

Abstract
We used a tumour necrosis factor (TNF)-α resistant breast adenocarcinoma MCF-7 cell line to investigate the involvement of the actin cytoskeleton in the mechanism of cell resistance to this cytokine. We found that TNF resistance correlates with the loss of cell epithelial properties and the gain of a mesenchymal phenotype, reminiscent of an epithelial-to-mesenchymal transition (EMT). Morphological changes were associated with a profound reorganization of the actin cytoskeleton and with a change in the repertoire of expressed actin cytoskeleton genes and EMT markers, as revealed by DNA microarray-based expression profiling. L-plastin, an F-actin cross-linking and stabilizing protein, was identified as one of the most significantly up-regulated genes in TNF-resistant cells. Knockdown of L-plastin in these cells revealed its crucial role in conferring TNF resistance. Importantly, overexpression of wild-type L-plastin in TNF-sensitive MCF-7 cells was sufficient to protect them against TNF-mediated cell death. Furthermore, we found that this effect is dependent on serine-5 phosphorylation of L-plastin and that non-conventional protein kinase C isoforms and the ceramide pathway may regulate its phosphorylation state. The protective role of L-plastin was not restricted to TNF-α resistant MCF-7 cells because a correlation between the expression of L-plastin and the resistance to TNF-α was observed in other breast cancer cell lines. Together, our study discloses a novel unexpected role of the actin bundling protein L-plastin as a cell protective protein against TNF-cytotoxicity.
Keywords: TNF-α, L-plastin, cytoskeleton, cell death
Introduction
Tumour necrosis factor (TNF)-α is a cytokine that elicits a wide range of biological responses. This cytokine plays an important role in breast tumour biology by regulating their oestrogen receptor (ER) activities, responsible for cell proliferation. Recently, it has been reported that TNF-α treatment in MCF-7 induces an inhibition of ER-α gene transcription through the PI3K/Akt signalling pathway, leading to a decrease in ER-α-dependent cell survival [1]. From this evidence, it seems reasonable to assume that the acquisition of resistance to TNF-α in breast cancer cells could compromise the clinical response to anti-estrogenic drugs as well as to other structurally and functionally unrelated drugs. This hypothesis is supported by clinical data revealing that TNF-resistant breast cancer cells isolated from patient samples develop cross-resistance to other chemotherapeutic drugs, such as doxorubicin [2]. In addition, a combined TNF-α/doxorubicin treatment has been reported to alleviate the resistance of MCF-7 cells to cytotoxic treatment [3]. Accordingly, low-dose of TNF-α increases the anti-tumour activity of doxorubicin in B16BL6 melanoma and soft tissue sarcoma-bearing mice [4, 5]. Understanding the molecular mechanism of resistance to TNF-α represents a significant challenge for more effective therapy not only in breast cancer but also in other cancers treated by TNF-α.
Several mechanisms have been reported to contribute to cellular resistance to TNF-induced cell killing, including the constitutive expression of several protective proteins in resistant tumour cells, such as MnSOD [6], endogenous TNF [7], major heat shock protein hsp70 [8], A20 zinc finger protein [9], However, these proteins confer only partial protection against TNF-cytotoxicity, suggesting that additional resistance mechanisms exist.
The actin cytoskeleton proteins appear to be regulated in response to several cell death signals including TNF [10]. Thus, gelsolin and α-fodrin were identified as caspase-3 substrate in cells stimulated by Fas (TNF receptor superfamily, member 6) [11] and TNF-α[12] respectively. Moreover, treatment of cells with cisplatin increased phosphorylation of α-adducin, its dissociation from the cytoskeleton and its cleavage by caspase-3 [13].
Koss et al. provided evidence that the response of endothelial cells to TNF-α may involve the phosphorylation of cytoskeleton proteins Ezrin, Radixin and Moesin [14]. More recently, report from Bieler et al. revealed that an intact actin cytoskeleton was required for PKB/Akt to prevent TNF-induced death [15]. Taken together these studies suggest that the cytoskeleton can no longer be considered as a simple structural framework playing a role in cell shape and motile events. Rather, the actin cytoskeleton seems to play an important role in the execution phase of cell death. Thus, identification of novel cytoskeleton genes which contribute to the regulation of the response to TNF-α may provide new perspective to the therapeutic use of TNF as an anticancer agent.
Here, we used DNA microarray methodology to investigate the cytoskeleton gene expression profile associated with the acquisition of cell resistance to TNF-α in breast carcinoma cell line. We provided evidence that TNF-α resistance correlates with the acquisition of mesenchymal cell phenotype and a reorganization of the actin cytoskeleton. Our data highlight the up-regulation of a group of F-actin stabilizing genes in resistant cells. Interestingly, we found that the F-actin bundling protein L-plastin is necessary and sufficient to confer, in a phosphorylation-dependent manner, cell resistance to TNF-α. This study revealed a novel unexpected function for the L-plastin in the mechanism of cell resistance to TNF-α.
Materials and methods
Reagents and antibodies
Recombinant human TNF-α was from R&D Systems (Oxon, UK). GF 109203X, H89, Gö6976 and Fostriecin were from Calbiochem (Leuven, Belgium). C2-ceramide was from Sigma (Bornem, Belgium). Rabbit anti-L-plastin and anti-serine-5 phosphorylated L-plastin (ser-5P) antibodies have been previously described [16]. Mouse monoclonal anti-L-plastin antibody (Clone LPL4A.1) was from Abcam (Cambridge, UK). Mouse anti-E-cadherin and anti-β-catenin were from BD-Biosciences (Erembodegem, Belgium). Mouse anti-cytokeratin-18 (DC-10) and anti-vimentin (V9) were from Santa-Cruz Biotechnology (Heidelberg, Germany). Mouse anti-b-actin was from Sigma. Rabbit anti- protein kinase C (PKC)-δ and –ζ were from Cell Signaling (Bioke, Leiden, The Netherlands). Mouse anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH), Alexa-Fluor 488- and 594-conjugated goat antimouse IgG and Alexa Fluor 488-coupled phalloidin were from Invitrogen (Merelbeke, Belgium). 4′,6-Diamidino-2-phenylindole, dihydrochloride (DAPI) was from MP Biomedicals (Illkirch, France). Horseradish peroxidase-conjugated antimouse and anti-rabbit IgGs were from GE Healthcare Life Sciences (Diegem, Belgium).
DNA constructs
pEGFP-C vectors (Clontech, Leusden, The Netherlands) containing wild-type (WT) or unphosphorylatable (S5/A)-L-plastin were generated from previously described pGEX-2T-WT-L-plastin and pGEX-2T-S5/A-L-plastin vectors, respectively [16]. Briefly, WT- or S5/A-L-plastin 1880-bp EcoRI-restricted cDNA fragments were inserted into pEGFP-C EcoRI-cut vectors. The 5′–3′ orientation of inserts was verified by sequencing.
Cell culture, treatment and transfection
TNF-sensitive MCF-7 cells and TNF-resistant MCF-7 derived clone (1001) and T47D cells were grown in RPMI-1640 medium (Lonza, Verviers, Belgium) supplemented with 10% (v/v) foetal calf serum. Tamoxifen resistant MCF-7 derived clone (MCF-7/TamR) (Kindly provided by Dr. A.E. Lykkesfeldt, Danish Cancer Society) was growth in phenol red free DME/F12 medium containing glutamax, 1% foetal calf serum, 6 ng/ml insulin and 10−6 M Tamoxifen (Sigma). MDA-MB-435s cells were grown in DME medium supplemented with 10% (v/v) foetal calf serum. Cells were transiently transfected with cDNA (10 μg) encoding WT- or S5/A-L-plastin-GFP. For TNF-treatment, cells were allowed to attach overnight before treatment with 75 ng/ml TNF-α for 72 hrs. Inhibition of kinase activities was performed by 1 hr treatment of cells with 5 μM of either of H89 (for protein kinase A [PKA] inhibition) or GF109203X (for PKC inhibition) or with 0.5 μM Gö6979 (for specific non-conventional PKC inhibition). Co-treatment of cells with both PKA and PKC inhibitor was also performed using the concentration described above. For ceramide and Fostriecin treatment, cells were pre-treated with 100 nM of Fostriecin as previously described [17] prior stimulation with 10 μM of C2-ceramide for 45 min. For knockdown experiments, cells were seeded at a density of 5 × 104 cells per well in a 6-well plate 1 day prior to transfection. siRNAs transfection was performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. 5 μl of Lipofectamine 2000 with 100 pmol of siRNA were used for each transfection. Double-stranded 21-mers validated siRNA for PKC-δ and -ζ and GFP were purchased from Qiagen (Venlo, The Netherlands). Sequences of custom-designed L-plastin siRNA are: 5′-AAATGCAGGCTGCAACAAAATdTdT-3′ and 5′-AAGTAGCCTCTCCTGTATTTAdTdT-3′. The mRNA and/or protein levels of siRNA targeted genes were analysed 48 hrs after transfection.
Indirect immunofluorescence
Cells were fixed with 3% paraformaldehyde, detergent permeabilized with 0.4% Triton X-100, and labelled as previously described [16]. Alexa Fluor 488-coupled phalloidin was used to visualize F-actin. Labelled cells were analysed by epifluorescence microscopy (LeicaDMRX microscope, HCX PL APO X40 or X63, Howard, Luxembourg). Images were acquired with a CCD camera (micromax, Princeton Instruments, Every, France) and analysed with Metaview software (Universal Imaging Corporation, Vianen, The Netherlands). In some experiments, cells were analysed with a Zeiss laser scanning confocal microscope (LSM-510-Meta, Zaventum, Belgium).
Immunoblotting analysis
Protein extracts from cells were prepared as previously described [16] and the protein concentration was determined using the Bradford assay kit from BioRad (Nazareth, Belgium). Proteins were separated by SDS-PAGE, transferred onto a nitrocellulose membrane which was incubated with a primary antibody followed with a secondary antibody coupled to horseradish peroxidase. Protein bands were revealed by enhanced chemiluminescence.
Oligonucleotide microarray
Total RNA was extracted using the TRIzol reagent (Invitrogen) followed by polyA purist purification (Ambion, Huntingdon, UK) according to the manufacturer’s instructions. RNA integrity and concentration were evaluated by the Agilent Bioanalyser 2100 capillary electrophoresis RNA 6000 nano assay (Agilent Biotechnologies, Diegem, Belgium). Gene profiling experiments were performed using the Actichip microarray and the significance level of gene expression data was evaluated as previously described [18]. Clustering and graphical analysis of the remaining gene expression data were performed using the Acuity 4.0 software (Molecular Devices, Sunnyval, CA, USA). Microarray data were in compliance with the standards proposed by the Microarray Gene Expression Data Society (http://www.mged.org), and were submitted to the ArrayExpress public repository [http://www.ebi.ac.uk/arrayexpress]. Reviewer’s user account (Username: Reviewer_E-MEXP-1927; Password: 1228312000130).
Cell viability assay
Cells were cultured in 96-well plates (5 × 103 cells/well) in the absence or presence of 75 ng/ml TNF-α. After 72 hrs, cell viability was assessed using the MTT assay. The absorbance at 620 nm of formazan product was measured using a microplate reader FLUOstar OPTIMA (BMG LABTECH (de Meem, The Netherlands)). The percentage of cell viability was calculated as previously described [19].
RT–PCR and quantitative RT–PCR (qRT–PCR)
RT–PCR and qRT–PCR analysis of L-pastin mRNA were performed using SuperScript III One-Step RT-PCR kit (Invitrogen). L-plastin primers were: forward 5′-ttggcacccaacactccta-3′ and reverse 5′-ccagggctttgtttatccag-3′ primers. GAPDH gene was used as an endogenous control. GAPDH primers were: forward 5′- agccacatcgctcagacac-3′ and reverse 5′-gcccaatacgaccaaatcc-3′.
Results and discussion
Cell resistance to TNF-α correlates with a reorganization of the actin cytoskeleton and the acquisition of a mesenchymal phenotype
The TNF-α resistant 1001 clone was established from the parental TNF-α sensitive MCF-7 cell line. We have previously reported that MCF-7 and 1001 cells express similar level of TNF receptors and trigger an active NF-κB signalling. The resistance of 1001 clone to TNF-α was not directly related to MnSOD and A20 gene expression but rather to a defect in de novo ceramide generation. However, TNF-dependent cell death occurred by apoptosis in MCF-7 cells [19, 20]. Here, we show that 1001 cells exhibited a mesenchymal phenotype when compared with epithelial MCF-7 cells (Fig. 1A). Phenotypic changes correlated with the loss of epithelial marker proteins (E-cadherin, and cytokeratin-18) and the gain of the mesenchymal marker vimentin in 1001 compared to MCF-7 cells (Fig. 1B and C). Although a moderate decrease in β-catenin protein expression level was detected in 1001 cells by immunoblot (Fig. 1C), its cell surface localization was dramatically altered and restricted to the cell-cell adherent junctions (Fig. 1B enlarged boxes).
Fig 1.
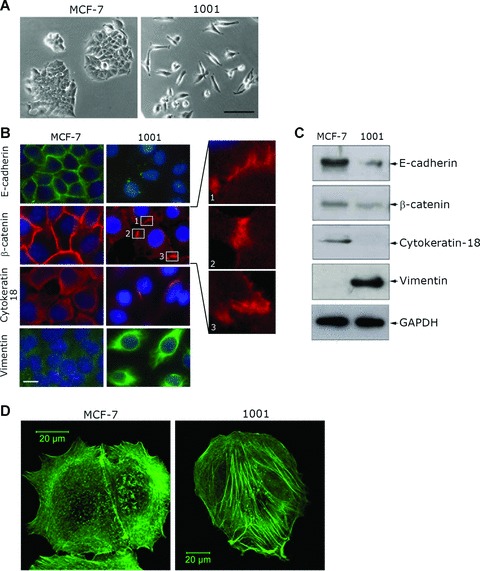
Acquisition of cell resistance to TNF-α is accompanied by morphological changes and actin cytoskeleton reorganization. (A) The morphology of TNF-sensitive MCF-7 and TNF-resistant 1001 cells by phase contrast microscopy. Bar: 100 μm. (B) Immunofluorescence analysis of epithelial and mesenchymal markers. Cells were labelled with E-cadherin, β-catenin, cytokeratin-18 or vimentin primary antibody. Secondary antibodies were Alexa-Fluor 488-conjugated goat antimouse IgG for E-cadherin and vimentin (green) and Alexa-Fluor-594-conjugated goat antimouse IgG for β-catenin and cytokeratin-18 (red). Nuclei were stained with DAPI (blue). Cells were analysed by epifluorescence microscopy (LeicaDMRX microscope). Three enlarged regions of β-catenin staining in 1001 cells are shown. Bar: 10 μm. (C) Expression of epithelial and mesenchymal marker proteins in MCF-7 and 1001 cells. Immunoblot analysis was performed on total protein extracts (50 μg) using E-cadherin-, β-catenin-, cytokeratin-18-, vimentin- or GAPDH-specific antibody as previously described [16]. (D) Actin cytoskeleton organization in MCF-7 and 1001 cells. Cells were stained with Alexa-Fluor 488-coupled phalloidin to visualize F-actin and analysed using a Zeiss laser scanning confocal microscope (LSM-510 Meta). Bar: 20 μm.
Because changes in cell morphology are frequently associated with changes in cytoskeletal organization, we analysed the organization of the actin cytoskeleton in these cells. MCF-7 cells contained sub-membranous cortical actin whereas 1001 cells exhibited well-organized actin stress fibres (Fig. 1D). This result suggests that changes in the repertoire of actin cytoskeleton-associated proteins involved in cytoskeletal reorganization might also contribute to TNF-α resistance.
TNF-α resistance is accompanied by changes in the repertoire of expressed genes involved in epithelial cell plasticity and actin cytoskeleton organization
To determine differentially expressed actin cytoskeleton-related genes in 1001 and MCF-7 cells, we performed gene expression profiling experiments using an actin cytoskeleton dedicated microarray termed Actichip [18]. Microarray analysis yielded a list of significant 106 down-regulated and 113 up-regulated genes in 1001 when compared to MCF-7 cells as determined by SAM analysis (Δ= 0.802; FDR = 5.62 %) (Supporting Information data 1). The 57 most significantly and differentially expressed genes (log2 expression ratio >1) were classified into 17 gene function categories, highly enriched in genes linked to actin dynamics, signalling and tissue- or cell-specific markers (Table 1). As expected from their mesenchymal phenotype, 1001 cells expressed mesenchymal marker genes (vimentin, fibronectin precursor) and genes involved in stress fibres assembly (talin-1 and -2 and vinculin), whereas the epithelial marker genes E-cadherin, desmoplakin, cytokeratin-8 and -18 were repressed in these cells. Basically, the expression profile of 1001 cells highlighted genes implicated in the stabilization of the cytoskeleton. Genes encoding actin cross-linking or capping proteins were overexpressed whereas those involved in actin filament depolymerization or monomer sequestration were down-regulated in 1001 cells. Although several regulated genes listed in Table 1 (e.g. drebrin, actinin α1, moesin) are known to be involved in actin-dependent cell death pathways [21, 22], other have so far no established function in cell death, prompting us to investigate their role in this process.
Table 1.
Actin cytoskeleton genes differentially expressed in TNF-sensitive MCF-7 and TNF-resistant 1001 cells classified by gene function categories
| Gene | Score (d) | Log2 ratio | |
|---|---|---|---|
| Actin bundling, cross-linking | |||
| L-plastin | LCP-1 | 18.18 | 2.19 |
| T-plastin | PLS3 | 6.46 | 1.79 |
| Actinin, α1 | ACTN1 | 6.10 | 1.49 |
| Drebrin 1 | DBN1 | 5.80 | 1.08 |
| Actinin, α4 | ACTN4 | 5.51 | 1.08 |
| Coronin, actin binding protein, 1C | CORO1C | 3.27 | 1.70 |
| Anillin | ANLN | 2.58 | 1.57 |
| Calponin 3, acidic | CNN3 | 1.89 | 2.78 |
| Growth arrest-specific 6 | GAS6 | −7.35 | −1.40 |
| Transgelin 2 | TAGLN2 | −3.18 | −1.64 |
| Calicin | CCIN | −2.29 | −1.03 |
| Desmosome marker | |||
| Tight junction protein 1 (Zonula occludens 1) | TJP1 | 8.10 | 1.08 |
| Desmocollin 1 | DSC1 | 7.34 | 1.12 |
| Junction plakoglobin | JUP | 5.63 | 1.99 |
| Desmoglein 1 | DSG1 | −4.50 | −1.14 |
| Cytokeratin 8 | KRT8 | −14.51 | −3.31 |
| Cytokeratin 18 | KRT18 | −12.11 | −3.76 |
| Catenin, delta 2 | CTNND2 | −7.80 | −1.44 |
| Desmoplakin | DSP | −7.59 | −1.47 |
| Epithelial marker | |||
| Dysadherin | FXYD5 | 5.82 | 1.66 |
| Matrix metalloproteinase-1 | MMP1 | 4.20 | 3.17 |
| Matrix metallopeptidase-14 | MT-MMP1 | 1.47 | 1.29 |
| E-cadherin | CDH1 | −3.47 | −1.51 |
| Cell adhesion associated protein | |||
| Talin 1 | TLN1 | 17.39 | 1.41 |
| Vinculin | VCL | 9.08 | 1.20 |
| Talin 2 | TLN2 | 6.53 | 1.07 |
| Catenin, α-like 1 | CTNNAL1 | 3.60 | 2.02 |
| Signalling | |||
| Rho GDP-dissociation inhibitor 2 | ARHGDIB | 4.16 | 2.37 |
| Cdc42-interacting protein 4 | TRIP10 | 3.32 | 1.27 |
| Rho-GTPase-activating protein 6 | ARHGAP6 | 3.07 | 1.70 |
| Rho-related GTP-binding protein RhoF | RHOF | 2.84 | 1.66 |
| Calcium signalling, phospholipid binding family | |||
| Formyl peptide receptor 1 | FPR1 | 15.00 | 2.22 |
| Annexin A2 | ANXA2 | 6.43 | 1.33 |
| Annexin A3 | ANXA3 | 5.09 | 1.77 |
| Annexin A1 | ANXA1 | 3.96 | 5.06 |
| Plasma membrane-cortical cytoskeleton Linkers/scaffold | |||
| Merlin | NF2 | 5.78 | 1.40 |
| Moesin | MSN | 3.28 | 3.32 |
| Thyroid hormone receptor interactor 6 | TRIP6 | 3.16 | 1.48 |
| Erythrocyte membrane protein band 4.1-like 2 | EPB41L2 | 2.25 | 1.41 |
| Actin depolymerisation | |||
| Destrin (actin depolymerizing factor) | DSTN | −17.00 | −1.34 |
| HSP27 estrogen response element-TATA box-binding protein | SAFB | −2.41 | −1.02 |
| Protein tyrosine kinase 9-like (A6-related protein) | PTK9L | −1.99 | −1.08 |
| Actin binding protein | |||
| Epidermal growth factor receptor | ERBB1 | 4.80 | 3.15 |
| Eukaryotic translation elongation factor 1 α2 | EEF1A2 | −12.32 | −1.97 |
| Eukaryotic translation elongation factor 2 | EEF2 | −2.35 | −1.24 |
| Mesenchymal marker | |||
| Vimentin | VIM | 10.39 | 3.88 |
| Fibronectin 1 | FN1 | 5.39 | 1.94 |
| Actin-dependent motors | |||
| Myosin IA | MYO1A | 2.48 | 1.08 |
| Myosin regulatory light chain | MRLC2 | −1.66 | −1.01 |
| Regulation of muscle contraction | |||
| Smoothelin | SMTN | 4.09 | 1.05 |
| Cortactin (Amplaxin) | CTTN | −9.48 | −1.34 |
| Transcription factor | |||
| Transcription factor 7-like 2 | TCF7L2 | 12.58 | 1.19 |
| Lymphoid enhancer binding factor 1 | LEF1 | 6.88 | 1.35 |
| Actin nucleator | |||
| Actin related protein 2/3 complex, subunit 1B | ARPC1B | −9.83 | −1.98 |
| Actin sequestrating | |||
| Profilin 2 | PFN2 | −2.66 | −1.21 |
| Barbed end actin capping | |||
| Macrophage capping protein (actin filament), gelsolin-like | CAPG | 5.94 | 2.20 |
| Cytoskeletal protein | |||
| LIM domain and actin binding 1 | LIMA1 | 7.09 | 2.68 |
Gene profiling experiments were performed using the Actichip microarray as previously described in ‘Material and methods’. A score (d) value is assigned to each gene based on its change in expression relative to the standard deviation obtained by repeated measurements for the gene. Genes with high score are deemed as significantly regulated. A log2 ratio of 1 corresponds to a two-fold change in the expression level of a transcript. Positive log ratios correspond to up-regulated genes and negative log ratios (highlighted in grey) correspond to down-regulated genes in 1001 cells when compared to MCF-7.
L-plastin confers resistance to TNF-mediated cell death
The major group of cytoskeleton genes that were up-regulated in 1001 cells comprised genes encoding proteins involved in actin filament cross-linking. One of the most significantly overexpressed genes in resistant 1001 cells (Table 1) encodes L-plastin which is a representative member of the CH-domain family of actin-cross-linking proteins [23]. L-plastin is expressed in haematopoietic cells where it is phosphorylated via cytokine pathways, including TNF-α[24]. We previously demonstrated that the phosphorylation of L-plastin on serine-5 residue (ser5) increases its F-actin-binding activity and promotes its targeting to actin assembly sites in cells [16]. Although the expression level of L-plastin was correlated with tumour stages in colorectal and prostate cancer, such a correlation remained controversial in breast cancer. This controversy could be due to the fact that some studies did not consider the phosphorylation status of L-plastin which is the only L-plastin form involved in tumour progression (reviewed in [25]).
The higher expression of L-plastin has been confirmed at mRNA and protein levels in 1001 cells (Fig. 2A). Interestingly, immunofluorescence experiments demonstrated that a small fraction of MCF-7 cells highly expressed L-plastin and that only this fraction of cells remained refractory to the cytotoxic effect of TNF-α (Fig. 2B). Accordingly, as resulted by the cell viability assay, L-plastin siRNA transfection in 1001 cells yielded in a 40% decrease of cells viability after TNF-α treatment when compared to that of untrasfected or GFP-siRNA transfected 1001 cells. The TNF resistance of the remaining 60% cells would be related to either the efficiency of transfection and/or the transient effect of the L-plastin silencing during the viability assay (Fig. 2C).
Fig 2.
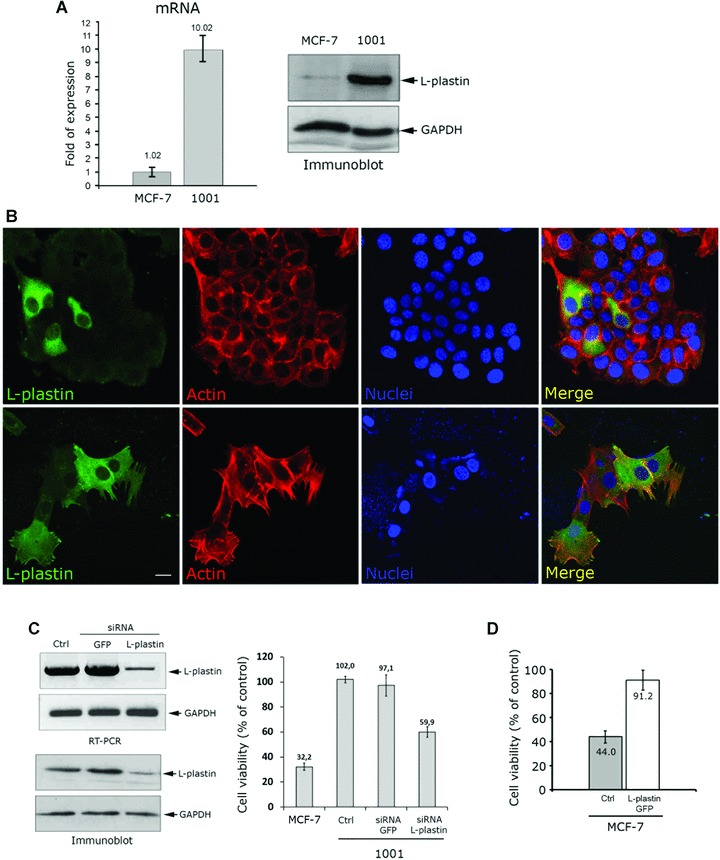
Expression of L-plastin is sufficient to protect cells against TNF-induced cell death. (A) L-plastin mRNA level was assessed by real time PCR using L-plastin and GAPDH primers. L-plastin mRNA is given as relative fold of expression of GAPDH mRNA. Results are given as the mean ± S.D. of three independent experiments. The protein level was detected by immunoblot using an anti-L-plastin antibody (upper panel). The anti-GAPDH antibody (lower panel) was used as a loading control. (B) Immunofluorescence analysis of L-plastin expression in MCF-7 cells. Control untreated cells (upper row) and TNF-treated cells (lower row) were labelled with mouse monoclonal anti-L-plastin antibody coupled to Alexa-Fluor-488-goat antimouse IgG (green), Alexa-Fluor-594 phalloidin to monitor actin filaments (red) and with DAPI to stain the nuclei (blue). The merged image of actin, L-plastin and nuclei is shown on the right. Cells were treated with 75 ng/ml of TNF-α for 72 hrs, as described in ‘Materials and methods’, and analysed by epifluorescence microscopy (LeicaDMRX microscope). Bar: 10 μm. (C) Knockdown of L-plastin in 1001 cells. siRNAs (100 pmol) transfection was performed using L-plastin siRNA. GFP siRNA was used as a control. The mRNA (upper panels) and protein levels (lower panels) of L-plastin in control untransfected cells (Ctrl) and GFP- or L-plastin- siRNA-transfected cells were assessed by RT-PCR and immunoblot. On the right: The effect of L-plastin knockdown on TNF-α resistance of 1001 cells. Control untransfected MCF-7 or 1001 cells and siRNA GFP or siRNA L-plastin transfected 1001 cells were treated with TNF-α (75 ng/ml) for 72 hrs. Cell viability was determined using the MTT assay and calculated as described in [19]. Results are given as the mean ± S.D. of three independent experiments. (D) Effect of L-plastin overexpression on MCF-7 cell sensitivity to TNF-α. Cells were transfected with WT L-plastin-GFP cDNA encoding pEGFP-C vector. Cell viability of control untransfected (Ctrl) or L-plastin-GFP expressing MCF-7 cells in the presence of TNF-α was determined as described in (C). Results are expressed as the mean ± S.D. of three independent experiments.
To further confirm the role of L-plastin in the regulation of TNF-mediated cell death, we overexpressed L-plastin-GFP in MCF-7 cells. The high efficiency of transfection was monitored by fluorescence microscopy and the overexpression of L-plastin GFP was confirmed by immunoblot (Supporting Information data 2). Interestingly, L-plastin overexpression was sufficient to significantly increase (50%) viability of MCF-7 cells after TNF-treatment (Fig. 2D). It is worthy to note that 44% of control untransfected cells were resistant to TNF-α. We could therefore assume that those cells represent a sub-population of MCF-7 cells expressing high level of L-plastin. This hypothesis is supported by our data in the Fig. 2B demonstrating that L-plastin expressing MCF-7 cells resist to the cytotoxic effect of TNF-α.
Notably, neither knockdown of L-plastin in resistant 1001 cells nor its overexpression in MCF-7 cells caused detectable phenotypic changes and epithelial-to-mesenchymal transition markers regulations (Supporting Information data 3). Based on this result, we may speculate that L-plastin acts independently of the EMT process. This result is in agreement with previous findings suggesting that the expression of L-plastin does not induce profound cytoskeleton reorganization but rather contributes to changes of the cortical cytoskeleton linked to membrane bound signalling complexes [16]. In this respect the mesenchymal phenotype could confer 1001 cells features other than TNF-α resistance, such as a high tumorigenic phenotype. It is worth noting that L-plastin also contributes to cell invasion and, hence, this protein could play a dual role in tumour progression and protection against cell death signals.
Acquisition of resistance to TNF-α requires L-plastin phosphorylation
It has been reported that the activity of L-plastin is regulated by phosphorylation on Ser5 residue [16]. Here, we found that transfected L-plastin-GFP in MCF-7 and endogenous L-plastin in 1001 cells are highly phosphorylated on ser5 (Fig. 3A), suggesting a critical role for this phosphorylation in TNF resistance. To test this hypothesis, we transfected MCF-7 cells with WT or ser5 mutated to Alanine (S5/A) unphosphorylable-L-plastin-GFP and performed TNF-α cell viability assays. Expression of S5/A-L-plastin-GFP in TNF-sensitive MCF-7 cells had no effect on their resistance to TNF-mediated cell death (Fig. 3B). In contrast, a significant increase in TNF-α resistance was observed in MCF-7 cells expressing WT-L-plastin-GFP. Altogether, our data provide evidence that phosphorylation of L-plastin on ser5 is critical for the acquisition of resistance to TNF-α. Previous findings have demonstrated that L-plastin may contribute to the stabilization of the cell cortex, or alternatively, contribute to the assembly of receptor mediated complexes [26]. In this regard, the involvement of L-plastin in TNF-receptors internalization seems to be unlikely, because we have previously reported that 1001 and MCF-7 cells displayed comparable levels of either p55 TNF receptor expression or NF-κB activation [19].
Fig 3.
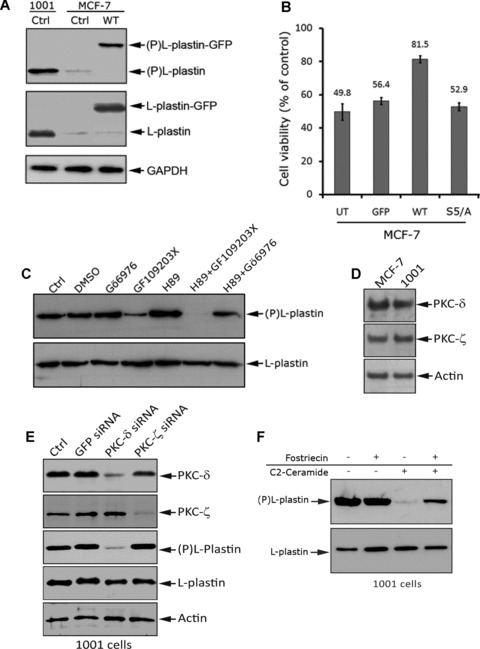
Overexpression of phosphorylated L-plastin in MCF-7 cells confers resistance to TNF-α. (A) Ser5 phosphorylation level of endogenous L-plastin in control 1001 and MCF-7 cells (Ctrl) and L-plastin-GFP-transfected MCF-7 cells (WT). Immunoblot analysis was performed using an anti-ser5 phosphorylated L-plastin specific antibody (Ser5-P) [16] (upper panel), an anti-L-plastin (middle panel) or an anti-GAPDH (lower panel). The higher band (upper and middle panels) corresponds to the transfected L-plastin fused to GFP (L-plastin-GFP) and the lower band corresponds to endogenous L-plastin (L-plastin). (B) Unphosphorylable L-plastin does not confer resistance to TNF-α in MCF-7 cells. Cell viability assay was performed after 72 hrs of TNF-α treatment (75 ng/ml) on untransfected (UT) and transfected MCF-7 cells with either GFP (GFP), WT L-plastin-GFP (WT) or unphosphorylable L-plastin-GFP (S5/A). The transfection efficiency was nearly identical in cells (data not shown). Results are expressed as the mean ± S.D. of three independent experiments. (C) High baseline phosphorylation of L-plastin in 1001 cells involves non-conventional PKC isoforms. Cells were treated for 3 hrs with vehicle (DMSO), 0.5 μM of Gö6976 (conventional PKC inhibitor), 5 μM of GF109203X (conventional and non-conventional-PKC inhibitor), 5 μM of H-89 (PKA inhibitor). Cells were also treated with H-89 (5 μM) combined to either GF109203X (5 μM) or Gö6976 (0.5 μM).Immunoblot analysis was performed using anti-Ser5-P (upper panel) or anti-L-plastin (lower panel) antibody. (D) Expression of PKC-δ and –ζ in MCF-7 and 1001 cells. Immunoblot analysis was performed on total protein extracts (50 μg) using an anti-PKC-δ (Upper panel), an anti-PKC-ζ (middle panel), or an anti-actin (lower panel) antibody. (E) Knockdown of PKC-δ decreased L-plastin phosphorylation in 1001 cells. 1001 cells were transfected with either PKC-δ or PKC-ζ siRNAs (100 pmol) as well as with GFP siRNA used as a control. The protein expression levels of PKC-δ and ζ as well as L-plastin and phosphor-L-plastin were assessed by immunoblot in control untransfected cells (Ctrl), GFP-, PKC-δ or PKC-ζ siRNA transfected 1001 cells. The anti-actin antibody was used as a loading control. (F) Exogenous cell permeable C2-ceramide induces a decrease in L-plastin phosphorylation in 1001 cells by a mechanism involving the activity of the PP2A. Cells were untreated (–) or pre-treated (+) with 100 nM of Fostriecin. Cells were further incubated in the absence (–) or presence (+) of 10 μM C2-ceramide for 45 min. Immunoblot analysis was performed using anti-Ser5-P (upper panel) or anti-L-plastin (lower panel) antibody.
The nature of kinases involved in phosphorylation of L-plastin is still controversial. Although several studies support the direct phosphorylation of L-plastin by PKA [16, 27], the implication of PKC in L-plastin phosphorylation remains to be confirmed [24, 28]. Moreover, Basu et al. provided evidence that a differential sensitivity of breast cancer cells to TNF-α could be related to a differential expression level of PKCs [29]. Consistent with these studies, we investigated the relative contribution of PKA and PKC in L-plastin phosphorylation, by using H89 and Gö6976, agents which selectively inhibit PKA and conventional PKC isozymes, respectively. Interestingly, even at high concentrations, (5 μM H-89 and 0.5 μM Gö6976) these drugs did not inhibit phosphorylation of L-plastin in 1001 cells. This observation makes unlikely the involvement of PKA or conventional PKC isoforms in the phosphorylation L-plastin in 1001 cells. Surprisingly, treatment of cells with 5 μM of GF109203X, an inhibitor of the three PKC isoforms (conventional, novel and atypical), significantly decreased L-plastin phosphorylation without affecting its total expression level (Fig. 3C). This result supports the involvement of non-conventional PKC isozymes in the phosphorylation of L-plastin in 1001 cells. Interestingly, a maximal dephosphorylation of L-plastin was observed in 1001 cells co-treated with both PKA and PKC inhibitors H89 and GF109203X respectively (5 μM each). So far, we have no explanation as why, when used alone, H89 did not have an inhibitory effect on L-plastin phosphorylation. Nevertheless, our results highlight the involvement of non-conventional PKC isozymes in L-plastin phosphorylation, without precluding a possible role for both PKA in the phosphorylation of L-plastin. This will be the subject of further investigation because a cooperative activity between PKA and PKA pathways has been previously described [30, 31].
PKC-δ is the major kinase involved in L-plastin phosphorylation
Non-conventional PKC represents a family of seven isozymes that have been categorized into two groups: the new or novel PKCs (, δ, η and τ) and the atypical PKCs (-ζ, -ι and -λ). Although MCF-7 cells expressed the majority of non-conventional PKCs isozymes [29], PKC-δ and-ζ have been described as regulators of actin cytoskeleton and TNF-α signalling [32–35]. Immunoblot analysis showed no difference in the expression level of PKC-δ and -ζ isozymes in both MCF-7 and 1001 cells (Fig. 3D). However, knockdown of PKC-δ, but not PKC-ζ, by siRNA, induced a significant decrease of the L-plastin phosphorylation, suggesting that PKC-δ is the major PKC isozyme involved in basal L-plastin phosphorylation in 1001 cells (Fig. 3E).
Regulation of L-plastin phosphorylation via the sphingomyelin/ceramide pathway
High baseline phosphorylation level of L-plastin may also be related to a reduced phosphatase activity in 1001 cells. We have previously reported that resistance of 1001 cells to TNF-α correlated with a defect in the sphingomyelin/ceramide pathway and treatment of 1001 cells with exogenous ceramide restores their sensitivity to TNF-α. [19]. Moreover, it is well established that ceramide activates the protein phosphatase 2A (PP2A) [36]. Here we demonstrate that exogenous cell-permeable C2-ceramide rapidly decreased L-plastin phosphorylation level in 1001 cells (Fig. 3F). Interestingly, pre-treatment of cells with Fostriecin, a potent and specific inhibitor of protein phosphatase 2A (PP2A), alleviates the effect of ceramide on L-plastin dephosphorylation (Fig. 3F). Collectively, these results provided evidence that ceramide induces L-plastin dephosphorylation by a mechanism involving, at least in part, the activity of PP2A. Our data are consistent with those recently described by Zeidan et al. demonstrating that the activation of the acid-sphingomyelinase (ASMase)/ceramide pathway by cisplatin in MCF7 cells induces the dephosphorylation of ezrin, leading to cytoskeleton rearrangements and increased cell motility [37]. We, and others, have previously shown that L-plastin participates in cancer cell invasion [16, 38]. The present finding raises the exciting possibility that in cells with a defect in ceramide pathway, L-plastin may, in addition to protection against cell death, participate in early ceramide-triggered events such as tumour cell invasion.
Although the precise molecular mechanisms by which L-plastin regulates the cellular sensitivity to TNF-α remain to be addressed, this study discloses a novel unexpected role of the actin bundling protein L-plastin as a cell protective protein against TNF-α cytotoxicity. It is noteworthy that the protective role of L-plastin observed in the present study does not seem to be specific to TNF-α resistant MCF-7 cells. Indeed, a high expression and phosphorylation levels of L-plastin was detected in other MCF-7 derived clones displaying resistance to Tamoxifen (MCF-7/TamR) as well as in MDA-MB-435s cell line (Fig. 4A). Interestingly, MCF-7/TamR and MDA-MB435s are both resistant to TNF-α (Fig. 4B). In contrast, L-plastin expression was not detected in the T47D cell line which is sensitive to TNF-α (Fig. 4A and B). Such a role for L-plastin has also been identified in radiotherapy resistant clones derived from MCF-7, T47D and MDA-MB-231 breast cancer cell lines [39], although the resistance mechanism to TNF-α and radiotherapy are fundamentally different.
Fig 4.
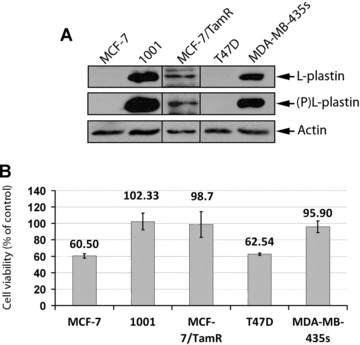
The high expression and the phosphorylation level of L-plastin correlates with the resistance of breast cancer cell lines to the cytotoxic effect of TNF-α. (A) Expression of L-plastin and Ser5-phosphorylated L-plastin in MCF-7, 1001, MCF-7 TamR, T47D and MDA-MB-435s cells. Immunoblot analysis was performed on total protein extracts (50 μg) using an anti-L-plastin (Upper panel), an anti-ser5 phosphorylated L-plastin specific antibody (middle panel), or an anti-actin (lower panel) antibody. (B) The cytotoxic effect of TNF-α in breast cancer cell lines. Cell viability of MCF-7, 1001, MCF-7/TamR, T47D and MDA-MB-435s cells treated with TNF-α (75 ng/ml) for 72 hrs was determined as described in Fig. 2C. Results are expressed as the mean ± S.D. of three independent experiments.
Together, our findings suggest that the status of L-plastin expression and phosphorylation may be a useful molecular marker for predicting the responsiveness of cancer cells to treatment with cytotoxic agents. Considering this new role of L-plastin in breast cancer cells, its targeting may represent a novel strategy in cancer therapy.
Acknowledgments
We thank Arnaud Muller and Mikalai Yatskou for their support in statistical analysis in microarray experiments as well as Marie Catillon for her outstanding technical assistance. We are grateful to Victoria El-Khoury and Olivia Roland for discussion and critical reading of the manuscript. Z.A. is a fellowship from the ‘Ministère de la Culture, de l’Enseignement Supérieur et de la Recherche’, Luxembourg. This work was supported by the ‘Centre National de Recherche Scientifique’, France, and by a grant of the Human Frontier Science Organisation (RGP0058/2005).
Supporting Information
List of all actin cytoskeleton genes mainlyexpressed in MCF-7 and 1001 cells as identified through one classSAM analysis of Actichip data. Genes mainly regulated in MCF-7 and1001 are sorted by their significance (d) score.
A, Transfection efficiency ofL-plastin GFP in MCF-7 cells. Untransfected (left) or L-plastin GFPtransfected (right) MCF-7 cells were plated on cover slips andanalysed by fluorescence microscopy. Bar, 100 μm.
Expression level of L-plastin GFP in MCF-7 cells. Cell extracts from untransfected and L-plastin GFP transfected MCF-7 cells were analysed by immunoblot using an anti-L-plastin (upper) or an anti-GAPDH (lower) antibody. Band at 97 kD, corresponding to L-plastin GFP fused protein, was detected in transfected cells.
data 3. Overexpression and knockdown L-plastin in MCF-7 and 1001 cells, respectively, did not affect cell phenotype and EMT markers expression.
A, Phase contrast microscopy of the morphology ofMCF-7 cells untransfected (Ctrl) and L-plastin GFP transfected(L-plastin-GFP) cells as well as of 1001 cells untransfected (Ctrl)and L-plastin siRNA transfected cells (siRNA L-plastin). Bar: 50μm.
B, Expression of epithelial and mesenchymal markerproteins in MCF-7 and 1001 cells described in A. Immunoblotanalysis was performed using L-plastin, E-cadherin-,β-catenin-, cytokeratin-18-, vimentin- or GAPDH-specificantibody as previously described [16].
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Lee SH, Nam HS. TNF alpha-induced down-regulation of estrogen receptor alpha in MCF-7 breast cancer cells. Mol Cells. 2008;26:285–90. [PubMed] [Google Scholar]
- 2.Dollbaum C, Creasey AA, Dairkee SH, et al. Specificity of tumor necrosis factor toxicity for human mammary carcinomas relative to normal mammary epithelium and correlation with response to doxorubicin. Proc Natl Acad Sci USA. 1988;85:4740–4. doi: 10.1073/pnas.85.13.4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cao W, Ma SL, Tang J, et al. A combined treatment TNF-alpha/doxorubicin alleviates the resistance of MCF-7/Adr cells to cytotoxic treatment. Biochim Biophys Acta. 2006;1763:182–7. doi: 10.1016/j.bbamcr.2005.12.008. [DOI] [PubMed] [Google Scholar]
- 4.Brouckaert P, Takahashi N, van Tiel ST, et al. Tumor necrosis factor-alpha augmented tumor response in B16BL6 melanoma-bearing mice treated with stealth liposomal doxorubicin (Doxil) correlates with altered Doxil pharmacokinetics. Int J Cancer. 2004;109:442–8. doi: 10.1002/ijc.11703. [DOI] [PubMed] [Google Scholar]
- 5.Ten Hagen TL, Van Der Veen AH, Nooijen PT, et al. Low-dose tumor necrosis factor-alpha augments antitumor activity of stealth liposomal doxorubicin (DOXIL) in soft tissue sarcoma-bearing rats. Int J Cancer. 2000;87:829–37. doi: 10.1002/1097-0215(20000915)87:6<829::aid-ijc12>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 6.Wong GH, Elwell JH, Oberley LW, Goeddel DV. Manganous superoxide dismutase is essential for cellular resistance to cytotoxicity of tumor necrosis factor. Cell. 1989;58:923–31. doi: 10.1016/0092-8674(89)90944-6. [DOI] [PubMed] [Google Scholar]
- 7.Vanhaesebroeck B, Decoster E, Van Ostade X, et al. Expression of an exogenous tumor necrosis factor (TNF) gene in TNF-sensitive cell lines confers resistance to TNF-mediated cell lysis. J Immunol. 1992;148:2785–94. [PubMed] [Google Scholar]
- 8.Jaattela M, Wissing D, Bauer PA, et al. Major heat shock protein hsp70 protects tumor cells from tumor necrosis factor cytotoxicity. EMBO J. 1992;11:3507–12. doi: 10.1002/j.1460-2075.1992.tb05433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Opipari AW, Jr, Hu HM, Yabkowitz R, et al. The A20 zinc finger protein protects cells from tumor necrosis factor cytotoxicity. J Biol Chem. 1992;267:12424–7. [PubMed] [Google Scholar]
- 10.Mathew SJ, Haubert D, Kronke M, Leptin M. Looking beyond death: a morphogenetic role for the TNF signalling pathway. J Cell Sci. 2009;122:1939–46. doi: 10.1242/jcs.044487. [DOI] [PubMed] [Google Scholar]
- 11.Kothakota S, Azuma T, Reinhard C, et al. Caspase-3-generated fragment of gelsolin: effector of morphological change in apoptosis. Science. 1997;278:294–8. doi: 10.1126/science.278.5336.294. [DOI] [PubMed] [Google Scholar]
- 12.Janicke RU, Ng P, Sprengart ML, Porter AG. Caspase-3 is required for alpha-fodrin cleavage but dispensable for cleavage of other death substrates in apoptosis. J Biol Chem. 1998;273:15540–5. doi: 10.1074/jbc.273.25.15540. [DOI] [PubMed] [Google Scholar]
- 13.van de Water B, Tijdens IB, Verbrugge A, et al. Cleavage of the actin-capping protein alpha -adducin at Asp-Asp-Ser-Asp633-Ala by caspase-3 is preceded by its phosphorylation on serine 726 in cisplatin-induced apoptosis of renal epithelial cells. J Biol Chem. 2000;275:25805–13. doi: 10.1074/jbc.M001680200. [DOI] [PubMed] [Google Scholar]
- 14.Koss M, Pfeiffer GR, 2nd, Wang Y, et al. Ezrin/radixin/moesin proteins are phosphorylated by TNF-alpha and modulate permeability increases in human pulmonary microvascular endothelial cells. J Immunol. 2006;176:1218–27. doi: 10.4049/jimmunol.176.2.1218. [DOI] [PubMed] [Google Scholar]
- 15.Bieler G, Hasmim M, Monnier Y, et al. Distinctive role of integrin-mediated adhesion in TNF-induced PKB/Akt and NF-kappaB activation and endothelial cell survival. Oncogene. 2007;26:5722–32. doi: 10.1038/sj.onc.1210354. [DOI] [PubMed] [Google Scholar]
- 16.Janji B, Giganti A, De Corte V, et al. Phosphorylation on Ser5 increases the F-actin-binding activity of L-plastin and promotes its targeting to sites of actin assembly in cells. J Cell Sci. 2006;119:1947–60. doi: 10.1242/jcs.02874. [DOI] [PubMed] [Google Scholar]
- 17.Sheth P, Samak G, Shull JA, et al. Protein phosphatase 2A plays a role in hydrogen peroxide-induced disruption of tight junctions in Caco-2 cell monolayers. Biochem J. 2009;421:59–70. doi: 10.1042/BJ20081951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muller J, Mehlen A, Vetter G, et al. Design and evaluation of Actichip, a thematic microarray for the study of the actin cytoskeleton. BMC Genomics. 2007;8:294–310. doi: 10.1186/1471-2164-8-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cai Z, Bettaieb A, Mahdani NE, et al. Alteration of the sphingomyelin/ceramide pathway is associated with resistance of human breast carcinoma MCF7 cells to tumor necrosis factor-alpha-mediated cytotoxicity. J Biol Chem. 1997;272:6918–26. doi: 10.1074/jbc.272.11.6918. [DOI] [PubMed] [Google Scholar]
- 20.Zyad A, Branellec D, Mahe Y, et al. The development of human tumor-cell resistance to TNF-alpha does not confer resistance to cytokine-induced cellular cytotoxic mechanisms. Int J Cancer. 1992;52:953–8. doi: 10.1002/ijc.2910520621. [DOI] [PubMed] [Google Scholar]
- 21.Hebert M, Potin S, Sebbagh M, et al. Rho-ROCK-dependent ezrin-radixin-moesin phosphorylation regulates Fas-mediated apoptosis in Jurkat cells. J Immunol. 2008;181:5963–73. doi: 10.4049/jimmunol.181.9.5963. [DOI] [PubMed] [Google Scholar]
- 22.Klaiman G, Petzke TL, Hammond J, et al. Targets of caspase-6 activity in human neurons and Alzheimer disease. Mol Cell Proteomics. 2008;7:1541–55. doi: 10.1074/mcp.M800007-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lapillonne A, Coue O, Friederich E, et al. Expression patterns of L-plastin isoform in normal and carcinomatous breast tissues. Anticancer Res. 2000;20:3177–82. [PubMed] [Google Scholar]
- 24.Shiroo M, Matsushima K. Enhanced phosphorylation of 65 and 74 kDa proteins by tumor necrosis factor and interleukin-1 in human peripheral blood mononuclear cells. Cytokine. 1990;2:13–20. doi: 10.1016/1043-4666(90)90038-u. [DOI] [PubMed] [Google Scholar]
- 25.Samstag Y, Klemke M. Ectopic expression of L-plastin in human tumor cells: diagnostic and therapeutic implications. Adv Enzyme Regul. 2007;47:118–26. doi: 10.1016/j.advenzreg.2006.12.008. [DOI] [PubMed] [Google Scholar]
- 26.Chen H, Mocsai A, Zhang H, et al. Role for plastin in host defense distinguishes integrin signaling from cell adhesion and spreading. Immunity. 2003;19:95–104. doi: 10.1016/s1074-7613(03)00172-9. [DOI] [PubMed] [Google Scholar]
- 27.Wang J, Brown EJ. Immune complex-induced integrin activation and L-plastin phosphorylation require protein kinase A. J Biol Chem. 1999;274:24349–56. doi: 10.1074/jbc.274.34.24349. [DOI] [PubMed] [Google Scholar]
- 28.Shinomiya H, Hagi A, Fukuzumi M, et al. Complete primary structure and phosphorylation site of the 65-kDa macrophage protein phosphorylated by stimulation with bacterial lipopolysaccharide. J Immunol. 1995;154:3471–8. [PubMed] [Google Scholar]
- 29.Basu A, Mohanty S, Sun B. Differential sensitivity of breast cancer cells to tumor necrosis factor-alpha: involvement of protein kinase C. Biochem Biophys Res Commun. 2001;280:883–91. doi: 10.1006/bbrc.2000.4209. [DOI] [PubMed] [Google Scholar]
- 30.Motzkus D, Maronde E, Grunenberg U, et al. The human PER1 gene is transcriptionally regulated by multiple signaling pathways. FEBS Lett. 2000;486:315–9. doi: 10.1016/s0014-5793(00)02315-2. [DOI] [PubMed] [Google Scholar]
- 31.Fricke K, Heitland A, Maronde E. Cooperative activation of lipolysis by protein kinase A and protein kinase C pathways in 3T3-L1 adipocytes. Endocrinology. 2004;145:4940–7. doi: 10.1210/en.2004-0803. [DOI] [PubMed] [Google Scholar]
- 32.Vemuri B, Singh SS. Protein kinase C isozyme-specific phosphorylation of profilin. Cell Signal. 2001;13:433–9. doi: 10.1016/s0898-6568(01)00164-4. [DOI] [PubMed] [Google Scholar]
- 33.Liedtke CM, Hubbard M, Wang X. Stability of actin cytoskeleton and PKC-delta binding to actin regulate NKCC1 function in airway epithelial cells. Am J Physiol Cell Physiol. 2003;284:C487–96. doi: 10.1152/ajpcell.00357.2002. [DOI] [PubMed] [Google Scholar]
- 34.Kilpatrick LE, Sun S, Mackie D, et al. Regulation of TNF mediated antiapoptotic signaling in human neutrophils: role of delta-PKC and ERK1/2. J Leukoc Biol. 2006;80:1512–21. doi: 10.1189/jlb.0406284. [DOI] [PubMed] [Google Scholar]
- 35.LaVallie ER, Chockalingam PS, Collins-Racie LA, et al. Protein kinase Czeta is up-regulated in osteoarthritic cartilage and is required for activation of NF-kappaB by tumor necrosis factor and interleukin-1 in articular chondrocytes. J Biol Chem. 2006;281:24124–37. doi: 10.1074/jbc.M601905200. [DOI] [PubMed] [Google Scholar]
- 36.Dobrowsky RT, Kamibayashi C, Mumby MC, et al. Ceramide activates heterotrimeric protein phosphatase 2A. J Biol Chem. 1993;268:15523–30. [PubMed] [Google Scholar]
- 37.Zeidan YH, Jenkins RW, Hannun YA. Remodeling of cellular cytoskeleton by the acid sphingomyelinase/ceramide pathway. J Cell Biol. 2008;181:335–50. doi: 10.1083/jcb.200705060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klemke M, Rafael MT, Wabnitz GH, et al. Phosphorylation of ectopically expressed L-plastin enhances invasiveness of human melanoma cells. Int J Cancer. 2007;120:2590–9. doi: 10.1002/ijc.22589. [DOI] [PubMed] [Google Scholar]
- 39.Smith L, Qutob O, Watson MB, et al. Proteomic screening of 725 antibodies simultaneously using antibody microarray technology to identify proteins associated with radiotherapy resistance in breast cancer cells. Breast Cancer Res. 2008;10:S1–49. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of all actin cytoskeleton genes mainlyexpressed in MCF-7 and 1001 cells as identified through one classSAM analysis of Actichip data. Genes mainly regulated in MCF-7 and1001 are sorted by their significance (d) score.
A, Transfection efficiency ofL-plastin GFP in MCF-7 cells. Untransfected (left) or L-plastin GFPtransfected (right) MCF-7 cells were plated on cover slips andanalysed by fluorescence microscopy. Bar, 100 μm.
Expression level of L-plastin GFP in MCF-7 cells. Cell extracts from untransfected and L-plastin GFP transfected MCF-7 cells were analysed by immunoblot using an anti-L-plastin (upper) or an anti-GAPDH (lower) antibody. Band at 97 kD, corresponding to L-plastin GFP fused protein, was detected in transfected cells.
data 3. Overexpression and knockdown L-plastin in MCF-7 and 1001 cells, respectively, did not affect cell phenotype and EMT markers expression.
A, Phase contrast microscopy of the morphology ofMCF-7 cells untransfected (Ctrl) and L-plastin GFP transfected(L-plastin-GFP) cells as well as of 1001 cells untransfected (Ctrl)and L-plastin siRNA transfected cells (siRNA L-plastin). Bar: 50μm.
B, Expression of epithelial and mesenchymal markerproteins in MCF-7 and 1001 cells described in A. Immunoblotanalysis was performed using L-plastin, E-cadherin-,β-catenin-, cytokeratin-18-, vimentin- or GAPDH-specificantibody as previously described [16].