

Abstract
Toll-like receptor 4 (TLR4), a proximal signalling receptor in innate immune responses to lipopolysaccharide of gram-negative pathogens, is expressed in the heart. Accumulating evidence have consolidated the notion that TLR4 plays an essential role in the pathogenesis of cardiac dysfunction. However, the molecular mechanisms of TLR4 responsible for ischemia-induced cardiac dysfunction remain unclear. To address the signalling mechanisms of TLR4-deficiency cardioprotection against ischemic injury, in vivo regional ischemia was induced by occlusion of the left anterior descending coronary artery in wild-type (WT) C3H/HeN and TLR4-mutated C3H/HeJ mice. The results demonstrated that blunted ischemic activation of p38 mitogen-activated protein kinase and JNK signalling occurred in C3H/HeJ hearts versus C3H/HeN hearts, while ERK and AMP-activated protein kinase (AMPK) signalling pathways were augmented during ischemia in C3H/HeJ hearts versus C3H/HeN hearts. Intriguingly, ischemia-stimulated endoplasmic reticulum stress was higher in C3H/HeN hearts than that in C3H/HeJ as demonstrated by up-regulation of Grp78/BiP, Gadd153/CHOP and IRE-1α. Myocardial infarct, caspase-3 activity and terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) staining demonstrated that C3H/HeN hearts suffered more damage than those of C3H/HeJ hearts during ischemia. Moreover, isolated cardiomyocytes from C3H/HeJ hearts showed resistance to hypoxia-induced contractile dysfunction compared to those from C3H/HeN hearts, which are associated with greater hypoxic activation of AMPK and ERK signalling, better intracellular Ca2+ handling in C3H/HeJ versus C3H/HeN cardiomyocytes. These findings suggest that the cardioprotective effects against ischemic injury of hearts with deficiency in TLR4 signalling may be mediated through modulating AMPK and ERK signalling pathway during ischemia.
Keywords: TLR4, AMPK, contractile dysfunction, MAPK, ischemia
Introduction
Myocardial ischemia resulted from blockade in the coronary arteries caused by atherosclerosis can impair oxygen delivery [1]. Toll-like receptor 4 (TLR4), a proximal signalling receptor in innate immune response, is also expressed in heart and vasculature [2]. TLR4 binds the complex of protein MD2 and lipopolysaccharide from the gram-negative bacteria, and autogenous ligands such as heat shock proteins and fibronectin which are released during oxidative stress, following ischemic injury [3]. Innate immune and inflammatory pathways have been implicated in myocardial ischemic injury and congestive heart failure [4]. Increased TLR4 expression has been observed in human heart failure and ischemic hearts [2]. TLR4 also plays a role in myocardial dysfunction during bacterial sepsis [5] and pressure overload-induced cardiac hypertrophy [6]. Mitogen-activated protein kinases (MAPKs) such as p38, ERK and JNK are located downstream of TLR4 [7]. Ischemia activates p38, which demonstrated negative inotropic and restrictive diastolic effects [8]. We have reported that p38 contributed to increased glycolysis and inhibition of glycogen synthesis during ischemia [9]. However, the mechanisms by which modulation of TLR4-mediated signalling protects the myocardium from ischemic injury remain unclear.
The AMP-activated protein kinase (AMPK) is an energy-sensing enzyme that can be activated by ischemia [1, 9, 10], and we have found that AMPK plays an important role in cardioprotection against ischemic injury [11, 12]. AMPK mediates several metabolic changes such as decreasing fatty acid and cholesterol synthesis, inhibiting hepatic glucose production and increasing fatty acid oxidation and muscle glucose uptake [13, 14]. In cardiac muscle, AMPK activity is increased by stimuli such as exercise, hypoxia, ischemia and stress [11]. It plays a central role in monitoring the cellular energy status and controlling energy production and consumption [13, 15, 16]. Intriguingly, it has been reported that AMPK protects cardiomyocytes against hypoxic injury by attenuation of endoplasmic reticulum (ER) stress [17]. In eukaryotic cells, the ER plays a vital role in the maturation, processing and transporting secretory- and membrane-associated proteins [8]. The ER is exquisitely sensitive to alteration in homeostasis, with perturbations in the environment resulting in a condition known as ER stress [18].
Here we examined whether the cardioprotection of TLR4-mutated hearts against ischemic injury is mediated by AMPK signalling pathways and what's the role of TLR4 downstream, the MAPK signalling pathway in the cardioprotective effect. We also try to address the relationship between ischemic stress and ER stress in the heart. Interestingly, the TLR4-mutated C3H/HeJ hearts displayed resistance to ischemic stress than wild-type (WT) C3H/HeN hearts, which is linked to higher AMPK and ERK activation and lower p38 and JNK activation. C3H/HeJ hearts also demonstrated less ER stress status than C3H/HeN hearts, as indicated by ER stress markers such as GRP78, Gadd153 and IRE1α up-regulation in response to ischemic stress. Moreover, the adaptive abilities of single cardiomyocyte isolated from C3H/HeJ hearts were significantly improved compared to that of cardiomyocytes from C3H/HeN hearts in the hypoxic condition. C3H/HeJ cardiomyocytes displayed greater hypoxic activation of AMPK and ERK signalling pathways, and better intracellular Ca2+ handling than C3H/HeN cardiomyocytes.
Materials and methods
Animals and surgical procedures
Male WT (C3H/HeN) mice and TLR4-mutated mice (C3H/HeJ, naturally occurring mutations in the TLR4 gene) were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). All animal procedures used in this study were approved by the Animal Care and Use Committees at the University of Wyoming (Laramie, WY, USA). All animals were kept in our institutional animal facility at the University of Wyoming with free access to standard laboratory chow and tap water. C3H/HeN and C3H/HeJ Mice were anesthetized with ketamine (95 mg/kg) and xylazine (40 mg/kg) intraperitoneally, incubated and ventilated with a respirator [9]. After thoracotomy, a suture was placed to ligate the proximal left anterior descending coronary artery for different time. Control mice underwent sham thoracotomy. Hearts were then rapidly excised, and the ischemic region of the left ventricle was freeze clamped in liquid nitrogen.
Myocardial infarct size measurement
Mice were anesthetized, intubated and ventilated with respirator [9, 19]. The body temperature was maintained at 37°C with a heating pad. After left lateral thoracotomy, the left anterior descending was occluded for 20 min. with an 8–0 nylon suture and polyethylene tubing to prevent arterial injury, and then reperfused for 3 hrs. Electrocardio gram (ECGs) confirmed the ischemic hallmark of ST-segment elevation during coronary occlusion (ADInstruments, Colorado Springs, CO, USA). The heart was then excised and stained to delineate the extent of myocardial necrosis as a percentage of non-perfused ischemic area at risk. Viable tissue in the ischemic region was stained red by 2,3,5,-triphenyltetrazolium chloride (TTC) and the non-ischemic region was stained blue with Evan's blue dye. Hearts was fixed and sectioned into 1-mm slices, photographed using a Leica microscope (Wheat Ridge, CO, USA) and analyzed using NIH Image software.
Isolation of mouse cardiomyocytes
Cardiomyocytes were enzymatically isolated as described previously [20]. In brief, hearts were removed and perfused with oxygenated (5% CO2/95% O2) Krebs-Henseleit bicarbonate (KHB) buffer containing (in mmol/l) 118 NaCl, 4.7 KCl, 1.25 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3, 10 HEPES and 11.1 glucose. Hearts were then perfused with a Ca2+-free KHB containing Liberase Blendzyme 4 (Hoffmann-La Roche, Inc., Indianapolis, IN, USA) for 10 min. After perfusion, left ventricles were removed and minced to disperse cardiomyocytes in Ca2+-free KHB buffer. Extracellular Ca2+ was added incrementally back to 1.25 mmol/l. Only rod-shaped myocytes with clear edges were selected for mechanical and intracellular Ca2+ transient studies.
Cardiomyocyte shortening/relengthening measurement
The mechanical properties of cardiomyocytes were assessed using a SoftEdge MyoCam system (IonOptix Corporation, Milton, MA, USA) [21]. In brief, left ventricular myocytes were placed in a chamber mounted on the stage of an inverted microscope (Olympus, IX-70, Center Valley, PA, USA) and superfused at 25°C with a buffer containing (in mmol/l): 131 NaCl, 4 KCl, 1 CaCl2, 1 MgCl2, 10 glucose and 10 HEPES, at pH 7.4. The cells were field stimulated with suprathreshold voltage at a frequency of 0.5 Hz, 3 msec. duration, using a pair of platinum wires placed on opposite sides of the chamber and connected to an electrical stimulator (FHC, Inc. Brunswick, NE, USA). The myocyte being studied was displayed on a computer monitor using an IonOptix MyoCam camera. An IonOptix SoftEdge software was used to capture changes in cell length during shortening and relengthening. Cell shortening and relengthening were assessed using the following indices: peak shortening (PS), the amplitude myocytes shortened on electrical stimulation, indicative of peak ventricular contractility; time-to-PS (TPS), the duration of myocyte shortening, an indicative of systolic duration; time-to-90% relengthening (TR90), the duration to reach 90% relengthening, an indicative of diastolic duration (90% rather 100% relengthening was used to avoid noisy signal at baseline concentration); and maximal velocities of shortening/relengthening, maximal slope (derivative) of shortening and relengthening phases, indicative of maximal velocities of ventricular pressure increase/decrease.
Intracellular Ca2+ transient measurement
Myocytes were loaded with fura-2/AM (0.5 μmol/l) for 10 min. and fluorescence measurements were recorded with a dual-excitation fluorescence photomultiplier system (IonOptix). Myocytes were placed on an Olympus IX-70 inverted microscope and imaged through a Fluor (St. Louis, MO, USA) ×40 oil objective. Cells were exposed to light emitted by a 75 W lamp and passed through either a 360 or a 380 nm filter, while being stimulated to contract at 0.5 Hz. Fluorescence emissions were detected between 480 and 520 nm by a photomultiplier tube after first illuminating the cells at 360 nm for 0.5 sec. then at 380 nm for the duration of the recording protocol (333 Hz sampling rate). The 360 nm excitation scan was repeated at the end of the protocol and qualitative changes in intracellular Ca2+ concentration were inferred from the ratio of fura-2 fluorescence intensity at two wavelengths (360/380). Fluorescence decay time was measured as an indication of the intracellular Ca2+ clearing rate. Both single and bi-exponential curve fit equations were applied to calculate the intracellular Ca2+ decay constant [22].
AMPK activity and phosphorylation
AMPK activity was measured with the synthetic SAMS peptide [9, 19] after immuno-isolation with AMPK-α subunit antibody [9, 19, 23] coupled to protein G/A sepharose. AMPK phosphorylation was assessed by immunoblotting with an antibody to α subunits containing phosphorylated Thr172 (Cell Signaling, Beverly, MA, USA) [9, 19].
Immunoblotting
Immunoblots were performed as previously described [19, 24]. Heart homogenate or cell lysis proteins were resolved by SDS-PAGE and transferred onto polyvinylidene difluoride membranes. For reprobing, membranes were stripped with 50 mmol/l Tris-HCl, 2% SDS and 0.1 mol/l β-mercaptoethanol (pH6.8). Rabbit polyclonal antibodies against phosphor-AMPK, total AMPK-α, phosphor-p38, total p38, phosphor-ERK, total ERK, phosphor-JNK, total JNK, IRE-1α, eIF2α and GAPDH were purchased from Cell Signaling. Goat polyclonal antibodies against GRP78, Gadd153, IRE1α, TLR4 and β-tubulin were from Santa Cruz (Santa Cruz, CA, USA). Rabbit polyclonal antibodies against phosphor-acetyl-CoA carboxylase (p-ACC) and total ACC were from Millipore (Billerica, MA, USA). The intensity of bands was measured with a scanning densitometer (model GS-800; Bio-Rad, Hercules, CA, USA) coupled with Bio-Rad personal computer analysis software.
Hypoxia treatment
Isolated mouse cardiomyocytes were subjected to two groups. Hypoxic groups were kept at 37°C in a humidified sealed chamber under a humidified atmosphere of 5% CO2 and 95% N2 for 15 min. Normoxic groups were placed into a water-jacketed incubator at 37°C during the same period.
Analysis of myocyte apoptosis
The degree of apoptotic activity was assessed by measuring caspase-3 activity in tissue homogenates using a commercially available fluorimetric assay (Calbiochem, Madison, WI, USA). In addition, TUNEL staining using fluorescein-labelled dUTP (Roche Applied Science, Indianapolis, IN, USA) and counterstaining with 0.5 μg/ml propidium iodide was performed on 5-μm sections from hearts fixed by paraformaldehyde as previously described [12]. Slides were examined in a blinded fashion for apoptotic nuclei using a Leica TSC SP2 confocal microscope. To determine the percentage of apoptotic cells, the TUNEL+ nuclei and TUNEL– cells were counted using the ImagePro image analysis software (Media Cybernetics, Bethesda, MD, USA).
Statistical analysis
Data were mean ± S.E.M. difference among groups were assessed using anova followed by Newman–Keuls post hoc test. A P-value less than 0.05 was considered statistically significant.
Results
Deficiency in TLR4 signalling reduces infarct size after ischemia/reperfusion
The immunoblotting results demonstrated the dramatically decreased cardiac TLR4 expression level in TLR4-mutated C3H/HeJ mice compared to WT C3H/HeN mice (Fig. 1A), and ischemic treatment did not affect the TLR4 expression in both C3H/HeN and C3H/HeJ hearts (Fig. 1A). In order to examine whether deficiency in TLR4 signal transduction affects the cardiac tolerance to ischemia/reperfusion injury, C3H/HeN and C3H/HeJ mice were subjected to a 20-min. ligation of the left coronary artery followed by 3 hrs reperfusion (Fig. 1B). The extent of myocardial infarction was measured with dual staining to define the degree of necrosis within the ischemic region at risk [19, 25]. C3H/HeJ hearts demonstrated significantly smaller infarct size than C3H/HeN hearts (Fig. 1B). These results indicate that deficiency in TLR4 signalling leads to a cardioprotective effect to prevent ischemic injury.
Fig. 1.
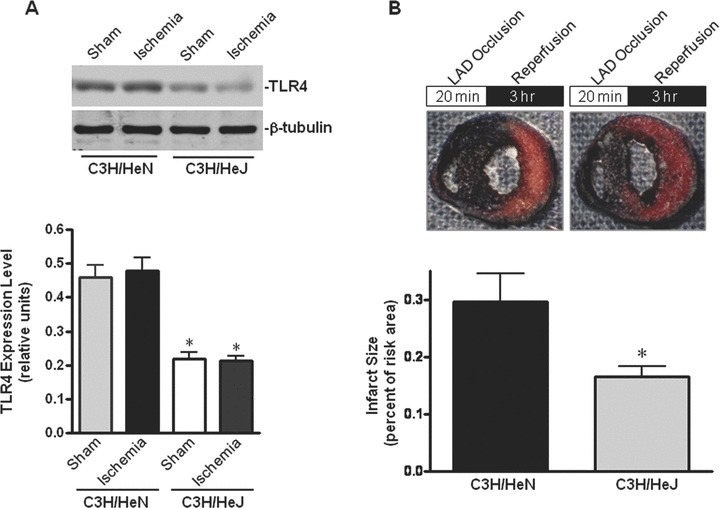
Deficiency in TLR4 signal transduction mitigates cardiac injury caused by ischemia/reperfusion. (A) The expression levels of TLR4 in WT C3H/HeN and TLR4-mutated C3H/HeJ hearts. Immunoblots were performed with antibodies for TLR4 or β-tubulin. The graph shows TLR4 relative to β-tubulin. Values are mean ± S.E.M. for five experiments. *P< 0.01 versus C3H/HeN sham and ischemia, respectively. (B) Myocardial infarction induced by 20 min. of left coronary occlusion in vivo followed by 3 hrs of reperfusion. Viable myocardium stained red with TTC; infracted tissue, white and normal non-ischemic tissue, blue. The infarct area was quantified and expressed as a per cent of the ischemic area at risk. *P < 0.05 versus C3H/HeN. n= 6 hearts per genotype. Values are mean ± S.E.M.
Augmented AMPK activation in TLR4-mutated mice during ischemia
To assess whether AMPK signalling pathway is involved in the resistance to ischemic injury in C3H/HeJ mice, we first examined the AMPK phosphorylation of C3H/HeJ and C3H/HeN hearts during in vivo regional ischemia. The phosphorylation at Thr172 and activation of AMPK was significantly augmented in C3H/HeJ hearts as compared to that in C3H/HeN hearts (Fig. 2A and B). Moreover, the phosphorylation of the AMPK downstream targets, ACC and eukaryotic elongation factor 2 (eEF2), were also enhanced in C3H/HeJ hearts compared to C3H/HeN hearts (Fig. 2C and D).
Fig. 2.
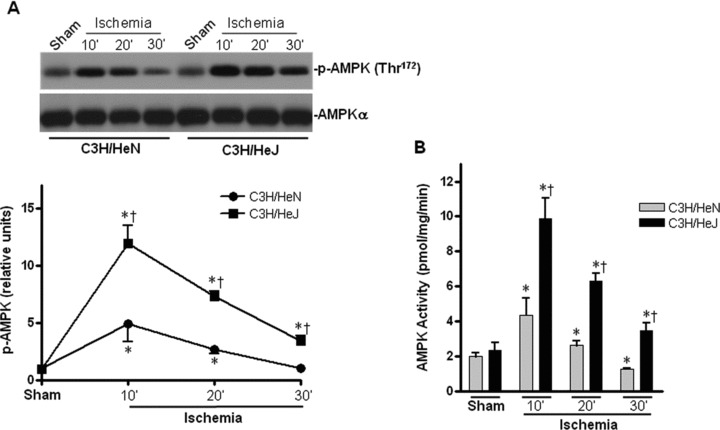
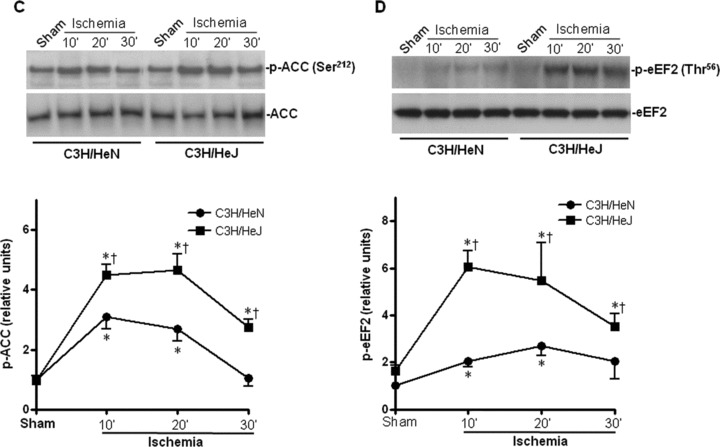
Differential cardiac AMPK activation in C3H/HeN versus C3H/HeJ mice during ischemia. (A) Time course of AMPK phosphorylation induced by in vivo regional ischemia or control sham operation. Immunoblots were performed with antibodies for phosphor-AMPK (Thr172) (p-AMPK) or pan-α AMPK (AMPK-α). The graph shows phosphorylated AMPK relative to the total amount of kinase. (B) AMPK activity in C3H/HeN and C3H/HeJ hearts after sham operations or in vivo regional ischemia. (C) Phosphorylation of AMPK downstream ACC and (D) Phosphorylation of AMPK downstream eEF2 induced by in vivo regional ischemia or control sham operation. Values are mean ± S.E.M. for five experiments. *P< 0.05 versus sham, †P< 0.05 versus C3H/HeN ischemic time-points, respectively. Immunoblots were performed with antibodies for phosphor-ACC (Ser212) (p-ACC), total ACC (ACC), phosphor-eEF2 (Thr56) or total eEF2 (eEF2).
Impaired activation of MAPK signalling in C3H/HeN versus C3H/HeJ during ischemia
In order to determine the role of the TLR4 downstream effectors, such as MAPKs, in ischemic injury, we measured the activation of p38, JNK and ERK. The results demonstrated that ischemia time-dependently stimulated p38, JNK and ERK activation in both C3H/HeN and C3H/HeJ hearts as manifested by phosphorylation immunoblots (Fig. 3A–C). Intriguingly, p38 and JNK signalling were blunted in C3H/HeJ hearts compared to C3H/HeN hearts during ischemia (Fig. 3A and B). In contrast, ERK signalling was enhanced in C3H/HeJ hearts compared to C3H/HeN hearts during ischemia (Fig. 3C).
Fig. 3.
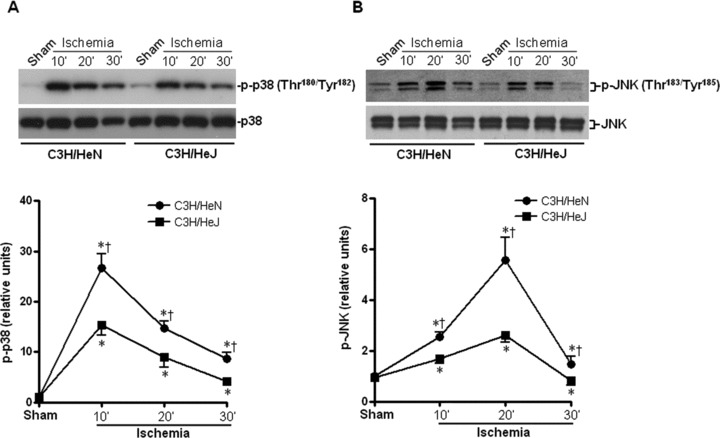
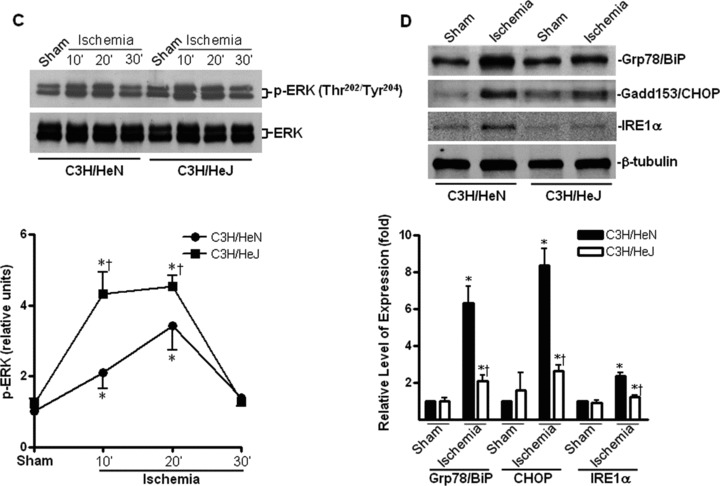
Effects of TLR4 functional deficiency on ischemia-induced mitogen-activated protein kinases (MAPKs) and cardiac ER stress. Time course of p38 (A), JNK (B) and ERK (C) phosphorylation induced by in vivo regional ischemia or sham operation, in C3H/HeN and C3H/HeJ hearts. Immunoblots were performed with antibodies for phosphor-p38 (Thr180/Tyr182) (p-p38), total p38 (p38), phosphor-JNK (Thr183/Tyr185) (p-JNK), total JNK (JNK), phosphor-ERK (Thr202/Tyr204) (p-ERK) or total ERK (ERK). The graph shows phosphorylated p38 MAPK (A), phosphorylated JNK (B) and phosphorylated ERK (C) relative to the total amount of respective kinases. (D) The representative immunoblots show that ischemic stress (30 min.) stimulated the up-regulation of ER chaperons, Grp78/BiP and Gadd153/CHOP and ER integral membrane protein, IRE-1α. Immunoblots were performed with antibodies for Grp78/BiP, Gadd153/CHOP, IRE-1α or GAPDH. The graph shows the relative expression levels of Grp78/BiP, Gadd153/CHOP or IRE-1α to the GAPDH expression. Values are mean ± S.E.M. for five experiments. *P< 0.05 versus sham, †P< 0.05 versus C3H/HeN ischemic time-points, respectively.
Resistance to cardiac ER stress in TLR4-mutated heart during ischemia
It has been reported that ischemia leads to the accumulation of misfolded proteins in the ER, causing ER stress [17]. Under conditions of ER stress, inhibition of protein synthesis and up-regulation of ER chaperone expression reduce the misfolded proteins in the ER. It has been shown that AMPK activation is associated with inhibition of protein synthesis via phosphorylation of eEF2 and attenuation of hypoxia-induced ER stress in cardiomyocytes [17]. We therefore examined whether deficiency in TLR4 signalling affected ischemia-induced ER stress in the hearts. The results demonstrated that ischemic treatment promoted ER stress, as shown by up-regulation of the ER chaperone, Gadd153/CHOP andGrp78/BiP and the integral protein of ER membrane, IRE-1α (Fig. 3D), while up-regulation in all these ER stress makers was blunted in C3H/HeJ hearts during ischemia (Fig. 3D).
TLR4-mutated mitigates ischemic contractile dysfunction of cardiomyocytes
To explore whether the augmented AMPK signalling in C3H/HeJ hearts contributes to the resistance of C3H/HeJ hearts to ischemic injury, we determined the single cardiomyocyte contractile function in response to hypoxic treatment. Isolated cardiomyocytes from C3H/HeN and C3H/HeJ hearts were subjected to an anoxic environment to mimic the ischemic condition. Mechanical properties were evaluated under an extracellular Ca2+ concentration of 1.0 mmol/l and a stimulus frequency of 0.5 Hz [20]. Our results demonstrated comparable resting cell length in cardiomyocytes among all four groups (Fig. 4A). However, cardiomyocytes from C3H/HeN hearts displayed significantly reduced PS (Fig. 4B) and maximal velocity of shortening/ relengthening (±dl/dt) (Fig. 4C and D) in response to hypoxia compared with those from C3H/HeJ hearts. Reduced ±dl/dt was associated with prolonged duration of shortening/relengthening duration (TPS90 and TR90) in C3H/HeN cardiomyocytes (Fig. 4E and F). Interestingly, the hypoxia-induced mechanical dysfunction in C3H/HeN cardiomyocytes was significantly attenuated by TLR4 mutation in C3H/HeJ cardiomyocytes (Fig. 4), suggesting a beneficial role of TLR4 deficiency in cardiac protection against hypoxia-induced cardiac dysfunction.
Fig. 4.
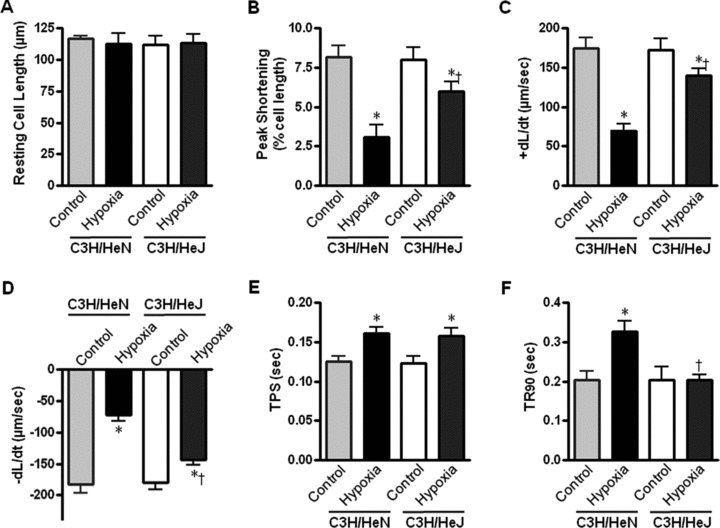
Contractile properties of cardiomyocytes from C3H/HeN and C3H/HeJ hearts during hypoxia (30 min.). (A) Resting cell length; (B) Peak shortening (PS, normalized to cell length)l; (C) and (D), Maximal velocity of shortening (+dl/dt) and relengthening (−dl/dt); (E) Time-to-PS (TPS); (F) Time-to-90% relengthening (TR90). Values are mean ± S.E.M., n= 60–90 cells per group, *P < 0.05 versus control, respectively, †P< 0.05 versus C3H/HeN hypoxia.
Augmented hypoxic activation of AMPK and ERK in C3H/HeJ cardiomyocytes
To assess whether AMPK and ERK signalling pathways are also involved in resistance to hypoxic contractile dysfunction of C3H/HeJ cardiomyocytes, we examined the hypoxic phosphorylation of AMPK and ERK in C3H/HeJ and C3H/HeN cardiomyocytes. The phosphorylation of both AMPK and ERK were significantly augmented in C3H/HeJ cardiomyocytes as compared to those in C3H/HeN cardiomyocytes (Fig. 5A and B), which indicated that AMPK and ERK signalling may also play a role in resistance to hypoxic contractile dysfunction in cardiomyocytes.
Fig. 5.
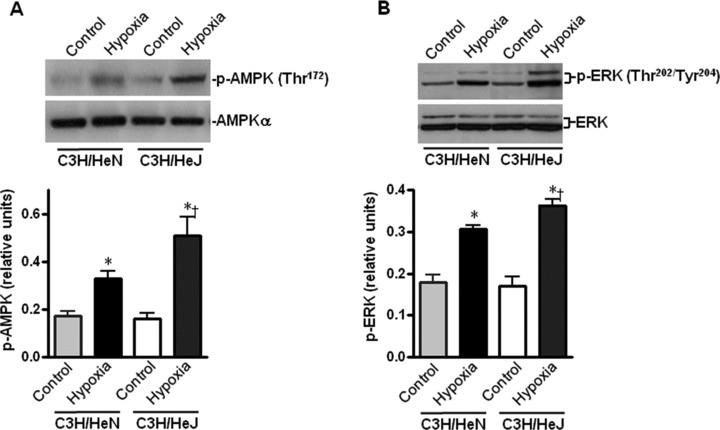
Effects of TLR4 functional deficiency on hypoxia-induced phosphorylation of AMPK and ERK in cardiomyocytes. The representative immunoblots show that hypoxia (30 min.) stimulated phosphorylation of AMPK (A) and ERK (B) in C3H/HeN and C3H/HeJ cardiomyocytes. Immunoblots were performed with antibodies for phosphor-AMPK (Thr172) (p-AMPK), pan-α AMPK (AMPK-α), phosphor-ERK (Thr202/Tyr204) (p-ERK) or total ERK (ERK). The graph shows phosphorylated AMPK (A), phosphorylated ERK (B) relative to the total amount of respective kinases. Values are mean ± S.E.M. for four experiments. *P< 0.05 versus control, †P< 0.05 versus C3H/HeN hypoxia.
The intracellular Ca2+ properties of C3H/HeN and C3H/HeJ cardiomyocytes
To explore the potential mechanism(s) involved in TLR4 deficiency-elicited protection against hypoxic cardiomyocyte contractile defect and augmented hypoxic activation of AMPK signalling, intracellular Ca2+ homeostasis was evaluated using the fluorescence dye fura-2/AM [21]. The results revealed that hypoxia elevated the resting intracellular Ca2+ levels in cardiomyocytes from both C3H/HeN and C3H/HeJ groups (Fig. 6A), with a greater response in C3H/HeJ cardiomyocytes (Fig. 6A). Hypoxia reduced intracellular Ca2+ clearance (both single and bi-exponential decays, Fig. 6C and D) associated with unchanged electrically stimulated rise in intracellular Ca2+ levels (Fig. 6B) in C3H/HeN cardiomyocytes, the effect of which was ablated in C3H/HeJ group (Fig. 6C and D). Moreover, TLR4 deficiency itself did not elicit any overt effect on intracellular Ca2+ properties in non-hypoxic conditions, suggesting that genetic modification of the TLR4 gene does not affect the innate intracellular Ca2+ handling (Fig. 6). The greater resting intracellular Ca2+ levels in hypoxic C3H/HeJ cardiomyocytes may contribute to the stronger hypoxic AMPK activation (Fig. 5A) via activation of calcium/calmodulin-dependent protein kinase kinase 2 [26–30], while the increase of ischemic AMP concentrations, one of the regulators for AMPK activation [11, 13, 31–35], does not appear to be a contributor of this difference of AMPK activation between C3H/HeN and C3H/HeJ hearts (data not shown). The reduced intracellular Ca2+ clearing rate of C3H/HeN cardiomyocytes might be a factor leading to prolonged TR90 in these cardiomyocytes (Fig. 4F).
Fig. 6.
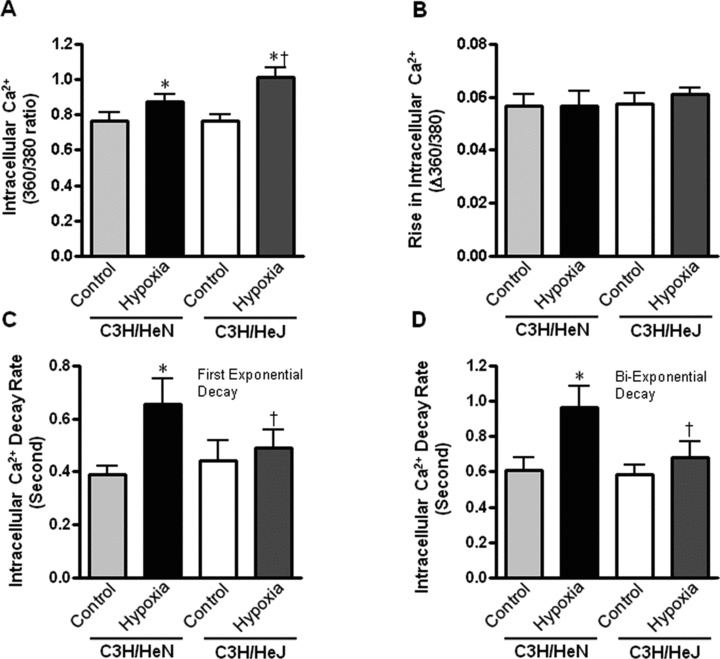
Intracellular Ca2+ properties of cardiomyocytes from C3H/HeN and C3H/HeJ hearts. (A) The intracellular Ca2+ levels; (B) The rise in intracellular Ca2+ levels in response to electrical stimulus; (C) The first exponential decay constant of intracellular Ca2+ and (D) The bi-exponential decay constant of intracellular Ca2+ of C3H/HeN and C3H/HeJ in response to hypoxia (30 min.). Values are mean ± S.E.M. for six experiments, *P< 0.05 versus control, respectively. †P< 0.05 versus C3H/HeN hypoxia.
Reduced apoptosis in TLR4-mutated hearts during ischemia
To determine whether impaired contractile function of cardiomyocytes from C3H/HeN hearts as opposed to C3H/HeJ hearts during ischemia/hypoxia was due to more damage in C3H/HeN hearts, caspase-3 activity and TUNEL staining assays were performed. The data clearly demonstrated that ischemia facilitated caspase-3 activity (Fig. 7A) in both C3H/HeN and C3H/HeJ hearts with a greater caspase-3 activity in C3H/HeN hearts (Fig. 7A). TLR4 mutation did not affect the baseline caspase-3 activity in the absence of ischemic injury. In addition, data using confocal microscopy revealed a greater number of TUNEL+ nuclei following ischemia in C3H/HeN hearts compared with C3H/HeJ hearts (Fig. 7B and C). Therefore, TLR4 mutation leads to cardiac resistance to ischemic injury via improvement of intracellular Ca2+ handling, and augmentation of AMPK and ERK signalling pathways.
Fig. 7.
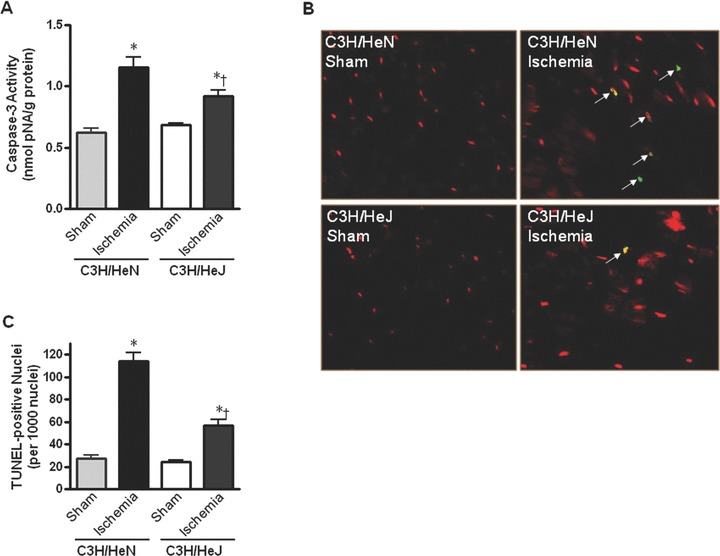
Myocardial apoptotic activity following in vivo regional ischemia. (A) Caspase-3 activity in hearts from C3H/HeN and C3H/HeJ mice. (B) Representative composite confocal photomicrographs of sections from C3H/HeN and C3H/HeJ hearts following ischemia. Hearts underwent TUNEL staining with fluorescein-labelled dUTP and counterstaining with propidium iodide. TUNEL+ nuclei are stained yellow (arrows). (C) The graph shows the quantification of apoptotic nuclei from C3H/HeN and C3H/HeJ hearts following ischemia. Values are mean ± S.E. for four experiments, *P< 0.05 versus control. †P < 0.05 versus C3H/HeN.
Discussion
In this study, we found a significantly augmented activation of the cellular fuel gauge AMPK following ischemia in TLR4-mutated C3H/HeJ hearts compared to WT C3H/HeN hearts, which may be a contributor for the resistance to ischemic injury of C3H/HeJ hearts. Mechanical function analysis of isolated cardiomyocytes from C3H/HeN and C3H/HeJ hearts suggested that TLR4 deficiency protected against hypoxia-/ischemia-induced cardiac damage. Interestingly, TLR4 deficiency significantly blunted ischemic activation of p38 and JNK inflammatory-associated signalling pathways [7], while it enhanced ischemic activation of the ERK survival signalling pathway [7]. Moreover, TLR4 deficiency also decreased ischemia-triggered ER stress status, as shown by down-regulation of ER chaperons in C3H/HeJ hearts versus C3H/HeN hearts. Caspase-3 activity, TUNEL staining and myocardial infarct analysis clearly demonstrated that TLR4-deficient hearts restrained the hearts from ischemic injury. The improvement of intracellular Ca2+ handling in the hypoxic C3H/HeJ cardiomyocytes may enhance the capacity of AMPK signalling in response to ischemic stress. These data favour the notion of an involvement of AMPK and MAPK in TLR4 mediated responses to ischemic stress and tend to establish an important role for TLR4 signalling in ischemia-induced myocardial injury.
The results also revealed that cardiomyocytes from C3H/HeN exhibited depressed PS, reduced maximal velocity of shortening/ relengthening, and prolonged duration of contraction in response to hypoxia treatment. These data are somewhat consistent with the previous findings using Langendorff apparatus on whole heart function after ischemia [36]. Cardiomyocytes from C3H/HeJ hearts displayed significantly improved PS, and maximal velocity of shortening/relengthening compared with C3H/HeN mice. Nonetheless, TLR4 deficiency failed to fully restore hypoxia-induced cardiomyocyte mechanical impairment, indicating that a cardiac remodelling process was initiated in response to hypoxia. The explanation of mechanical defects observed in our study may be the impaired intracellular Ca2+ handling in C3H/HeN hearts. The reduction of intracellular Ca2+ clearance is likely responsible for prolonged relaxation duration (TR90) and reduced PS in C3H/HeN hypoxic cardiomyocytes.
It has been shown that TLR4 signalling may play an important role in the development of heart failure after acute myocardial infarct [37]. Our group and others have reported that ischemic activation of p38, one of the TLR4 downstream effectors, plays an important role in regulation of glucose uptake [9] and cardioprotection against ischemic injury [38, 39]. Though there were several studies support the view that p38 is a downstream of AMPK signalling [9, 11], controversy of their relationship also can be considered [40]. There were studies which demonstrated that although AMPK and p38 were both activated during myocardial ischemia, the activation of p38 occurs independently of AMPK [40]. It has also been shown that p38 is not a downstream component involved in AMPK-mediated signalling pathway in mouse skeletal muscle [41]. Capano et al. reported that ischemia lead to rapid activation of AMPK which then phosphorylates and activates p38 MAPK [42]. FR167653, a highly specific inhibitor of p38 MAPK, selectively inhibits p38 activation during regional myocardial ischemia-reperfusion injury and significantly reduces infarct size [38]. The potential linkage between AMPK and p38 during ischemia needs to be addressed in the future.
Our previous studies have shown that activation of JNK is linked to ER stress mediated apoptosis and caspase-12 cleavage [18, 24]. JNK activation has a pro-apoptotic effect under various cell death-inducing stimuli, such as oxidative stress and DNA damaging agents [43, 44]. Also, using a highly specific peptide inhibitor of JNK, Milano et al.[45] showed that inhibition of JNK reduces myocardial ischemic-reperfusion injury and infarct size in anesthetized rats in vivo. On the other hand, ERK activation is anti-apoptotic in most tissues [46]. Our data showed the lower activation of p38 and JNK, but higher ERK activation in TLR4-deficient C3H/HeJ hearts compared to WT C3H/HeN hearts during ischemia, which indicates that MAPKs may contribute to the cardioprotection of TLR4 deficiency. However, further studies are needed to investigate the pathophysiological significance and complex interactions among the various apoptotic and anti-apoptotic pathways.
Recently, ER was reported as a third compartment that participates in apoptosis, in addition to the two known apoptotic pathways, the death receptor and the mitochondrial pathways [8]. Accumulated evidence have indicated that ER stress contributes to several diseases such as neurodegenerative disorders, diabetes and ischemia reperfusion-induced heart damage [47]. Our result revealed that cardiac ER stress was induced in both ischemic C3H/HeN and C3H/HeJ hearts, but C3H/HeJ hearts appears to be resistant to ischemia-induced ER stress, which may be associated with the augmentation of AMPK activation and downstream eEF2 phosphorylation. This is in line with a study that activation of AMPK contributes to the protection of heart against hypoxic injury through attenuation of ER stress and of protein synthesis via eEF2 phosphorylation [17]. The better intracellular Ca2+ handling capacity of TLR4 deficiency cardiomyocytes may be associated with greater hypoxic activation of AMPK in C3H/HeJ, even though there was no difference in increase of AMP concentrations in both hypoxic C3H/HeN and hypoxic C3H/HeJ cardiomyocytes.
Taken together, the results indicate that TLR4 mediates cardioprotection against ischemic injury, through modulating AMPK and MAPK signalling pathways. In addition, TLR4 might play an important role in ischemia-stimulated cardiac ER stress. These data have convincingly demonstrated the clinical potential target of TLR4 in the ischemia-reduced cardiac dysfunction.
Acknowledgments
The authors greatly appreciate Dr. Zhaojie Zhang and Dr. Qun Li for technical assistance. This work was presented in part at 2008 Scientific Session of American Heart Association (AHA) in New Orleans on 9 November. This work was supported by grants from R03AG028163, a pilot grant from University of Wyoming Northern Rockies Regional IDeA Networks for Biomedical Research Excellence (INBRE), 5P20RR016474, American Heart Association (AHA) National SDG0835168N, American Federation for Aging Research (AFAR) 08007 and a pilot grant of 1UL1RR025014-01 from the National Center for Research Resources (NCRR).
References
- 1.Young LH, Li J, Baron SJ, et al. AMP-activated protein kinase: a key stress signaling pathway in the heart. Trends Cardiovasc Med. 2005;15:110–8. doi: 10.1016/j.tcm.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 2.Frantz S, Kobzik L, Kim YD, et al. Toll4 (TLR4) expression in cardiac myocytes in normal and failing myocardium. J Clin Invest. 1999;104:271–80. doi: 10.1172/JCI6709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oyama J, Blais C, Jr, Liu X, et al. Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation. 2004;109:784–9. doi: 10.1161/01.CIR.0000112575.66565.84. [DOI] [PubMed] [Google Scholar]
- 4.Li C, Ha T, Kelley J, et al. Modulating Toll-like receptor mediated signaling by (1–>3)-beta-D-glucan rapidly induces cardioprotection. Cardiovasc Res. 2004;61:538–47. doi: 10.1016/j.cardiores.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 5.Baumgarten G, Knuefermann P, Nozaki N, et al. In vivo expression of proinflamma-tory mediators in the adult heart after endotoxin administration: the role of tolllike receptor-4. J Infect Dis. 2001;183:1617–24. doi: 10.1086/320712. [DOI] [PubMed] [Google Scholar]
- 6.Ha T, Li Y, Hua F, et al. Reduced cardiac hypertrophy in toll-like receptor 4-deficient mice following pressure overload. Cardiovasc Res. 2005;68:224–34. doi: 10.1016/j.cardiores.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 7.Chen P, Li J, Barnes J, et al. Restraint of proinflammatory cytokine biosynthesis by mitogen-activated protein kinase phos-phatase-1 in lipopolysaccharide-stimulated macrophages. J Immunol. 2002;169:6408–16. doi: 10.4049/jimmunol.169.11.6408. [DOI] [PubMed] [Google Scholar]
- 8.Ellgaard L, Helenius A. Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 2003;4:181–91. doi: 10.1038/nrm1052. [DOI] [PubMed] [Google Scholar]
- 9.Li J, Miller EJ, Ninomiya-Tsuji J, et al. AMP-activated protein kinase activates p38 mitogen-activated protein kinase by increasing recruitment of p38 MAPK to TAB1 in the ischemic heart. Circ Res. 2005;97:872–9. doi: 10.1161/01.RES.0000187458.77026.10. [DOI] [PubMed] [Google Scholar]
- 10.Xing Y, Musi N, Fujii N, et al. Glucose metabolism and energy homeostasis in mouse hearts overexpressing dominant negative alpha2 subunit of AMP-activated protein kinase. J Biol Chem. 2003;278:28372–7. doi: 10.1074/jbc.M303521200. [DOI] [PubMed] [Google Scholar]
- 11.Young LH. AMP-activated protein kinase conducts the ischemic stress response orchestra. Circulation. 2008;117:832–40. doi: 10.1161/CIRCULATIONAHA.107.713115. [DOI] [PubMed] [Google Scholar]
- 12.Russell RR, 3rd, Li J, Coven DL, et al. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apopto-sis, and injury. J Clin Invest. 2004;114:495–503. doi: 10.1172/JCI19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hardie DG. Role of AMP-activated protein kinase in the metabolic syndrome and in heart disease. FEBS Lett. 2008;582:81–9. doi: 10.1016/j.febslet.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 14.Hue L, Rider MH. The AMP-activated protein kinase: more than an energy sensor. Essays Biochem. 2007;43:121–37. doi: 10.1042/BSE0430121. [DOI] [PubMed] [Google Scholar]
- 15.Hardie DG. Minireview: the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology. 2003;144:5179–83. doi: 10.1210/en.2003-0982. [DOI] [PubMed] [Google Scholar]
- 16.Hardie DG, Hawley SA, Scott JW. AMP-activated protein kinase–development of the energy sensor concept. J Physiol. 2006;574:7–15. doi: 10.1113/jphysiol.2006.108944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Terai K, Hiramoto Y, Masaki M, et al. AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol Cell Biol. 2005;25:9554–75. doi: 10.1128/MCB.25.21.9554-9575.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J, Holbrook NJ. Elevated gadd153/chop expression and enhanced c-Jun N-terminal protein kinase activation sensitizes aged cells to ER stress. Exp Gerontol. 2004;39:735–44. doi: 10.1016/j.exger.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 19.Miller EJ, Li J, Leng L, et al. Macrophage migration inhibitory factor stimulates AMP-activated protein kinase in the ischaemic heart. Nature. 2008;451:578–82. doi: 10.1038/nature06504. [DOI] [PubMed] [Google Scholar]
- 20.Dong F, Zhang X, Yang X, et al. Impaired cardiac contractile function in ventricular myocytes from leptin-deficient ob/ob obese mice. J Endocrinol. 2006;188:25–36. doi: 10.1677/joe.1.06241. [DOI] [PubMed] [Google Scholar]
- 21.Li Q, Ceylan-lsik AF, Li J, et al. Deficiency of Insulin-like growth factor (IGF-1) leads to resistance against aging-associated cardiomyocyte dysfunction. Rejuvenation Res. 2008;11:725–33. doi: 10.1089/rej.2008.0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ren J, Privratsky JR, Yang X, et al. Metallothionein alleviates glutathione depletion-induced oxidative cardiomyopathy in murine hearts. Crit Care Med. 2008;36:2106–16. doi: 10.1097/CCM.0b013e31817bf925. [DOI] [PubMed] [Google Scholar]
- 23.Mu J, Brozinick JT, Jr, Valladares O, et al. A role for AMP-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol Cell. 2001;7:1085–94. doi: 10.1016/s1097-2765(01)00251-9. [DOI] [PubMed] [Google Scholar]
- 24.Zhao P, Xiao X, Kim AS, et al. c-Jun inhibits thapsigargin-induced ER stress through up-regulation of DSCR1/Adapt78. Exp Biol Med. 2008;233:1289–300. doi: 10.3181/0803-RM-84. [DOI] [PubMed] [Google Scholar]
- 25.Li J, Coven DL, Miller EJ, et al. Activation of AMPK alpha- and gamma-isoform complexes in the intact ischemic rat heart. Am J Physiol Heart Circ Physiol. 2006;291:H1927–34. doi: 10.1152/ajpheart.00251.2006. [DOI] [PubMed] [Google Scholar]
- 26.Hawley SA, Pan DA, Mustard KJ, et al. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2:9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 27.Hurley RL, Anderson KA, Franzone JM, et al. The Ca2+/calmodulin-dependent protein kinase kinases are AMP-activated protein kinase kinases. J Biol Chem. 2005;280:29060–6. doi: 10.1074/jbc.M503824200. [DOI] [PubMed] [Google Scholar]
- 28.Woods A, Dickerson K, Heath R, et al. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2:21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 29.Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res. 2007;100:328–41. doi: 10.1161/01.RES.0000256090.42690.05. [DOI] [PubMed] [Google Scholar]
- 30.Horie T, Ono K, Nagao K, et al. Oxidative stress induces GLUT4 translocation by activation of PI3-K/Akt and dual AMPK kinase in cardiac myocytes. J Cell Physiol. 2008;215:733–42. doi: 10.1002/jcp.21353. [DOI] [PubMed] [Google Scholar]
- 31.Baron SJ, Li J, Russell RR, 3rd, et al. Dual mechanisms regulating AMPK kinase action in the ischemic heart. Circ Res. 2005;96:337–45. doi: 10.1161/01.RES.0000155723.53868.d2. [DOI] [PubMed] [Google Scholar]
- 32.Ruderman NB, Cacicedo JM, Itani S, et al. Malonyl-CoA and AMP-activated protein kinase (AMPK): possible links between insulin resistance in muscle and early endothelial cell damage in diabetes. Biochem Soc Trans. 2003;31:202–6. doi: 10.1042/bst0310202. [DOI] [PubMed] [Google Scholar]
- 33.Zou MH, Wu Y. AMP-activated protein kinase activation as a strategy for protecting vascular endothelial function. Clin Exp Pharmacol Physiol. 2008;35:535–45. doi: 10.1111/j.1440-1681.2007.04851.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tian R, Balschi JA. Interaction of insulin and AMPK in the ischemic heart: another chapter in the book of metabolic therapy? Circ Res. 2006;99:3–5. doi: 10.1161/01.RES.0000233142.26369.f6. [DOI] [PubMed] [Google Scholar]
- 35.Taha M, Lopaschuk GD. Alterations in energy metabolism in cardiomyopathies. Ann Med. 2007;39:594–607. doi: 10.1080/07853890701618305. [DOI] [PubMed] [Google Scholar]
- 36.Wang M, Tsai BM, Crisostomo PR, et al. Tumor necrosis factor receptor 1 signaling resistance in the female myocardium during ischemia. Circulation. 2006;114:I282–9. doi: 10.1161/CIRCULATIONAHA.105.001164. [DOI] [PubMed] [Google Scholar]
- 37.Satoh M, Shimoda Y, Maesawa C, et al. Activated toll-like receptor 4 in monocytes is associated with heart failure after acute myocardial infarction. Int J Cardiol. 2006;109:226–34. doi: 10.1016/j.ijcard.2005.06.023. [DOI] [PubMed] [Google Scholar]
- 38.Yada M, Shimamoto A, Hampton CR, et al. FR167653 diminishes infarct size in a murine model of myocardial ischemia-reperfusion injury. J Thorac Cardiovasc Surg. 2004;128:588–94. doi: 10.1016/j.jtcvs.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 39.Valouskova E, Modriansky M. Modulation of UCP2 expression by p38 – a link to cardioprotection. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2008;152:3–7. doi: 10.5507/bp.2008.001. [DOI] [PubMed] [Google Scholar]
- 40.Jacquet S, Zarrinpashneh E, Chavey A, et al. The relationship between p38 mitogen-activated protein kinase and AMP-activated protein kinase during myocardial ischemia. Cardiovasc Res. 2007;76:465–72. doi: 10.1016/j.cardiores.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 41.Ho RC, Fujii N, Witters LA, et al. Dissociation of AMP-activated protein kinase and p38 mitogen-activated protein kinase signaling in skeletal muscle. Biochem Biophys Res Commun. 2007;362:354–9. doi: 10.1016/j.bbrc.2007.07.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Capano M, Crompton M. Bax translocates to mitochondria of heart cells during simulated ischaemia: involvement of AMP-activated and p38 mitogen-activated protein kinases. Biochem J. 2006;395:57–64. doi: 10.1042/BJ20051654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deng Y, Ren X, Yang L, et al. A JNK-dependent pathway is required for TNFalpha-induced apoptosis. Cell. 2003;115:61–70. doi: 10.1016/s0092-8674(03)00757-8. [DOI] [PubMed] [Google Scholar]
- 44.Kamata H, Honda S, Maeda S, et al. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–61. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 45.Milano G, Morel S, Bonny C, et al. A peptide inhibitor of c-Jun NH2-terminal kinase reduces myocardial ischemia-reperfusion injury and infarct size in vivo. Am J Physiol Heart Circ Physiol. 2007;292:H1828–35. doi: 10.1152/ajpheart.01117.2006. [DOI] [PubMed] [Google Scholar]
- 46.Whitlock BB, Gardai S, Fadok V, et al. Differential roles for alpha(M)beta(2) integrin clustering or activation in the control of apoptosis via regulation of akt and ERK survival mechanisms. J Cell Biol. 2000;151:1305–20. doi: 10.1083/jcb.151.6.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ozcan U, Yilmaz E, Ozcan L, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–40. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]