

Abstract
Angiogenesis is controlled by a balance between pro- and anti-angiogenic factors. Studies in mice and human beings have shown that this balance, as well as the general sensitivity of the endothelium to these factors, is genetically pre-determined. In an effort to dissect this genetic basis, different types of genetic variability have emerged: mutations and translocations in angiogenic factors have been linked to several vascular malformations and haemangiomas, whereas SNPs have been associated with complex genetic disorders, such as cancer, neurodegeneration and diabetes. In addition, copy number alterations of angiogenic factors have been reported in several tumours. More recently, epigenetic changes caused by aberrant DNA methylation or histone acetylation of anti-angiogenic molecules have been shown to determine angiogenesis as well. Initial studies also revealed a crucial role for microRNAs in stimulating or reducing angiogenesis. So far, most of these genetic studies have focused on tumour angiogenesis, but future research is expected to improve our understanding of how genetic variants determine angiogenesis in other diseases. Importantly, these genetic insights might also be of important clinical relevance for the use of anti-angiogenic strategies in cancer or macular degeneration.
Keywords: angiogenesis, mutation, translocation, single nucleotide polymorphism, DNA-metylation, histone acetylation, microRNA, anti-angiogenic therapy
Introduction
Angiogenesis is genetically pre-determined
-
Mutations causing vascular anomalies
- Venous anomalies
- Haemangiomas
- The transforming growth factor-ß in vascular anomalies
- Cerebral cavernous malformations
Translocations reveal novel angiogenic genes
-
Single nucleotide polymorphisms shape the angio-genome
- SNPs in VEGF and their association with cancer
- SNPs in VEGF pathway genes associated with other diseases
- Genetic variability in VEGFR-2
- Genetic variability in HIF-1α
- SNPs in VEGFR-1 integrate angiogenesis within the P53 pathway
- Variations in angiogenic genes are linked with neurodegeneration
- Angiogenic factors in genome-wide association studies
Copy number variability affects angiogenesis
-
Epigenetic regulation of angiogenesis
- Methylation of anti-angiogenic factors
- Methylation as a second hit event in cancer
- Histone modifications determine angiogenesis
Micromanagers of angiogenesis
Perspectives
Introduction
Already two millennia ago, Aristotle described the vital importance of the vascular system as follows: ‘the system of blood vessels can be compared with those of watercourses in gardens: they start from one source and branch off into numerous channels, so as to carry a supply to every part of the garden‘. After Aristotle, the study of the vascular system, and in particular its growth - a phenomenon generally referred to as angiogenesis - continued to attract interest from scientists. Yet, it took until 1971, when Judah Folkman proposed that interference with angiogenic factors might impede vessel growth and starve tumours [1], before interest from the scientific community became fully primed. This led to a tremendous effort on the part of academic and industry researchers to define the molecular mechanisms driving angiogenesis.
Today, angiogenesis is considered to be a complex and dynamic biological process [2]. A large body of evidence shows that angiogenesis is essential during embryonic development, but becomes notably quiescent in adulthood [3, 4]. Remarkably, however, endothelial cells retain their ability of dividing rapidly in response to physiological stimuli, such as for instance during normal female reproductive function. Angiogenesis is also rapidly reactivated during wound healing to promote tissue repair. In many disorders, the balance between angiogenic stimulators or inhibitors is tilted over, resulting in an excessive or defective angiogenic stimulus. It is also well established that the resulting pro- or anti-angiogenic disease state contributes to several cancers and various ischaemic and inflammatory diseases, where it importantly determines disease progression [2, 5, 6].
Angiogenesis is genetically pre-determined
As with most biological processes, angiogenesis varies considerably between individuals. Tantalizing examples of how genetic variability contributes to phenotypic variability exist, raising the question whether such genetic variability may also determine the degree of angiogenesis and contribute to disease.
Quantitative genetic analysis of circulating angiogenic factors, including epidermal growth factor (EGF) and angiogenin, has shown that variation in the expression of these molecules can be attributed to genetic effects [7]. For the vascular endothelial growth factor (VEGF), heritability estimates were even responsible for 80% of the genetic variability, whereas environmental factors determined only 20% (ref. [7]). Similar effects were seen in nuclear families, where significant correlations between VEGF plasma levels were observed in all pairs of relatives, except in spouses [8]. This clearly indicates that genetic factors influence the inter-individual variability in angiogenic responsiveness. Understanding the genetic basis of such phenotypic variation is currently one of the major goals and challenges of human genetic studies. Surprisingly, however, relatively few studies designed to dissect the genetic basis of angiogenesis (i.e.the ‘angio-genome’) have been performed so far.
In a seminal study, Rohan et al. analysed different inbred strains of mice for their relative ability to trigger a Vegf-mediated angiogenic response in the corneal neovascular micropocket assay. Variations of up to 10-fold were observed in some strains, such as C57BL/6J being relatively deficient, and others, such as 129/SvImJ showing strong angiogenic responsiveness [9]. The response to Vegf was subsequently mapped to regions on chromosomes 2 and 10, which contained candidate angiogenesis-related genes including those for endostatin, matrix metalloproteinase 11, integrin-ß2 (ref. [10]). Because genetic susceptibility may vary according to the angiogenic stimulator, a similar mapping approach to identify loci that influence basic fibroblast growth factor (Fgf-2)-induced neovascularization was undertaken [11]. Four chromosomal areas were identified, which remarkably differed from the previously identified Vegf loci. The genes responsible for modulating the angiogenic response in these chromosomal regions have, unfortunately, not been identified yet. This may be due to the fact that various types of genetic variability can mediate gene expression. Indeed, a myriad of genetic and molecular modifications, such as genetic mutations or single nucleotide variations (SNPs) [12], translocations, copy number variations (CNVs) [13], epigenetic changes (DNA methylation and histone modifications) [14] and non-coding RNAs (microRNAs) [15] are found to importantly regulate gene expression (Fig. 1). Each of these modifications has been shown to induce a variety of complex human traits and diversity among diseases, and consequently, may also determine the angiogenic response.
Fig. 1.

Different types of genetic variability affecting angiogenesis. Left panel:Reduced or enhanced binding of transcription factors to the gene promo-tor can, respectively, decrease or increase gene transcription. DNA promotor methylation, histone and chromatine folding modifications, SNPs and CNVs are all capable of promoting or reducing the interaction between the transcription factor and its promotor. Translocations can change the chromosal environment of a gene leading to increased or decreased mRNA levels. MiRNAs reduce gene expression by translational repression or by inducing the degradation of the targeted mRNA. SNPs or mutations can induce differences in mRNA folding, thereby altering translation efficacy of the mRNA. Right panel:Mutations and non-synonymous SNPs (mostly located in exonic sequences) can cause a mutated or truncated protein with altered biological activities. Some mutations also cause complete deficiency of the gene product. Green boxes denote increased, whereas red boxes denote reduced gene expression. CNV, copy number variation; miRNAs, microRNAs; SNPs, single nucleotide polymorphisms; UTR, untranslated region; TF, transcription factor.
We will now briefly describe the precise nature of these different types of genetic variability and systematically review how they may shape the ‘angio-genome’, affecting angiogenesis in both health and disease.
Mutations causing vascular anomalies
Vascular anomalies can be categorized into vascular malformations and tumours of endothelial cells (or haemangiomas), and often grow commensurately with the child affecting only selected areas of the body. Sometimes, vascular anomalies may also expand suddenly following trauma, sepsis or hormonal changes [16]. Little is known about their genetic basis. In the majority of the cases, vascular anomalies occur sporadically, but the extent to which these sporadic forms are actually caused by a complex interaction between environmental factors and susceptibility genes is still unclear. In some cases, a clear Mendelian inheritance pattern is observed, facilitating genetic mapping studies and the identification of molecules implicated in vasculogenesis and angiogenesis, as discussed below [17, 18].
Venous anomalies
Venous anomalies present as bluish lesions on skin and mucosa and are referred to as cutaneomucosal venous malformations or VMCMs. They are caused by mutations in the endothelial cell-specific receptor TIE2, which acts as a receptor for the angiopoietins, ANG1 and ANG2. Originally, TIE2 was described as the second member of an orphan receptor tyrosine kinase subfamily expressed predominantly in the embryonic endothelium [19, 20]. Consistent with its expression pattern, disruption of Tie2 function in mice resulted in early embryonic lethality due to vascular abnormalities [21]. Ultrastructurally, vessels of Tie2-deficient embryos showed a decreased number of endothelial cells and a reduced contact between endothelial and underlying perivascular cells (pericytes and smooth muscle cells), suggesting a role in the maturation and stabilization of the embryonic vasculature for this receptor.
Haemangiomas
Infantile haemangioma, the most prevalent of all vascular tumours, mostly occurs sporadically after birth and has a characteristic clinical course marked by early proliferation, followed by spontaneous clinical resolution by the age of 9 years. Most haemangiomas are medically insignificant, but may occasionally impinge on vital structures and cause ulcerations, bleeding, cardiac failure or significant structural abnormalities. Rare familial occurrence exists, and in these cases, linkage has been established to chromosome 5q, with three potential candidate genes involved in angiogenesis mapped within this region, i.e. fibroblast growth factor receptor-4 (FGFR-4), platelet-derived growth factor receptor-ß (PDGFR-ß), and VEGF receptor-3 (VEGFR-3) [22]. No autosomal mutations in the first two genes have been described yet. A number of somatic mutations in VEGF receptor-2 and -3 (VEGFR-2or VEGFR-3), have, however, been found in sporadic haemangioma specimens: one missense mutation in the kinase domain of the VEGFR-2 and a second missense mutation in the kinase insert of VEGFR-3[23]. In each case, the mutation was detected in tumour tissue but not in adjacent normal tissue, indicating that one potential mechanism involved in haemangioma formation is the alteration of the VEGF signalling pathway in endothelial and/or pericyte cells. Mouse genetic studies indeed revealed that embryos lacking Vegfr- 2 die around 10 days of gestation due to the complete absence of organized blood vessels [24]. Loss of Vegfr- 3 also impaired remodelling of the expanding embryonic vasculature [25], thereby confirming the involvement of Vegfr-2 and -3 in the development of the vascular system. Although these knockout studies clearly highlight the importance of Vegfr-2 and -3 during angiogenesis, conditional knockout studies of these receptors are required to study haemangioma formation in mouse models.
Another intriguing concept is that tissues derived from the same neural crest or mesodermal migrating cells, which therefore share the same metameric origin, are all similarly affected in certain anomalies [26]. This suggests that some developmental processes have gone awry in patients with such metameric lesions, and that possibly, a somatic mutation, occurring in the region of the neural crest or the adjacent cephalic mesoderm, triggers the venous or arterial metameric defects. Examples of such metameric diseases are the PHACES (Posterior fossa malformations, Haemangioma, Arterial anomalies, Cardiac defects, Eye abnormalities and Sternal defects) and Sturge-Weber syndrome or some of the large facial haemangiomas [27]. Although each of these diseases may be rare, their relationships among each other and their postulated link with the development of the neural crest may shed light on the complex aetiology of various cerebral vascular disorders. The observation that some large facial haemangiomas are metameric is further also intriguing, because the angioblasts that form blood vessels are not unique for segment-specific boundaries, but contribute to blood vessel formation throughout the face [28, 29]. The proliferation of endothelial cells and the growth of these haemangiomas thus occur in response to signals derived from cell types exhibiting segmental or regional identity, such as cranial neural crest cells. Indeed, a direct relationship between endothelial cells and neural crest cells exists, as pericytes, which provide connective tissue support to the blood vessels, are derived from cranial neural crest cells [30], and interactions between pericytes and endothelial cells influence maintenance, maturation and remodelling of blood vessels [31].
The transforming growth factor-ß in vascular anomalies
Similarly, genetic discoveries of vascular malformations have helped to establish a firm association with transforming growth factor-ß (or TGFß) signalling defects. Indeed, mutations in various genes involved in the TGFß signalling pathway, including SMAD4[32], endoglin[33] and ALK1[34], have been identified in hereditary haemorrhagic telangiectasia or HHT. This disease is characterized by arteriovenous malformations, which are direct connections between arterioles and venules without intervening capillaries. Clinically, the most common symptoms of HHT are recurrent nosebleeds and mucutaneous telangiectasias, but the most severe and life-threatening defects are found in the lungs, brain, liver and gastrointestinal tract, where they cause hypoxemia, stroke, brain abscess, heart failure and/or fatal haemorrhage [35].
Despite the identification of genes responsible for HHT, its underlying pathogenetic mechanisms remain obscure. A plausible reason is the complexity of the TGFß transduction pathway. Traditionally, TGFß signalling in endothelial cells has been considered to occur via the TGFß type II receptors (such as the TGFßR-2) and the type I receptors, ALK1 and ALK5, which act downstream of the TGFßR-2 and determine signalling specificity of the receptor complex [36]. An accessory type III coreceptor called endoglin is also expressed by endothelial cells, and appears to potentiate TGFß signalling. Activation of the ALKs then leads to phosphorylation of the Sma- and Mad-related proteins (SMADs), which regulate specific gene expression responses [37]. In endothelial cells, contradictory results have been obtained about signalling of ALK1.
One study showed, for instance, that ALK1 signalling inhibited the proliferation and migration of endothelial cells [38], whereas another study reported the opposite [39]. The role of endoglin in TGFß and ALK1 signalling is another conflicting issue. First, it was demonstrated that endoglin promotes TGFß/ALK1 signalling and endothelial cell proliferation [40], but subsequently it was reported that loss of endoglin function exerted the same effect [41]. According to some studies in endothelial cells, ALK5 also seems to be required for TGFß signalling viaALK1. Others reported, however, that ALK1 is predominantly detected in the endothelium and ALK5 in vascular smooth muscle layers, suggesting that ALK1 is the sole TGFß type I receptor in endothelial cells [36]. Mice, in which the Alk1, Alk5or Tgfßr-2gene were conditionally deleted in the vascular endothelium further revealed that Alk1 deletion mimicked pathological features of HHT, whereas Alk5 or Tgfßr- 2 deletion did not affect endothelial cells. This suggests that TGFßR-2 and ALK5 are perhaps not even required for ALK1 signalling in endothelial cells [36]. Taken together, these data paint still an unclear picture on the role of TGFß signalling in endothelial cells.
Another vascular anomaly is the arterial tortuosity syndrome (ATS), an autosomal recessive condition characterized by tortuous large and medium-sized arteries, often resulting in death at young age. Other typical features include aneurysms of large arteries and stenosis of the pulmonary artery, in association with facial features and connective tissue manifestations. Recently, the causal ATS genetic defect has been identified in the SLC2A10 gene, which encodes the facilitative glucose transporter GLUT10 (ref. [42]). It has been proposed that loss-of-function of GLUT10 results in diminished glucose-responsive transcription of decorin, a known inhibitor of the TGFß signalling pathway, and leads to up-regulation of the TGFß responsive elements, connective tissue growth factor, and versican, thereby also inhibiting proper extracellular matrix formation [43].
Cerebral cavernous malformations
Cerebral cavernous (or capillary-venous) malformations (CCMs) consist of dilated capillary-like vessels and large saccular vessels with thickened walls in the brain parenchyme, in which endothelial cells lack tight junctions [44]. The disease follows an autosomal dominant inheritance pattern to which four loci have been mapped: CCM1 with mutations in KRIT1 (KREV1 interaction trapped-1) [45]; CCM2 with mutations in malcaverin[46]; CCM3 with mutations in PDCD10[47] and CCM4, which has been mapped to chromosome 3q26.3–27.2. The function of the CCM disease-causing genes is starting to become unravelled, as mice lacking Krit1 have been reported to die in mid-gestation. The first detectable defects in these mice were exclusively vascular in nature, with dilated precursor vessels of the brain, reminiscent of the intracranial vascular defects observed in the human disease [46, 48]. These defects in mice were associated with early down-regulation of artery-specific markers, including Ephrin B2, Notch4 and Dll4 [49].
Mutations in a number of other genes, such as glomulin, RASA1, NOTCH3, etc. have been identified to cause other vascular anomalies, respectively, glomuvenous malformations, capillary malformations, cerebral autosomal dominant arteriopathy with subcortical infarcts and leucoencephalopathy or CADASIL [50]. For a complete overview of the genes contributing to these vascular anomalies, we refer to a more specialized review (ref. [50]). Mutations in von Hippel-Lindau (VHL) disease, which cause highly vascularized tumours in multiple organs as well as haemangiomas of the retina and central nervous system (CNS), will be discussed below.
On a more general level, we can conclude that the study of inheritable vascular anomalies generates important knowledge on factors important for vasculo- and angiogenesis.
Translocations reveal novel angiogenic genes
The sporadic nature of many vascular anomalies has hampered the identification of the underlying genetic defects. In some cases, however, vascular malformations can be caused by chromosomal anomalies, involving a translocation or deletion of entire genomic regions. Although such chromosomal anomalies are relatively sparse, they are particularly useful in identifying disease-causing genes, as it is generally assumed that haploinsufficiency of one or more genes at the translocation breakpoint plays a pivotal role in causing the vascular defect.
Haemangioendothelioma, for instance, is a relatively rare vascular tumour that is considered to be a low- to intermediate-grade malignant neoplasm. Cytogenetic reports of haemangioendothelioma are rare, but recently, a case was identified with an epitheloid haemangioendothelioma and a reciprocal translocation between the chromosomal regions 10p13 and 14q24. As the expression of genes located in these chromosomal regions may change due to this translocation, the expression of genes was further investigated. Intriguingly, the expression of placental growth factor (PlGF) was strongly up-regulated in the tumour tissue, suggesting that increased PlGF levels could be responsible for the haemangioendothelioma, at least in this patient [51].
Another vascular disorder possibly caused by chromosomal translocation is the Klippel-Trenaunay syndrome (KTS), which consists of capillary malformations, venous malformations or varicose veins with bony and soft tissue hypertrophy [52]. Chromosomal breakpoints in a small subset of KTS patients have identified AGGF1(previously known as VG5Q) on chromosome 5q13 as a potential culprit gene causing KTS [53]. Subsequently, mutations in AGGF1 have also been identified in KTS patients. Intriguingly, the mutant AGGF1 protein increased angiogenesis in the chick chorioallantoic membrane (CAM) and matrigel assays, consistent with histological studies showing an increase in the number and diameter of venules in the dermis of KTS-affected tissues [53]. The reported nucleotide change in patients without a chromosomal translocation was later shown to be a common polymorphism in other populations, casting some doubt about the association of this mutation with the sporadic disease phenotype [54, 55].
Single nucleotide polymorphisms shape the angio-genome
Single nucleotide polymorphisms or SNPs are the most common type of genetic variation among people. Each SNP represents a difference in a nucleotide, occurring normally throughout a person's DNA. On average, there is one SNP every 300 nucleotides, which means that there are roughly 10 million SNPs in the human genome. SNPs are well known to influence gene expression and to determine phenotypic variability, either through direct causal effects or by serving as proxies for other causal variants in linkage disequilibrium [56].
Traditionally, VEGF has been considered as the most important and most-specific growth factor for endothelial cells. The milestone achievement, showing that a monoclonal anti-VEGF antibody (bevacizumab or Avastin®, Genentech, South San Francisco, CA, USA) in combination with chemotherapy improved the survival of metastatic colorectal cancer patients, further stimulated the search for genetic variability in VEGF expression and its downstream angiogenic effectors [57]. By now, numerous associations with SNPs located in the VEGF gene have been reported, particularly in the field of oncology. VEGF is a highly polymorphic gene, in which >100 polymorphisms have been described so far (dbSNP or http://www.ncbi.nlm.nih.govprojectsSNP). Baseline VEGF expression levels in plasma are highly variable, and typically range between 9 and 150 pg of VEGF per ml [58]. Most genetic studies performed so far focused on four SNPs, i.e. the −2578C/A (rs699947), −1154G/A (rs1570360), −634C/G (rs2010963) and +936C>T (rs3025039), which were shown to affect the expression of VEGF, either in vitro or in vivo (Table 1 and Fig. 2). A number of SNPs in other angiogenic factors have been studied as well - their effects will be discussed below.
Table 1.
Overview of functional SNPs in VEGF pathway genes
| Gene | Gene region | SNP position | SNP ID | Risk allele | Risk allele frequency |
|---|---|---|---|---|---|
| VEGF | Promotor | −2578C/A | rs699947 | A | 0.41 |
| 5′UTR | −1154G/A | rs150360 | A | 0.34 | |
| 5′UTR | −634C/G | rs2010963 | C | 0.20 | |
| 3′UTR | +936C/T | rs3025039 | T | 0.35 | |
| VEGFR-2 | Promotor | −604C/T | rs2071559 | C | 0.52 |
| Exon 7 | +1192A/G | rs2305948 | A | 0.07 | |
| Exon 11 | +1719A/T | rs1870377 | A | 0.28 | |
| VEGFR-1 | Promotor | +519C/T | N/A | T | 0.06 |
| HIF-1α | Exon 12 | +1772C/T | rs11549465 | T | 0.09 |
| Exon 12 | +1790G/A | rs11549467 | A | 0.02 |
The gene region, SNP position, SNP ID as well as the risk allele, frequency and functional effect of each gene are listed. N/A means not available.
Fig. 2.
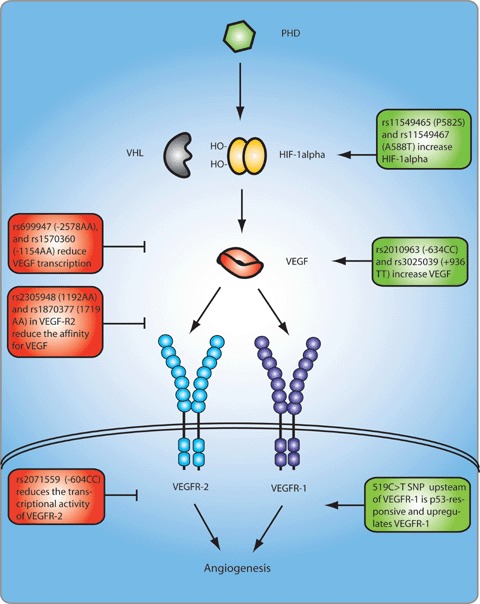
SNPs affecting expression of genes in the VEGF signalling pathway. Green boxes denote increased, whereas red boxes denote reduced expression of the respective protein levels or function. The A allele of the two coding VEGFR-2 SNPs, i.e. rs2305948 and rs1870377, induces a valine to isoleucine and histidine to glutamine substitution, respectively, at positions 297 and 472, thereby reducing the affinity for VEGF. In HIF-1α, the T allele of rs11549465 induces a proline to serine substitution at position 582, and the A allele in rs11549467 induces an alanine to threonine substitution at position 588, thereby increasing HIF-1α stability. PHD, prolyl hydroxylation domain protein; VHL, von Hippel-Lindau protein; HIF, hypoxia inducible factor; VEGF-A, vascular endothelial growth factor-A; VEGFR, VEGF receptor.
SNPs in VEGF and their association with cancer
Recent genome-wide studies in large populations of breast, colorectal, lung and prostate cancer have started to uncover the genetic basis for cancer. However, none of these studies identified a genetic variation in VEGF or a VEGF pathway-related gene. This suggests that angiogenic factors are no major initiators of cancer. Instead, VEGF might be more relevant in determining disease progression and metastasis once the tumour has developed.
Indeed, numerous associations of VEGF polymorphisms with clinical variables in cancer have been established. For instance, the −2578C/A polymorphism in the VEGF promoter, which is in full disequilibrium (LD) with an 18bp deletion/insertion located in a putative upstream estrogen response enhancer, alters VEGF promoter activity and responsiveness. Shahbazi et al. were the first to show that mononuclear cells from −2578CC carriers produce significantly higher VEGF levels [59]. This was subsequently confirmed by Lambrechts et al., who found that −2578AA carriers were associated with reduced VEGF in plasma and serum [60]. In line with these findings, −2578CC carriers were associated with increased lymph node metastasis and decreased survival in renal cancer [61], and with an increased risk for invasive breast cancer [62]. Studies in colorectal cancer failed, however, to observe an effect on survival [63]. Similar studies showed that carriers of the −1154AA genotype have lower circulating VEGF, whereas mononuclear cells from −1154GG individuals produce more VEGF [59]. The −1154AA genotype was associated with a lower stage of cutaneous malignant melanoma [64], reduced tumour size in renal cell carcinoma [61], whereas the −1154GG genotypes were associated with an increased risk for invasive but not in situ cancer [62]. No effects were seen in ovarian cancer [65]. In the 5′-untranslated region (UTR) of the VEGF gene, the −634CC homozygotes correlated with higher circulating VEGF [66]. In vitro, the −634C allele also disrupted the 5′UTR RNA structure and increased the expression of a long VEGF isoform (L-VEGF) [60]. In breast cancer, −634CC carriers exhibited larger tumours and increased histologic grading [67] and in prostate cancer −634CC carriers had a higher-grade carcinoma [68]. Other studies reported, however, that the −634C allele was associated with lower VEGF and prolonged survival in colorectal [63] and non-small-cell lung cancer [69]. No effects were seen on breast [62], renal cell [61] and ovarian carcinoma [65]. Another SNP that reduces VEGF expression is the +936C/T SNP, located in the 3′UTR of the VEGF gene [70, 71]. Several associations have been reported for this SNP, e.g.+936CC genotypes correlated with reduced survival in non-small-cell lung cancer patients [69], but have not been replicated in other studies on other types of cancer.
The reasons why several of these studies have failed to establish clinically relevant correlations are not entirely clear. Possibly current studies are still under-powered. Much larger populations are therefore needed to reliably determine the effect of VEGFSNPs on disease outcome. Genetic consortia, which integrate sample collections from several centres, may represent a feasible approach to assess rapidly the effects of these VEGFSNPs on tumour progression, as recently demonstrated by the analysis of >10,000 breast cancer DNA samples, assessing the association of breast cancer susceptibility genes with clinical variables [72, 73].
SNPs in VEGF pathway genes associated with other diseases
Many other SNPs in the VEGF gene or in other genes involved in the VEGF pathway have been studied in various other diseases, and numerous SNPs contributing significantly to disease risk have been reported. Most of these genetic studies suffer, however, from a number of shortcomings, as they have typically been hampered by small sample sizes (up to 100 or 200 patients). Given the modest effect sizes of genetic associations in general, and the typically small sample sizes employed for these studies, it is not surprising that many of these genetic reports in relatively small populations, originally claiming a positive association, have subsequently been challenged by reports lacking any association or even contradicting it. The same applies for studies focusing on the main inducer of VEGF expression, the hypoxia-inducible factor-1α or HIF-1α, and the VEGF receptors, VEGFR-1 and VEGFR-2. In order to provide a comprehensive overview, we have here tabulated all significant associations reported for the VEGF gene in Table S1. Below, we discuss, however, a selection of only the most relevant and solid associations reported for these genes.
Genetic variability in VEGFR-2
Atherosclerotic plaque instability is directly involved in triggering acute coronary syndromes in patients with coronary artery disease (CAD). Considerable damage of the vascular endothelium has been reported and genetic variability in molecules maintaining endothelial integrity has also been proposed to underlie CAD. VEGF and its receptor VEGFR-2 have therefore been considered as potential disease genes for CAD. Studies in approximately 1000 CAD patients revealed that carriers of the −2578AA genotype were more common in patients with coronary vessel stenosis [74]. Genetic variability in the VEGFR-2 gene has also been investigated, and a significant association with three polymorphisms was established in a first and then independently replicated in a second population (both analysing >1000 individuals in total) [75]. Notably, the −604T/C SNP (rs2071559) in the binding site for the transcriptional factor E2F reduced transcriptional activity of the VEGFR-2 promoter by 68% and was associated with reduced VEGFR-2 antigen levels in serum (Fig. 2). The mutant alleles of two exonic SNPs, located in exon 7 and 11 (1192G/A or rs2305948 and 1719A/T or rs1870377), also reduced binding efficacy of VEGF to VEGFR-2 by approximately 15%, suggesting that these variations in VEGFR-2 eventually may serve as novel markers determining the risk of CAD (Table 1 and Fig. 2) [75].
Genetic variability in HIF-1α
Hypoxia-inducible factor-1α or HIF-1α is a transcriptional factor mediating the cellular response to hypoxia and regulating the transcription of multiple genes, including VEGF. HIF-1α itself is hydrox-ylated - and thereby protected from degradation - in the presence of oxygen by a family of three prolyl hydroxylase domain (PHD) proteins. During normal oxygen tension, the HIF-1α is enzymatically hydroxylated at proline residues, which are recognized by the VHL protein and targeted for degradation. During hypoxic stress, HIF-1α is not hydroxylated and fails to interact with the VHL complex and escapes ubiquitin-mediated proteolysis.
HIF-1α is overexpressed in many human cancers such as kidney, breast, colon, ovary and prostate carcinoma [76]. Recently two SNPs in the HIF-1α, i.e. P582S (rs11549465) and A588T (rs11549467), resulting in proline to serine and alanine to threo-nine amino acid substitutions, respectively, were found within the oxygen-dependent degradation domain of HIF-1α (Table 1 and Fig. 2) [77]. The presence of these variants, especially the P582S variant, was shown to cause a significantly higher transcriptional activity correlating with higher mutant HIF-1α protein expression and increased vessel density [77]. The P582S mutant genotype also correlates with accelerated tumour progression in head and neck squamous carcinoma [77] and with more severe ulcerative growth pattern in colorectal cancer [78] or survival in transitional cell cancer of the bladder [79].
Mutations in EGLN1, encoding the PHD2 enzyme, which lead to a marked decrease in enzyme activity and increased HIF-1α expression, have been described in patients with familial erythrocytosis. Remarkably, however, patients carrying EGLN1 mutations did not exhibit any obvious angiogenic abnormalities [80].
SNPs in VEGFR-1 integrate angiogenesis within the P53 pathway
Somatic P53 missense mutations are found in approximately 50% of human cancers. An increasing number of studies indicate that a subset of these P53-mutant proteins are oncogenic and actively participate in neoplastic transformation [81]. It is further also assumed that the P53 pathway is inactivated in wild-type P53-carrying tumours via indirect mechanisms, for instance, via genetic variability in genes associated with the P53 pathway [81]. By performing a global genome analysis, Menendez et al. identified a C-to-T SNP in a putative P53 response element in the promoter of the VEGFR-1 gene [82]. This SNP was detected in approximately 6% of the people examined, and in-depth functional analyses showed that only one allele, i.e. the mutant T-allele, was P53-responsive (Table 1 and Fig. 2) [82]. The inclusion of the VEGFR-1 gene and, therefore, the VEGF pathway into the P53 transcriptional network, adds complexity to the spectrum of biological outcomes associated with P53 mutations. Indeed, VEGFR-1 overexpression has been associated with pathological angiogenesis, tumour progression, cell survival, proliferation, migration, invasion and metastasis [83]. Although no clinical information about the functional consequences of this functional SNP in the VEGFR-1 promoter is available yet, the intriguing possibility arises that this SNP modulates the heterogeneous P53 response in cancer patients.
Variations in angiogenic genes are linked with neurodegeneration
In the midst of the excitement over the therapeutic effects of neutralizing VEGF in cancer, there emerged, a few years ago, an unexpected discovery - the existence of a novel link between VEGF and neurodegenerative disease. Transgenic mice with a targeted deletion of the hypoxia-responsive element in the Vegf promoter resulting in reduced Vegf expression, developed adult-onset, progressive motor neurodegeneration, with pathologic features consistent with amyotrophic lateral sclerosis (ALS) [84]. Vegf also exhibited a huge therapeutic potential in animal models for ALS. The connection between Vegf and ALS in rodents was also confirmed in human beings: the low VEGF-producing alleles of the -2578C/A, - 1154G/A and -634G/C variations increased the risk of ALS in some - but not all - populations [60]. In a meta-analy-sis involving >7000 individuals from all populations studied, subgroup analyses by gender revealed that the -2578AA genotype, which lowers VEGF expression, consistently increased the risk of ALS in males [85]. Interestingly, the -2578A VEGF allele, which correlates with reduced VEGF expression levels in plasma and serum, also confers an increased risk for patients with Alzheimer disease in one study [86]. A similar association study assessing the role of SNPs in the VEGF gene as a determinant of longevity was also performed. The -2578AA genotype was detected significantly more often in long-lived (85–99 years old) subjects than in young/elderly (25–84 years old) subjects, suggesting that VEGF gene variability is a potential genetic factor influencing the lifespan in human beings [87].
The discovery of the link between VEGF and ALS has stimulated interest in evaluating the role of other angiogenic factors in ALS. Recently and most surprisingly, mutations in angiogenin (ANG) and progranulin (PGRN) genes were identified in patients with ALS and frontotemporal dementia, respectively. Greenway and colleagues focused their attention on ANG and identified seven different missense mutations in the ANG gene from ALS patients, six of which were in evolutionary conserved regions [88]. Initially, these ANG mutations were only detected in ALS patients who shared a common ancient haplotype from Scottish/Irish ancestry, but more recent findings revealed that a limited number of mutations is also present in ALS patients from other ancestries (e.g. from Hispanic [89] or Italian ancestry [89, 90]). Mutations in PGRN typically cause a form of frontotemporal dementia with ubiquitine-positive tau-negative inclusions [91], but more recent findings indicate that PGRN mutations can be associated with motor neuron degeneration and various clinical subtypes of dementia. PGRN is a pluripo-tent secreted growth factor that mediates cell cycle progression, stimulates neovascularization and is highly expressed in aggressive cancer cell lines. PGRN also plays a major role in breast tumourigenesis by conferring resistance to tamoxifen [92]. At least part of the angiogenic and tumour-proliferative effects of PGRN and ANG seem to be mediated through VEGF: ANG appears to be a downstream effector of VEGF in endothelial cells [93], whereas PGRN up-regulates expression of VEGF in breast cancer cells. Little is known, however, about the functions of PGRN and ANG in the CNS [94]. One of the outstanding questions yet to be answered is to what degree the angiogenic activity of these molecules might also trigger vascular defects in neurodegenerative disease, thereby contributing to disease development.
Angiogenic factors in genome-wide association studies
The recent unbiased identification of SNPs on a genome-wide level has become a fruitful enterprise in identifying novel genes in complex diseases and numerous SNPs have been unambiguously identified to underlie disease susceptibility [12]. Intriguingly, a recent meta-analysis of genome-wide association (GWA) data in type 2 diabetes revealed increased susceptibility for carriers of an SNP located nearby the VEGF gene, i.e. rs9472138 (ref. [95]). The minor, at-risk T-allele of this SNP increased the risk for type 2 diabetes in 63,537 individuals by only 1.06-fold, which is however similar in size to the risk effects of other SNPs in diabetes (Table 1). Because the rs9472138 SNP is located ±500 kb upstream of the VEGF gene, it will be interesting to find out if - and how - this SNP affects VEGF expression. Because genome-wide studies often focus on tagging SNPs, it is also possible that the effects of the actual common causal variants responsible for the observed association might be larger.
Other GWA studies in breast cancer also identified SNPs in five loci, including one SNP in the fibroblast growth receptor 2 (FGFR-2) gene [96, 97]. The variable allele of the SNP in FGFR-2 was more strongly related to estrogen receptor-positive than oestrogen receptor-negative cancer, and to a low rather than high tumour grade, but did not significantly influence survival [72]. Functional studies also revealed that the at-risk SNPs allele specifically increased FGFR-2 expression [98]. Historically, the FGF-1 and -2 ligands of FGFR-2 were the first angiogenic factors to be identified [99]. However, FGFs are also potent mitogens and morphogens and therefore also play a direct role in cancer cell proliferation. It is thus unclear whether the effect of this FGFR-2 SNP affects tumour cells, vessels or both, and additional studies are required to answer this question.
Copy number variability affects angiogenesis
It has traditionally been considered that two copies of a gene are present per genome. This has been refuted by recent discoveries revealing that large segments of DNA can vary in their copy number, implicating that entire genes can be present in more - or less - than two copies. It has also become apparent that these CNVs are quite common and can have dramatic phenotypic consequences as a result of altered gene dosage, disrupted coding sequences, or perturbed long-range gene regulation [13]. Research studying the effects of CNVs on gene expression is, however, a relatively young research field, and very little is known about the effect of CNVs in angiogenic processes. Yet, it is expected that CNVs might determine angiogenesis, because, for instance, a significant overlap exists between the QTL loci for angiogenesis and known CNVs in mice (e.g. on chromosome 4, 13 and 15) [11].
To determine whether genomic DNA copy number variations contribute to global gene expression pattern changes in hepato-cellular carcinoma (HCC), array-based comparative genomic hybridization (CGH) data studying CNVs were compared to microarray expression data [100]. Approximately 542 genes were shown to have a significant correlation between expression values and CNVs. Among these genes was Sprouty2, a key antagonistic regulator of receptor tyrosine kinases, whose suppression of expression or function facilitates tumour proliferation and angiogenesis [101]. When stably expressed in mouse hepatocytes, an inhibitor of Sprouty2 also conferred a neoplas-tic phenotype in mice, by inducing high expression levels of phospho-extracellular signal-regulated kinase (ERK) and triggering deregulation of genes involved in cell proliferation, apop-tosis and angiogenesis [100]. Notably, a change in the expression of endothelial cell markers, such as CD34 and angiopoi-etin-2, was also noticed as an indicator of an altered endothelial phenotype in the cancerous liver tissues. Similar studies focusing on a large number of hepatocellular and primary lung ade-nocarcinomas revealed a number of chromosomal amplifications involving chromosomal region 6p21. Remarkably, the VEGF gene was located in the amplified regions and VEGF expression levels were also dramatically up-regulated. Overexpression of VEGF via 6p21 thus illustrates the potential role of CNVs as a modulator of angiogenesis [102, 103].
Epigenetic regulation of angiogenesis
In multicellular organisms, the identity of each cell is determined by its unique gene-expression pattern. This identity is remembered and passed on to daughter cells by epigenetic mechanisms, which are heritable changes that do not involve changes in the DNA sequence [14]. Several epigenetic defects are known to contribute to disease, such as for instance changes in the localized or global density of DNA methylation.
In mammals, DNA methylation is confined to the addition of a methyl group to the cytosine in a CpG dinucleotide. This covalent modification is known to have an essential role in genome function and has been implicated in processes such as gene regulation, chromosomal stability and parental imprinting [104]. Aberrant DNA methylation commonly occurs in human cancers in the form of regional hypermethylation and genome-wide hypomethylation: hypermethylation of CpG islands located in the promoter regions of tumour suppressor genes results in tran-scriptional silencing, whereas global DNA hypomethylation (also known as demethylation) is associated with activation of proto-oncogenes and generation of genomic instability [105, 106].
Methylation of anti-angiogenic factors
Various examples of aberrant methylation in angiogenic genes exist. Hypermethylation of the thrombospondin 1(THBS-1) gene, an angiogenesis inhibitor with tumour suppressor properties, was first found in glioblastoma multiforme [107], and has now been confirmed in various other cancers, including gastric carcinoma [108], penile squamous cell carcinoma [109] and neuroblastomas [110]. In addition, THBS-1 hypermethylation was found to be significantly associated with distal location, vascular invasion, distant metastasis and worse prognosis in gastric cancer [111]. In primary colorectal cancers, loss of THBS-1 expression also correlated with impaired TGFß signalling, as indicated by decreased SMAD2 phosphorylation and nuclear localization [112]. Furthermore, methylation-induced silencing of THBS-1 expression reduced the concentration of secreted TGFß, thereby attenuating TGFß signalling [112]. Aberrant methylation of THBS-1thus represents an epigenetic mechanism for suppressing TGFß signalling in cancer. Extensive hypermethylation of a large CpG island in Sprouty4 has been observed in high-grade prostate cancer, whereas benign prostate hyperplasia tissues were predominantly unmethylated [113]. Furthermore, suppression of Sprouty4 expression correlated with the methylation status of this CpG region in clinical samples [113]. Hypermethylation of ADAMTS-8, a secreted protease with antiangiogenic properties, and concomitant down-regulation of ADAMTS-8 expression in brain tumours [114], and of tissue inhibitor of metalloproteinase-2 and -3 (TIMP-2 and -3) in various tumours [106], have also been observed. Hypermethylation of anti-angiogenic factors in cancer is thus a commonly used mechanism by the tumour to promote a pro-angiogenic state.
Intriguingly, the opposite situation, whereby the expression of pro-angiogenic genes is stimulated by epigenetic mechanisms, has also been observed. Indeed, overexpression of the lymph-angiogenic molecule, VEGF-C, due to demethylation was reported in gastric cancer cell lines [115]. Notably, overexpression of VEGF-C is well known to correlate with lymphatic vessel density and lymph node metastasis [116, 117]. Studies with the rodent carcinogen, phenobarbital, have also shown that methylation patterns are altered to a greater extent in liver tumour-susceptible as compared to resistant mice. Uniquely, some of the hypo-methylated genes in the susceptible mice are involved in angiogenic processes: e.g. chymase 1, tyrosine kinase non-receptor 2 and ephrin B2[118].
Methylation as a second hit event in cancer
Mutations in cancer typically consist of a localized intragenic mutation (e.g.point mutation, microdeletion, etc.) followed by a second hit event, frequently being a large deletion or mitotic recombination, and resulting in loss of heterozygosity of the tumour suppressor gene. An equivalent cause of second hit has been recognized by the phenomenon of hypermethylation of tumour-suppressor genes [105].
A well-established example of how aberrant methylation serves as a second hit event in cancer and affects angiogenesis is provided by VHL disease. VHL disease is an autosomal dominantly inherited familial cancer syndrome predisposing to a variety of benign and malignant neoplasms. The most common of these are retinal and CNS haemangiomas, renal cell carcinoma (RCC), pheochromocytoma and pancreatic tumours [119]. Patients with VHL disease are typically characterized by the presence of somatic germ-line deletions, frameshifts, and nonsense mutations in the VHL gene [120]. The VHL gene functions in several pathways linked with carcinogenesis, most notably the hypoxia-inducible pathway. Similarly, when there is loss of VHL function, HIF-1α accumulates within cells, allowing for the constitutive activation of target genes, including VEGF, platelet-derived growth factor (PDGF) B chain, transforming growth factor-α (TGFα) resulting in increased angiogenesis, cell growth and survival [121]. Analysis of VHL haemangioblastomas has revealed that somatic mutations in the VHL gene or hypermethylation of tumour-suppressor genes leading to inactivation of both VHL alleles are two equivalent mechanisms in the development of VHL-associated haeman-giomas [122, 123]. A recent study investigating the methylation patterns of 96 patients with renal cell carcinoma revealed that loss of heterozygosity of the VHL gene was present in 78.4% of the patients. A second hit mutation in VHL was seen in 71% of those, whereas promoter methylation of the VHL gene was present in 20.4% of the patients [124].
Histone modifications determine angiogenesis
Eukaryotic DNA is wrapped around histone octamers and forms nucleosomes that are themselves folded into higher-ordered chro-matin structures. Histones have N-terminal tails extending from the compact nucleosomal cores that affect histone interaction and gene regulation. Histone acetyltransferases (HATs) acetylate, and histone/protein deacetylases (HDACs) deacetylate, ε-acetyllysine residues of these histone tails. HAT acetylation generally increases accessibility and promotes gene transcription, whereas HDAC deacetylation typically dampens histone-DNA and histone-nonhi-stone protein interactions [104].
Recent findings have revealed an unexpected role for HDACs in the maintenance of vascular integrity during cardiovascular development and disease. Mice lacking the deacetylase Hdac7 die during mid-gestation from vascular dilatation and rupture caused by loss of adhesion between endothelial cells [125]. Consistent with this phenotype, Hdac7 is expressed specifically in the developing vascular endothelium, where it functions as a repressor of matrix metalloproteinase 10 (MMP-10), a secreted endoproteinase that degrades the extracellular matrix and perturbs vascular integrity. In Hdac7 mutant embryos, Mmp10 expression is dramatically up-regulated in the vascular endothelium, and its antagonist, tissue inhibitor of metalloproteinase 1 (Timp-1), is down-regulated [125]. An independent study by Mottet et al. found that HDAC7 silencing also leads to an increase in PDGF-B expression, as well as inhibition of endothelial cell migration [126]. Another member of the HDAC gene family, SIRT1, plays a key role in angiogenic signalling: SIRT1 is highly expressed in the vasculature during blood vessel growth, where it controls the angiogenic activity of endothelial cells [127]. Loss of SIRT1 function blocks sprouting angiogenesis and branching morphogenesis of endothelial cells with consequent down-regulation of genes involved in blood vessel development and vascular remodelling [127]. Disruption of Sirt1 gene expression in zebrafish and mice results in defective blood vessel formation and blunts ischaemia-induced neovascu-larization. These effects are mediated, at least in part, by deacetylation of the forkhead transcription factor Foxo1, an essential negative regulator of blood vessel development [127].
It has further been established that hypoxia enhances HDAC activity and that HDACs are closely involved in tumour angiogenesis through suppression of hypoxia-responsive tumour suppressor genes. Indeed, Kim et al. have shown that HDAC is induced under hypoxic conditions and that overexpression of HDAC1 down-regulated expression of P53 and von Hippel-Lindau tumour suppressor genes, and stimulated angiogenesis of human endothelial cells [128]. A specific HDAC inhibitor, tri-chostatin A (TSA), also up-regulated the expression of P53 and von Hippel-Lindau and down-regulated HIF-1α and VEGF [128]. TSA also specifically inhibited hypoxia-induced angiogenesis in the Lewis lung carcinoma model in mice, thereby suggesting that HDAC inhibitors have a potential therapeutic value for cancer therapy [128]. A pertinent problem with the interpretation of these in vivo effects of HDAC inhibitors in cancer, and in particular with their effect on HIF-1α and VEGF suppression, is that these effects might occur indirectly - via the tumour cells. Indeed, the reactivation of epigenetically silenced tumour suppressor genes, such as VHL, in the tumour cells could reduce HIF-1α and VEGF in tumour cells, and therefore also tumour angiogenesis indirectly [129].
To study the mechanisms behind the epigenetic regulation of tumour angiogenesis more specifically, Hellebrekers et al. performed a microarray analysis to identify genes down-regulated in tumour-conditioned versus quiescent endothelial cells. Various genes were identified to be significantly down-regulated in tumour-conditioned endothelial cells and by inhibiting HDAC activity, the expression of these genes was restored [129]. A number of these genes, including clusterin, fibrillin and quiescin Q6 was also down-regulated in human tumour endothelium and functional analysis using RNA interference confirmed that these were negative regulators of endothelial cell growth in a collagen gel assay [129]. This proves that HDAC inhibitors also directly affect endothelial cell biology and angiogenesis. Additional studies have also reported that HDAC inhibitors influence the expression of the angiogenesis-related genes angiopoietin-2, TIE-2, survivin and eNOS [130, 131], and attenuated VEGF-induced phosphorylation of AKT and ERK1/2 in endothelial cells [132]. Angiopoietin-2, survivin and CXCR4 up-regulation by VEGF in endothelial cells was also inhibited [132]. However, decreased expression of these genes is not due to direct effects of these compounds on epigenetic promoter modifications, because direct effects of HDAC inhibition would result in increased promoter histone acetylation and thus transcriptional activation. Further studies are thus required to unravel the effects of HDAC inhibitors on (tumour) endothelial cell gene expression, and to relate these effects with epigenetic promoter modifications.
Micromanagers of angiogenesis
Over the past few years, microRNAs (miRNAs) have emerged as a prominent class of gene regulators and have been implicated in diverse biological processes [133]. MicroRNAs are short, single-stranded RNAs transcribed from non-coding genes and are generated in a two-step processing pathway mediated by two enzymes belonging to the class of RNAse III endonucleases, Dicer and Drosha. After being processed into approximately 22 base sequences, miRNAs bind to identical or similar target sequences in the 3'-UTRs of genes, thereby inhibiting translation or cleaving the mRNA target [134]. Numerous publications attest to the importance of the developmental effects of miRNA-mediated regulation of gene expression. For example, miRNAs participate in the maternal-to-zygotic transition during early embryogenesis and affect the differentiation of many tissue types [133]. In human beings, miRNA disruption has been described in association with several cancers – a finding with important implications for cancer aetiology, diagnosis and potentially for cancer treatment [135].
Evidence for the importance of miRNAs in the regulation of angiogenesis comes from observations that Dicer is required for embryonic angiogenesis and that its deficiency results in severely compromised embryos and yolk sacs, as well as decreased expression of several important positive regulators of angiogenesis [136]. More recently, it has been observed that silencing Dicer or, to a lesser extent Drosha, in adult endothelial cells results in the down-regulation of positive regulators of the angiogenic phe-notype and impaired tube formation on Matrigel [137, 138]. Overall, these observations suggest that miRNAs are necessary during angiogenesis. Consistent with this observation, changes in miRNA expression profiles in response to angiogenesis have been reported and a few individual miRNAs have been shown to importantly regulate angiogenic processes. Intriguingly, both pro-angiogenic (targeting anti-angiogenic mRNAs) and anti-angio-genic miRNAs (targeting pro-angiogenic mRNAs) exist.
The microRNA cluster miR-17–92 is up-regulated in colono-cytes co-expressing the proto-oncogenes K-rasand c-Myc [139]. Notably, miR-17–92-transduced cells also formed larger, better-perfused tumours, thereby establishing a role for the miR-17–92 in augmenting tumour angiogenesis [139]. The tumour-promoting and pro-angiogenic functions of the miRNAs encoded by this cluster have been attributed to miR-18 and miR-19, which inhibit apoptosis of tumour cells and promote tumour angiogenesis by suppressing the release of soluble anti-angiogenic factors, including Thbs-1 by tumour cells, thereby affecting endothelial cells in a paracrine manner. The miR-130a, which is expressed in proliferating endothelial cells, down-regulates the expression of the diverged homeodomain gene GAx (also known as MEOx2) - a well-established inhibitor of angiogenesis. Another microRNA, the miR-378, has been demonstrated to function as an oncogene, enhancing both tumour cell survival and blood vessel expansion through repression of the expression of two tumour suppressors [140]. Other miRNAs implicated in stimulating angiogenic processes are Let-7b and miR-27b [138]. Altogether, this indicates that specific miRNAs can be pro-angiogenic by repressing the expression of anti-angiogenic factors.
Other miRNAs were shown to exert anti-angiogenic effects as well. For instance, the miR-221 and -222 inhibit angiogenic processes by blocking tube formation and endothelial cell migration [141]. Moreover, the impairment in angiogenesis in Dicer-deficient mice was associated with a lack of miR17–5p and let7b miRNAs, which negatively regulate the expression of the anti-angiogenic TIMP1 (ref. [142]). Injection of miR17–5p and let7b miRNAs into the ovaries of Dicer-deficient mice normalized the expression of TIMP-1 and restored corpus luteum angiogenesis [142]. In addition, miR-15 and miR-16 have been shown to be down-regulated by hypoxia in a carcinoma cell line and to control the expression of VEGF [143]. Very recently, miRNA-126 was shown to regulate the response of endothelial cells to VEGF, in part by repressing negative regulators of VEGF, including the Sprouty-related SPRED1 and the phospoinositol-3 kinase regulatory subunit 2(PIK3R2/p85-ß) [144, 145]. The ability of these miRNAs to antagonize the angiogenic activity of various factors is striking, offering potential approaches as an anti-angiogenic strategy in cancer.
Perspectives
Emerging evidence shows that various levels of genetic variability, including mutations, SNPs, methylation patterns and miRNAs, influence the expression of angiogenic factors, and hence, determine the pro- or anti-angiogenic balance associated with disease. In the past, genetic studies have been crucial in identifying mutations in angiogenic factors, as evidenced by the discovery of disease-causing mutations in HHT and other vascular anomalies. Novel genetic approaches have now been developed, allowing the unbiased identification of genetic variants at other genetic levels. For instance, GWA studies have demonstrated their potential to identify SNPs associated with complex diseases such as diabetes, inflammatory bowel disease and cancer. Likewise, arrays allowing the methylation state of CpG islands or expression of microRNAs to be determined on a genome-wide scale have only recently become available within the scientific community. An obvious question is whether the availability of these novel technologies will further contribute to our understanding of how genetic factors in angiogenesis contribute to disease.
At first sight, relatively few angiogenic genes, if any at all, have been identified in GWA studies. This can be due to the fact that angiogenic factors do not cause complex diseases, but rather contribute to disease progression and heterogeneity. The cost of enrolling very large DNA cohorts needed to discover genes in GWA studies has so far precluded the collection of rich clinical follow-up data necessary for such analysis. It is expected, however, that many more clinical variables will be available in the future, in comparison to the phenotypically crude studies reported so far, allowing future GWA studies to identify novel or relevant vascular factors through the correlation of SNP genotypes with disease progression. Complex disease phenotypes, in which angiogenic factors will undoubtedly play a major role, include recovery from myocardial infarction, vascularization of solid tumours, etc. It is thus anticipated that unbiased genome-wide approaches at the different genetic levels will further contribute to our understanding of how the angio-genome contributes to disease.
An area of substantial future interest for the pharmaceutical industry is to perform pharmacogenomic GWA studies and to identify genetic markers for patient selection and stratification. Expensive or chronic medications for life-threatening conditions with narrow therapeutic index or frequent adverse events would be most suitable for pharmacogenomics. A prototype example of such a therapy in the vascular field is anti-VEGF treatment in cancer: indeed, although improving survival, only 45%, 35% and 30% of the bevacizumab-treated colorectal, lung and breast cancer patients, respectively, respond to angiogenesis inhibition [57, 146, 147]. A substantial number of treated patients also exhibit side effects, such as hypertension, thrombosis and proteinuria [57, 146, 147]. The underlying mechanisms determining which individuals respond to bevacizumab, or develop side effects, are yet unknown, but could possibly be determined by genetic variability in the germline DNA sequence. For instance, genetic variability in VEGFor its downstream receptors could render the tumour vas-culature more sensitive or resistant to VEGF inhibition. The observation that anti-VEGF treated tumours compensatorily up-regulate the expression of other angiogenic factors, thereby rendering the tumour vasculature less responsive to VEGF inhibition, is another intriguing observation which might be determined genetically. Copy number gains of the VEGF gene in tumours may further also represent a mechanism of resistance to VEGF inhibition. Insights into the genetic basis contributing to the efficacy of anti-VEGF therapy might also be of great value for patients with age-related macular degeneration.
Evidence, so far, has indicated that epigenetic changes also regulate angiogenic processes. Clinical trials with demethylating agents and inhibitors of histone deacetylation have yielded promising results, especially against haematologic malignancies. The findings that these drugs target both tumour cells as well as tumour endothelial cells make them suitable combinatorial cancer therapeutics. An intriguing question is whether epigenetic changes or chromosomal alterations in response to bevacizumab treatment contribute to the development of resistance against bevacizumab. The relative extent to which these epigenetic changes in (anti)-angiogenic factors also contribute to complex diseases other than cancer, has - to the best of our knowledge - not been investigated yet. In addition, the contribution of miRNAs modulating angio-genic processes to disease phenotypes is far from clear. Due to the lack of basic information about the miRNA gene structure, SNPs or methylation patterns in promotors or other regulatory regions might also represent an efficient way to determine the pro-or anti-angiogenic balance in complex diseases. Many pertinent and unanswered questions about which types of - and how - genetic variability modulate the angio-genome in diseases thus remain, emphasizing the need for further research into this field.
Acknowledgments
I.B. is supported by the Institute for the Promotion of Innovation by Science and Technology in Flanders (IWT), T.S by the Deutsche Forschungsgemeinschaft (DFG), C.R. by the Leducq foundation and D.L. by the Research Foundation Flanders (FWO), Belgium.
Supporting Information
Additional Supporting Information may be found in the online version of this article
Table S1 Overview of SNPs located in the VEGF gene and associated with various complex disorders. The SNP ID, SNP position, minor allele, minor allele frequency, risk allele and associated conditions are listed.
This material is available as part of the online article from: http://www.blackwell-synergy.com/doi/abs/10.1111/j.1582-4934.2008.00515.x (This link will take you to the article abstract).
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Folkman J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–6. doi: 10.1056/NEJM197111182852108. [DOI] [PubMed] [Google Scholar]
- 2.Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–6. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 3.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–9. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 4.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O'Shea KS, Powell-Braxton L, Hillan KJ, Moore MW. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–42. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 5.Jain RK, Carmeliet PF. Vessels of death or life. Sci Am. 2001;285:38–45. doi: 10.1038/scientificamerican1201-38. [DOI] [PubMed] [Google Scholar]
- 6.Carmeliet P, Jain RK. Angiogenesis in cancer and other diseases. Nature. 2000;407:249–57. doi: 10.1038/35025220. [DOI] [PubMed] [Google Scholar]
- 7.Pantsulaia I, Trofimov S, Kobyliansky E, Livshits G. Heritability of circulating growth factors involved in the angiogene-sis in healthy human population. Cytokine. 2004;27:152–8. doi: 10.1016/j.cyto.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 8.Berrahmoune H, Herbeth B, Lamont JV, Masson C, Fitzgerald PS, Visvikis-Siest S. Heritability for plasma VEGF concentration in the Stanislas family study. Ann Hum Genet. 2007;71:54–63. doi: 10.1111/j.1469-1809.2006.00298.x. [DOI] [PubMed] [Google Scholar]
- 9.Rohan RM, Fernandez A, Udagawa T, Yuan J, D'Amato RJ. Genetic heterogeneity of angiogenesis in mice. FASEB J. 2000;14:871–6. doi: 10.1096/fasebj.14.7.871. [DOI] [PubMed] [Google Scholar]
- 10.Rogers MS, Rohan RM, Birsner AE, D'Amato RJ. Genetic loci that control vascular endothelial growth factor-induced angiogenesis. FASEB J. 2003;17:2112–4. doi: 10.1096/fj.03-0246fje. [DOI] [PubMed] [Google Scholar]
- 11.Rogers MS, Rohan RM, Birsner AE, D'Amato RJ. Genetic loci that control the angiogenic response to basic fibroblast growth factor. FASEB J. 2004;18:1050–9. doi: 10.1096/fj.03-1241com. [DOI] [PubMed] [Google Scholar]
- 12.McCarthy MI, Abecasis GR, Cardon LR, Goldstein DB, Little J, Ioannidis JP, Hirschhorn JN. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet. 2008;9:356–69. doi: 10.1038/nrg2344. [DOI] [PubMed] [Google Scholar]
- 13.Beckmann JS, Estivill X, Antonarakis SE. Copy number variants and genetic traits: closer to the resolution of phenotypic to genotypic variability. Nat Rev Genet. 2007;8:639–46. doi: 10.1038/nrg2149. [DOI] [PubMed] [Google Scholar]
- 14.Schones DE, Zhao K. Genome-wide approaches to studying chromatin modifications. Nat Rev Genet. 2008;9:179–91. doi: 10.1038/nrg2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Couzin J. MicroRNAs make big impression in disease after disease. Science. 2008;319:1782–4. doi: 10.1126/science.319.5871.1782. [DOI] [PubMed] [Google Scholar]
- 16.Dubois J, Garel L. Imaging and therapeutic approach of hemangiomas and vascular malformations in the pediatric age group. Pediatr Radiol. 1999;29:879–93. doi: 10.1007/s002470050718. [DOI] [PubMed] [Google Scholar]
- 17.Garzon MC, Huang JT, Enjolras O, Frieden IJ. Vascular malformations: Part I. J Am Acad Dermatol. 2007;56:353–70. doi: 10.1016/j.jaad.2006.05.069. quiz 71–4. [DOI] [PubMed] [Google Scholar]
- 18.Garzon MC, Huang JT, Enjolras O, Frieden IJ. Vascular malformations. Part II: associated syndromes. J Am Acad Dermatol. 2007;56:541–64. doi: 10.1016/j.jaad.2006.05.066. [DOI] [PubMed] [Google Scholar]
- 19.Davis S, Aldrich TH, Jones PF, Acheson A, Compton DL, Jain V, Ryan TE, Bruno J, Radziejewski C, Maisonpierre PC, Yancopoulos GD. Isolation of angiopoi-etin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell. 1996;87:1161–9. doi: 10.1016/s0092-8674(00)81812-7. [DOI] [PubMed] [Google Scholar]
- 20.Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell. 1996;87:1171–80. doi: 10.1016/s0092-8674(00)81813-9. [DOI] [PubMed] [Google Scholar]
- 21.Dumont DJ, Gradwohl G, Fong GH, Puri MC, Gertsenstein M, Auerbach A, Breitman ML. Dominant-negative and targeted null mutations in the endothelial receptor tyrosine kinase, tek, reveal a critical role in vasculogenesis of the embryo. Genes Dev. 1994;8:1897–909. doi: 10.1101/gad.8.16.1897. [DOI] [PubMed] [Google Scholar]
- 22.Walter JW, Blei F, Anderson JL, Orlow SJ, Speer MC, Marchuk DA. Genetic mapping of a novel familial form of infantile heman-gioma. Am J Med Genet. 1999;82:77–83. doi: 10.1002/(sici)1096-8628(19990101)82:1<77::aid-ajmg15>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 23.Walter JW, North PE, Waner M, Mizeracki A, Blei F, Walker JW, Reinisch JF, Marchuk DA. Somatic mutation of vascular endothelial growth factor receptors in juvenile hemangioma. Genes Chromosomes Cancer. 2002;33:295–303. doi: 10.1002/gcc.10028. [DOI] [PubMed] [Google Scholar]
- 24.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–6. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 25.Dumont DJ, Jussila L, Taipale J, Lymboussaki A, Mustonen T, Pajusola K, Breitman M, Alitalo K. Cardiovascular failure in mouse embryos deficient in VEGF receptor-3. Science. 1998;282:946–9. doi: 10.1126/science.282.5390.946. [DOI] [PubMed] [Google Scholar]
- 26.Waner M, North PE, Scherer KA, Frieden IJ, Waner A, Mihm MC., Jr The nonrandom distribution of facial hemangiomas. Arch Dermatol. 2003;139:869–75. doi: 10.1001/archderm.139.7.869. [DOI] [PubMed] [Google Scholar]
- 27.Krings T, Geibprasert S, Luo CB, Bhattacharya JJ, Alvarez H, Lasjaunias P. Segmental neurovascular syndromes in children. Neuroimaging Clin N Am. 2007;17:245–58. doi: 10.1016/j.nic.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 28.Noden DM. Origins and patterning of avian outflow tract endocardium. Development. 1991;111:867–76. doi: 10.1242/dev.111.4.867. [DOI] [PubMed] [Google Scholar]
- 29.Noden DM. Origins and assembly of avian embryonic blood vessels. Ann N Y Acad Sci. 1990;588:236–49. doi: 10.1111/j.1749-6632.1990.tb13214.x. [DOI] [PubMed] [Google Scholar]
- 30.Korn J, Christ B, Kurz H. Neuroectodermal origin of brain pericytes and vascular smooth muscle cells. J Comp Neurol. 2002;442:78–88. doi: 10.1002/cne.1423. [DOI] [PubMed] [Google Scholar]
- 31.Allt G, Lawrenson JG. Pericytes: cell biology and pathology. Cells Tissues Organs. 2001;169:1–11. doi: 10.1159/000047855. [DOI] [PubMed] [Google Scholar]
- 32.Gallione CJ, Repetto GM, Legius E, Rustgi AK, Schelley SL, Tejpar S, Mitchell G, Drouin E, Westermann CJ, Marchuk DA. A combined syndrome of juvenile polyposis and hereditary haemorrhagic telangiectasia associated with mutations in MADH4 (SMAD4) Lancet. 2004;363:852–9. doi: 10.1016/S0140-6736(04)15732-2. [DOI] [PubMed] [Google Scholar]
- 33.McAllister KA, Grogg KM, Johnson DW, Gallione CJ, Baldwin MA, Jackson CE, Helmbold EA, Markel DS, McKinnon WC, Murrell J, McCormick MK, Pericak-Vance MA, Heutink P, Oostra BA, Haitjema T, Westerman CJJ, Porteous ME, Guttmacher AE, Letarte M, Marchuk DA. Endoglin, a TGF-beta binding protein of endothelial cells, is the gene for hereditary haemorrhagic telangiectasia type 1. Nat Genet. 1994;8:345–51. doi: 10.1038/ng1294-345. [DOI] [PubMed] [Google Scholar]
- 34.Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, Stenzel TT, Speer M, Pericak-Vance MA, Diamond A, Guttmacher AE, Jackson CE, Attisano L, Kucherlapati R, Porteous ME, Marchuk DA. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996;13:189–95. doi: 10.1038/ng0696-189. [DOI] [PubMed] [Google Scholar]
- 35.Lebrin F, Mummery CL. Endoglin-mediated vascular remodeling: mechanisms underlying hereditary hemorrhagic telangiectasia. Trends Cardiovasc Med. 2008;18:25–32. doi: 10.1016/j.tcm.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 36.Park SO, Lee YJ, Seki T, Hong KH, Fliess N, Jiang Z, Park A, Wu X, Kaartinen V, Roman BL, Oh SP. ALK5- and TGFBR2-independent role of ALK1 in the pathogen-esis of hereditary hemorrhagic telangiecta-sia type 2. Blood. 2008;111:633–42. doi: 10.1182/blood-2007-08-107359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ten Dijke P, Goumans MJ, Pardali E. Endoglin in angiogenesis and vascular diseases. Angiogenesis. 2008;11:79–89. doi: 10.1007/s10456-008-9101-9. [DOI] [PubMed] [Google Scholar]
- 38.Lamouille S, Mallet C, Feige JJ, Bailly S. Activin receptor-like kinase 1 is implicated in the maturation phase of angiogenesis. Blood. 2002;100:4495–501. doi: 10.1182/blood.V100.13.4495. [DOI] [PubMed] [Google Scholar]
- 39.Goumans MJ, Valdimarsdottir G, Itoh S, Rosendahl A, Sideras P, ten Dijke P. Balancing the activation state of the endothelium viatwo distinct TGF-beta type I receptors. EMBO J. 2002;21:1743–53. doi: 10.1093/emboj/21.7.1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lebrin F, Goumans MJ, Jonker L, Carvalho RL, Valdimarsdottir G, Thorikay M, Mummery C, Arthur HM, ten Dijke P. Endoglin promotes endothelial cell proliferation and TGF-beta/ALK1 signal trans-duction. EMBO J. 2004;23:4018–28. doi: 10.1038/sj.emboj.7600386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pece-Barbara N, Vera S, Kathirkamathamby K, Liebner S, Di Guglielmo GM, Dejana E, Wrana JL, Letarte M. Endoglin null endothelial cells proliferate faster and are more responsive to transforming growth factor beta1 with higher affinity receptors and an activated Alk1 pathway. J Biol Chem. 2005;280:27800–8. doi: 10.1074/jbc.M503471200. [DOI] [PubMed] [Google Scholar]
- 42.Coucke PJ, Willaert A, Wessels MW, Callewaert B, Zoppi N, De Backer J, Fox JE, Mancini GM, Kambouris M, Gardella R, Facchetti F, Willems PJ, Forsyth R, Dietz HC, Barlati S, Colombi M, Loeys B, De Paepe A. Mutations in the facilitative glucose transporter GLUT10 alter angio-genesis and cause arterial tortuosity syndrome. Nat Genet. 2006;38:452–7. doi: 10.1038/ng1764. [DOI] [PubMed] [Google Scholar]
- 43.Huang R, Merrilees MJ, Braun K, Beaumont B, Lemire J, Clowes AW, Hinek A, Wight TN. Inhibition of versican synthesis by antisense alters smooth muscle cell phenotype and induces elastic fiber formation in vitro and in neointima after vessel injury. Circ Res. 2006;98:370–7. doi: 10.1161/01.RES.0000202051.28319.c8. [DOI] [PubMed] [Google Scholar]
- 44.Revencu N, Vikkula M. Cerebral cavernous malformation: new molecular and clinical insights. J Med Genet. 2006;43:716–21. doi: 10.1136/jmg.2006.041079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Laberge-le Couteulx S, Jung HH, Labauge P, Houtteville JP, Lescoat C, Cecillon M, Marechal E, Joutel A, Bach JF, Tournier-Lasserve E. Truncating mutations in CCM1, encoding KRIT1, cause hereditary cavernous angiomas. Nat Genet. 1999;23:189–93. doi: 10.1038/13815. [DOI] [PubMed] [Google Scholar]
- 46.Liquori CL, Berg MJ, Siegel AM, Huang E, Zawistowski JS, Stoffer T, Verlaan D, Balogun F, Hughes L, Leedom TP, Plummer NW, Cannella M, Maglione V, Squitieri F, Johnson EW, Rouleau GA, Ptacek L, Marchuk DA. Mutations in a gene encoding a novel protein containing a phosphotyrosine-binding domain cause type 2 cerebral cavernous malformations. Am J Hum Genet. 2003;73:1459–64. doi: 10.1086/380314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bergametti F, Denier C, Labauge P, Arnoult M, Boetto S, Clanet M, Coubes P, Echenne B, Ibrahim R, Irthum B, Jacquet G, Lonjon M, Moreau JJ, Neau JP, Parker F, Tremoulet M, Tournier-Lasserve E. Mutations within the programmed cell death 10 gene cause cerebral cavernous malformations. Am J Hum Genet. 2005;76:42–51. doi: 10.1086/426952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Plummer NW, Gallione CJ, Srinivasan S, Zawistowski JS, Louis DN, Marchuk DA. Loss of p53 sensitizes mice with a mutation in Ccm1 (KRIT1) to development of cerebral vascular malformations. Am J Pathol. 2004;165:1509–18. doi: 10.1016/S0002-9440(10)63409-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Whitehead KJ, Plummer NW, Adams JA, Marchuk DA, Li DY. Ccm1 is required for arterial morphogenesis: implications for the etiology of human cavernous malformations. Development. 2004;131:1437–48. doi: 10.1242/dev.01036. [DOI] [PubMed] [Google Scholar]
- 50.Brouillard P, Vikkula M. Genetic causes of vascular malformations. Hum Mol Genet. 2007;16:R140–9. doi: 10.1093/hmg/ddm211. [DOI] [PubMed] [Google Scholar]
- 51.He M, Das K, Blacksin M, Benevenia J, Hameed M. A translocation involving the placental growth factor gene is identified in an epithelioid hemangioendothelioma. Cancer Genet Cytogenet. 2006;168:150–4. doi: 10.1016/j.cancergencyto.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 52.Oduber CE, van der Horst CM, Hennekam RC. Klippel-Trenaunay syndrome: diagnostic criteria and hypothesis on etiology. Ann Plast Surg. 2008;60:217–23. doi: 10.1097/SAP.0b013e318062abc1. [DOI] [PubMed] [Google Scholar]
- 53.Tian XL, Kadaba R, You SA, Liu M, Timur AA, Yang L, Chen Q, Szafranski P, Rao S, Wu L, Housman DE, DiCorleto PE, Driscoll DJ, Borrow J, Wang Q. Identification of an angiogenic factor that when mutated causes susceptibility to Klippel-Trenaunay syndrome. Nature. 2004;427:640–5. doi: 10.1038/nature02320.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barker KT, Foulkes WD, Schwartz CE, Labadie C, Monsell F, Houlston RS, Harper J. Is the E133K allele of VG5Q associated with Klippel-Trenaunay and other overgrowth syndromes? J Med Genet. 2006;43:613–4. doi: 10.1136/jmg.2006.040790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gutierrez S, Magano L, Delicado A, Mori MA, de Torres ML, Fernandez L, Palomares M, Fernandez E, Tarduchy GR, Molano J, Gracia R, Pajares IL, Lapunzina P. The G397A (E133K) change in the AGGF1 (VG5Q) gene is a single nucleotide polymorphism in the Spanish population. Am J Med Genet A. 2006;140:2832–3. doi: 10.1002/ajmg.a.31532. [DOI] [PubMed] [Google Scholar]
- 56.Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, Pasternak S, Wheeler DA, Willis TD, Yu F, Yang H, Zeng C, Gao Y, Hu H, Hu W, Li C, Lin W, Liu S, Pan H, Tang X, Wang J, Wang W, Yu J, Zhang B, Zhang Q, Zhao H, Zhao H, Zhou J, Gabriel SB, Barry R, Blumenstiel B, Camargo A, Defelice M, Faggart M, Goyette M, Gupta S, Moore J, Nguyen H, Onofrio RC, Parkin M, Roy J, Stahl E, Winchester E, Ziaugra L, Altshuler D, Shen Y, Yao Z, Huang W, Chu X, He Y, Jin L, Liu Y, Shen Y, Sun W, Wang H, Wang Y, Wang Y, Xiong X, Xu L, Waye MM, Tsui SK, Xue H, Wong JT, Galver LM, Fan JB, Gunderson K, Murray SS, Oliphant AR, Chee MS, Montpetit A, Chagnon F, Ferretti V, Leboeuf M, Olivier JF, Phillips MS, Roumy S, Sallee C, Verner A, Hudson TJ, Kwok PY, Cai D, Koboldt DC, Miller RD, Pawlikowska L, Taillon-Miller P, Xiao M, Tsui LC, Mak W, Song YQ, Tam PK, Nakamura Y, Kawaguchi T, Kitamoto T, Morizono T, Nagashima A, Ohnishi Y, Sekine A, Tanaka T, Tsunoda T, Deloukas P, Bird CP, Delgado M, Dermitzakis ET, Gwilliam R, Hunt S, Morrison J, Powell D, Stranger BE, Whittaker P, Bentley DR, Daly MJ, de Bakker PI, Barrett J, Chretien YR, Maller J, McCarroll S, Patterson N, Pe'er I, Price A, Purcell S, Richter DJ, Sabeti P, Saxena R, Schaffner SF, Sham PC, Varilly P, Altshuler D, Stein LD, Krishnan L, Smith AV, Tello-Ruiz MK, Thorisson GA, Chakravarti A, Chen PE, Cutler DJ, Kashuk CS, Lin S, Abecasis GR, Guan W, Li Y, Munro HM, Qin ZS, Thomas DJ, McVean G, Auton A, Bottolo L, Cardin N, Eyheramendy S, Freeman C, Marchini J, Myers S, Spencer C, Stephens M, Donnelly P, Cardon LR, Clarke G, Evans DM, Morris AP, Weir BS, Tsunoda T, Mullikin JC, Sherry ST, Feolo M, Skol A, Zhang H, Zeng C, Zhao H, Matsuda I, Fukushima Y, Macer DR, Suda E, Rotimi CN, Adebamowo CA, Ajayi I, Aniagwu T, Marshall PA, Nkwodimmah C, Royal CD, Leppert MF, Dixon M, Peiffer A, Qiu R, Kent A, Kato K, Niikawa N, Adewole IF, Knoppers BM, Foster MW, Clayton EW, Watkin J, Gibbs RA, Belmont JW, Muzny D, Nazareth L, Sodergren E, Weinstock GM, Wheeler DA, Yakub I, Gabriel SB, Onofrio RC, Richter DJ, Ziaugra L, Birren BW, Daly MJ, Altshuler D, Wilson RK, Fulton LL, Rogers J, Burton J, Carter NP, Clee CM, Griffiths M, Jones MC, McLay K, Plumb RW, Ross MT, Sims SK, Willey DL, Chen Z, Han H, Kang L, Godbout M, Wallenburg JC, L'Archeveque P, Bellemare G, Saeki K, Wang H, An D, Fu H, Li Q, Wang Z, Wang R, Holden AL, Brooks LD, McEwen JE, Guyer MS, Wang VO, Peterson JL, Shi M, Spiegel J, Sung LM, Zacharia LF, Collins FS, Kennedy K, Jamieson R, Stewart J. A second generation human haplotype map of over 3.1 million SNPs. Nature. 2007;449:851–61. doi: 10.1038/nature06258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, Berlin J, Baron A, Griffing S, Holmgren E, Ferrara N, Fyfe G, Rogers B, Ross R, Kabbinavar F. Bevacizumab plus irinote-can, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004;350:2335–42. doi: 10.1056/NEJMoa032691. [DOI] [PubMed] [Google Scholar]
- 58.Jelkmann W. Pitfalls in the measurement of circulating vascular endothelial growth factor. Clin Chem. 2001;47:617–23. [PubMed] [Google Scholar]
- 59.Shahbazi M, Fryer AA, Pravica V, Brogan IJ, Ramsay HM, Hutchinson IV, Harden PN. Vascular endothelial growth factor gene polymorphisms are associated with acute renal allograft rejection. J Am Soc Nephrol. 2002;13:260–4. doi: 10.1681/ASN.V131260. [DOI] [PubMed] [Google Scholar]
- 60.Lambrechts D, Storkebaum E, Morimoto M, Del-Favero J, Desmet F, Marklund SL, Wyns S, Thijs V, Andersson J, van Marion I, Al-Chalabi A, Bornes S, Musson R, Hansen V, Beckman L, Adolfsson R, Pall HS, Prats H, Vermeire S, Rutgeerts P, Katayama S, Awata T, Leigh N, Lang-Lazdunski L, Dewerchin M, Shaw C, Moons L, Vlietinck R, Morrison KE, Robberecht W, Van Broeckhoven C, Collen D, Andersen PM, Carmeliet P. VEGF is a modifier of amyotrophic lateral sclerosis in mice and humans and protects motoneurons against ischemic death. Nat Genet. 2003;34:383–94. doi: 10.1038/ng1211. [DOI] [PubMed] [Google Scholar]
- 61.Kawai Y, Sakano S, Korenaga Y, Eguchi S, Naito K. Associations of single nucleotide polymorphisms in the vascular endothelial growth factor gene with the characteristics and prognosis of renal cell carcinomas. Eur Urol. 2007;52:1147–55. doi: 10.1016/j.eururo.2007.01.073. [DOI] [PubMed] [Google Scholar]
- 62.Jacobs EJ, Feigelson HS, Bain EB, Brady KA, Rodriguez C, Stevens VL, Patel AV, Thun MJ, Calle EE. Polymorphisms in the vascular endothelial growth factor gene and breast cancer in the Cancer Prevention Study II cohort. Breast Cancer Res. 2006;8:R22. doi: 10.1186/bcr1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim JG, Chae YS, Sohn SK, Cho YY, Moon JH, Park JY, Jeon SW, Lee IT, Choi GS, Jun SH. Vascular endothelial growth factor gene polymorphisms associated with prognosis for patients with colorectal cancer. Clin Cancer Res. 2008;14:62–6. doi: 10.1158/1078-0432.CCR-07-1537. [DOI] [PubMed] [Google Scholar]
- 64.Howell WM, Bateman AC, Turner SJ, Collins A, Theaker JM. Influence of vascular endothelial growth factor single nucleotide polymorphisms on tumour development in cutaneous malignant melanoma. Genes Immun. 2002;3:229–32. doi: 10.1038/sj.gene.6363851. [DOI] [PubMed] [Google Scholar]
- 65.Hefler LA, Mustea A, Konsgen D, Concin N, Tanner B, Strick R, Heinze G, Grimm C, Schuster E, Tempfer C, Reinthaller A, Zeillinger R. Vascular endothelial growth factor gene polymorphisms are associated with prognosis in ovarian cancer. Clin Cancer Res. 2007;13:898–901. doi: 10.1158/1078-0432.CCR-06-1008. [DOI] [PubMed] [Google Scholar]
- 66.Awata T, Inoue K, Kurihara S, Ohkubo T, Watanabe M, Inukai K, Inoue I, Katayama S. A common polymorphism in the 5'-untranslated region of the VEGF gene is associated with diabetic retinopathy in type 2 diabetes. Diabetes. 2002;51:1635–9. doi: 10.2337/diabetes.51.5.1635. [DOI] [PubMed] [Google Scholar]
- 67.Jin Q, Hemminki K, Enquist K, Lenner P, Grzybowska E, Klaes R, Henriksson R, Chen B, Pamula J, Pekala W, Zientek H, Rogozinska-Szczepka J, Utracka-Hutka B, Hallmans G, Forsti A. Vascular endothelial growth factor polymorphisms in relation to breast cancer development and prognosis. Clin Cancer Res. 2005;11:3647–53. doi: 10.1158/1078-0432.CCR-04-1803. [DOI] [PubMed] [Google Scholar]
- 68.Sfar S, Hassen E, Saad H, Mosbah F, Chouchane L. Association of VEGF genetic polymorphisms with prostate carcinoma risk and clinical outcome. Cytokine. 2006;35:21–8. doi: 10.1016/j.cyto.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 69.Heist RS, Zhai R, Liu G, Zhou W, Lin X, Su L, Asomaning K, Lynch TJ, Wain JC, Christiani DC. VEGF polymorphisms and survival in early-stage non-small-cell lung cancer. J Clin Oncol. 2008;26:856–62. doi: 10.1200/JCO.2007.13.5947. [DOI] [PubMed] [Google Scholar]
- 70.Renner W, Kotschan S, Hoffmann C, Obermayer-Pietsch B, Pilger E. A common 936 C/T mutation in the gene for vascular endothelial growth factor is associated with vascular endothelial growth factor plasma levels. J Vasc Res. 2000;37:443–8. doi: 10.1159/000054076. [DOI] [PubMed] [Google Scholar]
- 71.Krippl P, Langsenlehner U, Renner W, Yazdani-Biuki B, Wolf G, Wascher TC, Paulweber B, Haas J, Samonigg H. A common 936 C/T gene polymorphism of vascular endothelial growth factor is associated with decreased breast cancer risk. Int J Cancer. 2003;106:468–71. doi: 10.1002/ijc.11238. [DOI] [PubMed] [Google Scholar]
- 72.Garcia-Closas M, Hall P, Nevanlinna H, Pooley K, Morrison J, Richesson DA, Bojesen SE, Nordestgaard BG, Axelsson CK, Arias JI, Milne RL, Ribas G, Gonzalez-Neira A, Benitez J, Zamora P, Brauch H, Justenhoven C, Hamann U, Ko YD, Bruening T, Haas S, Dork T, Schurmann P, Hillemanns P, Bogdanova N, Bremer M, Karstens JH, Fagerholm R, Aaltonen K, Aittomaki K, von Smitten K, Blomqvist C, Mannermaa A, Uusitupa M, Eskelinen M, Tengstrom M, Kosma VM, Kataja V, Chenevix-Trench G, Spurdle AB, Beesley J, Chen X, Australian Ovarian Cancer Management G, Kathleen Cuningham Foundation Consortium For Research Into Familial Breast C, Devilee P, van Asperen CJ, Jacobi CE, Tollenaar RA, Huijts PE, Klijn JG, Chang-Claude J, Kropp S, Slanger T, Flesch-Janys D, Mutschelknauss E, Salazar R, Wang-Gohrke S, Couch F, Goode EL, Olson JE, Vachon C, Fredericksen ZS, Giles GG, Baglietto L, Severi G, Hopper JL, English DR, Southey MC, Haiman CA, Henderson BE, Kolonel LN, Le Marchand L, Stram DO, Hunter DJ, Hankinson SE, Cox DG, Tamimi R, Kraft P, Sherman ME, Chanock SJ, Lissowska J, Brinton LA, Peplonska B, Klijn JG, Hooning MJ, Meijers-Heijboer H, Collee JM, van den Ouweland A, Uitterlinden AG, Liu J, Lin LY, Yuqing L, Humphreys K, Czene K, Cox A, Balasubramanian SP, Cross SS, Reed MW, Blows F, Driver K, Dunning A, Tyrer J, Ponder BA, Sangrajrang S, Brennan P, McKay J, Odefrey F, Gabrieau V, Sigurdson A, Doody M, Struewing JP, Alexander B, Easton DF, Pharoah PD. Heterogeneity of breast cancer associations with five susceptibility Loci by clinical and pathological characteristics. PLoS Genet. 2008;4:e1000054. doi: 10.1371/journal.pgen.1000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Antoniou AC, Spurdle AB, Sinilnikova OM, Healey S, Pooley KA, Schmutzler RK, Versmold B, Engel C, Meindl A, Arnold N, Hofmann W, Sutter C, Niederacher D, Deissler H, Caldes T, Kampjarvi K, Nevanlinna H, Simard J, Beesley J, Chen X, Neuhausen SL, Rebbeck TR, Wagner T, Lynch HT, Isaacs C, Weitzel J, Ganz PA, Daly MB, Tomlinson G, Olopade OI, Blum JL, Couch FJ, Peterlongo P, Manoukian S, Barile M, Radice P, Szabo CI, Pereira LH, Greene MH, Rennert G, Lejbkowicz F, Barnett-Griness O, Andrulis IL, Ozcelik H, Gerdes AM, Caligo MA, Laitman Y, Kaufman B, Milgrom R, Friedman E, Domchek SM, Nathanson KL, Osorio A, Llort G, Milne RL, Benitez J, Hamann U, Hogervorst FB, Manders P, Ligtenberg MJ, van den Ouweland AM, Peock S, Cook M, Platte R, Evans DG, Eeles R, Pichert G, Chu C, Eccles D, Davidson R, Douglas F, Godwin AK, Barjhoux L, Mazoyer S, Sobol H, Bourdon V, Eisinger F, Chompret A, Capoulade C, Bressac-de Paillerets B, Lenoir GM, Gauthier-Villars M, Houdayer C, Stoppa-Lyonnet D, Chenevix-Trench G, Easton DF. Common breast cancer-predisposition alleles are associated with breast cancer risk in BRCA1 and BRCA2 mutation carriers. Am J Hum Genet. 2008;82:937–48. doi: 10.1016/j.ajhg.2008.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Howell WM, Ali S, Rose-Zerilli MJ, Ye S. VEGF polymorphisms and severity of atherosclerosis. J Med Genet. 2005;42:485–90. doi: 10.1136/jmg.2004.025734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang Y, Zheng Y, Zhang W, Yu H, Lou K, Zhang Y, Qin Q, Zhao B, Yang Y, Hui R. Polymorphisms of KDR gene are associated with coronary heart disease. J Am Coll Cardiol. 2007;50:760–7. doi: 10.1016/j.jacc.2007.04.074. [DOI] [PubMed] [Google Scholar]
- 76.Pouyssegur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approaches to enforce tumour regression. Nature. 2006;441:437–43. doi: 10.1038/nature04871. [DOI] [PubMed] [Google Scholar]
- 77.Tanimoto K, Yoshiga K, Eguchi H, Kaneyasu M, Ukon K, Kumazaki T, Oue N, Yasui W, Imai K, Nakachi K, Poellinger L, Nishiyama M. Hypoxia-inducible factor-1 alpha polymorphisms associated with enhanced transactivation capacity, implying clinical significance. Carcinogenesis. 2003;24:1779–83. doi: 10.1093/carcin/bgg132. [DOI] [PubMed] [Google Scholar]
- 78.Fransen K, Fenech M, Fredrikson M, Dabrosin C, Soderkvist P. Association between ulcerative growth and hypoxia inducible factor-1alpha polymorphisms in colorectal cancer patients. Mol Carcinog. 2006;45:833–40. doi: 10.1002/mc.20209. [DOI] [PubMed] [Google Scholar]
- 79.Nadaoka J, Horikawa Y, Saito M, Kumazawa T, Inoue T, Narita S, Yuasa T, Satoh S, Nishiyama H, Ogawa O, Tsuchiya N, Habuchi T. Prognostic significance of HIF-1 alpha polymorphisms in transitional cell carcinoma of the bladder. Int J Cancer. 2008;122:1297–302. doi: 10.1002/ijc.23256. [DOI] [PubMed] [Google Scholar]
- 80.Percy MJ, Zhao Q, Flores A, Harrison C, Lappin TR, Maxwell PH, McMullin MF, Lee FS. A family with erythrocytosis establishes a role for prolyl hydroxylase domain protein 2 in oxygen homeostasis. Proc Natl Acad Sci USA. 2006;103:654–9. doi: 10.1073/pnas.0508423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Soussi T, Wiman KG. Shaping genetic alterations in human cancer: the p53 mutation paradigm. Cancer Cell. 2007;12:303–12. doi: 10.1016/j.ccr.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 82.Menendez D, Krysiak O, Inga A, Krysiak B, Resnick MA, Schonfelder G. A SNP in the flt-1 promoter integrates the VEGF system into the p53 transcriptional network. Proc Natl Acad Sci USA. 2006;103:1406–11. doi: 10.1073/pnas.0508103103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shibuya M. Vascular endothelial growth factor receptor-1 (VEGFR-1/Flt-1): a dual regulator for angiogenesis. Angiogenesis. 2006;9:225–30. doi: 10.1007/s10456-006-9055-8. [DOI] [PubMed] [Google Scholar]
- 84.Oosthuyse B, Moons L, Storkebaum E, Beck H, Nuyens D, Brusselmans K, Van Dorpe J, Hellings P, Gorselink M, Heymans S, Theilmeier G, Dewerchin M, Laudenbach V, Vermylen P, Raat H, Acker T, Vleminckx V, Van Den Bosch L, Cashman N, Fujisawa H, Drost MR, Sciot R, Bruyninckx F, Hicklin DJ, Ince C, Gressens P, Lupu F, Plate KH, Robberecht W, Herbert JM, Collen D, Carmeliet P. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat Genet. 2001;28:131–8. doi: 10.1038/88842. [DOI] [PubMed] [Google Scholar]
- 85.Lambrechts D, Poesen K, Robberecht W, Fernandez-Santiago R, Al-Chalabi A, Del Bo R, Van Wj Vught P, Carmeliet P, Thijs V, Siddique T. Meta-analysis of VEGF variations in ALS: increased susceptibility in male carriers of the -2578AA genotype. J Med Genet. 2008 doi: 10.1136/jmg.2008.058222. (in press) [DOI] [PubMed] [Google Scholar]
- 86.Del Bo R, Scarlato M, Ghezzi S, Martinelli Boneschi F, Fenoglio C, Galbiati S, Virgilio R, Galimberti D, Galimberti G, Crimi M, Ferrarese C, Scarpini E, Bresolin N, Comi GP. Vascular endothelial growth factor gene variability is associated with increased risk for AD. Ann Neurol. 2005;57:373–80. doi: 10.1002/ana.20390. [DOI] [PubMed] [Google Scholar]
- 87.Del Bo R, Ghezzi S, Scarlato M, Albani D, Galimberti D, Lucca U, Tettamanti M, Scarpini E, Forloni G, Bresolin N, Comi GP. Role of VEGF gene variability in longevity: a lesson from the Italian population. Neurobiol Aging. 2008;29:1917–22. doi: 10.1016/j.neurobiolaging.2007.05.003. [DOI] [PubMed] [Google Scholar]
- 88.Greenway MJ, Andersen PM, Russ C, Ennis S, Cashman S, Donaghy C, Patterson V, Swingler R, Kieran D, Prehn J, Morrison KE, Green A, Acharya KR, Brown RH, Jr, Hardiman O. ANG mutations segregate with familial and ‘sporadic’ amy-otrophic lateral sclerosis. Nat Genet. 2006;38:411–3. doi: 10.1038/ng1742. [DOI] [PubMed] [Google Scholar]
- 89.Wu D, Yu W, Kishikawa H, Folkerth RD, Iafrate AJ, Shen Y, Xin W, Sims K, Hu GF. Angiogenin loss-of-function mutations in amyotrophic lateral sclerosis. Ann Neurol. 2007;62:609–17. doi: 10.1002/ana.21221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Conforti FL, Sprovieri T, Mazzei R, Ungaro C, La Bella V, Tessitore A, Patitucci A, Magariello A, Gabriele AL, Tedeschi G, Simone IL, Majorana G, Valentino P, Condino F, Bono F, Monsurro MR, Muglia M, Quattrone A. A novel Angiogenin gene mutation in a sporadic patient with amyotrophic lateral sclerosis from southern Italy. Neuromuscul Disord. 2008;18:68–70. doi: 10.1016/j.nmd.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 91.Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, Kumar-Singh S, Van Broeckhoven C. Null mutations in progranulin cause ubiquitin-posi-tive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–4. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- 92.Eriksen JL, Mackenzie IR. Progranulin: normal function and role in neurodegener-ation. J Neurochem. 2008;104:287–97. doi: 10.1111/j.1471-4159.2007.04968.x. [DOI] [PubMed] [Google Scholar]
- 93.Kishimoto K, Liu S, Tsuji T, Olson KA, Hu GF. Endogenous angiogenin in endothelial cells is a general requirement for cell proliferation and angiogenesis. Oncogene. 2005;24:445–56. doi: 10.1038/sj.onc.1208223. [DOI] [PubMed] [Google Scholar]
- 94.Tangkeangsirisin W, Serrero G. PC cell-derived growth factor (PCDGF/GP88, pro-granulin) stimulates migration, invasive-ness and VEGF expression in breast cancer cells. Carcinogenesis. 2004;25:1587–92. doi: 10.1093/carcin/bgh171. [DOI] [PubMed] [Google Scholar]
- 95.Zeggini E, Scott LJ, Saxena R, Voight BF, Marchini JL, Hu T, de Bakker PI, Abecasis GR, Almgren P, Andersen G, Ardlie K, Bostrom KB, Bergman RN, Bonnycastle LL, Borch-Johnsen K, Burtt NP, Chen H, Chines PS, Daly MJ, Deodhar P, Ding CJ, Doney AS, Duren WL, Elliott KS, Erdos MR, Frayling TM, Freathy RM, Gianniny L, Grallert H, Grarup N, Groves CJ, Guiducci C, Hansen T, Herder C, Hitman GA, Hughes TE, Isomaa B, Jackson AU, Jorgensen T, Kong A, Kubalanza K, Kuruvilla FG, Kuusisto J, Langenberg C, Lango H, Lauritzen T, Li Y, Lindgren CM, Lyssenko V, Marvelle AF, Meisinger C, Midthjell K, Mohlke KL, Morken MA, Morris AD, Narisu N, Nilsson P, Owen KR, Palmer CN, Payne F, Perry JR, Pettersen E, Platou C, Prokopenko I, Qi L, Qin L, Rayner NW, Rees M, Roix JJ, Sandbaek A, Shields B, Sjogren M, Steinthorsdottir V, Stringham HM, Swift AJ, Thorleifsson G, Thorsteinsdottir U, Timpson NJ, Tuomi T, Tuomilehto J, Walker M, Watanabe RM, Weedon MN, Willer CJ, Illig T, Hveem K, Hu FB, Laakso M, Stefansson K, Pedersen O, Wareham NJ, Barroso I, Hattersley AT, Collins FS, Groop L, McCarthy MI, Boehnke M, Altshuler D. Meta-analysis of genome-wide association data and large-scale replication identifies additional susceptibility loci for type 2 diabetes. Nat Genet. 2008;40:635–45. doi: 10.1038/ng.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Easton DF, Pooley KA, Dunning AM, Pharoah PD, Thompson D, Ballinger DG, Struewing JP, Morrison J, Field H, Luben R, Wareham N, Ahmed S, Healey CS, Bowman R, Meyer KB, Haiman CA, Kolonel LK, Henderson BE, Le Marchand L, Brennan P, Sangrajrang S, Gaborieau V, Odefrey F, Shen CY, Wu PE, Wang HC, Eccles D, Evans DG, Peto J, Fletcher O, Johnson N, Seal S, Stratton MR, Rahman N, Chenevix-Trench G, Bojesen SE, Nordestgaard BG, Axelsson CK, Garcia-Closas M, Brinton L, Chanock S, Lissowska J, Peplonska B, Nevanlinna H, Fagerholm R, Eerola H, Kang D, Yoo KY, Noh DY, Ahn SH, Hunter DJ, Hankinson SE, Cox DG, Hall P, Wedren S, Liu J, Low YL, Bogdanova N, Schurmann P, Dork T, Tollenaar RA, Jacobi CE, Devilee P, Klijn JG, Sigurdson AJ, Doody MM, Alexander BH, Zhang J, Cox A, Brock IW, MacPherson G, Reed MW, Couch FJ, Goode EL, Olson JE, Meijers-Heijboer H, van den Ouweland A, Uitterlinden A, Rivadeneira F, Milne RL, Ribas G, Gonzalez-Neira A, Benitez J, Hopper JL, McCredie M, Southey M, Giles GG, Schroen C, Justenhoven C, Brauch H, Hamann U, Ko YD, Spurdle AB, Beesley J, Chen X, Mannermaa A, Kosma VM, Kataja V, Hartikainen J, Day NE, Cox DR, Ponder BA. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature. 2007;447:1087–93. doi: 10.1038/nature05887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hunter DJ, Kraft P, Jacobs KB, Cox DG, Yeager M, Hankinson SE, Wacholder S, Wang Z, Welch R, Hutchinson A, Wang J, Yu K, Chatterjee N, Orr N, Willett WC, Colditz GA, Ziegler RG, Berg CD, Buys SS, McCarty CA, Feigelson HS, Calle EE, Thun MJ, Hayes RB, Tucker M, Gerhard DS, Fraumeni JF, Jr, Hoover RN, Thomas G, Chanock SJ. A genome-wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat Genet. 2007;39:870–4. doi: 10.1038/ng2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Meyer KB, Maia AT, O'Reilly M, Teschendorff AE, Chin SF, Caldas C, Ponder BA. Allele-specific up-regulation of FGFR2 increases susceptibility to breast cancer. PLoS Biol. 2008;6:e108. doi: 10.1371/journal.pbio.0060108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Esch F, Baird A, Ling N, Ueno N, Hill F, Denoroy L, Klepper R, Gospodarowicz D, Bohlen P, Guillemin R. Primary structure of bovine pituitary basic fibroblast growth factor (FGF) and comparison with the amino-terminal sequence of bovine brain acidic FGF. Proc Natl Acad Sci USA. 1985;82:6507–11. doi: 10.1073/pnas.82.19.6507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lee SA, Ho C, Roy R, Kosinski C, Patil MA, Tward AD, Fridlyand J, Chen X. Integration of genomic analysis and in vivo transfection to identify sprouty 2 as a candidate tumor suppressor in liver cancer. Hepatology. 2008;47:1200–10. doi: 10.1002/hep.22169. [DOI] [PubMed] [Google Scholar]
- 101.Mason JM, Morrison DJ, Basson MA, Licht JD. Sprouty proteins: multifaceted negative-feedback regulators of receptor tyrosine kinase signaling. Trends Cell Biol. 2006;16:45–54. doi: 10.1016/j.tcb.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 102.Weir BA, Woo MS, Getz G, Perner S, Ding L, Beroukhim R, Lin WM, Province MA, Kraja A, Johnson LA, Shah K, Sato M, Thomas RK, Barletta JA, Borecki IB, Broderick S, Chang AC, Chiang DY, Chirieac LR, Cho J, Fujii Y, Gazdar AF, Giordano T, Greulich H, Hanna M, Johnson BE, Kris MG, Lash A, Lin L, Lindeman N, Mardis ER, McPherson JD, Minna JD, Morgan MB, Nadel M, Orringer MB, Osborne JR, Ozenberger B, Ramos AH, Robinson J, Roth JA, Rusch V, Sasaki H, Shepherd F, Sougnez C, Spitz MR, Tsao MS, Twomey D, Verhaak RG, Weinstock GM, Wheeler DA, Winckler W, Yoshizawa A, Yu S, Zakowski MF, Zhang Q, Beer DG, Wistuba II, Watson MA, Garraway LA, Ladanyi M, Travis WD, Pao W, Rubin MA, Gabriel SB, Gibbs RA, Varmus HE, Wilson RK, Lander ES, Meyerson M. Characterizing the cancer genome in lung adenocarcinoma. Nature. 2007;450:893–8. doi: 10.1038/nature06358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chiang DY, Villanueva A, Hoshida Y, Peix J, Newell P, Minguez B, LeBlanc AC, Donovan DJ, Thung SN, Sole M, Tovar V, Alsinet C, Ramos AH, Barretina J, Roayaie S, Schwartz M, Waxman S, Bruix J, Mazzaferro V, Ligon AH, Najfeld V, Friedman SL, Sellers WR, Meyerson M, Llovet JM. Focal gains of VEGFA and molecular classification of hepatocellular carcinoma. Cancer Res. 2008;68:6779–88. doi: 10.1158/0008-5472.CAN-08-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sasaki H, Matsui Y. Epigenetic events in mammalian germ-cell development: repro-gramming and beyond. Nat Rev Genet. 2008;9:129–40. doi: 10.1038/nrg2295. [DOI] [PubMed] [Google Scholar]
- 105.Feinberg AP. Phenotypic plasticity and the epigenetics of human disease. Nature. 2007;447:433–40. doi: 10.1038/nature05919. [DOI] [PubMed] [Google Scholar]
- 106.Hellebrekers DM, Griffioen AW, van Engeland M. Dual targeting of epigenetic therapy in cancer. Biochim Biophys Acta. 2007;1775:76–91. doi: 10.1016/j.bbcan.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 107.Li Q, Ahuja N, Burger PC, Issa JP. Methylation and silencing of the Thrombospondin-1 promoter in human cancer. Oncogene. 1999;18:3284–9. doi: 10.1038/sj.onc.1202663. [DOI] [PubMed] [Google Scholar]
- 108.Oue N, Matsumura S, Nakayama H, Kitadai Y, Taniyama K, Matsusaki K, Yasui W. Reduced expression of the TSP1 gene and its association with promoter hypermethylation in gastric carcinoma. Oncology. 2003;64:423–9. doi: 10.1159/000070302. [DOI] [PubMed] [Google Scholar]
- 109.Guerrero D, Guarch R, Ojer A, Casas JM, Ropero S, Mancha A, Pesce C, Lloveras B, Garcia-Bragado F, Puras A. Hypermethylation of the thrombospondin-1 gene is associated with poor prognosis in penile squamous cell carcinoma. BJU Int. 2008;102:747–55. doi: 10.1111/j.1464-410X.2008.07603.x. [DOI] [PubMed] [Google Scholar]
- 110.Yang QW, Liu S, Tian Y, Salwen HR, Chlenski A, Weinstein J, Cohn SL. Methylation-associated silencing of the thrombospondin-1 gene in human neu-roblastoma. Cancer Res. 2003;63:6299–310. [PubMed] [Google Scholar]
- 111.Miyamoto N, Yamamoto H, Taniguchi H, Miyamoto C, Oki M, Adachi Y, Imai K, Shinomura Y. Differential expression of angiogenesis-related genes in human gastric cancers with and those without high-frequency microsatellite instability. Cancer Lett. 2007;254:42–53. doi: 10.1016/j.canlet.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 112.Rojas A, Meherem S, Kim YH, Washington MK, Willis JE, Markowitz SD, Grady WM. The aberrant methylation of TSP1 suppresses TGF-beta1 activation in colorectal cancer. Int J Cancer. 2008;123:14–21. doi: 10.1002/ijc.23608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang J, Thompson B, Ren C, Ittmann M, Kwabi-Addo B. Sprouty4, a suppressor of tumor cell motility, is down regulated by DNA methylation in human prostate cancer. Prostate. 2006;66:613–24. doi: 10.1002/pros.20353. [DOI] [PubMed] [Google Scholar]
- 114.Dunn JR, Reed JE, du Plessis DG, Shaw EJ, Reeves P, Gee AL, Warnke P, Walker C. Expression of ADAMTS-8, a secreted protease with antiangiogenic properties, is downregulated in brain tumours. Br J Cancer. 2006;94:1186–93. doi: 10.1038/sj.bjc.6603006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Matsumura S, Oue N, Mitani Y, Kitadai Y, Yasui W. DNA demethylation of vascular endothelial growth factor-C is associated with gene expression and its possible involvement of lymphangiogenesis in gastric cancer. Int J Cancer. 2007;120:1689–95. doi: 10.1002/ijc.22433. [DOI] [PubMed] [Google Scholar]
- 116.Dadras SS, Lange-Asschenfeldt B, Velasco P, Nguyen L, Vora A, Muzikansky A, Jahnke K, Hauschild A, Hirakawa S, Mihm MC, Detmar M. Tumor lymphangio-genesis predicts melanoma metastasis to sentinel lymph nodes. Mod Pathol. 2005;18:1232–42. doi: 10.1038/modpathol.3800410. [DOI] [PubMed] [Google Scholar]
- 117.Stacker SA, Achen MG, Jussila L, Baldwin ME, Alitalo K. Lymphangiogenesis and cancer metastasis. Nat Rev Cancer. 2002;2:573–83. doi: 10.1038/nrc863. [DOI] [PubMed] [Google Scholar]
- 118.Phillips JM, Goodman JI. Identification of genes that may play critical roles in pheno-barbital (PB)-induced liver tumorigenesis due to altered DNA methylation. Toxicol Sci. 2008;104:86–99. doi: 10.1093/toxsci/kfn063. [DOI] [PubMed] [Google Scholar]
- 119.Kaelin WG. Von Hippel-Lindau disease. Annu Rev Pathol. 2007;2:145–73. doi: 10.1146/annurev.pathol.2.010506.092049. [DOI] [PubMed] [Google Scholar]
- 120.Richards FM, Webster AR, McMahon R, Woodward ER, Rose S, Maher ER. Molecular genetic analysis of von Hippel-Lindau disease. J Intern Med. 1998;243:527–33. doi: 10.1046/j.1365-2796.1998.00334.x. [DOI] [PubMed] [Google Scholar]
- 121.Kim WY, Kaelin WG. Role of VHL gene mutation in human cancer. J Clin Oncol. 2004;22:4991–5004. doi: 10.1200/JCO.2004.05.061. [DOI] [PubMed] [Google Scholar]
- 122.Herman JG, Latif F, Weng Y, Lerman MI, Zbar B, Liu S, Samid D, Duan DS, Gnarra JR, Linehan WM, Baylin SB. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc Natl Acad Sci USA. 1994;91:9700–4. doi: 10.1073/pnas.91.21.9700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Prowse AH, Webster AR, Richards FM, Richard S, Olschwang S, Resche F, Affara NA, Maher ER. Somatic inactivation of the VHL gene in Von Hippel-Lindau disease tumors. Am J Hum Genet. 1997;60:765–71. [PMC free article] [PubMed] [Google Scholar]
- 124.Banks RE, Tirukonda P, Taylor C, Hornigold N, Astuti D, Cohen D, Maher ER, Stanley AJ, Harnden P, Joyce A, Knowles M, Selby PJ. Genetic and epige-netic analysis of von Hippel-Lindau (VHL) gene alterations and relationship with clinical variables in sporadic renal cancer. Cancer Res. 2006;66:2000–11. doi: 10.1158/0008-5472.CAN-05-3074. [DOI] [PubMed] [Google Scholar]
- 125.Chang S, Young BD, Li S, Qi X, Richardson JA, Olson EN. Histone deacetylase 7 maintains vascular integrity by repressing matrix metalloproteinase 10. Cell. 2006;126:321–34. doi: 10.1016/j.cell.2006.05.040. [DOI] [PubMed] [Google Scholar]
- 126.Mottet D, Bellahcene A, Pirotte S, Waltregny D, Deroanne C, Lamour V, Lidereau R, Castronovo V. Histone deacetylase 7 silencing alters endothelial cell migration, a key step in angiogenesis. Circ Res. 2007;101:1237–46. doi: 10.1161/CIRCRESAHA.107.149377. [DOI] [PubMed] [Google Scholar]
- 127.Potente M, Ghaeni L, Baldessari D, Mostoslavsky R, Rossig L, Dequiedt F, Haendeler J, Mione M, Dejana E, Alt FW, Zeiher AM, Dimmeler S. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev. 2007;21:2644–58. doi: 10.1101/gad.435107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kim MS, Kwon HJ, Lee YM, Baek JH, Jang JE, Lee SW, Moon EJ, Kim HS, Lee SK, Chung HY, Kim CW, Kim KW. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat Med. 2001;7:437–43. doi: 10.1038/86507. [DOI] [PubMed] [Google Scholar]
- 129.Hellebrekers DM, Melotte V, Vire E, Langenkamp E, Molema G, Fuks F, Herman JG, Van Criekinge W, Griffioen AW, van Engeland M. Identification of epigenetically silenced genes in tumor endothelial cells. Cancer Res. 2007;67:4138–48. doi: 10.1158/0008-5472.CAN-06-3032. [DOI] [PubMed] [Google Scholar]
- 130.Qian DZ, Wang X, Kachhap SK, Kato Y, Wei Y, Zhang L, Atadja P, Pili R. The his-tone deacetylase inhibitor NVP-LAQ824 inhibits angiogenesis and has a greater antitumor effect in combination with the vascular endothelial growth factor receptor tyrosine kinase inhibitor PTK787/ZK222584. Cancer Res. 2004;64:6626–34. doi: 10.1158/0008-5472.CAN-04-0540. [DOI] [PubMed] [Google Scholar]
- 131.Rossig L, Li H, Fisslthaler B, Urbich C, Fleming I, Forstermann U, Zeiher AM, Dimmeler S. Inhibitors of histone deacety-lation downregulate the expression of endothelial nitric oxide synthase and compromise endothelial cell function in vasorelaxation and angiogenesis. Circ Res. 2002;91:837–44. doi: 10.1161/01.res.0000037983.07158.b1. [DOI] [PubMed] [Google Scholar]
- 132.Qian DZ, Kato Y, Shabbeer S, Wei Y, Verheul HM, Salumbides B, Sanni T, Atadja P, Pili R. Targeting tumor angio-genesis with histone deacetylase inhibitors: the hydroxamic acid derivative LBH589. Clin Cancer Res. 2006;12:634–42. doi: 10.1158/1078-0432.CCR-05-1132. [DOI] [PubMed] [Google Scholar]
- 133.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-tran-scriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9:102–14. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 134.Kuehbacher A, Urbich C, Dimmeler S. Targeting microRNA expression to regulate angiogenesis. Trends Pharmacol Sci. 2008;29:12–5. doi: 10.1016/j.tips.2007.10.014. [DOI] [PubMed] [Google Scholar]
- 135.Blenkiron C, Miska EA. miRNAs in cancer: approaches, aetiology, diagnostics and therapy. Hum Mol Genet. 2007:R106–13. doi: 10.1093/hmg/ddm056. 16 Spec No 1. [DOI] [PubMed] [Google Scholar]
- 136.Yang WJ, Yang DD, Na S, Sandusky GE, Zhang Q, Zhao G. Dicer is required for embryonic angiogenesis during mouse development. J Biol Chem. 2005;280:9330–5. doi: 10.1074/jbc.M413394200. [DOI] [PubMed] [Google Scholar]
- 137.Suarez Y, Fernandez-Hernando C, Pober JS, Sessa WC. Dicer dependent microRNAs regulate gene expression and functions in human endothelial cells. Circ Res. 2007;100:1164–73. doi: 10.1161/01.RES.0000265065.26744.17. [DOI] [PubMed] [Google Scholar]
- 138.Kuehbacher A, Urbich C, Zeiher AM, Dimmeler S. Role of Dicer and Drosha for endothelial microRNA expression and angiogenesis. Circ Res. 2007;101:59–68. doi: 10.1161/CIRCRESAHA.107.153916. [DOI] [PubMed] [Google Scholar]
- 139.Dews M, Homayouni A, Yu D, Murphy D, Sevignani C, Wentzel E, Furth EE, Lee WM, Enders GH, Mendell JT, Thomas-Tikhonenko A. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet. 2006;38:1060–5. doi: 10.1038/ng1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Chen Y, Gorski DH. Regulation of angio-genesis through a microRNA (miR-130a) that down-regulates antiangiogenic home-obox genes GAX and HOXA5. Blood. 2008;111:1217–26. doi: 10.1182/blood-2007-07-104133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Poliseno L, Tuccoli A, Mariani L, Evangelista M, Citti L, Woods K, Mercatanti A, Hammond S, Rainaldi G. MicroRNAs modulate the angiogenic properties of HUVECs. Blood. 2006;108:3068–71. doi: 10.1182/blood-2006-01-012369. [DOI] [PubMed] [Google Scholar]
- 142.Otsuka M, Zheng M, Hayashi M, Lee JD, Yoshino O, Lin S, Han J. Impaired microRNA processing causes corpus luteum insufficiency and infertility in mice. J Clin Invest. 2008;118:1944–54. doi: 10.1172/JCI33680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Hua Z, Lv Q, Ye W, Wong CK, Cai G, Gu D, Ji Y, Zhao C, Wang J, Yang BB, Zhang Y. MiRNA-directed regulation of VEGF and other angiogenic factors under hypoxia. PLoS ONE. 2006;1:e116. doi: 10.1371/journal.pone.0000116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, Richardson JA, Bassel-Duby R, Olson EN. The endothe-lial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev Cell. 2008;15:261–71. doi: 10.1016/j.devcel.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Fish JE, Santoro MM, Morton SU, Yu S, Yeh RF, Wythe JD, Ivey KN, Bruneau BG, Stainier DY, Srivastava D. miR-126 regulates angiogenic signaling and vascular integrity. Dev Cell. 2008;15:272–84. doi: 10.1016/j.devcel.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Miller KD, Chap LI, Holmes FA, Cobleigh MA, Marcom PK, Fehrenbacher L, Dickler M, Overmoyer BA, Reimann JD, Sing AP, Langmuir V, Rugo HS. Randomized phase III trial of capecitabine compared with bevacizumab plus capecitabine in patients with previously treated metastatic breast cancer. J Clin Oncol. 2005;23:792–9. doi: 10.1200/JCO.2005.05.098. [DOI] [PubMed] [Google Scholar]
- 147.Sandler A, Gray R, Perry MC, Brahmer J, Schiller JH, Dowlati A, Lilenbaum R, Johnson DH. Paclitaxel-carboplatin alone or with bevacizumab for non-small-cell lung cancer. N Engl J Med. 2006;355:2542–50. doi: 10.1056/NEJMoa061884. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.