

Abstract
We have shown cardiac protection by metallothionein (MT) in the development of diabetic cardiomyopathy (DCM) via suppression of cardiac cell death in cardiac-specific MT-overexpressing transgenic (MT-TG) mice. The present study was undertaken to define whether diabetes can induce cardiac endoplasmic reticulum (ER) stress and whether MT can prevent cardiac cell death via attenuating ER stress. Diabetes was induced by streptozotocin in both MT-TG and wild-type (WT) mice. Two weeks, and 2 and 5 months after diabetes onset, cardiac ER stress was detected by expression of ER chaperones, and apoptosis was detected by CCAAT/enhancer-binding protein (C/EBP) homologous protein (CHOP) and cleaved caspase-3 and caspase-12. Cardiac apoptosis in the WT diabetic mice, but not in MT-TG diabetic mice, was significantly increased 2 weeks after diabetes onset. In parallel with apoptotic effect, significant up-regulation of the ER chaperones, including glucose-regulated protein (GRP)78 and GRP94, cleaved ATF6 and phosporylated eIF2α, in the hearts of WT, but not MT-TG diabetic mice. Infusion of angiotensin II (Ang II) also significantly induced ER stress and apoptosis in the hearts of WT, but not in MT-TG mice. Direct administration of chemical ER stress activator tunicamycin significantly increased cardiac cell death only in WT mice. Pre-treatment with antioxidants completely prevented Ang II-induced ER stress and apoptosis in the cultured cardiac cells. These results suggest that ER stress exists in the diabetic heart, which may cause the cardiac cell death. MT prevents both diabetes- and Ang II-induced cardiac ER stress and associated cell death most likely via its antioxidant action, which may be responsible for MT's prevention of DCM.
Keywords: diabetic cardiomyopathy, metallothionein, ER stress, apoptosis, ATF6, caspase-3
Introduction
The endoplasmic reticulum (ER) is recognized as an organelle that participates in the folding of secretory and membrane proteins [1, 2]. A variety of conditions, including infection, nutrient deprivation (such as the absence of glucose) as well as nutrient excess (such as the overabundance of lipids or glucose), alterations in ER lumenal Ca2+ or redox status, and toxic chemicals, can disrupt protein folding reactions in the ER. These conditions are collectively referred to as ER stress [1–3]. ER stress leads to accumulation of unfolded proteins in the ER, which in turn evokes the unfolded protein response (UPR). This response attempts to reduce the amount of unfolded proteins by inducing production of the ER chaperones, such as 78 kD and 94 kD glucose-regulated protein (GRP78 and GRP94) that promote protein folding, and by reducing general protein synthesis and enhancing the degradation of misfolded proteins via a ubiquitin–proteasome system termed ER-associated degradation [1–4]. The persistent accumulation of these misfolded proteins beyond the capacity of ER function causes cellular dysfunction and cell death in several human disorders [1–4]. Emerging evidence indicates that a variety of diseases, such as diabetes mellitus, is related to ER stress [5–7]. Furthermore, there were reportedly tissue differences for ER stress induction by insulin resistance [8].
Several studies indicated the involvement of ER stress in heart diseases [5, 9–11], but whether ER stress is also involved in diabetic cardiomyopathy (DCM) remains unclear. DCM, a leading cause of death for diabetic patients, is caused by multiple pathogenic factors including hyperglycaemia, hyperlipidaemia, hypertension and systemic and cardiac inflammation [12–14]. However, a pivotal contributor to the pathogenic changes of DCM was considered as the oxidative stress [12, 14]. Metallothionein (MT) as a potent antioxidant was found to protect the heart from type 1 and type 2 diabetes [15–18]. In terms of the cellular mechanisms involved in the prevention of MT against DCM, we found that MT protected the heart from diabetes mainly through inhibition of diabetic cardiac cell death [17, 19]. However, it is unclear how diabetes can induce cardiac cell death.
Studies have demonstrated the involvement of oxidative stress in ER stress [6, 20, 21]. As a potent antioxidant MT is up-regulated in response to ER stress [22], suggesting that MT may act as an adaptive mechanism to protect cells from ER stress. Therefore, we suggested that ER stress may occur in the diabetic hearts and contribute to cardiac cell death that initiates DCM as observed in our previous studies [17, 19, 23]. The aim of this study was to investigate whether ER stress involves in the development of DCM with streptozotocin (STZ)-induced diabetic models in cardiac-specific MT-overexpressing transgenic (MT-TG) and their wild-type (WT) mice. Because the reninangiotensin system plays an important role in the development of DCM [23], we further examined whether angiotensin II (Ang II) induces cardiac ER stress along with cell death and whether MT protects the heart from Ang II-induced ER stress and cell death. We also have demonstrated a direct protection by MT against ER stress-induced apoptotic cell death and shown that MT protection against ER stress-mediated cardiac cell death caused by diabetes and Ang II is most likely mediated by its antioxidant property.
Materials and methods
Animal models
MT-TG were produced from WT (Friend virus B, FVB) mice originally obtained from Harlan Bioproducts for Science (Indianapolis, IN, USA) and characterized in previous studies [16, 17]. Both MT-TG and WT mice were housed in the University of Louisville Research Resources Center at 22°C with a 12-hr light/dark cycle and were provided free access to standard rodent chow and tap water. All animal procedures were approved by the Institutional Animal Case and Use Committee, which is certified by the American Association for Accreditation of Laboratory Animal Care. Eight-week-old, male mice were used for the following studies.
The first study was to observe whether there was ER stress in the diabetic hearts. STZ (Sigma Chemical Co., St. Louis, MO, USA) was dissolved in sodium citrate buffer (pH 4.5), and given intraperitoneally to both WT and MT-TG mice at 40 mg/kg body weight daily for 5 days, i.e. multiple low-dose STZ (MLD-STZ) model. Five days after last injection of STZ, whole blood glucose obtained from mouse tail-vein was detected using a SureStep complete blood glucose monitor (LifeScan, Milpitas, CA, USA). STZ-treated mice with whole blood glucose higher than 12 mmol/l were considered diabetic [16, 17]. Animals were killed at 2 weeks, and 2 and 5 months after the onset of diabetes.
The second study was to determine whether Ang II was also able to induce cardiac ER stress, and whether MT protects Ang II-induced cardiac ER stress and associated apoptotic cell death. Ang II (Sigma Chemical Co.) was freshly prepared in a 154 mmol NaCl vehicle immediately prior to be used. MT-TG and WT mice received a single subcutaneous injection of Ang II at 1 mg/kg body weight. Control animals were given an equal volume of vehicle. Both control and Ang II-treated animals were killed at 7 and 24 hrs later. The selection of the Ang II dose and animal killing time was based on previous studies showing that administration of 1 mg Ang II/kg body weight to rats for 7 hrs induced a high incidence of apoptosis in the heart [23, 24].
The third study was to define the direct link of ER stress to apoptotic cell death in the heart and its prevention by MT. Both MT-TG and WT mice were intraperitoneally given a single injection of chemical ER stress activator, tunicamycin (Sigma Chemical Co.) at 1.5 mg/kg body weight. The tunicamycin was freshly prepared as a 0.05 mg/ml suspension in 150 mM dextrose. The dose selection and preparation of tunicamycin was based on a published study [25]. Twelve hours after tunicamycin treatment, animals were killed.
Cell culture
Embryonic rat heart derived cells (H9c2), purchased from (ATCC CLR-1446; Rockville, MD, USA), were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% foetal bovine serum from Atlanta Biologicals (Norcross, GA, USA) and antibiotics (50 U/ml penicillin and 50 μg/ml streptomycin) at 37°C in an atmosphere of 95% air and 5% CO2. H9c2 cells were stably transfected with a vector in which the human MT-IIA gene was placed under the control of the constitutively active [J-actin promoter. Transfected cells were cultured in DMEM with 10% foetal bovine serum and G418. G418-resistant colonies were selected 10–14 days after transfection and propagated. The expression of human MT-IIA mRNA and protein was confirmed by RT-PCR and Western blotting. Among all MT-IIA expressing clones, the clone H9c2MT7 exhibited relative high MT level and maximal preservation of the WT morphology. Correspondingly, H9c2MT7 cells showed a marked reduction in reactive oxygen species production when exposed to hydrogen peroxide or to hypoxia/reoxygenation evaluated by dihydroethidium staining. In addition, this cell line was also resistant to cadmium toxicity [26]. Both H9c2 and H9c2MT7 cells were treated with Ang II at 100 nM for 24 hrs. In some experiments, H9c2 cells were pre-treated with MnTMPyP, a cell-permeable superoxide dismutase mimic, at 50 μM for 30 min. [16] or an antioxidant N-acetyl-cysteine (NAC) at 100 μM for 24 hrs [27, 28] and then exposed to Ang II at 100 nM in the presence of either MnTMPyP or NAC for 24 hrs. MnTMPyP can directly act on superoxide while NAC needs to be converted into glutathione (GSH) to act on hydrogen peroxide; therefore, their pre-treatment times are different [16, 27, 28].
Western blotting assay
As ER stress chaperions, cleaved activating transcription factor 6 (ATF6) GRP78, GRP94 and phosphorylation of eukaryotic initiation factor 2α (p-eIF2α) were examined by Western blotting assay. As one of the components of the ER stress-mediated apoptosis pathway is CCAAT/enhancer-binding protein (C/EBP) homologous protein (CHOP), also known as growth arrest- and DNA damage-inducible gene 153 (GADD153), and cleaved caspase-12 as well as cleaved caspase-3 were also examined by Western blotting assay, based on published methods [9–11, 17, 23]. Briefly, cardiac tissue homogenates were lysed and fractionated electrophoretically on sodium dodecyl (lauryl) sulphate polyacrylamide gel electrophoresis (10–15% gradient gels), and proteins were transferred to a nitrocellulose membrane. The membrane was blocked with a 5% non-fat, dried milk for 1 hr. The following antibodies were used: anti-ATF6 (1:2000, Abcam, Cambridge, MA, USA), anti-GRP78 (1:4000, Abcam), anti-GRP94 (1:1000, Abcam), anti-p-eIF2α and anti-CHOP (1:200 and 1:500, respectively, Santa Cruz, Santa Cruz, CA, USA), anti-cleaved caspase 3 (1:2000, Calbiochem, La Jolla, CA, USA) and anti-cleaved cas-pase-12 (1:1000, Exalpha Biologicals, Shirley, MA, USA). Membranes were incubated with the primary antibodies overnight at 4°C. After the unbound antibodies were removed with Tris-buffered saline (pH 7.2) containing 0.05% Tween 20, membranes were incubated with the secondary antibody for 1 hr at room temperature. Antigen-antibody complexes were visualized with enhanced chemiluminescence (ECL) system.
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labelling (TUNEL) assay
Heart tissue was fixed in 10% formalin, embedded in paraffin and sectioned at 3–4 μm. The slides were stained for TUNEL with the ApopTag Peroxidase In Situ Apoptosis Detection Kit (Chemicon, Temecula, CA, USA). Each slide was deparaffinized and rehydrated, and treated with pro-teinase K (20 mg/l) for 15 min. The endogenous peroxidase was inhibited with 3% hydrogen peroxide for 5 min. and then incubated with the TUNEL reaction mixture containing terminal deoxynucleotidyl transferase (TdT) and digoxigenin-11-dUTP for 1 hr at 37°C. The TdT reaction was carried out in a humidified chamber at 37°C for 1 hr, and then 3, 3-diaminobenzidine chromogen was applied. Methyl green was used as counterstaining. Mouse testicular tissue was used as positive control. For negative control, TdT was omitted from the reaction mixture. The apoptotic cell death was quantitatively analysed by counting the TUNEL+ cells randomly selected from five fields from each of the three slides for each mouse [17, 19].
MT measurement
MT expression was detected by a modified Western blotting protocol [29]. Briefly, cardiac proteins were treated with DTT at a final concentration of 20 mM at 56°C for 30 min., and then added with iodoacetamide (Sigma Chemical Co.) at 50 mM in room temperature for 1 hr in the dark. The supernatant was collected by centrifuging at 800 × g for 5 min., and mixed with loading buffer and heated at 95°C for 5 min. and then subjected to electrophoresis on 18% SDS-PAGE gel at 120 V. Gel was incubated in transfer buffer for 10–20 min. The electrophoresed proteins were transferred to nitrocellulose membrane at 40 V for 2.5 hrs at 4°C in the above transfer buffer modified by addition of 2 mM CaCl2. After transfer, the membrane was incubated in 2.5% glutaraldehyde (Fisher Scientific, Pittsburgh, PA, USA) at room temperature for 1 hr in the dark. The membrane was then blocked in blocking buffer [3% bovine serum albumin (BSA)] at room temperature for at least 1 hr and incubated with anti-MT monoclonal antibody (Dako North America, Carpinteria, CA, USA) diluted 1:1000 in 3% BSA at 4°C for overnight. The membrane was then washed and reacted with secondary horseradish peroxidase-conjugated antibody at room temperature for 1 hr. Antigen-antibody complexes were visualized with ECL system. However, since in the transfer conditions CaCl2 was utilized for MT protein analysis, these blots could not be stripped and reprobed for GAPDH. Thus, two parallel gels were run under the same conditions. One gel (MT gel) was transferred to a membrane in transfer buffer containing CaCl2, while another (GAPDH gel) was transferred to membrane in buffer without CaCl2, as described in previous studies [29, 30].
Statistical analysis
Data were collected from repeated experiments and were presented as mean ± S.D. One-way anova and Student's t-test was used for statistical analysis. Origin 7.5 (OriginLab data analysis and graphing software) was used for all statistical tests. Statistical significance was considered as P< 0.05.
Results
Diabetes-induced cardiac ER stress and prevention by MT
Both MT-TG and WT mice were made diabetic by MLD-STZ, and showed similar patterns of persistent increase in blood glucose levels (Data not shown). Our previous studies showed cardiac cell death in the heart of WT diabetic mice, but not MT-TG diabetic mice, at 2 weeks after the onset of diabetes [17], and also significantly pathological and functional changes in WT diabetic, but not MT-TG diabetic mice, at 2 and 5 months [16, 17, 30]. Therefore, we examined whether these pathological changes are related to cardiac ER stress by analysing the expression of ER chaperones from diabetic and age-matched control mice at 2 weeks, and 2 and 5 months after the onset of diabetes (Fig. 1). Western blotting assay showed that both phosphorylated eIF2α (p-eIF2α) and GRP94 proteins were significantly increased in the heart of WT diabetic mice at 2 weeks, but not 2 and 5 months, after diabetes onset (Fig. 1A, B). Cleaved ATF6 protein was significantly increased both at 2 weeks and 2 months, but not at 5 months (Fig. 1C). In contrast to the above changes, GRP78 protein level was increased in the heart of diabetic mice at all three time-points with the highest expression at 2 weeks after diabetes (Fig. 1D). More importantly, none of these increased ER chaperones in the hearts of WT diabetic mice were observed in the heart of MT-TG diabetic mice (Fig. 1).
Fig. 1.
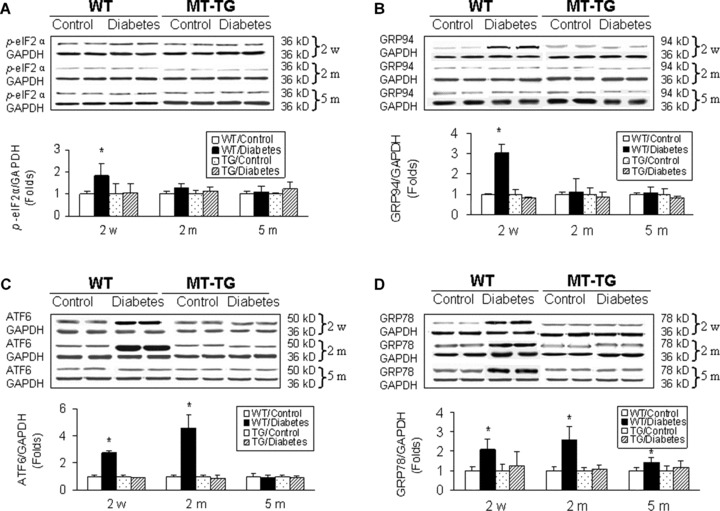
Diabetes-induced cardiac up-regulation of ER chaperones. Both MT-TG and WT mice were made diabetic by MLD-STZ, and cardiac ER stress was examined by Western blotting analysing the expression of ER chaperones p-eIF2α (A), GRP94 protein (B), cleaved ATF6 (C) and GRP78 (D) from the control and diabetic mice at indicated time-points. Experimental data were presented as means ± S.D. (n= 5 at least for each group). *, P < 0.05 versus control.
Diabetes-induced cardiac MT up-regulation
Kondoh et al. has shown the induction of hepatic MT in response to ER stress [22]. We next examined whether diabetic ER stress also induced MT synthesis in the heart. Western blotting assay revealed that diabetes induced a significant increase in MT contents of heart from 2 weeks to 5 months with a rapid increase at 2 weeks and then gradually decreased until 5 months after diabetes, but it remained significantly higher than control levels (Fig. 2). Interestingly, diabetes-induced cardiac MT synthesis in the heart of WT mice was not observed in the heart of MT-TG diabetic mice, suggesting that pre-existed MT prevents diabetes-induced stress and MT synthesis. The prevention of diabetes-induced MT synthesis in MT-TG heart implies that ER stress is responsible for MT synthesis and there is no diabetes-induced cardiac ER stress in the heart of MT-TG diabetic mice.
Fig. 2.
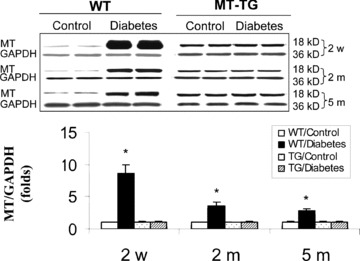
Diabetes-induced cardiac up-regulation of MT protein. Animal treatment and tissue collections are same as those described in Fig. 1. Cardiac MT contents were examined by Western blotting assay. It should be noted that in order to see whether there was MT induction in the hearts of MT-TG diabetic mice, the exposure time for the films to gels was significantly shorter for samples from MT-TG mice than those from WT mice; Therefore, MT expressions in MT-TG samples looks significantly low than diabetes-induced cardiac MT levels. Experimental data were presented as means ± S.D. (n= 5 at least for each group). *, P < 0.05 versus control.
Diabetes-induced cardiac cell death and prevention by MT
Although we have reported the induction of cardiac cell death in the WT diabetic mice, it was only examined for the short time period (within 1 month) [17, 19]. Therefore, we extended the examination of cardiac cell death from 2 weeks to 2 and 5 months after diabetes onset with TUNEL assay for apoptotic cells (Fig. 3A) and Western blotting assay for the activated form of caspase-3 (Fig. 3B). Both assays delineated that diabetes mainly induced cardiac cell death at the early stage of diabetes (2 weeks), but not in the late stages (2 and 5 months), which is consistent with previous studies [17, 19]. However, there was no apoptotic cell death in the hearts of MT-TG diabetic mice (Fig. 3). To determine whether the cardiac cell death is ER stress-mediated, CHOP protein expression (Fig. 3C) and caspase-12 cleavage (Fig. 3D) were examined by Western blotting assay, which showed that significant increases in CHOP expression and cleaved caspase-12 were observed only in the heart of WT diabetic mice at 2 weeks, but not at 2 and 5 months after diabetes onset, suggesting the association of diabetes-induced cardiac cell death with ER stress. The ER-stress-mediated cardiac cell death was not seen in the heart of MT-TG diabetic mice (Fig. 3C, D).
Fig. 3.
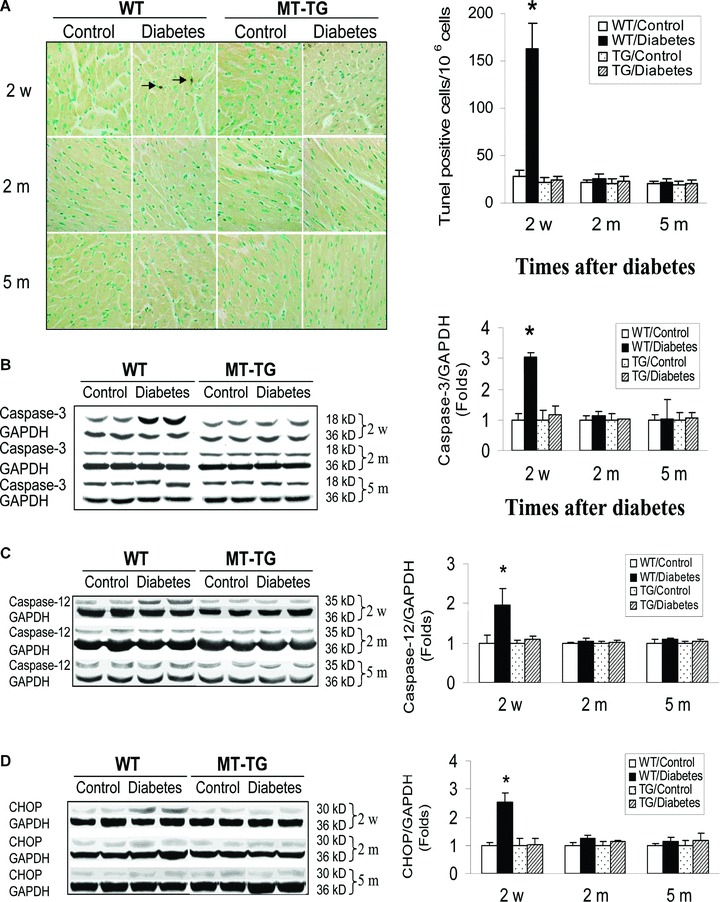
Diabetes-induced cardiac cell death. Animal treatments and tissue collections are same as those described in Fig. 1. Cardiac apoptotic cell death was examined by TUNEL staining for apoptotic cells (A) and also by Western blotting analysis for cleaved caspase-3 (B) and caspase-12 (C) and CHOP expression (D). Experimental data were presented as means ± S.D. (n= 5 at least for each group). *, P < 0.05 versus control.
MT protection against Ang II-induced cardiac ER stress, MT synthesis and cell death
To examine whether Ang II as an important pathogenic factor for DCM is also able to induce ER stress, MT induction and cardiac cell death, both MT-TG and WT mice were intraperitoneally given Ang II at 1 mg/kg. Seven and 24 hrs after Ang II infusion, cardiac ER chaperone proteins were examined (Fig. 4). Treatment of WT mice with Ang II significantly increased the expression of p-eIF2α (Fig. 4A), cleaved ATF6 (Fig. 4C) and GRP78 (Fig. 4D), but not GRP94 (Fig. 4B). The up-regulated ER chaperone proteins by Ang II were not observed in the heart of MT-TG mice (Fig. 4). Correspondingly, Ang II also significantly induced cardiac MT synthesis and cardiac cell death only in the WT mice (Fig. 5A, B), as observed in the hearts of diabetic mice (Figs. 2, 3). The association of Ang II-induced cardiac cell death with ER stress was also favoured by significant increases in CHOP protein expression and caspase-12 activation (Fig. 5C, D).
Fig. 4.
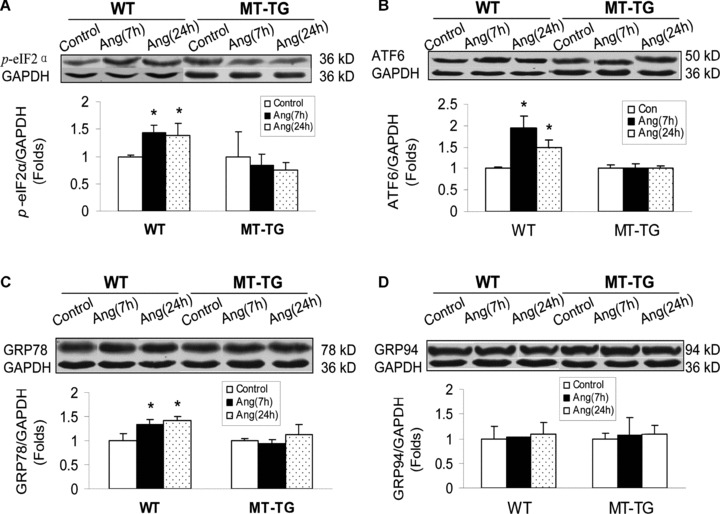
Ang II-induced cardiac up-regulation of ER chaperones. Both MT-TG and WT mice were administered Ang II at 1 mg/kg and 7 and 24 hrs later cardiac ER stress were examined by Western blotting analysing the expression of ER chaperones p-eIF2α (A), cleaved ATF6 (B), GRP78 (C) and GRP94 protein (D). Experimental data were presented as means ± S.D. (n= 5 at least for each group). *, P< 0.05 versus control.
Fig. 5.
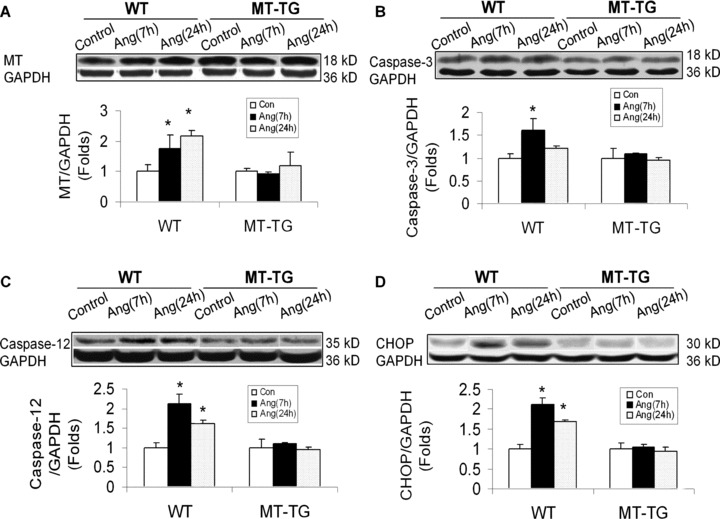
Ang II-induced cardiac up-regulation of MT protein and cell death. Animal treatments and tissue collections are same as those described in Fig. 4. Cardiac MT contents were examined by Western blotting assay (A). Cardiac apoptotic cell death was examined by Western blotting analysis for the cleaved caspase-3 (B) and caspase-12 (C) and CHOP expression (D). Again in order to see whether there was MT induction in the hearts of MT-TG diabetic mice, the exposure time for the films to gels was significantly shorter for samples from MT-TG mice than those from WT mice; Therefore, MT expressions in MT-TG samples looks significantly low than diabetes-induced cardiac MT levels. Experimental data were presented as means ± S.D. (n= 5 at least for each group). *, P< 0.05 versus control.
MT protection against chemical ER stressor-induced cardiac ER stress, MT synthesis and cell death
To define the direct protection by MT from ER stress-mediated apoptotic cell death, both WT and MT-TG mice were administrated with tunicamycin (Tu) at 1.5 mg/kg, and 12 hrs later, cardiac ER chaperone proteins were examined. Western blotting assay showed that proteins of p-eIF2α (Fig. 6A), cleaved ATF6 (Fig. 6B) and GRP78 (Fig. 6C) all were significantly up-regulated, except for GRP94 protein (Fig. 6D). Similar to the above experiments in diabetic and Ang II-infusion models, MT significantly prevented chemical ER stressor-up-regulated ER chaperone proteins (Fig. 6).
Fig. 6.
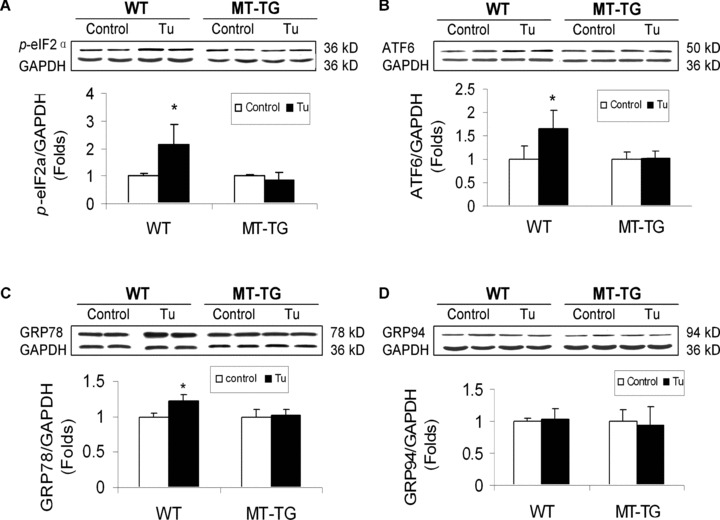
Tunicamycin-induced cardiac up-regulation of ER chaperones. Both WT and MT-TG mice were directly administrated with tunicamycin at 1.5 mg/kg, and 12 hrs later cardiac ER stress were examined by Western blotting analysing the expression of ER chaperones p-eIF2α (A), cleaved ATF6 (B), GRP78 (C) and GRP94 protein (D). Experimental data were presented as means ± S.D. (n = 6 at least for each group). Tu: tunicamycin. *, P< 0.05 versus control.
It is noted that chemical ER stressor also significantly induced cardiac MT synthesis (Fig. 7A), as shown in the hearts of other ER-stress models (Figs 2 and 5A). Cardiac cell death was significantly increased in the heart of Tu-treated WT mice, but not Tu-treated MT-TG mice, examined with Western blotting for caspase-3 activation (Fig. 7B) and TUNEL assay (Fig. 7C), and confirmed by CHOP protein expression (Fig. 7D) and caspase-12 cleavage (Fig. 7E).
Fig. 7.
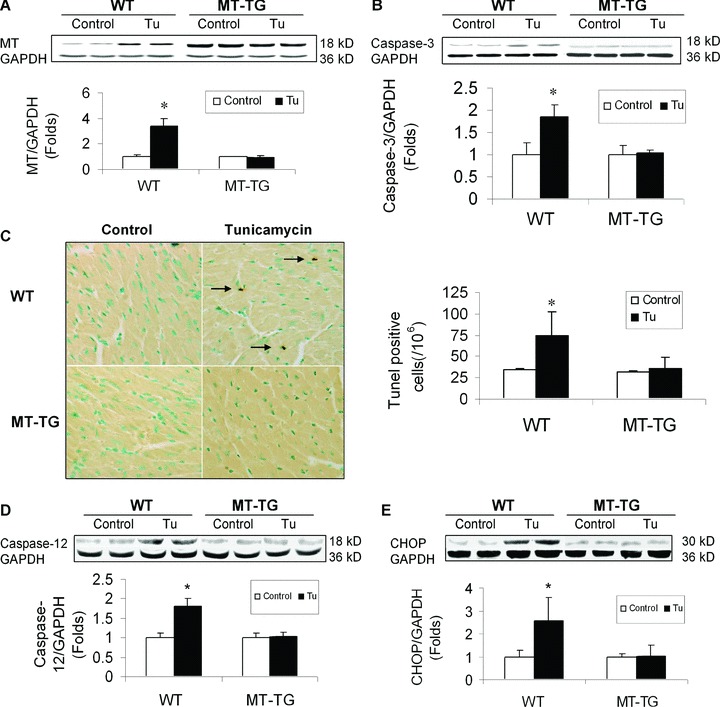
Tunicamycin-induced cardiac up-regulation of MT protein and cell death. Animal treatments and tissue collections are same as those described in Fig. 6. Cardiac MT contents were examined by Western blotting assay (A). Cardiac apoptotic cell death was examined for the cleaved caspase-3 by Western blotting analysis (B) and apoptotic cells with TUNEL staining (C). Cleaved caspase-12 (D) and CHOP protein expression (E) were also examined by Western blotting assay. Experimental data were presented as means ± S.D. (n= 6 at least for each group). Tu: tunicamycin.*, P< 0.05 versus control.
Ang II-induced ER stress and associated cell death was attenuated by MT and antioxidants in cultured cardiac cells
In order to understand the molecular mechanisms underlying the protective role of MT in the heart against Ang II-induced ER stress-mediated apoptotic cell death, we used stable MT-IIA over-expressing H9c2MT7 cells that exhibited similar growth kinetics and morphology, but had a remarkable increased MT protein level as compared to the parent H9c2 cells [26]. Treatment with Ang II at 100 nM for 24 hrs significantly induced ER stress, shown by increased expressions of cleaved ATF6 (Fig. 8A), p-eIF2α (Fig. 8B), GRP78 (Fig. 8C) and GRP94 (Fig. 8D). All these ER-stress effects were not observed in H9c2MT7 cells, suggesting the protection of cardiac cells by MT from Ang II-induced ER stress. Treatment with Ang II at 100 nM for 24 hrs also significantly induced apoptotic cell death in H9c2 cells, measured by caspase-12 activation (Fig. 9A), and CHOP protein expression (Fig. 9B), but not in H9c2MT7 cells. This in vitro finding is consistent with those of in vivo findings (Fig. 5C, D).
Fig. 8.
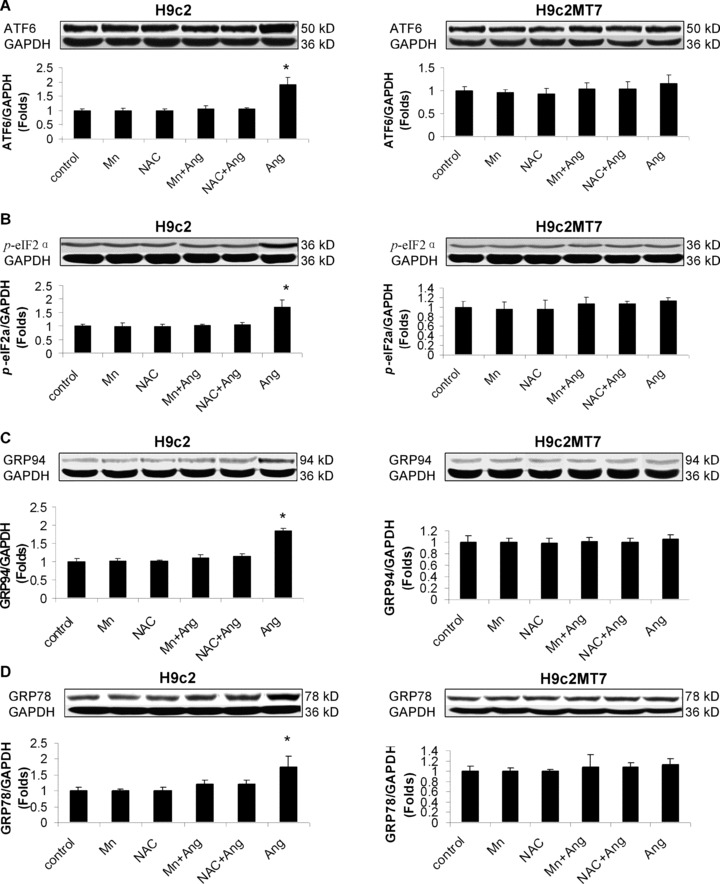
Protective effect of antioxidants and MT against Ang II-induced ER stress. H9c2 and H9c2MT7 were exposed to Ang II at 100 nM for 24 hrs with or without pre-existence of MnTMPyP at 50 μM for 30 min. or NAC at 50 μM for 24 hrs. ER stress was examined by measuring the cleaved ATF6 (A), p-eIF2α (8B), GRP78 (C) and GRP94 (D) with Western blotting assay. Experimental data was presented as means ± S.D. from three separate experiments with triple samples for each experiment. *, P< 0.05 versus control.
Fig. 9.
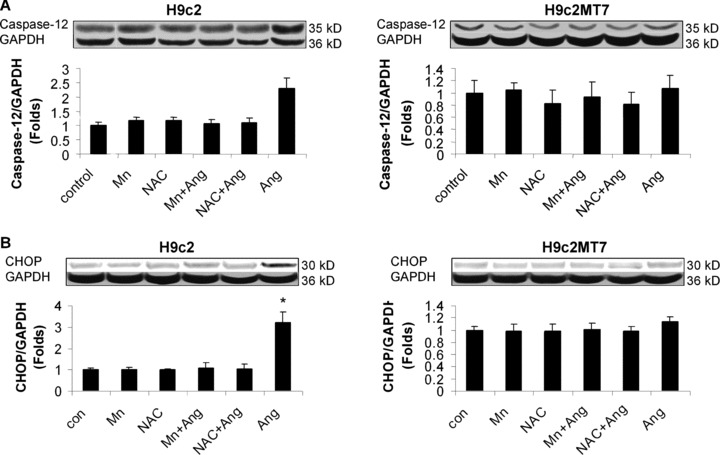
Protective effect of antioxidants and MT against Ang II-induced ER stress-mediated cell death. H9c2 and H9c2MT7 were treated with the same condition as described in Fig. 8. ER stress-mediated cell death was detected by measuring the cleaved caspase-12 (A) and CHOP expression (B) with Western blotting assay. Experimental data was presented as means ± S.D. from three separate experiments with triple samples for each experiment. *, P< 0.05 versus control.
Considering that Ang II apoptotic effect was related to oxidative stress since exposure of H9c2 cells to Ang II directly induced peroxynitrite formation due to over-generation of superoxide [23], whether MT protection against Ang II-induced RE stress and associated apoptosis was also attributed to its antioxidant action was evaluated. Pre-exposure of H9c2 cells to MnTMPyP at 50 μM for 30 min. or NAC at 50 μM for 24 hrs significantly prevented Ang II-induced ER stress and associated apoptosis (Fig. 9A, B), while same pretreatments did not have any effect on normal H9c2 cells and H9c2MT7 cells (Fig. 9A, B). These results suggest that both ER stress and apoptotic effects caused by Ang II are mediated by oxidative stress, and also that MT protection against Ang II-induced ER stress and associated apoptotic effects is most likely mediated by its antioxidant action.
Discussion
The present study provided the following innovative findings: (1) Diabetes induces cardiac ER stress along with induction of cardiac cell death; (2) As one of the major pathogenic factors for DCM, Ang II also induces cardiac ER stress and cell death; (3) Cardiac overexpression of MT rescues diabetes-, Ang II- and even chemical ER stressor-induced cardiac cell death via suppression of cardiac ER stress; (4) Oxidative stress plays a pivotal role in Ang II-induced both ER stress and associated apoptotic effects and (5) MT protection against Ang II-induced ER stress and associated apoptotic effects are most likely mediated by its antioxidant action.
DCM is a chronic and complex pathogenesis, caused by abnormal cellular metabolism and defects in organelles such as myofibrils, mitochondria and sarcolemma [12–14]. Apoptotic cell death as an early cellular event in response to diabetes has been reported to play a critical role in the development of DCM [13, 17, 19]. Although emerging evidence has indicated that ER stress-mediated apoptosis is involved in the pathogenesis of diabetic eye and kidney as well as non-diabetic heart failure [31–36], there was no direct evidence for the involvement of ER stress in diabetes-induced cardiac cell death and DCM. The present study shows an association of diabetes- and Ang II-induced cardiac cell death with cardiac ER stress, and also MT protection against cardiac ER stress and associated cell death caused by diabetes, Ang II and chemical ER stressor in mouse model.
Cardiac cells contain abundant ER, which is a central organelle of each eukaryotic cell as the place of protein folding, maturation and secretion [4]. Properly folded proteins are trafficked from the ER to the Golgi, lysosomes or to the plasma membrane. Conditions interfering with the function of ER are collectively called ER stress. ER stress is induced by UPR accumulation. The UPR deals with the adverse effects of ER stress and enhances cell survival through three stress-sensing proteins found on the ER membrane: PKR-like eIF2α kinase (PERK), inositol-requiring kinase-1α (IRE-1α) and ATF-6 [4, 37]. Chaperone GRP78 binds the N-termini of IRE-1α, PERK and ATF6, preventing their activation. Unfolded proteins in the ER cause GRP78 to release IRE-1α, PERK and ATF6, leading to their oligomerization and activation in ER membranes. Among these events, the activated IRE-1α activates its downstream kinases that activate TRAF2, ASK1 and JNK, leading to activation of caspase-9 as mitochondrial cell death pathway. Failure of GRP78 binding to PERK causes activation of caspase-12 and CHOP-dependent ER stress-mediated apoptosis as a mitochondrial independent cell death pathway [4, 37]. Therefore, these chaperons are up-regulated to activate both mitochondria-dependent and independent cell death pathways in response to different stresses [38–40].
Consistent with the above concept, we have shown the induction of apoptotic cell death via mitochondria-cytochrome c release pathway in the heart of diabetic mice [19, 41]. In the present study, we further showed the up-regulated two of the three arms of ER stress signalling, PERK- and ATF6-mediated pathways. We found that one of PERK signalling members, eIF2α phosphorylation, was significantly increased in the heart of WT diabetic mice at 2 weeks after diabetes onset (Fig. 1A), and cleaved ATF6 expression was also significantly increased in the heart of WT diabetic mice at both 2 weeks and 2 months after diabetes onset (Fig. 1C). The increased ER stress was accompanied with a significant increase in cardiac cell death, measured by TUNEL assay and Western blotting for the activated caspase-3 and caspase-12, and CHOP as the markers of ER stress-mediated apoptotic cell death (Fig. 3). We also demonstrated the significant increases in GRP78 and GRP94 (Fig. 1). Several studies have indicated the involvement of either eIF2α or ATF6, along with up-regulation of GRP78 and/or GRP94 in the induction of apoptotic cell death caused by several pathogenic conditions [42–44]. However, our finding is the first time to systemically examine the cardiac ER stress and ER-stress-mediated cardiac cell death under diabetic and Ang II-treated conditions. Our studies also indicate that diabetes induces cardiac cell death via both ER-stress-dependent and mitochondria-dependent pathways ([19, 41] and present study).
Although both GRP78 and GRP94 protein up-regulation can be considered as an index of ER stress, these proteins can also act as an adaptive mechanism to protect the cells against stress-induced death [45–47]. We found that cardiac cell death mainly occur at 2 weeks after diabetes (Fig. 3) along with significant increases in eIF2α phosphorylation only at 2 weeks (Fig. 1). We found that cleaved ATF6 was increased in the hearts of diabetic mice (Fig. 1). Although increase in cleaved ATF6 was also observed in the heart of WT diabetic mice at 2 months after diabetes, there was no significant increase in apoptotic cell death, suggesting that ATF6 cleavage may not be the direct cause for cardiac cell death although it is an ER stress response.
We also demonstrated that a significant and persistent up-regulation of GRP78 as an adaptive and protective regulator was also significantly increase in the hearts of diabetic mice from 2 weeks until 5 months (Fig. 1C). Cardiac adaptive response in the hearts of diabetic mice, particularly in the early stage, was mirrored by the up-regulation of cardiac expression of MT as an antioxidant and ER stress protein (Fig. 2). The response pattern of cardiac MT is exactly parallel with that of GRP78 (Fig. 1). Consistent with this finding, MT and GRP78 were up-regulated in the heart of WT mice with Ang II infusion (Fig. 5A) or with chemical ER stressor (Fig. 7A) although there was induction of apoptosis (Figs. 5B and 7B) that may be mainly due to the induction of eIF2α phosphorylation (Figs. 4A, B and 6A, B). Interestingly, there was no increase in GRP94 protein in the heart of WT mice treated with either Ang II (Fig. 4D) or chemical stressor (Fig. 6D). We do not have any explanation for this discrepancy in these three models for the up-regulation of GRP94, and complicated functions of GRP94 have been documented although it also acts as protective factor in certain cells [48–51].
It could be questioned that if MT or GRP78 expression is up-regulated in response to diabetes as an adaptive response to protect the heart from ER stress-mediated cell death, why it does not prevent the occurrence of diabetic or Ang II-induced ER stress and cell death. We assume that the up-regulated MT or GRP78 expression observed in the WT mice is secondary to diabetes- or Ang II-induced ER stress. This stress induces MT or GRP78 expression but also simultaneously initiates the signalling of ER stressmediated cardiac cell death. Therefore, the ER stress-induced MT or GRP78 expression in the heart does not prevent ER stress-initiated apoptotic cell death, but prevents ER stress further-induced cell death; therefore, there was no significant increase in apoptotic cell death at the late stages of 2 and 5 months. To prevent the initial occurrence of ER stress and associated apoptosis, pre-enhanced cardiac MT content is requested. To support this notion, MT-TG mice were significantly resistant to diabetes-, Ang II- and Tu-induced cardiac ER stress and associate cell death (Figs 3, 5 and 7). H9c2MT7 cells are highly resistant to Ang II-induced ER stress-associated cell death (Fig. 8). Fu et al. also showed that proteasome inhibition induced ATF6 activation and cardiomyocyte death along increases in CHOP expression and capase-12 activation. Supplement and/or pharmacological induction of GRP78 could attenuate cardiac cell death by proteasome inhibition [52].
Regarding the mechanisms by which MT prevents cardiac cell death under different ER stresses, we assume that anti-oxidative action of MT plays a critical role in preventing diabetes- and Ang II-induced cardiac ER stress and associated cell death. Our results showed that pre-treatment with antioxidant significantly attenuated Ang II-induced ER stress (Fig. 8) and ER stress-mediated cell death, measured by CHOP expression and caspase-12 activation (Fig. 9). Both oxidative stress and ER stress were implicated in the diabetic complications and a few studies have reported that accumulation of reactive oxygen species as the cause of ER stress [6, 20, 21]. For instance, Xue et al. demonstrated the induction of UPR by tumour necrosis factor-α in L929 cells via inducing reactive oxygen species accumulation [53], and Yan et al. also found that ER stress and UPR were attributable to oxidative stress [54]. To support these previous studies, a recent study showed that WT and MT-TG mice were received the GSH synthase inhibitor buthionine sulfoximine in drinking water for 2 weeks. Buthionine sulfoximine led to a robust decrease in the GSH and increased reactive oxygen species production, consolidating oxidative stress, along with cardiac ER stress and dysfunction in the WT mice, but not in the MT-TG mice [55]. Taken together, all these data suggested that MT is a potent antioxidant to protect cells and tissues from diabetes, Ang II and Buthionine sulfoximine (BOS) via suppression of reactive oxygen species accumulation [16, 17, 23, 55]. Therefore, MT protection against Ang II-induced ER stress and associated apoptotic effects is most likely mediated by its antioxidant property.
Acknowledgments
The work was supported, in part, by research grants from American Diabetes Association (05–07-CD-02, to L.C.). J.X. and Q.L. are recipients of Scholarship under State Scholarship Fund from China Scholarship Council. The authors thank Dr. Michelle Barati, Department of Medicine, University of Louisville, for her generosity in providing certain reagents. The authors are also grateful to Dr. M. George Cherian, Professor Emeritus, University of Western Ontario, Canada, for his insightful reading and editing.
References
- 1.Haynes CM, Titus EA, Cooper AA. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol Cell. 2004;15:767–76. doi: 10.1016/j.molcel.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 2.Momoi T. Caspases involved in ER stress-mediated cell death. J Chem Neuroanat. 2004;28:101–5. doi: 10.1016/j.jchemneu.2004.05.008. [DOI] [PubMed] [Google Scholar]
- 3.Sakaki K, Kaufman RJ. Regulation of ER stress-induced macroautophagy by protein kinase C. Autophagy. 2008;4:841–3. doi: 10.4161/auto.6607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. J Clin Invest. 2005;115:2656–64. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cai L. Alcoholic cardiomyopathy: acetaldehyde, insulin insensitization and ER stress. J Mol Cell Cardiol. 2008;44:979–82. doi: 10.1016/j.yjmcc.2008.03.018. [DOI] [PubMed] [Google Scholar]
- 6.Kaneto H, Matsuoka TA, Nakatani Y, et al. Oxidative stress, ER stress, and the JNK pathway in type 2 diabetes. J Mol Med. 2005;83:429–39. doi: 10.1007/s00109-005-0640-x. [DOI] [PubMed] [Google Scholar]
- 7.Sundar Rajan S, Srinivasan V, Balasubramanyam M, et al. Endoplasmic reticulum (ER) stress & diabetes. Indian J Med Res. 2007;125:411–24. [PubMed] [Google Scholar]
- 8.Yoshiuchi K, Kaneto H, Matsuoka TA, et al. Direct monitoring of in vivo ER stress during the development of insulin resistance with ER stress-activated indicator transgenic mice. Biochem Biophys Res Commun. 2008;366:545–50. doi: 10.1016/j.bbrc.2007.11.182. [DOI] [PubMed] [Google Scholar]
- 9.Li SY, Ren J. Cardiac overexpression of alcohol dehydrogenase exacerbates chronic ethanol ingestion-induced myocardial dysfunction and hypertrophy: role of insulin signaling and ER stress. J Mol Cell Cardiol. 2008;44:992–1001. doi: 10.1016/j.yjmcc.2008.02.276. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10.Liu J, Mao W, Iwai C, et al. Adoptive passive transfer of rabbit beta1-adrenoceptor peptide immune cardiomyopathy into the Rag2-/- mouse: participation of the ER stress. J Mol Cell Cardiol. 2008;44:304–14. doi: 10.1016/j.yjmcc.2007.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mao W, Iwai C, Liu J, et al. Darbepoetin alfa exerts a cardioprotective effect in autoimmune cardiomyopathy via reduction of ER stress and activation of the PI3K/Akt and STAT3 pathways. J Mol Cell Cardiol. 2008;45:250–60. doi: 10.1016/j.yjmcc.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cai L, Kang YJ. Oxidative stress and diabetic cardiomyopathy: a brief review. Cardiovasc Toxicol. 2001;1:181–93. doi: 10.1385/ct:1:3:181. [DOI] [PubMed] [Google Scholar]
- 13.Cai L, Kang YJ. Cell death and diabetic cardiomyopathy. Cardiovasc Toxicol. 2003;3:219–28. doi: 10.1385/ct:3:3:219. [DOI] [PubMed] [Google Scholar]
- 14.Harmancey R, Taegtmeyer H. The complexities of diabetic cardiomyopathy: lessons from patients and animal models. Curr Diab Rep. 2008;8:243–8. doi: 10.1007/s11892-008-0042-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liang Q, Carlson EC, Donthi RV, et al. Overexpression of metallothionein reduces diabetic cardiomyopathy. Diabetes. 2002;51:174–81. doi: 10.2337/diabetes.51.1.174. [DOI] [PubMed] [Google Scholar]
- 16.Cai L, Wang J, Li Y, et al. Inhibition of superoxide generation and associated nitrosative damage is involved in metal-lothionein prevention of diabetic cardiomyopathy. Diabetes. 2005;54:1829–37. doi: 10.2337/diabetes.54.6.1829. [DOI] [PubMed] [Google Scholar]
- 17.Cai L, Wang Y, Zhou G, et al. Attenuation by metallothionein of early cardiac cell death via suppression of mitochondrial oxidative stress results in a prevention of diabetic cardiomyopathy. J Am Coll Cardiol. 2006;48:1688–97. doi: 10.1016/j.jacc.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 18.Feng W, Wang Y, Cai L, et al. Metallothionein rescues hypoxia-inducible factor-1 transcriptional activity in car-diomyocytes under diabetic conditions. Biochem Biophys Res Commun. 2007;360:286–9. doi: 10.1016/j.bbrc.2007.06.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cai L, Li W, Wang G, et al. Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activation pathway. Diabetes. 2002;51:1938–48. doi: 10.2337/diabetes.51.6.1938. [DOI] [PubMed] [Google Scholar]
- 20.Yokouchi M, Hiramatsu N, Hayakawa K, et al. Involvement of selective reactive oxygen species upstream of proapoptotic branches of unfolded protein response. J Biol Chem. 2008;283:4252–60. doi: 10.1074/jbc.M705951200. [DOI] [PubMed] [Google Scholar]
- 21.Van Der Vlies D, Makkinje M, Jansens A, et al. Oxidation of ER resident proteins upon oxidative stress: effects of altering cellular redox/antioxidant status and implications for protein maturation. Antioxid Redox Signal. 2003;5:381–7. doi: 10.1089/152308603768295113. [DOI] [PubMed] [Google Scholar]
- 22.Kondoh M, Tsukada M, Kuronaga M, et al. Induction of hepatic metallothionein synthesis by endoplasmic reticulum stress in mice. Toxicol Lett. 2004;148:133–9. doi: 10.1016/j.toxlet.2003.12.066. [DOI] [PubMed] [Google Scholar]
- 23.Zhou G, Li X, Hein DW, et al. Metallothionein suppresses angiotensin II-induced nicotinamide adenine dinucleotide phosphate oxidase activation, nitrosative stress, apoptosis, and pathological remodeling in the diabetic heart. J Am Coll Cardiol. 2008;52:655–66. doi: 10.1016/j.jacc.2008.05.019. [DOI] [PubMed] [Google Scholar]
- 24.Burniston JG, Saini A, Tan LB, et al. Angiotensin II induces apoptosis in vivo in skeletal, as well as cardiac, muscle of the rat. Exp Physiol. 2005;90:755–61. doi: 10.1113/expphysiol.2005.030908. [DOI] [PubMed] [Google Scholar]
- 25.Zinszner H, Kuroda M, Wang X, et al. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–95. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xue W, Liu Q, Cai L, et al. Stable overexpression of human metallothionein-IIA in a heart-derived cell line confers oxidative protection. Toxicol Lett. 2009;188:70–6. doi: 10.1016/j.toxlet.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 27.Kulkarni JS, Khanna A. Functional hepatocyte-like cells derived from mouse embryonic stem cells: a novel in vitro hepatotoxicity model for drug screening. Toxicol In Vitro. 2006;20:1014–22. doi: 10.1016/j.tiv.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 28.Huang D, Zhang Y, Qi Y, et al. Global DNA hypomethylation, rather than reactive oxygen species (ROS), a potential facilitator of cadmium-stimulated K562 cell proliferation. Toxicol Lett. 2008;179:43–7. doi: 10.1016/j.toxlet.2008.03.018. [DOI] [PubMed] [Google Scholar]
- 29.Wang J, Song Y, Elsherif L, et al. Cardiac metallothionein induction plays the major role in the prevention of diabetic cardiomyopathy by zinc supplementation. Circulation. 2006;113:544–54. doi: 10.1161/CIRCULATIONAHA.105.537894. [DOI] [PubMed] [Google Scholar]
- 30.Song Y, Wang J, Li Y, et al. Cardiac metallothionein synthesis in streptozotocin-induced diabetic mice, and its protection against diabetes-induced cardiac injury. Am J Pathol. 2005;167:17–26. doi: 10.1016/S0002-9440(10)62949-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borradaile NM, Buhman KK, Listenberger LL, et al. A critical role for eukaryotic elongation factor 1A-1 in lipotoxic cell death. Mol Biol Cell. 2006;17:770–8. doi: 10.1091/mbc.E05-08-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oshitari T, Hata N, Yamamoto S. Endoplasmic reticulum stress and diabetic retinopathy. Vasc Health Risk Manag. 2008;4:115–22. doi: 10.2147/vhrm.2008.04.01.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun Y, Liu G, Song T, et al. Upregulation of GRP78 and caspase-12 in diastolic failing heart. Acta Biochim Pol. 2008;55:511–6. [PubMed] [Google Scholar]
- 34.Lindenmeyer MT, Rastaldi MP, Ikehata M, et al. Proteinuria and hyperglycemia induce endoplasmic reticulum stress. J Am Soc Nephrol. 2008;19:2225–36. doi: 10.1681/ASN.2007121313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Werstuck GH, Khan MI, Femia G, et al. Glucosamine-induced endoplasmic reticulum dysfunction is associated with accelerated atherosclerosis in a hyperglycemic mouse model. Diabetes. 2006;55:93–101. [PubMed] [Google Scholar]
- 36.Dong F, Ren J. Adiponectin improves cardiomyocyte contractile function in db/db diabetic obese mice. Obesity. 2009;17:262–8. doi: 10.1038/oby.2008.545. [DOI] [PubMed] [Google Scholar]
- 37.Szegezdi E, Fitzgerald U, Samali A. Caspase-12 and ER-stress-mediated apoptosis: the story so far. Ann N Y Acad Sci. 2003;1010:186–94. doi: 10.1196/annals.1299.032. [DOI] [PubMed] [Google Scholar]
- 38.Chien CM, Yang SH, Chang LS, et al. Involvement of both endoplasmic reticulum- and mitochondria-dependent pathways in car-diotoxin III-induced apoptosis in HL-60 cells. Clin Exp Pharmacol Physiol. 2008;35:1059–64. doi: 10.1111/j.1440-1681.2008.04968.x. [DOI] [PubMed] [Google Scholar]
- 39.Diwan A, Matkovich SJ, Yuan Q, et al. Endoplasmic reticulum-mitochondria crosstalk in NIX-mediated murine cell death. J Clin Invest. 2009;119:203–12. doi: 10.1172/JCI36445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li J, Xia X, Ke Y, et al. Trichosanthin induced apoptosis in HL-60 cells via mito-chondrial and endoplasmic reticulum stress signaling pathways. Biochim Biophys Acta. 2007;1770:1169–80. doi: 10.1016/j.bbagen.2007.04.007. [DOI] [PubMed] [Google Scholar]
- 41.Cai L. Suppression of nitrative damage by metallothionein in diabetic heart contributes to the prevention of cardiomyopa-thy. Free Radic Biol Med. 2006;41:851–61. doi: 10.1016/j.freeradbiomed.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 42.Gotoh T, Oyadomari S, Mori K, et al. Nitric oxide-induced apoptosis in RAW 264.7 macrophages is mediated by endoplasmic reticulum stress pathway involving ATF6 and CHOP. J Biol Chem. 2002;277:12343–50. doi: 10.1074/jbc.M107988200. [DOI] [PubMed] [Google Scholar]
- 43.Nakanishi K, Sudo T, Morishima N. Endoplasmic reticulum stress signaling transmitted by ATF6 mediates apoptosis during muscle development. J Cell Biol. 2005;169:555–60. doi: 10.1083/jcb.200412024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhao H, Liao Y, Minamino T, et al. Inhibition of cardiac remodeling by pravastatin is associated with amelioration of endoplasmic reticulum stress. Hypertens Res. 2008;31:1977–87. doi: 10.1291/hypres.31.1977. [DOI] [PubMed] [Google Scholar]
- 45.Dong D, Ni M, Li J, et al. Critical role of the stress chaperone GRP78/BiP in tumor proliferation, survival, and tumor angiogenesis in transgene-induced mammary tumor development. Cancer Res. 2008;68:498–505. doi: 10.1158/0008-5472.CAN-07-2950. [DOI] [PubMed] [Google Scholar]
- 46.Rao RV, Peel A, Logvinova A, et al. Coupling endoplasmic reticulum stress to the cell death program: role of the ER chaperone GRP78. FEBS Lett. 2002;514:122–8. doi: 10.1016/s0014-5793(02)02289-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reddy RK, Mao C, Baumeister P, et al. Endoplasmic reticulum chaperone protein GRP78 protects cells from apoptosis induced by topoisomerase inhibitors: role of ATP binding site in suppression of caspase-7 activation. J Biol Chem. 2003;278:20915–24. doi: 10.1074/jbc.M212328200. [DOI] [PubMed] [Google Scholar]
- 48.Bando Y, Katayama T, Kasai K, et al. GRP94 (94 kDa glucose-regulated protein) suppresses ischemic neuronal cell death against ischemia/reperfusion injury. Eur J Neurosci. 2003;18:829–40. doi: 10.1046/j.1460-9568.2003.02818.x. [DOI] [PubMed] [Google Scholar]
- 49.Kubota H, Suzuki T, Lu J, et al. Increased expression of GRP94 protein is associated with decreased sensitivity to X-rays in cervical cancer cell lines. Int J Radiat Biol. 2005;81:701–9. doi: 10.1080/09553000500434727. [DOI] [PubMed] [Google Scholar]
- 50.Dey A, Kessova IG, Cederbaum AI. Decreased protein and mRNA expression of ER stress proteins GRP78 and GRP94 in HepG2 cells over-expressing CYP2E1. Arch Biochem Biophys. 2006;447:155–66. doi: 10.1016/j.abb.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 51.Ito M, Onuki R, Bando Y, et al. Phosphorylated PKR contributes the induction of GRP94 under ER stress. Biochem Biophys Res Commun. 2007;360:615–20. doi: 10.1016/j.bbrc.2007.06.087. [DOI] [PubMed] [Google Scholar]
- 52.Fu HY, Minamino T, Tsukamoto O, et al. Overexpression of endoplasmic reticulum-resident chaperone attenuates cardiomyocyte death induced by protea-some inhibition. Cardiovasc Res. 2008;79:600–10. doi: 10.1093/cvr/cvn128. [DOI] [PubMed] [Google Scholar]
- 53.Xue X, Piao JH, Nakajima A, et al. Tumor necrosis factor alpha (TNFalpha) induces the unfolded protein response (UPR) in a reactive oxygen species (ROS)-dependent fashion, and the UPR counteracts ROS accumulation by TNFalpha. J Biol Chem. 2005;280:33917–25. doi: 10.1074/jbc.M505818200. [DOI] [PubMed] [Google Scholar]
- 54.Yan M, Shen J, Person MD, et al. Endoplasmic reticulum stress and unfolded protein response in Atm-deficient thymocytes and thymic lymphoma cells are attributable to oxidative stress. Neoplasia. 2008;10:160–7. doi: 10.1593/neo.07935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Guo R, Ma H, Gao F, et al. Metallothionein alleviates oxidative stress-induced endoplasmic reticulum stress and myocardial dysfunction. J Mol Cell Cardiol. 2009;47:228–37. doi: 10.1016/j.yjmcc.2009.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]