096. [DOI] [PubMed] [Google Scholar]
- 54.von Burstin J, Eser S, Paul MC, et al. E-cadherin regulates metastasis of pancreatic cancer in vivo and is suppressed by a SNAIL/HDAC1/HDAC2 repressor complex. Gastroenterology. 2009;137:361–71. doi: 10.1053/j.gastro.2009.04.004. [DOI] [PubMed] [Google Scholar]
- 55.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial-mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–42. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]