

Abstract
Human MTG16a (CBFA2T3), a chromatin repressor with nucleolar localization, was described to act as a suppressor of breast tumourigenesis. Here we show that MTG16a is a novel ribosomal gene repressor, which can counteract MYC-driven activation of ribosomal RNA (rRNA) transcription. We also show that either knocking down MTG16a by RNA interference, or sequestering MTG16a outside the nucleolus of human breast epithelial cells, hampers acinar morphogenesis concomitant with up-regulation of rRNA synthesis and increased ribogenesis. This is the first demonstration that loss of MTG16a function in the nucleolus of breast epithelial cells can induce morphological and molecular changes typical of breast cancer initiation.
Keywords: MTG16a (CBFA2T3), nucleolus, rDNA, acinar morphogenesis, breast cancer
Introduction
The nucleolus is the site of ribogenesis and therefore the functional site of proteins necessary for ribosomal RNA (rRNA) regulation, synthesis and processing [1]. Specific transcription factors and co-factors act as activators or repressors of rRNA transcription [2, 3]. Activators of ribosomal gene (rDNA) transcription facilitate the assembly of the RNA polymerase I (Pol I) transcription machinery on the rDNA promoter regions and trigger the recruitment of chromatin-modifying enzymes (e.g. histone acetyltransferases, HATs) that induce an active chromatin conformation. In contrast, repressors of rDNA transcription prevent the assembly of the Pol I transcription machinery, and lock the rDNA genes into a silent state by recruiting enzymes (e.g. histone deacetylases, HDACs) that induce a repressed chromatin conformation [4, 5].
We previously found that MTG16a/CBFA2T3 (hereafter referred to as MTG16a), a member of the Myeloid Translocation Gene (MTG) family of transcriptional repressors ([6], and references within), accumulates in the nucleolus and leads to the recruitment of specific HDACs to this compartment [7]. This observation made us hypothesize that MTG16a may play a role in rDNA transcriptional repression. Here we provide, for the first time, evidence that MTG16a can induce down-regulation of rRNA synthesis in human cells, and can significantly counteract the up-regulation of rRNA synthesis driven by the Pol I transcription activator MYC.
Increased rRNA synthesis/ribogenesis is a well-known hallmark of cancer cells, including breast cancer cells [8–11]. MTG16 was reported to be lost in a fraction of breast tumours as a consequence of either loss of the 16q chromosome region containing the MTG16 locus, or epigenetic silencing due to aberrant DNA methylation [12–15]. In this study, we show that either MTG16a knockdown by RNA interference, or the sequestration of MTG16a outside the nucleolus of human breast epithelial cells, leads to an increase of rRNA synthesis and ribogenesis. Further, this is to our knowledge the first study showing that depriving the nucleolus of the rRNA-regulatory MTG16a function concomitantly affects ribogenesis and breast acinar morphogenesis, as it happens in the initial stages of breast tumourigenesis [16, 17].
Materials and methods
Cells and cell cultures
The human primary breast epithelial cell line HME31 (kindly provided by Dr. JW Shay, University of Texas Southwestern Medical Center, Dallas, TX, USA) [18] as well as the HME31-derived telomerase-immortalized, untransformed, cell strain hTERT-HME1 (here referred to as HME1) (Clontech, Mountain View, CA, USA) were grown in Mammary Epithelial Growth Medium (MEGM) as per manufacturer’s instructions (Lonza, Walkersville, MD, USA). The human immortalized untransformed breast epithelial cell line MCF10A (ATCC, Manassas, VA, USA) was grown as previously described [19]. The Hela cell line and the breast cancer cell lines Hs578T, MDA-MB-468 and MDA-MB-231 (ATCC) were grown in Dulbecco Modified Eagle Medium (DMEM) plus 5% foetal bovine serum (FBS) (Invitrogen, Carlsbad, CA, USA). Human foreskin fibroblasts (HFF), stably infected with either pBabe-MYC (HFF-MYC) or the cognate empty vector (HFF-Ctrl) [20], were grown in DMEM plus 10% FBS and 0.5 μg/ml puromycin. Stable cell clones were grown under the conditions used for the parental cell lines, in the presence of the selective antibiotic, and periodically tested for the presence of the transfected construct by both PCR and DNA sequencing analyses.
Transient and stable transfections
Transient transfections were performed by using either Lipofectamine 2000 or Lipofectamine Plus (Invitrogen) with the following constructs: pcDNA3.1-MTG16a, pcDNA3.1-MTG16a-V5, pcDNA-MTG16a-GFP, pcDNA-MTG16Δ1-3-V5, all described in [7], and pBabe-MYC (kindly provided by Dr. C. Grandori, University of Washington, Seattle, WA, USA). To develop MTG16 knockdown clones, short hairpin RNA (shRNA) sequences targeting MTG16 mRNA (MTG16-1: 5′-CTTCCCTCTGCGGCCGTTT-3′; MTG16-2: 5′-GAGTGGAAGCACCTCAACA-3′) and a control scrambled sequence (5′-ACGTACGTACGTAGTGGGG-3′), which does not recognize any human mRNA, were cloned into the pSuper-retro vector (Oligoengine, Seattle, WA, USA). The silencing efficiency of the pSuper-MTG16-1 and p-Super-MTG16-2 constructs on the MTG16a protein was established by quantitating the cells expressing MTG16a-GFP protein by immunocytochemistry of either HME1 or MCF10A cells transiently cotransfected with pcDNA3.1-MTG16a-GFP and a five-fold amount of either pSuper-MTG16-1 or pSuper-MTG16-2. Cells transfected with DNA of either one of the MTG16 silencing constructs, or the control construct, were selected in growth medium containing 1 μg/ml puromycin. Single clones expanded in the selective medium were tested for MTG16 knockdown by real time RT-PCR and Western blotting with an anti-MTG16 antibody [7]. To develop clones expressing the MTG16Δ1-3 mutant, HME1 cells were transfected with either pLNCX2-MTG16Δ1-3-V5 [21], or the empty pLNCX2 vector, and selected with 1 mg/ml G418. Single clones positive for MTG16Δ1-3-V5 expression were identified by immunocytochemistry with an anti-V5 antibody (Invitrogen) followed by goat anti-mouse Alexa Fluor 546 antibody (Invitrogen). To develop HME1-MYC and HME1-ctrl cells, HME1 were transfected with either pBabe-MYC or the cognate empty vector, respectively, and subsequently selected with 1 μg/ml puromycin. MYC-positive cells were identified both by PCR amplification of the transfected construct and by immunocytochemistry with an anti-MYC N-262 antibody (Santa Cruz Biotechnologies, Santa Cruz, CA, USA) followed by goat anti-rabbit Alexa Fluor 488 (Invitrogen). MYC expression was quantified on five random images (acquired with the same exposure and magnification) by normalizing the intensity of the MYC signal to the number of nuclei with the Quantity One software (BioRad, Hercules, CA, USA).
Three-dimensional (3D) cultures on reconstituted basement membrane
HME1 cells and derived clones were grown in 3D cultures on reconstituted basement membrane to induce breast epithelial cell differentiation into acini-like structures, essentially as described [19]. Briefly, single cells were induced to form acini on chamber slides coated with Matrigel (BD Biosciences, San Jose, CA, USA) in growth medium plus 2% Matrigel for 10–15 days. After fixation with 4% paraformaldehyde, the Golgi apparatus was identified with anti-GM130 antibody (BD Biosciences) followed by goat anti-mouse Alexa Fluor 546 antibody (Invitrogen). Integrin was identified with anti-CD49f antibody (Chemicon, Temecula, CA, USA) followed by anti-rat Alexa Fluor 488 antibody (Invitrogen). Nuclei were counterstained with 300 nM DAPI (Sigma, St. Louis, MO, USA). At least 30 acini per each clone were analysed by confocal microscopy (SP2 Spectral Confocal Microscope, Leica, Bannockburn, IL, USA) to inspect for the status of the lumen and apicobasal polarity. The acinar morphology observed in 70%, or more, of the acini was considered to be the prevalent morphology.
RT-PCR
Total RNA obtained with the single-step method using Trizol (Invitrogen) was treated with DNase I (Ambion, Austin, TX, USA), retrotranscribed into cDNA with SuperScript™ First-Strand Synthesis System (Invitrogen), and used for either semiquantitative RT-PCR or real time RT-PCR. Semiquantitative RT-PCR was performed by using GoTaq Flexi DNA polymerase (Promega, Madison, WI, USA) and primers (sense: 5′-CCTCAGCGATGCCGGACTC-3′; antisense: 5′-GTGGCTGCTGCCGTTCATCA-3′) amplifying a 148 bp fragment diagnostic for MTG16b, and a 223 bp fragment diagnostic for MTG16a. Quantitative real time RT-PCR (qRT-PCR) was performed on an iCycler (Bio-Rad) using the iQ SYBR Green Supermix (Bio-Rad) with primers specific for MTG16a (sense: 5′-TACGTGCCTGAGGACATCTG-3′; antisense: 5′-CGCAGTTCCAGCAGCTCTCG-3′) or the pre-rRNA (sense: 5′-CTGAGGGAGCTCGTCGGTGT-3′, antisense: 5′-GCAGAGCGCCTCCGAAGTCA-3′). MTG16a and rDNA transcript levels were quantitated by the Delta-delta Ct method, using the housekeeping gene GAPDH (sense: 5′-GAAGGTGAAGGTCGGAGTC-3′, antisense: 5′-GAAGATGGTGATGGGATTTC-3′) for normalization. Three independent experiments were performed in triplicate, and significance was determined by the Student t-test.
[3H]Uridine incorporation
Cells were incubated with growth medium plus 10 μCi/ml [5-3H]Uridine (1 mCi/ml, GE Healthcare, Buckinghamshire, UK) for 2 hrs. Radio-labelled RNA was extracted by using Trizol (Invitrogen), and 2 μg of RNA were resolved on 1% agarose gel. The rRNA 28S, 18S, and 5.8S band intensities were quantified by using Quantity One (BioRad). The bands were excised from the gel, weighted, and dissolved in buffer QG (Qiagen, Valencia, CA, USA) to reach a total volume (excised band + buffer QG) of 300 μl. Fifty μl aliquots were spotted on filter paper and let dry.
[3H]Uridine incorporation was quantified by liquid scintillation with Ready Safe liquid scintillation cocktail (Beckman, Fullerton, CA, USA). The [3H]Uridine signal was normalized to the corresponding band intensity. For the incorporation experiments on transiently transfected cells, cells were labelled with [5-3H]Uridine 48 hrs after transfection. Significance was calculated by using the Student’s t-test on three independent determinations.
Nuclear extracts
Nuclear extracts were prepared as described (http://www.ihcworld.com/_protocols/lab_protocols/lamond-lab-protocols.htm). Briefly, 5–10 × 106 cells were lysed in lysis buffer (10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT) supplemented with Complete protease inhibitor cocktail (Roche, Indianapolis, IN, USA). Nuclei were released by using a Dounce homogenizer, collected by centrifugation at 300 ×g for 5 min. at 4°C. Nuclear pellets were resuspended in 0.25 mM Sucrose, 10 mM MgCl2 plus protease inhibitors and further collected after centrifugation at 3000 ×g for 10 min. at 4°C over a layer of 0.88 mM sucrose, 0.5 mM MgCl2 plus protease inhibitors. Nuclear pellets were then used for Western blot.
Western blotting
Proteins from nuclear extracts were resolved on a SDS-PAGE gel and blotted by using standard procedures. Endogenous MTG16 was detected with anti-MTG16 antibody [7] using Hela expressing exogenous MTG16a as a positive control, while exogenous MTG16a-V5 was detected with anti-V5 antibody (Invitrogen). Histone H3, detected with anti-pan histone H3, CT (Millipore, Billerica, MA, USA), was used as a loading control for nuclear extracts. Incubation with the primary antibody was followed by incubation with appropriate HRP-conjugated secondary antibodies (GE Healthcare, Buckinghamshire, UK) and ECL detection (GE Healthcare).
Immunocytochemistry
Cells were fixed in 4% paraformaldehyde for 7 min., permeabilized with PBS plus 0.1% Triton X100 for 5 min., blocked with PBS containing 1% BSA, 1% goat serum and 0.05% Tween 20 for 30 min., incubated with the primary antibody for 1 hr, rinsed, and incubated for 1 hr with the secondary antibody. MTG16-V5 or MTG1616Δ1-3-V5 was detected with either a rabbit anti-MTG16 antibody [7] or a mouse anti-V5 antibody (Invitrogen), B23 was detected with a mouse anti-B23 antibody (Santa Cruz Biotechnologies) and MYC was detected with a rabbit anti-MYC (N-262) antibody (Santa Cruz Biotechnologies). For detection of endogenous MTG16, cells were incubated with anti-MTG16 antibody over night at 4°C. After incubation with the appropriate fluorescent secondary antibodies (Alexa Fluor goat anti-rabbit 488 or Alexa Fluor goat anti-mouse 546, Invitrogen) and counterstaining with DAPI, cells were mounted with Vectashield (Vector Laboratories, Burlingame, CA, USA), and analysed with a fluorescence microscope (Axioskop, Zeiss, Thornwood, NY, USA).
NORs silver staining (AgNORs)
NORs were stained essentially as described [22] with minor modifications. Briefly, subconfluent cells grown onto 8-well chamber slides for 2–3 days were fixed with 95% Ethanol for 5 min. followed by incubation with Carnoy’s solution (absolute ethanol:glacial acetic acid 3:1, by Vol.) for 30 min. Slides were then hydrated through graded alcohol, and incubated in NOR silver-staining solution (obtained by mixing a 2% Gelatin solution in 1% formic acid with a 50% silver nitrate solution at a ration 1:2) for 12 min. at 37°C in the dark. Slides were washed with distilled water, let dry and mounted in Permount histological mounting medium (Fisher Scientific, Fair Lawn, NJ, USA). AgNORs size and number were analysed by using Image J (NIH) on a minimum of 250 cells per slide taken from random fields. Significance was calculated by using the Student’s t-test on three independent experiments performed in duplicate.
Quantitative chromatin immunoprecipitation (qChIP)
ChIP was performed using reagents purchased from Millipore, following the manufacturer’s protocol. Endogenous MTG16 occupancy at the rDNA promoter was assessed by ChIP with an antibody against the MTG16 C-terminus [7]. Exogenous MTG16a-V5 occupancy at the rDNA promoter was assessed by ChIP with an anti-V5 antibody (Invitrogen) in combination with anti-MTG16 antibody. Control ChIPs were performed with non-specific IgGs (Millipore). The immunoprecipitated DNA was amplified by real-time PCR with primers specific for either a region within the rDNA promoter (region ‘a’) (sense: 5′-GTGTGTGGCTGCGATGGTG-3′; antisense: 5′-CCAACCTCTCCAGCGACAGG-3′) or a control region in the 28S rDNA (region ‘c’) (sense: 5′-GGACCCGAAAGATGGTGAACTA-3′; antisense: 5′-GAGAGCGCCAGCTATCCTGAG-3′). Histone modifications were assessed by ChIP with either anti-Acetyl Histone H4 (AcH4) or anti-tri-methylated histone H3 lysine 9 (tri-met-H3K9) (Millipore). Control ChIPs were performed with non-specific IgGs (Millipore). The immunoprecipitated DNA was amplified by real-time PCR with primers specific for either the rDNA promoter region (region ‘a’) or a control region in the GAPDH promoter [23]. The DNA relative enrichment was calculated by using the Delta-delta Ct method. The PCR signals obtained for each gene region were normalized to the PCR signal obtained from the input DNA (total chromatin fraction). Significance was calculated by using the Student t-test on three independent determinations.
Results
MTG16a localizes in the nucleolus and represses rRNA synthesis in Hela cells
We previously found that MTG16a, one of the two proteins encoded by the human MTG16 gene, accumulates in the nucleolus because it is endowed with a novel nucleolar localization signal [7]. Our former observation that MTG16a colocalizes with histone deacetylases (HDACs) in the nucleolus led us to hypothesize that MTG16a plays a role in chromatin-mediated repression of rRNA synthesis [7]. Preliminary results obtained in a series of experiments on Hela cells corroborated our hypothesis. Specifically, we found that MTG16a not only localizes in the nucleolus of Hela cells (Fig. 1A), but also associates with the rDNA promoter region, and leads to a significant decrease of both rRNA synthesis and rRNA level. First, in chromatin immunoprecipitation (ChIP) experiments with an anti-MTG16 antibody capable of detecting the endogenous MTG16 protein (data not shown), followed by quantitative real time PCR with primers encompassing either ‘region a’ within the rDNA promoter, or ‘region c’ within the 28S region (Fig. 1B, top), we detected a significant (P < 0.01) enrichment of ‘region a’ relative to ‘region c’ in the chromatin immunoprecipitated with anti-MTG16 (Fig. 1B, bottom left). Further, after testing that the viability of Hela cells transiently transfected with MTG16a-V5 for 48 hrs was not affected (data not shown), we found by ChIP with a mixture of both anti-V5 and anti-MTG16 antibodies that MTG16a-V5 was associated significantly (P < 0.05) more with the rDNA promoter ‘region a’ than with the control ‘region c’ (Fig. 1B, bottom right). Second, by quantitating the incorporation of [3H]Uridine (2 hrs) into the rRNA of Hela cells transiently transfected with either MTG16a-V5, or the cognate empty vector, we assessed that MTG16a significantly (P < 0.05) decreased the level of newly synthesized [3H]Uridine-labelled rRNA (Fig. 1C, left). By real time RT-PCR with primers specifically annealing a region (region ‘b’) within the 5′ ETS of human pre-rRNA (hereafter referred to as rRNA) (Fig. 1B, top), we also found a significantly (P < 0.001) lower level of rRNA in Hela cells transiently transfected with MTG16a-V5 relative to control cells transfected with the cognate empty vector (Fig. 1C, right). Parallel quantitative ChIP analysis of the same cells showed that the down-regulation of rRNA synthesis was associated with a significant (P < 0.01) decrease of histone H4 acetylation (AcH4, a hallmark of active chromatin) and a significant (P < 0.01) increase of histone H3 lysine 9 tri-methylation (tri-met-H3K9, a hallmark of inactive chromatin) in the chromatin associated with the rDNA promoter ‘region a’ (Fig. 1D). Consistently, we also found that MTG16a can interact and co-localize with TIP5 (Fig. S1), a major component of the nucleolar remodelling complex (NoRC). NoRC is a well-known rDNA repressor complex able to recruit chromatin modifying activities [4, 5].
Fig 1.

MTG16a localizes in the nucleolus and represses rRNA synthesis in Hela cells. (A) Immunocytochemistry showing that exogenous MTG16a-V5 localizes mainly in the nucleolus of Hela cells. (B) ChIP with anti-MTG16 of untransfected Hela, followed by real time PCR with primers amplifying either a region in the rDNA promoter or a control region in the 28S rDNA (regions ‘a’ and ‘c’, respectively, in the scheme on top), shows that endogenous MTG16 is associated significantly (P < 0.01) more with the rDNA promoter than the control region (bottom, left). ChIP with both anti-MTG16 and anti-V5 of Hela transiently transfected with MTG16a-V5 shows that exogenous MTG16a is associated significantly (P < 0.05) more with the rDNA promoter than the control region (bottom, right). (C) [3H]Uridine incorporation analysis shows that transient expression of MTG16a-V5 significantly (P < 0.001) decreases rRNA synthesis in Hela cells; the right panel shows that equal amounts of RNA were loaded on agarose gel for quantification of the [3H]-labelled rRNA (left). qRT-PCR with primers amplifying a region in the 5′ ETS of pre-rRNA (region ‘b’) shows that transient expression of MTG16a-V5 significantly (P < 0.001) decreases the rRNA level in Hela cells (right). (D) ChIP analysis shows that transient expression of MTG16a-V5 significantly (P < 0.01) decreases H4 acetylation (AcH4, left) and significantly (P < 0.01) increases histone H3 lysine 9 tri-methylation (tri-met-H3K9, right) at the rDNA promoter. The bars in this figure represent the average of three determinations ± standard deviation.
These overall preliminary results strengthened our hypothesis that MTG16a has a function in the repression of rRNA synthesis, and prompted us to perform additional experiments to further test whether this is the case.
MTG16a counteracts MYC-induced up-regulation of rRNA synthesis in human cells
The action of rDNA repressor complexes at the rDNA promoter region is counteracted by activator complexes [2, 3]. Recently, the oncoprotein MYC was shown to be involved in the activation of rRNA synthesis [24–26]. Thus, to further investigate whether MTG16a is involved in rDNA repression, we set out to test whether MTG16a can counteract MYC-induced up-regulation of rRNA synthesis. To this end, we used the cell system developed by Benanti et al. [20] consisting of human foreskin fibroblasts stably infected with human MYC (HFF-MYC). HFF-MYC fibroblasts display both an increased rRNA synthesis (Fig. 2A, left) and an increased rRNA level (Fig. 2A, right) relative to HFF control (HFF-Ctrl) fibroblasts. MTG16a-V5 transiently transfected in HFF-MYC fibroblasts accumulates in the nucleolus (Fig. 2B) and induces a significant (P < 0.05) decrease in both rRNA synthesis (Fig. 2C, left) and rRNA level (Fig. 2C, right). The opposing action of MYC and MTG16a on rDNA transcription was further confirmed by an rDNA promoter-luciferase reporter assay in Hela cells (Fig. S2).
Fig 2.

MTG16a counteracts MYC-induced up-regulation of rRNA synthesis in human fibroblasts. (A) [3H]Uridine incorporation (left) and qRT-PCR analysis (right) show that human fibroblasts stably expressing exogenous MYC (HFF-MYC) display significantly (P < 0.05) higher rRNA synthesis and rRNA level relative to control fibroblasts carrying the cognate empty vector (HFF-ctrl). (B) Immunocytochemistry with anti-V5 antibody shows that MTG16a-V5 transiently transfected into HFF-MYC cells can localize in the nucleolus. (C) Transient transfection of HFF-MYC with MTG16a-V5 leads to a significant (P < 0.05) decrease of both rRNA synthesis (left) and rRNA level (right), as shown by [3H]Uridine incorporation and qRT-PCR, respectively. The bars in this figure represent the average of three determinations ± standard deviation.
These findings showing that MTG16a can counteract MYC-induced rRNA synthesis up-regulation, further implicate MTG16a as a repressor of rRNA synthesis.
MTG16a down-regulates rRNA synthesis in human breast epithelial cells
To further investigate the involvement of MTG16a as a repressor of rRNA synthesis, we focused our investigations on breast epithelial cells. We chose this cell model for two reasons: first, MTG16 was reported to act as a breast tumour suppressor [15], and second, nucleolar morphological changes that reflect defects in ribogenesis and rRNA synthesis are known to occur in the process of human breast tumourigenesis [16, 27].
We selected two immortalized, untransformed breast epithelial cells, HME1 and MCF10A, that express only MTG16a, but not MTG16b (Fig. 3A). By immunocytochemistry with an antibody specific for MTG16 [7], we found that in both cell lines endogenous MTG16a localizes both in the nucleoplasm and the nucleoli, which were identified with an antibody against the nucleolar protein B23 (Fig. 3B). Consistently, transiently expressed exogenous MTG16a-V5 also localized in the nucleolus, as shown here for HME1 cells (Fig. 3C). MTG16a-V5-transfected HME1 cells displayed both a significantly (P < 0.05) lower rRNA synthesis (Fig. 3D, left) and a significantly (P < 0.05) lower rRNA level (Fig. 3D, right) relative to cells transfected with the cognate empty vector.
Fig 3.

MTG16a down-regulates rRNA synthesis in human breast epithelial cells. (A) RT-PCR showing that the two human breast epithelial cell lines MCF10A and HME1 express only MTG16a but not MTG16b (top); Western blot of nuclear extracts using anti-MTG16 antibody confirms MTG16a expression in both cell lines (bottom). (B) Immuncytochemistry showing that endogenous MTG16a, detected with anti-MTG16 (green), can localize both in the nucleoplasm and in the B23-positive nucleoli (red) of both HME1 and MCF10A. Nuclei are counterstained with DAPI (blue). (C) Exogenous MTG16a-V5 can be detected both in the nucleoplasm and the nucleolus by IC with anti-V5 antibody. (D) Transient expression of MTG16a-V5 in HME1 cells induces a significant (P < 0.05) decrease of both rRNA synthesis (left) and rRNA level (right). The bars in this figure represent the average of three determinations ± standard deviation.
Based on these results, it seems that MTG16a plays a role as a repressor of rRNA synthesis in human cells, including breast epithelial cells.
MTG16a counteracts MYC-induced up-regulation of rRNA synthesis in human breast epithelial cells
Next, we tested whether MTG16a, as in the HFF-MYC fibroblast model, can counteract MYC-induced rRNA up-regulation in breast epithelial cells. We stably transfected HME1 cells with either human MYC (HME1-MYC cells) or the cognate empty vector (HME1-ctrl cells). MYC-overexpressing HME1 cells (Fig. 4A) showed a significant (P < 0.05) increase of both rRNA synthesis (Fig. 4B, left) and rRNA level (Fig. 4B, right) relative to control cells. MYC-induced up-regulation of rRNA synthesis in HME1 cells could be significantly (P < 0.05) decreased by transient expression of exogenous MTG16a (Fig. 4C), strengthening the conclusion that MTG16a can counteract the action of MYC on rRNA synthesis in different cell types.
Fig 4.

MTG16a counteracts MYC-induced up-regulation of rRNA synthesis in human breast epithelial cells. (A) Immunocytochemistry with anti-MYC antibody (left) followed by digital quantification (right) shows MYC overexpression in HME1 cells stably transfected with human MYC (HME1-MYC) relative to control HME1 cells transfected with the cognate empty vector. (B) HME1-MYC cells display significant (P < 0.05) up-regulation of both rRNA synthesis (left) and rRNA level (right) relative to control cells carrying the cognate empty vector (HME1-ctrl). (C) Transient expression of exogenous MTG16a-V5 significantly (P < 0.05) decreases both rRNA synthesis (left) and rRNA level (right) of HME1-MYC cells. The bars in this figure represent the average of three determinations ± standard deviation.
Low MTG16 expression is associated with an increased rRNA level in breast cancer cells
We tested the rRNA level of a few human breast cancer cell lines, including Hs578T, MDA-MB-468 and MDA-MB-231, previously reported to have a decreased MTG16 transcript level relative to normal breast tissue [15], and a primary human breast epithelial strain HME31 [18]. We found that a lower level of MTG16 transcript in the breast cancer lines (Fig. 5, left) correlated with a significantly higher rRNA level (Fig. 5, right) relative to the untransformed breast epithelial cells (HME31). Thus, there seems to be an association between loss of MTG16a and increased rRNA level.
Fig 5.
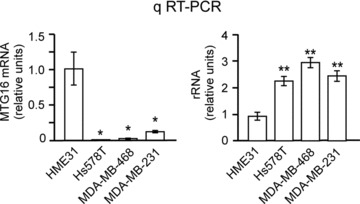
Low MTG16 expression is associated with an increased rRNA level in breast cancer cells. qRT-PCR showing that the breast cancer cell lines Hs578T, MDA-MB-231 and MDA-MB-468 have a significantly (P < 0.05) lower level of MTG16 (left) and a significantly (P < 0.01) higher rRNA level (right) than the primary breast epithelial cell line HME31. The bars in this figure represent the average of three determinations ± standard deviation.
MTG16a knockdown in human breast epithelial cells induces an increase of rRNA synthesis and NORs
To test whether there is a causal link between a lower level of MTG16a transcript and up-regulation of rRNA synthesis in breast epithelial cells, we knocked down MTG16a in HME1 cells by RNA interference (RNAi) with either one of two MTG16-specific short hairpin sequences. First, we found that the expression of exogenous MTG16a-GFP protein in the nucleus/nucleolus of HME1 (Fig. 6A, left) could be efficiently decreased with two distinct silencing sequences, shMTG16-1 and shMTG16-2 (Fig. 6A, right). Next, we selected stable clones with MTG16a knockdown greater than 70% (Fig. 6B). These clones displayed, in average, a significant increase in both rRNA synthesis (P < 0.01) (Fig. 6C, left) and rRNA level (P < 0.05) (Fig. 6C, right) relative to control clones. Similarly, stable MTG16a knockdown clones derived from MCF10A cells also showed a significantly higher rRNA level (Fig. S3A).
Fig 6.

MTG16a knockdown in human breast epithelial cells induces an increase of rRNA synthesis and NORs. (A) Exogenous MTG16a-GFP expression in HME1 cells (left) decreases significantly after RNA interference with shMTG16-1 and shMTG16-2 sequences (right). (B) HME1 clones carrying either pSuper-MTG16-1 (such as H16-1a and H16-1b) or pSuper-MTG16-2 (such as H16-2a and H16-2b) have a decreased MTG16a level relative to the HME1 parental cell line or a control clone (H-ctrl), as shown by Western blot with anti-MTG16 on nuclear extracts. Histone H3 was used as loading control. (C) HME1-derived MTG16a knockdown clones display both a significantly (P < 0.01) higher rRNA synthesis (left) and a significantly (P < 0.05) higher rRNA level (right) relative to control cells. The bars represent the average of at least three determinations on two independent clones ± standard deviation. (D) Digital analysis of silver stained nucleoli (right) shows a significant increase both in NORs number and NORs total area in HME1-derived MTG16a knockdown clones relative to the cognate control clones (left).
Next, we tested whether up-regulation of rRNA synthesis in the MTG16a knockdown clones was associated with an increase in the nucleolar organizer regions (NORs). The number and area of NORs, also known as the AgNOR value, can be detected by silver staining of argyrophilic proteins involved in rRNA transcription and processing. The AgNOR value reflects ribogenesis [28], and typically increases during breast tumourigenesis [16, 27]. We estimated the AgNOR value by digitally evaluating both the size and the number of the AgNORs of silver stained cells. As shown in Figure 6D, the HME1-derived MTG16a knockdown clones displayed a significantly increased AgNOR value relative to the AgNOR value of control clones.
These findings support the conclusion that MTG16a knockdown in breast epithelial cells promotes both up-regulation of rRNA synthesis and increased ribogenesis.
Up-regulation of rRNA synthesis in breast epithelial cells is associated with impaired acinar morphogenesis
Untransformed breast epithelial cell lines, such as HME1 and MCF10A, are able to form mammary gland-like acini in 3D culture on reconstituted basement membrane [17, 29]. Under these conditions, untransformed cells grow as hollow acini lined by a polarized epithelium, while transformed cells typically display defects in acinar morphogenesis. By using this method, we found that both MYC overexpression (Fig. 7A) and MTG16a knockdown (Fig. 7B) in HME1 cells resulted in impaired acinar morphogenesis in more than 70% of the acini. Similarly, MTG16a knockdown clones derived from MCF10A cells also showed aberrant acinar morphogenesis (Fig. S3B). Apparently, up-regulation of rRNA synthesis, either induced by MYC overexpression or MTG16a knockdown, correlates with morphological defects of the acini in 3D culture.
Fig 7.
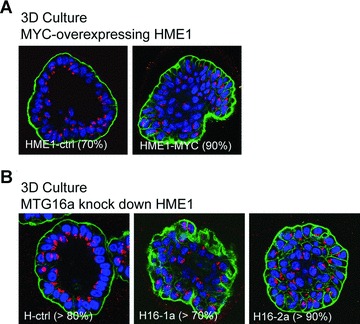
Up-regulation of rRNA synthesis in breast epithelial cells is associated with impaired acinar morphogenesis. (A) Three-dimensional (3D) culture on reconstituted basement membrane followed by confocal microscopy shows that HME1-MYC cells are unable of proper acinar morphogenesis in more than 90% of the acini, as indicated by the inability to form hollow, polarized acini, with organized arrangement of the nuclei (blue), presence of an outer layer of integrin (green) and apical localization of the Golgi apparatus (red). (B) HME1 clones (H16-1a and H16-2a) knocked down for MTG16a are also unable of proper acinar morphogenesis in 3D cultures in more than 70% of the acini, as opposed to control clones forming more than 80% hollow, polarized acini.
Sequestering MTG16a outside the nucleolus is sufficient to increase ribogenesis and disrupt 3D acinar morphogenesis
Whether there is a causal link between rRNA up-regulation and cell transformation, or vice versa is still debated [11]. To start to address this complex issue, we tested whether depriving HME1 cells of just the MTG16a nucleolar function is sufficient to recapitulate the two effects observed after MTG16a knockdown: increased rRNA level/ribogenesis and disruption of acinar morphogenesis. Based on our previous demonstration that MTG16a can be relocalized within the cell upon oligomerization with other MTG proteins [7], we set out to sequester MTG16a from the nucleolus into the nucleoplasm by expressing a mutant MTG16Δ1-3, which lacks the MTG16a nucleolar localization sequence, but maintains the MTG-MTG oligomerization domain [7]. By immunocytochemistry, we determined that while the wild-type MTG16a transfected in HME1 cells can localize both in the nucleoplasm and the nucleolus, the MTG16Δ1-3-GFP mutant cannot localize in the nucleolus (Fig. 8A, top). Based on immunocytochemistry of HME1 cells co-transfected with both MTG16Δ1-3-GFP and MTG16a-V5, MTG16Δ1-3 seemed capable of sequestering the wild-type MTG16a in the nucleoplasm (Fig. 8A, middle). By using an anti-MTG16 able to detect both endogenous MTG16a and exogenous MTG16Δ1-3, in combination with an antibody for the nucleolar marker B23, the nucleoli appeared to be depleted of endogenous MTG16a in cells transfected with MTG16Δ1-3-V5 (Fig. 8A, bottom).
Fig 8.
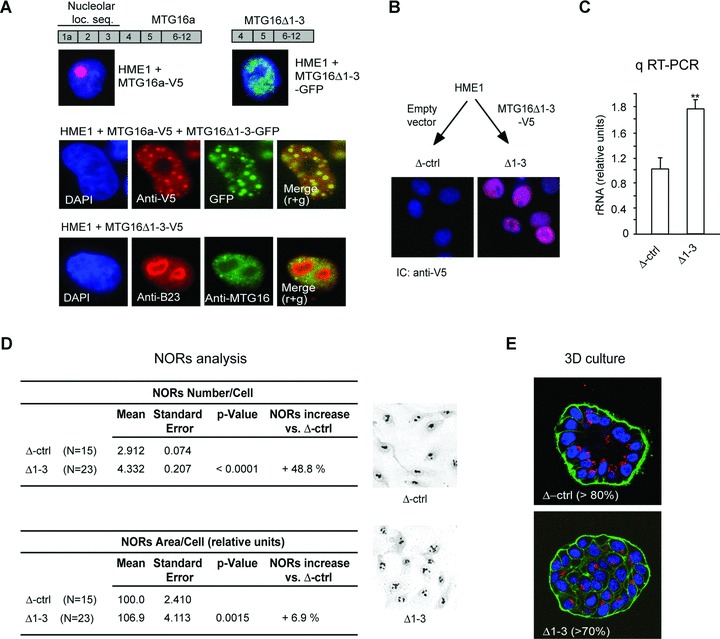
Sequestering MTG16a outside the nucleolus is sufficient to increase rRNA level and ribogenesis as well as disrupt acinar morphogenesis. (A) The V5-tagged wild-type MTG16a protein, containing the nucleolar localization sequence encoded by exons 1a-3, is targeted to the nucleolus of HME1 cells, while the GFP-tagged MTG16Δ1-3 deletion mutant protein, lacking the nucleolar localization sequence, cannot localize in the nucleolus (top). Immunocytochemistry of HME1 cells co-transfected with MTG16a-V5 and MTG16Δ1-3-GFP shows co-localization of the two exogenous proteins in the nucleoplasm (middle). Expression of MTG16Δ1-3-V5 in HME1 cells leads to depletion of endogenous MTG16a (detected with anti-MTG16, green) from the nucleoli (visualized with anti-B23, red). Nuclei are counterstained with DAPI (blue) (bottom). (B) Representative HME1 clone stably transfected with MTG16Δ1-3-V5 showing localization of the deletion mutant protein (red) in the nucleus (blue). (C) Δ1-3 cells display a significantly (P < 0.01) higher rRNA level than control cells (Δctrl). (D) Digital analysis of silver stained slides (right) shows a significant increase both in NORs number and NORs total area in Δ1-3 cells when compared to their cognate control cells. (E) Δ1-3 cells are unable of proper acinar morphogenesis when grown in 3D basement membrane culture.
Next, we developed stable HME1 clones carrying MTG16Δ1-3-V5 (Δ1-3) and control clones (Δ-Ctrl) carrying the cognate empty vector (Fig. 8B) to assess ribogenesis and 3D acinar morphogenesis. Δ1-3 cells displayed a significantly (P < 0.01) higher rRNA level compared to control cells (Fig. 8C). Consistently, Δ1-3 cells displayed also a significant increase (P < 0.001) in the AgNOR value (Fig. 8D). Finally, by growing Δ1-3 cells in 3D basement membrane culture, it was apparent that > 70% of the acini had an aberrant morphology (Fig. 8E). Even if it cannot be discounted that the observed changes are in part due to the accumulation of MTG16a/MTG16Δ1-3 oligomers in the nucleoplasm, it seems that keeping MTG16a from functioning in the nucleolus is sufficient to recapitulate the effects observed in HME1 cells after MTG16a knockdown: increased rRNA level, increased ribogenesis and aberrant acinar morphogenesis.
Discussion
In this study, we provide novel evidence that one of the functions of MTG16a, a member of the MTG family of transcriptional repressors, is to act as an rDNA repressor in human cells. Further, we show, for the first time, that loss of MTG16a rDNA repressor function in human breast epithelial cells leads to increased ribogenesis and disrupts acinar morphogenesis. This latter finding points at a potential mechanism at the basis of MTG16a tumour suppressor function in the breast.
We gathered evidence that MTG16a functions as a component of an rDNA repressor complex in different human cell types by using complementary strategies such as [3H]uridine incorporation into newly synthesized rRNA, and quantitatation of pre-rRNA. We do not know yet the features of the rDNA repressor complex of which MTG16a seems to be a component, but we found significant that MTG16a can interact and co-localize with TIP5, a component of the NoRC, known for playing a key role in chromatin-mediated rDNA repression [5]. The NoRC complex acts by recruiting chromatin-modifying enzymes (e.g. histone deacetylases, HDACs) to induce a repressed chromatin status at the rDNA promoter [4, 5]. Previously, we reported that MTG16a can recruit HDACs into the nucleolus [7]. In this study, we show that MTG16a-induced rDNA repression is marked by qualitative and quantitative histone changes at the chromatin associated with the rDNA promoter region. In addition to histone H4 deacetylation, MTG16a-induced rDNA repression is associated with an increased level of histone H3 K9 tri-methylation. Thus, nucleolar MTG16a may cooperate in a repressor complex by enabling the recruitment of histone modifying enzymes via the C-terminal domains conserved in the MTG proteins [30].
Regulation of rRNA synthesis is determined by the opposing action of transcriptional repressor and activator complexes [2, 3]. The oncoprotein MYC is a known activator that promotes Pol I-driven transcription of ribosomal genes by directly interacting with the rDNA promoter region [24–26]. In this study, we show that MTG16a can efficiently counteract MYC-induced rRNA activation in the same cell model, consisting of human fibroblasts stably overexpressing MYC, which led to the elegant demonstration that MYC is a component of an rDNA activator complex driving Pol I transcription [20, 25]. Apparently, MTG16a negatively interferes with MYC-mediated Pol I activation of rRNA synthesis. It is tempting to speculate that there is a competitive action between repressor and activator complexes involved in the regulation of rRNA synthesis. However, it remains to be demonstrated whether the MTG16a-containing repressor complex and the MYC-containing activator complex act on, and possibly compete for, the same rDNA regulatory regions (Fig. 9).
Fig 9.
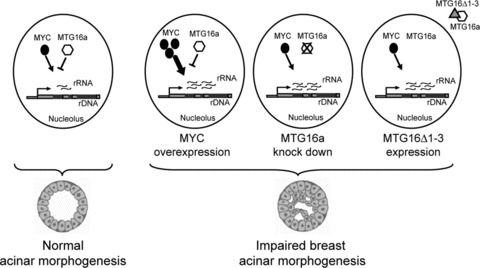
Scheme summarizing the potential relationship between rRNA synthesis level and breast acinar morphogenesis. A proper rRNA synthesis level, maintained by the opposing action of activator complexes (e.g. containing MYC) and repressor complexes (e.g. containing MTG16a), would associate with normal acinar morphogenesis (left). An increased rRNA synthesis level, due to different factors (e.g. MYC overexpression, MTG16a knockdown, or depletion of MTG16a from the nucleolus via the MTG16Δ1-3 mutant) able to alter the balance between rDNA activator and repressor complexes, would associate with impaired acinar morphogenesis (right).
Another relevant aspect of this study is that, for the first time, we show that there is a dual biological effect due to the abrogation of the MTG16a-mediated rDNA repression function in the nucleolus of human breast epithelial cells: an effect on ribogenesis, and an effect on acinar morphogenesis. Apparently, MTG16a acts as a repressor of rRNA synthesis not only in human fibroblasts but also in human breast epithelial cells, where it can counteract MYC-induced rRNA synthesis. Loss of MTG16a has been reported in breast cancer [14, 15], thus suggesting that MTG16 might act as a tumour suppressor [15]. By screening of a few breast cancer cell lines and untransformed primary breast epithelial cells, there seems to be an inverse association between the level of MTG16 transcript and the level of rRNA transcript. By knocking down MTG16a in untransformed breast epithelial cells by stable RNA interference, we induced not only an increase in both rRNA synthesis and rRNA level, but also an increase in the AgNOR value. The AgNOR value, which is a hallmark of increased ribogenesis, has been reported by several pathologists to increase in the course of breast tumourigenesis [27]. An increased AgNOR value can be detected at very early stages of breast tumourigenesis, in association with morphogenetic changes of atypia or ductal carcinoma in situ[16, 27]. Notably, we found that the increased AgNOR value in MTG16a knockdown breast epithelial cells correlated with impaired in vitro acinar morphogenesis, a phenotype indicative of breast epithelial transformation. Indeed, when grown in reconstituted basement membrane 3D culture, MTG16 knockdown HME1 cells formed acini with a filled lumen, altered apicobasal polarization, and presence of proliferating, Ki67-positive, cells (Fig. S4). Remarkably, by sequestering MTG16a outside the nucleolus of HME1 cells through oligomerization with a mutant MTG16a protein lacking the nucleolar localization domain, it seems that preventing MTG16a from performing its function in the nucleolus is sufficient to lead to an increase of both the rRNA level and ribogenesis, again in concomitance with the disruption of acinar morphogenesis.
In conclusion, this study demonstrates that MTG16a plays a role in the repression of rRNA synthesis in different human cells, and that the loss of MTG16a rDNA regulatory function in the nucleolus of human breast epithelial cells clearly is associated with increased rRNA synthesis/ribogenesis. Further, this study supports a role for MTG16a loss in the initiation of breast epithelial transformation. However, we do not know yet whether the increased rRNA synthesis/ribogenesis, consequent to the loss of MTG16a rDNA repressor function, is the cause, or the consequence, of the aberrant morphological reconfiguration of the acinar architecture of breast epithelial cells.
Acknowledgments
We wish to thank Dr. Carla Grandori, University of Washington, for kindly providing the HFF-MYC model and useful suggestions, Dr. Raffaella Santoro and Dr. Ingrid Grummt, German Cancer Research Center, Heidelberg, for kindly providing the FLAG-TIP5 construct and for critical advice, Dr. Massimo Derenzini, University of Bologna, for helpful discussion, Dr. Alan Hutson and Dr. Gaia Bistulfi, Roswell Park Cancer Institute, for assistance with the statistical analysis and assistance with the design of the MTG16 silencing sequences. We especially thank John Fischer, Roswell Park Cancer Institute, for valuable technical assistance. We are also indebted to the anonymous reviewers that, with their critical suggestions, enabled us to improve this paper. This work was supported by the Susan Komen Foundation (NS and SR), the Roswell Park Alliance Foundation (NS) and the Breast Cancer Coalition of Rochester (NS).
Supporting Information
Fig. S1 MTG16a interacts and co-localize with the NoRCcomponent TIP5. Immunoprecipitation with anti-FLAG antibodyfollowed by Western blotting with anti-MTG16 antibody on Hela cellsco-transfected with both MTG16a and FLAG-TIP5 showing invitro interaction between the two proteins (left). Immunocytochemistry on Hela cells transfected with either FLAG-TIP5 or MTG16a showing that both proteins can localize in the nucleus and the nucleolus (right, top and middle). Co-transfection with both FLAG-TIP5 and MTG16a followed by immunocytochemistry with both anti-MTG16 and anti-FLAG antibodies shows co-localization of the two proteins (right, bottom).
Fig. S2 MTG16a counteracts both physiological andMYC-induced rDNA transcription in Hela cells. Hela cells grown in a24 well plate were transfected with 100 ng pHrD-IRES-Luc,expressing luciferase under the control of the rDNA promoter(kindly provided by Dr. S. Majumer, Ohio State University), 10 ngpRL-TK, and the indicated amounts of pcDNA3.1-MTG16a and pBabe-MYC.Luciferase activity was measured 48 h after transfection by usingDual Glow Luciferase Assay System (Promega). MTG16a cansignificantly (P < 0.05) repress both physiological and MYC-induced pHrD-IRES-Luc expression in a dose-dependent fashion.
Fig. S3 MTG16a knockdown in MCF10A cells leads toincreased rRNA level and impaired acinar morphogenesis. (A)MCF10A-derived MTG16a knockdown clones display, in average, asignificantly (P < 0.05) higher rRNA level relative tocontrol cells. (B) Confocal microscopy showing thatMCF10A-derived MTG16a knockdown clones (clones M16-1 and M16-2 areshown) are unable of proper acinar morphogenesis in 3D basementmembrane cultures in more than 70% of the acini, asopposed to control clones (M-ctrl) forming more than 80%hollow, polarized acini. Nuclei are visualized in blue, integrin ingreen and the Golgi apparatus in red.
Fig. S4 Evidence of proliferating cells in morphologically aberrant acini derived from HMEI cells with stable MTG16a knockdown. Confocal microscopy of acini grown in 3D basement membrane culture, showing the presence of cells positive for the proliferation marker Ki67 in morphologically aberrant acini of H16-1a and H16-2a clones with stable MTG16a knockdown relative to a control clone (H-ctrl). Ki67 (green) was detected with anti-Ki67 (Zymed) and visualized with Alexa Flour 488 anti-rabbit, while nuclei (blue) were counterstained with DAPI.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Boisvert FM, van Koningsbruggen S, Navascues J, et al. The multifunctional nucleolus. Nat Rev Mol Cell Biol. 2007;8:574–85. doi: 10.1038/nrm2184. [DOI] [PubMed] [Google Scholar]
- 2.Russell J, Zomerdijk JC. The RNA polymerase I transcription machinery. Biochem Soc Symp. 2006:203–16. doi: 10.1042/bss0730203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grummt I. Life on a planet of its own: regulation of RNA polymerase I transcription in the nucleolus. Genes Dev. 2003;17:1691–702. doi: 10.1101/gad.1098503R. [DOI] [PubMed] [Google Scholar]
- 4.Grummt I. Different epigenetic layers engage in complex crosstalk to define the epigenetic state of mammalian rRNA genes. Hum Mol Genet. 2007:R21–7. doi: 10.1093/hmg/ddm020. 16 Spec No 1. [DOI] [PubMed] [Google Scholar]
- 5.Santoro R. The silence of the ribosomal RNA genes. Cell Mol Life Sci. 2005;62:2067–79. doi: 10.1007/s00018-005-5110-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rossetti S, Hoogeveen AT, Sacchi N. The MTG proteins: chromatin repression players with a passion for networking. Genomics. 2004;84:1–9. doi: 10.1016/j.ygeno.2004.02.011. [DOI] [PubMed] [Google Scholar]
- 7.Hoogeveen AT, Rossetti S, Stoyanova V, et al. The transcriptional corepressor MTG16a contains a novel nucleolar targeting sequence deranged in t (16; 21)-positive myeloid malignancies. Oncogene. 2002;21:6703–12. doi: 10.1038/sj.onc.1205882. [DOI] [PubMed] [Google Scholar]
- 8.Maggi LB, Jr, Weber JD. Nucleolar adaptation in human cancer. Cancer Invest. 2005;23:599–608. doi: 10.1080/07357900500283085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ruggero D, Pandolfi PP. Does the ribosome translate cancer. Nat Rev Cancer. 2003;3:179–92. doi: 10.1038/nrc1015. [DOI] [PubMed] [Google Scholar]
- 10.White RJ. RNA polymerases I and III, growth control and cancer. Nat Rev Mol Cell Biol. 2005;6:69–78. doi: 10.1038/nrm1551. [DOI] [PubMed] [Google Scholar]
- 11.Montanaro L, Trere D, Derenzini M. Nucleolus, ribosomes, and cancer. Am J Pathol. 2008;173:301–10. doi: 10.2353/ajpath.2008.070752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gong G, DeVries S, Chew KL, et al. Genetic changes in paired atypical and usual ductal hyperplasia of the breast by comparative genomic hybridization. Clin Cancer Res. 2001;7:2410–4. [PubMed] [Google Scholar]
- 13.Vos CB, ter Haar NT, Rosenberg C, et al. Genetic alterations on chromosome 16 and 17 are important features of ductal carcinoma in situ of the breast and are associated with histologic type. Br J Cancer. 1999;81:1410–8. doi: 10.1038/sj.bjc.6693372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bais AJ, Gardner AE, McKenzie OL, et al. Aberrant CBFA2T3B gene promoter methylation in breast tumors. Mol Cancer. 2004;3:22. doi: 10.1186/1476-4598-3-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kochetkova M, McKenzie OL, Bais AJ, et al. CBFA2T3 (MTG16) is a putative breast tumor suppressor gene from the breast cancer loss of heterozygosity region at 16q24.3. Cancer Res. 2002;62:4599–604. [PubMed] [Google Scholar]
- 16.Derenzini M, Betts CM, Trere D, et al. Diagnostic value of silver-stained interphasic nucleolar organizer regions in breast tumors. Ultrastruct Pathol. 1990;14:233–45. doi: 10.3109/01913129009076127. [DOI] [PubMed] [Google Scholar]
- 17.Debnath J, Brugge JS. Modelling glandular epithelial cancers in three-dimensional cultures. Nat Rev Cancer. 2005;5:675–88. doi: 10.1038/nrc1695. [DOI] [PubMed] [Google Scholar]
- 18.Shay JW, Van Der Haegen BA, Ying Y, et al. The frequency of immortalization of human fibroblasts and mammary epithelial cells transfected with SV40 large T-antigen. Exp Cell Res. 1993;209:45–52. doi: 10.1006/excr.1993.1283. [DOI] [PubMed] [Google Scholar]
- 19.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30:256–68. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- 20.Benanti JA, Wang ML, Myers HE, et al. Epigenetic down-regulation of ARF expression is a selection step in immortalization of human fibroblasts by c-Myc. Mol Cancer Res. 2007;5:1181–9. doi: 10.1158/1541-7786.MCR-06-0372. [DOI] [PubMed] [Google Scholar]
- 21.Rossetti S, Van Unen L, Touw IP, et al. Myeloid maturation block by AML1-MTG16 is associated with Csf1r epigenetic downregulation. Oncogene. 2005;24:5325–32. doi: 10.1038/sj.onc.1208651. [DOI] [PubMed] [Google Scholar]
- 22.Trere D. AgNOR staining and quantification. Micron. 2000;31:127–31. doi: 10.1016/s0968-4328(99)00069-4. [DOI] [PubMed] [Google Scholar]
- 23.Ren M, Pozzi S, Bistulfi G, et al. Impaired retinoic acid (RA) signal leads to RARbeta2 epigenetic silencing and RA resistance. Mol Cell Biol. 2005;25:10591–603. doi: 10.1128/MCB.25.23.10591-10603.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arabi A, Wu S, Ridderstrale K, et al. c-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nat Cell Biol. 2005;7:303–10. doi: 10.1038/ncb1225. [DOI] [PubMed] [Google Scholar]
- 25.Grandori C, Gomez-Roman N, Felton-Edkins ZA, et al. c-Myc binds to human ribosomal DNA and stimulates transcription of rRNA genes by RNA polymerase I. Nat Cell Biol. 2005;7:311–8. doi: 10.1038/ncb1224. [DOI] [PubMed] [Google Scholar]
- 26.Grewal SS, Li L, Orian A, et al. Myc-dependent regulation of ribosomal RNA synthesis during Drosophila development. Nat Cell Biol. 2005;7:295–302. doi: 10.1038/ncb1223. [DOI] [PubMed] [Google Scholar]
- 27.Ceccarelli C, Trere D, Santini D, et al. AgNORs in breast tumours. Micron. 2000;31:143–9. doi: 10.1016/s0968-4328(99)00071-2. [DOI] [PubMed] [Google Scholar]
- 28.Derenzini M. The AgNORs. Micron. 2000;31:117–20. doi: 10.1016/s0968-4328(99)00067-0. [DOI] [PubMed] [Google Scholar]
- 29.Bissell MJ. Modelling molecular mechanisms of breast cancer and invasion: lessons from the normal gland. Biochem Soc Trans. 2007;35:18–22. doi: 10.1042/BST0350018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amann JM, Nip J, Strom DK, et al. ETO, a target of t(8;21) in acute leukemia, makes distinct contacts with multiple histone deacetylases and binds mSin3A through its oligomerization domain. Mol Cell Biol. 2001;21:6470–83. doi: 10.1128/MCB.21.19.6470-6483.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 MTG16a interacts and co-localize with the NoRCcomponent TIP5. Immunoprecipitation with anti-FLAG antibodyfollowed by Western blotting with anti-MTG16 antibody on Hela cellsco-transfected with both MTG16a and FLAG-TIP5 showing invitro interaction between the two proteins (left). Immunocytochemistry on Hela cells transfected with either FLAG-TIP5 or MTG16a showing that both proteins can localize in the nucleus and the nucleolus (right, top and middle). Co-transfection with both FLAG-TIP5 and MTG16a followed by immunocytochemistry with both anti-MTG16 and anti-FLAG antibodies shows co-localization of the two proteins (right, bottom).
Fig. S2 MTG16a counteracts both physiological andMYC-induced rDNA transcription in Hela cells. Hela cells grown in a24 well plate were transfected with 100 ng pHrD-IRES-Luc,expressing luciferase under the control of the rDNA promoter(kindly provided by Dr. S. Majumer, Ohio State University), 10 ngpRL-TK, and the indicated amounts of pcDNA3.1-MTG16a and pBabe-MYC.Luciferase activity was measured 48 h after transfection by usingDual Glow Luciferase Assay System (Promega). MTG16a cansignificantly (P < 0.05) repress both physiological and MYC-induced pHrD-IRES-Luc expression in a dose-dependent fashion.
Fig. S3 MTG16a knockdown in MCF10A cells leads toincreased rRNA level and impaired acinar morphogenesis. (A)MCF10A-derived MTG16a knockdown clones display, in average, asignificantly (P < 0.05) higher rRNA level relative tocontrol cells. (B) Confocal microscopy showing thatMCF10A-derived MTG16a knockdown clones (clones M16-1 and M16-2 areshown) are unable of proper acinar morphogenesis in 3D basementmembrane cultures in more than 70% of the acini, asopposed to control clones (M-ctrl) forming more than 80%hollow, polarized acini. Nuclei are visualized in blue, integrin ingreen and the Golgi apparatus in red.
Fig. S4 Evidence of proliferating cells in morphologically aberrant acini derived from HMEI cells with stable MTG16a knockdown. Confocal microscopy of acini grown in 3D basement membrane culture, showing the presence of cells positive for the proliferation marker Ki67 in morphologically aberrant acini of H16-1a and H16-2a clones with stable MTG16a knockdown relative to a control clone (H-ctrl). Ki67 (green) was detected with anti-Ki67 (Zymed) and visualized with Alexa Flour 488 anti-rabbit, while nuclei (blue) were counterstained with DAPI.