

Abstract
Mesenchymal stem cells (MSCs) from adult somatic tissues may differentiate in vitro and in vivo into multiple mesodermal tissues including bone, cartilage, adipose tissue, tendon, ligament or even muscle. MSCs preferentially home to damaged tissues where they exert their therapeutic potential. A striking feature of the MSCs is their low inherent immunogenicity as they induce little, if any, proliferation of allogeneic lymphocytes and antigen-presenting cells. Instead, MSCs appear to be immunosuppressive in vitro. Their multi-lineage differentiation potential coupled to their immuno-privileged properties is being exploited worldwide for both autologous and allo-geneic cell replacement strategies. Here, we introduce the readers to the biology of MSCs and the mechanisms underlying immune tolerance. We then outline potential cell replacement strategies and clinical applications based on the MSCs immunological properties. Ongoing clinical trials for graft-versus-host-disease, haematopoietic recovery after co-transplantation of MSCs along with haematopoietic stem cells and tissue repair are discussed. Finally, we review the emerging area based on the use of MSCs as a target cell subset for either spontaneous or induced neoplastic transformation and, for modelling non-haematological mesenchymal cancers such as sarcomas.
Keywords: mesenchymal stem cells, immune tolerance, cell replacement therapies, differentiation, neoplastic cell transformation, clinical trials
Introduction
Mechanisms of immunological tolerance
MSCs and cell replacement strategies
-
Clinical applications based on MSCs immune modulatory properties: overview of ongoing clinical trials
- MSC in the HSCT setting
- Clinical use of MSCs in regenerative medicine to facilitate tissue repair
MSCs as a model to study cell transformation and disease
Concluding remarks
Introduction
Mesenchymal stem cells (MSCs) are multi-potent cells present in a variety of tissues during development and therefore can be isolated from several and perhaps most adult tissues, although bone marrow (BM) represents the source most often used. The prevailing consensus is that MSCs have similar biological characteristics regardless of origin, but differences are likely to be reported in the near future. It is now well accepted that MSCs constitute a source of progenitors of mesoderm-derived tissues such as bone, cartilage and fat. These multi-potent MSCs can be isolated based on plastic-adherence properties [1] and expanded in culture relatively easy.
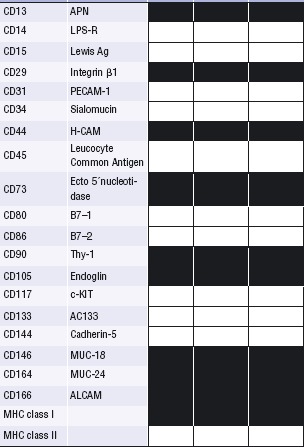
The definition of MSC generated ex vivois a composite of phe-notypical, morphological and functional characteristics, which clearly illustrate that these cells constitute a heterogeneous population. Expanded MSCs homogenously express a number of nonspecific markers, including CD105, CD73, CD166, CD44, CD90, CD271 and CD29, which are shared by cultured mature fibroblasts from any tissue. The most common surface antigens expressed in human MSCs from distinct sources including BM and adipose-tissue as well as mature human fibroblasts are displayed in Table 1. Important efforts are being undertaken by many groups in the field to identify suitable and reliable markers distinguishing MSCs from mature fibroblasts. Similar to haematopoietic stem cells, the absence of a unique specific marker represents a challenge for MSC isolation. MSCs can be isolated by flow cytometry or magnetic cell-sorters using fluorescence- or magnetic-labelled surface antigens. MSC-specific monoclonal antibodies against CD105, CD73, CD271 and CD90 are available directly conjugated to distinct fluorochromes for FACS-isolation or to microbeads for magnetic purification (http://www.miltenyi.com or http://www.stemcell.com). Importantly, MSC isolation may require a positive selection coupled to depletion steps. This is due to the complex immunopheno-type defining MSCs, which are positive for CD73, CD105, CD146, CD271 among other markers but lack expression of CD34, CD45, CD14, HLA-DR, and CD19. Although most, if not all, such markers are highly modulated in culture, other markers such as STRO-1 [2], SSEA-1/CD15 [3], SSEA-4 [4] or CD146 [5] have been proposed to label an in vivo mesenchymal precursor, though it is not clear yet whether they define a subset of MSCs or the ‘master’ very immature MSC. In addition to the phenotype, their differentiation capacity using clonal cultures of MSCs has provided much information about the in vitro heterogeneity of this population. While most of the cells within a given MSC population show a uni- or bipotential capacity of differentiation, there are only a small number of cells exhibiting tripotential differentiation capacity (osteogenesis, chondrogenesis and adipogenesis). These data suggest a possible hierarchical model where the tripotent cells can be considered as early mesenchymal progenitors within a heterogeneous cell culture that displays a sequential loss of lineage potential [6, 7].
Table 1.
Common surface markers used to characterize human MSCs
| Molecule | Alternative name | hMSCs (bone marrow) | hMSCs (fat tissue) | Mature fibroblasts (skin) |
|---|---|---|---|---|
 | ||||
▪ Positive □ Negative
Successful haematopoietic stem cell (HSC)-based therapies have been carried out for almost 50 years. Infusion of high numbers of HSCs is associated with a rapid haematopoietic recovery and low probability of graft failure [8] although it may be linked to an increased incidence of graft-versus-host-disease (GVHD) in an allogeneic setting [9]. Therefore, it is likely that future cell-based therapies will require a tight control of the cell dose to be transplanted in order to achieve a successful and safe outcome. In vitro expanded cells can overcome several problems associated with the ever-growing issue of insufficient stem cell availability. Unlike HSCs, which are prone to differentiation in vitro and therefore difficult to maintain in their stem cell potential, MSCs can be induced to proliferate extensively in vitro while maintaining their undiffer-entiated multi-potent stage.
From a clinical standpoint, MSCs as any other cell therapy products are considered drugs and thereby need to follow the same legal manufacturing requirements (i.e. Good Manufacturing Practice, GMP) if they are to be used into the clinic [10]. To date, most of the ongoing clinical trials using MSCs are developed with autologous cells generated in GMP facilities. Importantly, however, several studies have shown that MSCs are not inherently immunogenic and therefore escape from immune surveillance in vivo[11–13], opening up new promising avenues for the use of MSCs not only for autologous-but also for allogeneic-cell therapies. Despite being costly, the establishment of a stem-cell bank of well-characterized and ‘ready-to-use’ allogeneic MSCs could be an interesting approach towards facilitating the access of investigators and clinicians to this valuable material. Nevertheless, MSCs have different control points, which regulate their life span in vitro, probably resulting in limited cell growth. Accordingly, there is great controversy as to whether MSCs undergo in vitro senescence and/or genetic instability [14–16]. It is worth mentioning that the use of MSCs for clinical purposes will require the biosafety of these primary cells to be carefully investigated through appropriate and sensitive cellular, molecular and genetic tests. Long-term in vitro culture, culture medium conditions, microbiology and virology tests and phenotype should be controlled together with high-resolution molecular analysis and tumourogenesis in vivo assays [17, 18].
The recognition of the therapeutic potential of MSCs is likely the most exciting advance in cell therapy following the widespread use of HSC transplantation (HSCT). The potential clinical use of MSCs in tissue repair mainly involves bone, cartilage and tendon. As discussed below, proof-of-principle for MSC-based cell therapy has already been established for bone, as MSCs are currently being exploited to repair segmental bone defects of critical size in animals [19], to restore healing of non-union long bone fractures in humans (http://www.aastrom.com) or to treat bones of children with osteogenesis imperfecta [20]. Whether MSCs can generate any other tissue in vivo still remains to be elucidated. Due to their immunomodulatory properties, in addition to their regenerative potential, MSCs are currently being explored in other therapeutic approaches outlined in the present review: (i) to improve haematopoietic reconstitution after HSCT and (ii) to overcome GVHD upon allogeneic transplantation [21, 22] (clinical applications summarized in Table 2). Research efforts aimed at identifying factors and/or cell membrane molecules that control MSC fate decision are necessary to be able to determine the real potential of MSC in cell therapy. In this review, we discuss the biological properties of MSCs that render them as promising candidates for basic and clinical applications in cell replacement, tissue engineering, immune-modulation in an allogeneic HSCT setting and, as potential target cells to develop in vitro and in vivodisease models for human mesenchymal cancers.
Table 2.
Clinical applications based on the use of MSCs: advantages and pitfalls
| Potential clinical application | Advantages | Disadvantages/Pitfalls |
|---|---|---|
| Engineering cartilage, bone, muscle, fat and tendon for cell replacement strategies | -Existence of MSCs in multiple tissues - Relatively reproducible differentiation protocols- Low immunogeneicity -Opposite to HSCs, MSCs proliferate extensively in vitro while maintaining their undifferentiated multi-potent stage | -Need to optimize differentiation protocols depending on the source tissue-Biosafety/MSC senescence/transformation upon long-term culture-Homing to the damaged tissue is desired-Lack of a commercial GMP licenced MSC product-Optimal route of MSC delivery must be defined for individual indications-MSC purity and optimal dose remain to be specified |
| Vehicles for gene therapy | -Easy to transfect/transduce-Opposite to HSCs, MSCs proliferate extensively in vitro while maintaining their undifferentiated multi-potent stage | -Absence of clinical data-Confirmation is still required about potential transgene silencing upon differentiation-Risk of insertional mutagenesis |
| Enhance engraftment in HSCT | -MSCs are capable of homing to the BM and survive-MSCs are not rejected due to their low immunogeneicity-Encouraging preliminary pre-clinical/clinical data with low toxicity | -Allo-MSC may mount a T-cell memory response in non-myeloablated hosts-Logistic/timing issues in auto-HSCT-Lack of a commercial GMP licenced MSC product-Optimal route of MSC delivery must be defined for individual indications-MSC purity and optimal dose remain to be specified |
| Diminish GvHD | -Low immunogeneicity-Immunotolerance properties-Encouraging preliminary pre-clinical/clinical data with low toxicity | -Amount, timing and source of MSCs are crucial and must be considered-Multiple but not single MSC infusions may be required-Lack of a commercial GMP licenced MSC product-Optimal route of MSC delivery must be defined for individual-MSC purity and optimal dose remain to be specified indications.-Potentialincreased risk of relapse. |
Mechanisms of immunological tolerance
Immunological tolerance is often defined by the failure of the immune system to mount a response against a specific antigen [23, 24]. Although this conventional definition is useful from an experimental point of view, it is not always operational in clinical situations because under some circumstances, the immune system can recognize a specific antigen without mounting a clinically significant response causing tissue injury or graft rejection. For example, immune deviation towards a certain profile of cytokine production can protect from a pathogenic response mediated by an opposing profile of cytokine production, preventing an undesirable immune response [25]. In those latter situations, one may talk as clinical tolerance. The mechanisms of immune tolerance reviewed in this section are mostly mediating conventional tolerance but some may be at work also in clinical tolerance.
Immunological tolerance has been traditionally divided in central tolerance or peripheral tolerance depending on whether the operating mechanisms take place mostly on the primary lymphoid organs or in the periphery, in secondary lymphoid organs or ‘target’ tissues. Three main mechanisms (not mutually exclusive) are at work in the induction of central tolerance. One is clonal deletion. This is based on negative selection through the induction of apop-tosis of autoreactive clones during their development in the BM (for B cells) or in the thymus (for T cells) [26]. Another mechanism of central tolerance is the induction of a state of anergy, defined by the inability of B-cell or T-cell clones to mount a full response to a specific antigen upon rechallenge. The induction of anergy for immature B cells has been linked to the ligation of B-cell antigen receptors (a membrane-bound immunoglobulin) by soluble antigens or monovalent (non-cross-linking) antigens [27, 28]. For developing thymocytes, the molecular mechanism of anergy induction is unclear but may be linked to signalling through the antigen receptor (TCR) in a partial agonist mode [29–31]. A third mechanism that may lead to a state of clinical tolerance and is operational in the thymus is the development of antigen-specific, ‘natural’ regulatory T cells (Tregs) that have the capacity to suppress the development of immune responses in the periphery [32]. These cells are positively selected in the thymus and are characterized by the expression of transcription factor FoxP3. Most of these cells express CD4 and CD25 as well.
The induction of tolerance in the BM or the thymus is a highly efficient process. However, it does not guarantee the elimination of all the autoreactive clones of T or B lymphocytes. Additional mechanisms to prevent autoreactivity are operational in the periphery. The cellular and molecular basis of some of these mechanisms is the same as the one for mechanisms of central tolerance. For example, deletion of potential autoreactive T-cell clones can occur as a result of activation-induced cell death upon encounter with parenchymal cells that express ligands for death receptors (e.g. FasL for Fas) [33] or upon recirculation into the thymus of activated T cells [34, 35] or by the activity of veto cells that kill autoreactive cytotoxic T cells [36]. Anergy, particularly T-cell anergy, has also been linked to tolerance by peripheral mechanisms, mostly secondary to situations in which T cells recognize antigen in the absence of ligands (e.g. B7 family of molecules) for costimulatory receptors (e.g. CD28), TCR signalling in partial agonist mode, or after engagement of CTLA [37–41]. Administration of progenitors or immature dendritic cells may contribute to the development of peripheral tolerance in part through this mechanism [42]. At the biochemical level, the induction of T-cell anergy correlates with impairment of early TCR-dependent signalling along the ras-dependent signalling [43, 44].
Of increasing importance is the key role that Tregs play in the down-regulation of immune responses, and that may be critical for the maintenance of a state of clinical tolerance to allografts. In addition to thymic generation, Tregs can be induced in the periphery from CD4+ CD25−T cells, these Tregs being known as adaptive Tregs. These cells are heterogeneous in phenotype, with regulatory activity having been assigned to CD4−CD8− T cells and T cells producing high amounts of TGF-β and IL-10 (Tr1) [45–47]. The specific mechanism of down-regulation immune responses by Tregs remains unclear. For some of these subsets, cell-to-cell contact is required; however, under some conditions, regulatory activity may be secondary to the production of cytokines as IL-10 or TGF-β.
In recent years, there has been a re-emerging interest in the role that chimerism either macrochimerism or microchimerism may play in the generation of an immune tolerant state. In solid organ transplant recipients, such a state has likely emerged as a result of expansion of ‘passenger’ leukocytes of donor origin in the recipient [48]. The mechanisms of tolerance induction in chimerism may operate centrally in the thymus or BM, and in the periphery. Recent analysis of transplant recipients with long-term functional grafts and that have stopped immunosuppressive therapy have been useful to provide an emerging framework for chimerism. Under most conditions, these involve the administration of limited rounds of treatment with immunosuppressive drugs, including a pre-transplant period, to allow the cross-toler-ization of both immune systems. The mechanism that are operational under these circumstances are mostly deletional mechanisms, and stress the importance of dose, timing and targets of immunosuppressive therapies as variables for future optimization of tolerogenic protocols.
In the context of the mechanisms outlined above, administration of MSCs and HSCs stands as an attractive therapeutic candidate for the induction of immunological tolerance. It is of interest to note that, historically, the tolerogenic capacity of HSCs may have been operational in situations in which blood transfusions, and donor-specific blood transfusions in particular for living related kidney recipients, have been used [49, 50]. This strategy had a beneficial effect on graft survival (when not inducing sensitization of the recipient). The advances in our understanding of the tolerogenic mechanism lead us to consider several possibilities for the immunomodulatory and tolerogenic properties of MSCs [51–53]. First, one may consider the non-immunogenicity of MSCs due to the lack of expression, before differentiation, of MHC molecules (class II) on their surface. Of note, despite MHC class II molecules are commonly absent on the surface of MSCs, MSCs have been shown to contain intracellular deposits of MHC II alloantigens detectable at the protein level by Western Blot [54]. Second, MSCs may potentially induce T-cell anergy due to the lack of ligands for CD28 and other costimulatory molecules on their plasma membrane leading to a state of immunological ignorance. Third, stem cells may differentiate and facilitate the establishment of chimerism through thymic and extrathymic deletion of autoreactive T-cell clones or the induction of Treg subsets (either the conventional CD4+CD25+ T cells, or other cell subsets producing TGF-β and/or IL-10) that down-regulate effectors T-cell responses.
MSCs and cell replacement strategies
MSCs exhibit great potential for cell replacement strategies. This potential relies on three key properties: (i) their capacity to differentiate into several cell lineages; (ii) their immune-modulatory properties and (iii) their ability to secrete soluble factors which regulate crucial biological functions such as proliferation and differentiation over a broad spectrum of target cells. An important consideration is the fact that their immunomodulatory capacity facilitates allogeneic cell replacement therapies regardless the degree of HLA match between donor and recipient.
Initially, MSCs were assessed in vivo in cell replacement strategies by transplanting MSCs directly to the injured sites. Recently however, alternative strategies typically involve the generation of an engineered construct by seeding biocompatible scaffolds with these MSCs [55]. Moreover, current gene delivery methods offer the possibility of genetic modification of MSCs within these scaffolds to secrete the specific soluble signalling molecules expected to contribute to a specific tissue repair [56]. The MSCs incorporated into the construct will require a functional vasculature to receive the metabolic demands for survival, proliferation and differentiation. An alternative strategy for this type of tissue engineering would rely on the development of an in vitro vascularized scaffold, which is then seeded with MSCs [57].
Alternatively, successful cell replacement therapies might be achieved by harnessing the important intrinsic biological features of MSCs, which are capable of homing in vivo to sites of tissue injury, primarily as a result of local production of inflammatory mediators during tissue damage. If the MSCs are able to home to the damaged tissue and engraft there, they could be delivered intravenously. This strategy would be especially interesting in those scenarios where the damaged tissue is difficult to access (i.e. central nervous system) or when there are several systemic organs implicated (i.e. tumour metastasis) [58, 59]. In fact, this strategy has been widely used in patients with genetic disorders where transplantation of allogeneic MSCs resulted in partial response of the clinical manifestations in patients with Hurler syndrome, metachromatic leukodystrophy or osteogenesis imper-fecta [20, 60].
Among the different tissue candidates to take advantage of potential MSC-based regeneration, bone is probably at the forefront of current cell replacement and tissue repair applications. This may be, at least in part, due to better understanding of the cellular and molecular mechanisms implicated in MSC-mediated osteogenesis. MSCs may differentiate relatively easy in vitro into osteoblasts by treatment with dexamethasone that stimulates MSC proliferation and promotes osteogenic lineage differentiation. Other supplements such as ascorbic acid phosphate and 1, 25-dihydroxyvitamin D3 and members of the bone morphogenetic protein (BMP) family of growth factors are also commonly used for osteogenic induction from MSCs [61]. As aforementioned, the use of natural and synthetic biomaterials (mainly porous ceramics of hydroxyapatite and β-tricalcium phosphate) as carriers for MSC delivery holds the promise for orthopaedic therapeutic applications [55].
In addition to osteogenic differentiation capacity and their potential in bone regeneration, MSCs might also be used for restoration of function in individuals suffering from cartilage damage. The in vitro induction of chondrogenesis seems to depend on coordinated activities of many growth factors as well as cell density and adhesion. The transforming growth factor-β (TGFβ) superfamily of proteins and the BMPs are well-established regulatory factors in chondrogenesis [62]. Although TGF-ß1 was initially used for in vitro induction of chondrogenesis from MSCs, TGF-β3 has recently been shown to induce a more rapid and thorough expression of chondrogenic markers [62]. Interestingly, autolo-gous MSCs have been used in vivo dispersed in gels of collagen-type-I or alginate in order to enhance the chondrogenic differentiation for repairing defects on femoral condyles [63, 64]. Despite the very little information, the potential of MSC differentiation into other connective tissues, such as tendons and ligaments is currently being investigated.
Myogenic differentiation of MSCs is currently being applied to cardiac and skeletal muscle [65, 66]. MSCs have been used in myocardial infarction, resulting in improved remodelling and reduction of damage size. MSCs injected in the damaged myocardium disappear over time and there was no evidence of differentiation of injected MSCs into cardiomyocytes. Strikingly, however, there is a growing body of evidence suggesting that MSCs stimulate the repair of damaged tissue through endogenous mechanisms [65]. However, compared to the osteoblastic and chondrocytic lineages, very little is known about the signalling pathways and molecular determinants involved in myogenic differentiation of MSCs. Consequently, the clinical benefits of MSC-based cell therapies for damaged cardiac and skeletal muscle are still in their infancy [66].
Differentiation of MSCs into tissues derived from a germ layer other than mesoderm has also been reported. Thus, MSCs have been differentiated into neurons and glia, opening up new avenues to study tropism toward neuroectoderm tissues and long-term future cell replacement strategies for neurodegenerative diseases [67]. Recent data indicate that delivery of soluble factors directly into the tissue is likely to be the main mechanism of action [67]. In this regard, early intervention tends to be more effective because MSCs exert a paracrine influence during active tissue inflammation and therefore MSC grafts are much less effective once acute injury becomes chronic. A similar situation seems to occur when MSCs enhance wound healing. In this scenario, MSCs enhance migration of macrophages, keratinocytes and endothelial cells to the damaged tissue, inducing their proliferation and accelerating wound closure [68]. Worth mentioning, special attention should be paid to the differentiation of MSCs to unwanted tissues, which may also represent a serious concern as recently reported [69]. These authors reported that the developmental fate of BM-derived MSCs is not restricted by the surrounding tissue after myocardial infarction and that the MSC fraction may induce unde-sired bone formation in the infarcted myocardium. These findings question the biologic basis and clinical safety of using whole BM and in particular MSCs to treat non-haematopoietic disorders.
Premature clinical use of MSC-based therapies may put patients at risk, especially since this research field lacks robust preclinical data using suitable animal models. Therefore, despite the promise that MSCs hold in cell replacement strategies, caution is required. Essential precautions need to be taken and biosafety issues need to be addressed before designing prospective clinical trials [70]. Our suggestion would be to invest resources, time and scientific efforts in gaining fundamental knowledge about the molecular and cellular mechanisms underlying MSC isolation, maintenance and in vitro and in vivoproliferation and differentiation before moving from the bench to the bedside [16, 70]. The main advantages and disadvantages linked to the use of MSCs in the clinic are summarized in Table 2.
Clinical applications based on MSCs immune modulatory properties: overview of ongoing clinical trials
As described above, MSCs are multi-potent non-haematopoietic progenitor cells capable of differentiating into various mesenchy-mal lineages, being able to migrate to damaged tissues and favour tissue repair. Biologically, MSCs release cytokines and growth factors supporting the differentiation of the HSCs, which is based on an interaction between MSCs and haematopoietic cells through signalling pathways such as Notch, Tie2 and the axis CXCR4-CXCL12 [71–74]. From an immunological point of view, the inhibition of T-cell responses by MSCs correlates with cyclin D2 inhibition [75] and a p27kip1 up-regulation [76]. Both contact dependent and independent mechanisms have been involved. They also block the proliferation and differentiation of B lymphocytes, in an extracellular response kinase (ERK) and p38 MAP kinase pathway-dependent manner [77]. They also reduce the expression of co-stimulatory molecules and down-regulate IL12 secretion in mature antigen presenting cells [78, 79].
The immune-privileged properties attributed to MSCs make them a powerful tool that could be used in many inflammatory or autoimmune diseases. They are currently being explored within several clinical trials, which are summarized below. Most clinical trials are currently underway or in a participants' recruitment phase. The website http://www.clinicaltrial.gov displays a registry of federally and privately supported clinical trials conducted not only in the United States but also worldwide. As reported in this database, the clinical trials based on the immune modulatory properties of MSCs can be divided in five groups:
Immune protection against rejection after solid organ or HSCs transplantation. Examples are: (i) co-transplantation of MSCs along with pancreatic islets for type 1 diabetic patients (Fuzhou General Hospital, China) in order to protect the transplanted islets from inflammatory damage, thus facilitating islet engraft-ment and (ii) MSCs transplantation in recipients who have undergone a kidney transplant in order to suppress immune rejection and improve donor kidney survival (Fuzhou General Hospital, China).
Treatment of chronic inflammatory diseases. For instance, MSCs transplantation in decompensate cirrhosis (University of Tehran, Iran) or chronic allograft nephropathy (Fuzhou General Hospital, China).
Treatment of autoimmune diseases. Immunological properties of MSCs are being currently harnessed in multiple sclerosis clinical trials (University of Cambridge, UK) and in Lupus Nephritis clinical trials (Organ Transplant Institute, China).
Treatment of diseases of unknown aetiology but with a strong inflammatory response. MSCs are being used in Crohn's disease to decrease inflammation (Osiris Therapeutics) and in the treatment of moderate to severe Chronic Obstructive Pulmonary Disease (Osiris Therapeutics).
Treatment of GVHD after an allogeneic HSC transplantation (HSCT). Allogeneic HSC transplant is a well-established treatment for several haematopoietic and non-haematopoietic diseases. Recipients receiving an allogeneic HSCT are immunosuppressed in order to prevent rejection of the transfused HSCs. GVHD is induced by T cells either transplanted or generated from infused donor HSCs, which react with donor tissue. Because of their immunoregulatory capacity, ex vivoexpanded MSCs are being used in clinical trials to treat severe GVHD [80]. To date, GVHD is probably the more susceptible disease prone to be treated with MSCs. In fact, many multi-centre randomized trials have been started. Preliminary results about the use of MSCs in GVHD are discussed below in detail.
MSC in the HSCT setting
Allogeneic HSCT is a potentially curative treatment option for many patients diagnosed with haematologic malignancies. Its efficacy is based both on the high doses of chemotherapy and radiotherapy administered prior to transplantation and on the immune effect of T lymphocytes from the graft which may recognize and exert a cytotoxic effect against tumour cells from the host inducing a graft-versus-leukaemia (GVL) effect. An important clinical observation supporting the proof of principle of this anti-tumour effect is the fact that patients who relapse after allogeneic transplantation may reach complete remission after donor lymphocyte infusions [81]. Unfortunately, allogeneic T cells present in the graft also recognize normal host tissues, causing severe tissue damage in target organs such as skin, liver and gut, being the underlying mechanism for the development of GVHD [82, 83], which remains one of the most severe complications after allogeneic transplantation. The frequency of GVHD is around 30% and 80% among recipients of HLA-matched sibling or unrelated donor transplants, respectively. Steroids are still the first-line treatment for established GVHD with a response rate of 30–50%. Unfortunately, the outcome for patients with severe, steroid-resistant acute GVHD is very poor, and the overall survival is low [82, 83].
Based on their immune modulatory properties, MSCs have been used as a promising approach for the treatment of GVHD [84]. Le Blanc K et al. [13] transplanted haploidentical MSCs to a patient with severe treatment-resistant grade IV acute GVHD of the gut and liver. The clinical response was striking with normalization of stool and bilirubin on two separated occasions. Subsequently the same authors reported their experience on eight patients with steroid refractory grades III–IV acute GVHD and one patient with extensive chronic GVHD treated with MSCs [85]. Strikingly, acute GVHD disappeared completely in 75% of patients [85]. These studies have been further extended in Europe with 55 patients enrolled [86]. These patients have been treated for steroid-resistant acute GVHD who had previously failed to first line treatment (n= 55), second line (n= 33), third line (n= 14) or fourth or more line treatment (n= 6). The median dose of bone-marrow derived MSCs was 1.4 × 106 cells per kg bodyweight. Twenty-seven patients received one dose, 22 patients received two doses, and 6 patients received three to five doses of MSCs obtained from HLA-identical sibling donors (n= 5), haploidentical donors (n= 18) or third-party HLA-mismatched donors (n= 69). The overall response rate was 71% with 30 patients achieving a complete response and 9 patients a significant GVHD clinical improvement. Median time from first MSC infusion to clinical response was 18 days [86]. Interestingly, the HLA match of the allogeneic MSCs and the donor had no effect on response rate. No patients had side effects during or immediately after infusions of MSCs. Complete responders had lower transplantation-related mortality 1 year after infusion than did patients with partial or no response (37% versus 72%) and higher overall survival 2 years after HSCT (53% versus 16%) [16]. MSCs may be obtained from several sources in addition to BM [87]. Subsequently, in a series of 6 patients with steroid-resistant acute GVHD treated with adipose-tissue-derived MSCs complete response was documented in 83% of patients. Median dose of infused MSCs was 1 × 106/kg and no side effects were observed after the infusion [88]. Therefore, although the experience is limited, MSCs seem a promising alternative approach to treat severe steroid-resistant acute GVHD [86–89].
Because MSCs are immunosuppressive and may alleviate GVHD, it is unknown whether MSCs will increase the risk of leukaemic relapse by abrogating the GVL effect. In a prospective randomized trial, HLA-identical sibling-matched HSCs were transplanted alone (non-MSC group, n= 15) or co-transplanted with MSCs (MSC group, n= 10) in patients suffering from haematopoietic malignancies. The median number of MSCs infused was 3.4 × 105/kg. MSC infusions were well tolerated and the median time to neutrophil engraftment was 16 days for MSC group and 15 days for non-MSC group. The median time to platelet engraftment was 30 and 27 days, respectively. Grades II–IV acute GVHD was observed in 10% and 53.3% of patients from the MSC and non-MSC groups, respectively. Similarly, chronic GVHD was found in 14% and 29% of patients from the non-MSC and MSC groups, respectively. However, the proportion of patients who relapsed was 60% and 20%, and the 3-year disease-free survival was 30% and 67% in the MSC and non-MSC groups, respectively [89]. According to this study, co-transplantation of MSCs and HSCs may have prevented GVHD, but the relapse rate was significantly higher in the group where MSCs were co-infused along with HSCs. These results clearly indicate that MSCs should be handled with extreme caution before a large-scale prospective clinical trial is undertaken. However, as pointed out by Vianello et al. [90], there is an aspect that does not entirely fit with the data, that is, the absence of differences in the number of infectious events in the group of patients treated with MSCs, since a non-selective immunosuppression would have equally affected patient's antiviral immunity. Therefore, alternative mechanisms may be involved in the clinical consequences of MSC transplantation and, in this regard recent evidence supports the capability of the haematopoietic milieu to be the sole causative mechanism of cancer development [73, 91]. Accordingly, further investigations on MSC homing may help to clarify the mechanisms responsible for the clinical outcome. In this regard, 46 patients underwent co-transplantation of in vitro-expanded MSCs plus HSCs from their HLA-identical sibling donors [92]. Moderate to severe acute GVHD was observed in 28% of patients. Chronic GVHD was observed in 61% of patients and 2-year progression-free survival was 53%. No MSC-associated toxicities were reported. Similar to the percentage reported in previous series, stromal cell chimerism was demonstrated in 2 of 18 examined patients after transplantation [93]. These studies provide insights into the difficulties in detecting MSCs after infusion, even in immunocompromised patients who have undergone an HSCT. As far as the use of MSC in the GVHD prophylaxis or treatment is concerned, there are several clinically relevant questions that need to be addressed. What are the optimal timing, cell dose and additional immunosuppressive therapy to be used? Might the use of MSCs be better indicated for a specific disease or specific cohort of patients? Double-blind, randomized studies using MSC versus placebo have started in Europe and in the USA in patients with steroid-resistant acute GVHD.
In addition to their immunosuppressive effect, MSCs enhance haematopoietic engraftment and several clinical trials have been devoted to confirm this potential. In the previously mentioned study from Lazarus et al.[92], median days to reach >500/mm3 granulocytes and >20,000 platelets/mm3 were 14 and 20 days after transplantation, respectively. In this regard, haploidentical HSCT is associated with an increased risk of graft failure. In order to enhance haematopoietic recovery, MSCs were co-transplanted in 14 children undergoing transplantation of CD34+ HSCs from a haploidentical donor. While a 15% haematopoietic graft failure rate was observed in 47 historical controls, all patients given MSCs showed sustained haematopoietic engraftment, without any adverse reaction. Furthermore, children given MSCs experienced fewer infections than controls [94]. These data suggest that MSCs reduce the risk of graft failure in patients undergoing a haploidentical HSCT. Finally, in the autologous HSCT setting, 28 patients diagnosed with breast cancer received autologous MSCs in addition to HSC. Clonogenic MSCs were detected in peripheral blood up to 1 hr after infusion in 62% of patients. Median time to achieve >500 granulocytes/mm3 and >20,000 platelets/mm3 was 8 and 8.5 days, respectively, suggesting that infusion of MSCs after myeloablative conditioning seems to have a positive impact on haematopoiesis [21].
Clinical use of MSCs in regenerative medicine to facilitate tissue repair
Since MSCs are able to migrate to damaged tissues, this property can be exploited in regenerative medicine and tissue repair. Ten patients undergoing allogeneic HSCT were administered MSCs due to tissue toxicity. Five patients with severe haemorrhagic cystitis cleared after MSC infusion. Two patients were treated for pneumo-mediastinum, which also disappeared after MSC infusion and a patient with steroid-resistant GVHD of the gut experienced perforated diverticulitis and peritonitis that was also reversed after infusion of MSCs [95]. These preliminary data suggest that MSCs may also play a role in repairing HSCT-associated severe tissue toxicity.
Besides HSCT, clinical trials are underway to evaluate the potential use of MSCs in repairing damaged or lost tissue in osteoarthritis, steroid-induced avascular osteonecrosis, anal fistula and intervertebral discs [96–98]. Although these pre-clinical and clinical trials are still very preliminary, the results are promising and therefore efforts in this cell therapy field should be continued [96–98]. In conclusion, the use of MSCs in cellular therapy is already being explored in several settings, especially in HSCT with three goals: (i) to enhance HSC engraftment, (ii) to treat/alleviate GVHD and (iii) to promote tissue repair.
MSCs as a model to study cell transformation and disease
In agreement with the cancer stem cell theory [99], MSCs might constitute a target cell for some transforming mutations and chromosomal abnormalities, which may arise in an MSC or mesodermal precursor, giving rise to non-haematopoietic mesenchymal tumours such as sarcomas. For instance, it has been shown that the expression of FUS-DDIT3, a fusion oncoprotein previously involved in the development of myxoid liposarcoma [100], in mouse is capable of transforming MSCs recapitulating in vivothe development of such a tumour [101]. Likewise, Ewing's sarcoma has been reproduced in vivoby expression of EWS-FLI-1 in MSCs [102, 103]. In another work, the expression of EWS-FLI fusion protein in human MSCs induced a pattern of gene expression similar to that observed in human Ewing's sarcoma [104]. Different types of sarcomas can also be generated from transformed MSCs. This is the case of the malignant fibrous histiocytoma derived from MSCs inactivated for Wnt signalling [105]. Recently, a classification of liposarcomas based on the differentiation state of the MSCs that give rise to the tumours has been suggested [106]. Other groups have reported in vitro MSC transformation after ectopic expression of the telomerase hTERT [107] or the oncogenic H-ras [108] genes. It was also described that BM-derived cells (most likely MSCs) chemically transformed using 3methy-cholanthrene could form many types of tumours [109]. Together, these data further link cancer to development and support the idea that MSCs could play a relevant role and become an instrumental tool in studies about the aetiology and pathogenesis of sarcomas.
One should consider, though, that the role of MSCs in tumourogenesis could be an indirect phenomenon. MSCs are frequently recruited to the site of tissue injury and sometimes, in the appropriate and permissive environment and under stress conditions, this could also represent a potential source of malignancy. It was described that BM-derived MSCs recruited after infection with H. pilori could be the origin of gastric cancers [110]. Likewise, the presence of MSCs in the tumour stroma could facilitate breast cancer metastasis [111] or the growth of syngeneic tumours [112, 113] due, in part, to the high migration and mobility associated to a mesenchymal/fibroblastoid phenotype. Moreover, BM-MSCs seem to create in many cases an optimal microenvironment for the development of several tumours such as multiple myeloma [91, 114] or acute lymphoblastic leukaemia [115]. Activation of anti-apoptotic pathways seems to play a central role in this regard since BM-derived MSC are resistant to chemotherapy-induced apoptosis [116] and also contribute to generate drug resistance in tumour cells [115, 117].
It is worth mentioning that in addition to these MSC-based cell transformation models, human MSCs may transform spontaneously in vitro at higher frequency than other primary human stem cells. Human adipose tissue-derived MSCs have been shown to undergo spontaneous transformation after long-term in vitro culture [16]. These transformed MSCs can form carcinomas in mice through an unusual mesenchymal to epithelial transition [118]. It was also reported the outgrowth of a transformed population derived from cultured human BM derived-MSC [119]. The same phenomenon was also observed in murine BM-derived MSC [15, 120, 121]. Nevertheless, some controversy has recently arisen in the field, since Bernardo et al.[14] reported a lack of MSC transformation after extensive in vitro culture. Similarly, another study showed that despite MSCs from both human and rhesus macaques show cell cycle alterations after extended in vitro culture, only the monkey MSCs display chromosomal alterations and both types of MSCs fail to generate tumours in vivo[122]. The phenomenon of spontaneous transformation of MSCs has been very recently described for the first time and due to the controversy further investigation efforts are still required to ascertain whether or not MSC transformation is inherent to MSC-specific intrinsic properties or is an indirect effect of underlying confounding mechanisms or specific pathological conditions.
In contrast to the susceptibility of MSCs to transformation, there are studies claiming a potential role of these cells in anti-cancer therapy. Human MSCs can home to tumour sites and inhibit the growth of neoplastic cells as has been shown in animal models of glioma [123], Kaposi's sarcoma [124] or hepatoma [125, 126]. Due to the ability of MSCs to migrate and seed tumours, they have also been used as vehicles to deliver oncolytic viruses into tumours and metastasis [127, 128, and García-Castro J et al. (submitted)]. MSCs over-expressing the apoptotic inducer TRAIL [129] or IFN-β[58] were able to inhibit tumour growth in a lung cancer and pulmonary metastases models, respectively, supporting the potential of MSCs as anti-cancer therapeutic option to inhibit the growth of cancer cells. All of these studies reveal the importance of having in place appropriate biosafety, standardization and quality controls before translating MSC application into the clinic. The genetic stability of MSCs in the ex vivo culture process represents a key issue and the absence of transformation should be documented before infusion of MSCs into patients.
Despite the above evidence of MSC transformation, little is still known about its mechanistic basis. MSC transformation has often been linked to the accumulation of chromosome instability [15, 16, 119, 121]. This observation, together with the high resistance of MSCs to apoptosis [116], supports the relevance of an accurate cell cycle control in MSCs. In fact, alterations in cell cycle regulators such as p16INK4A[16, 108] or p53 [16, 120], have been detected in transformed MSCs. Furthermore, over-expression of c-myc [15, 16] and the acquisition of telomerase activity [16, 107, 119] are other common events associated to the transformation process. Characterization of small but biologically relevant molecular changes using high-resolution technologies including microarrays or spectral kariotyping displayed alterations in the expression of genes involved in cell cycle regulation, protein-ubiq-uitination and apoptosis after extended culture in vitro[16, 122]. A two sequential step model of MSC transformation has been proposed [130]. In the first step, cells would bypass senescence by up-regulation of c-myc and repression of p16INK4A. In a second step, cells would bypass senescence by gaining telomerase activity, Ink4a/Arf locus deletion and Rb hyperphosphorilation. These changes would be accompanied with alterations in other cell cycle regulators (CDK-1, 2 and 6) and DNA damage repair proteins (Rad51, XRCC4, etc.) [130].
Finally, the role of Wnt signalling, a key developmental signalling pathway, in MSC transformation requires some discussion. Wnt seems important in tumourogenesis induced by MSCs. The canonical Wnt/β-catenin signalling pathway plays a central role in modulating the balance between self-renewal and differentiation in stem cells [131]. In addition, Wnt signalling also regulates the invasion capacity and the proliferation of human MSC [132, 133]. Wnt signalling is also involved in the epithelial-to-mesenchymal transition. While these properties exerted by the Wnt pathway may be useful in tissue regeneration, an inadequate activation of this pathway may deregulate the balance between proliferation, differentiation and apoptosis and, most importantly the epithelial-to-mesenchymal transition seen in many carcinomas and strongly related with malignant behaviour [134], leading therefore to a malignant transformation [91, 105, 134].
In summary, in line with the well-established link between development, stem cells and cancer, MSCs are being exploited as a target cell for the origin of mesenchymal tumours, especially sarcomas. In addition, whether MSCs and stroma cells form part of the neoplastic clone and their potential contribution to the tumour microenvironment is under active investigation. Nevertheless, very little is still known about the mechanisms of MSC transformation and more research efforts are required to find target-specific therapies against these mesenchymal tumours. Further studies on MSC spontaneous transformation will also be of great value to ensure the quality and biosafety of MSCs in clinical trials.
Concluding remarks
MSCs exist in BM and other tissues and are one of the most promising adult stem cell types because of their availability and the relatively simple requirements for in vitro expansion. These cells contribute to the homeostasis of mesenchymal tissues and support the growth and differentiation of HSCs. A striking feature of the MSCs is their low inherent immunogenicity. Instead, MSCs appear to be immunosuppressive in vitro. Their multi-lineage differentiation potential coupled to their immuno-privileged properties is being exploited for both autologous and allogeneic cell therapies, tissue engineering and regenerative medicine. Additionally, the rapid evolution of experimental data has acknowledged the critical relevance of MSCs as the target cell for either spontaneous or induced cell transformation representing a potential platform for modelling non-haematological mesenchymal cancers such as sarcomas. In this review, we have briefly outlined the current status of MSCs, focusing on their biological characteristics and potential for clinical applications.
Stem cell therapy is rapidly developing and has generated excitement and promise as well as confusion and at times contradictory results in the lay and scientific literature. Despite the great promise held by MSCs and although several clinical trials are underway for a variety of malignant disorders, HSCT or regeneration of damaged tissues, it must be recognized that MSCs are poorly defined by a combination of physical, phenotypical and functional properties. As MSCs could be derived from different tissues as starting material, by diverse isolation protocols, cultured and expanded in different media and conditions, the MSC preparations from different laboratories are highly heterogeneous. All of these variables might have implications on (i) the selection of cell types and the composition of heterogeneous subpopulations, (ii) they can selectively favour expansion of different cell populations with totally different potentials and (iii) they might alter the long-term fate of adult stem cells upon in vitro culture. The recent controversy on the multi-lineage differentiation potentials of some specific MSC preparations might be attributed to this lack of common standards. In addition, the most appropriate source of MSCs, method of isolation and expansion, dose to be infused, timing and route of delivery, and adverse biosafety effects like the potential of MSCs to undergo spontaneous transformation still need to be determined. More precise genetic stability studies, molecular and cellular markers to define subsets of MSCs and the standardization of the protocols for expansion of MSC are urgently needed. Also, a recent study [135] suggest that MSCs may not be as immunoprivileged as initially thought and therefore further ‘in depth’ insights about the immunological mechanisms underlying the immunosuppressive effects of MSCs are still in high demand.
Acknowledgments
This work was funded by the Consejería de Salud de la Junta de Andalucía (grants # 0027/2006 to J. G-C, 0028/2006 and 0029/2006 to PM and 0108/2007 to R.R), The International Jose Carreras Foundation against the Leukemia to PM (EDThomas-05), The Junta de Castilla y León to JAPS (SACYL 2004 and HUS04A07), The Spanish Ministry of Health to PM (FIS PI070026) and to J.G-C (FIS PI052217) and Comunidad de Madrid MesenCAM Network (S-BIO-02042006). Research in the Madrenas Laboratory is funded by the Canadian Institutes of Health Research, the Kidney Foundation of Canada, and the Multi-Organ Transplant program at the London Health Sciences Centre. We apologize for the limitations in referencing all the authors that have contributed to this research area.
References
- 1.Friedenstein AJ, Piatetzky S, Petrakova KV. Osteogenesis in transplants of BM cells. J Embryol Exp Morphol. 1966;16:381–90. [PubMed] [Google Scholar]
- 2.Simmons PJ, Torok-Storb B. Identification of stromal cell precursors in human BM by a novel monoclonal antibody, STRO-1. Blood. 1991;78:55–62. [PubMed] [Google Scholar]
- 3.Anjos-Afonso F, Bonnet D. Nonhemato-poietic/endothelial SSEA-1 + cells define the most primitive progenitors in the adult murine BM mesenchymal compartment. Blood. 2007;109:1298–306. doi: 10.1182/blood-2006-06-030551. [DOI] [PubMed] [Google Scholar]
- 4.Gang EJ, Bosnakovski D, Figueiredo CA, Visser JW, Perlingeiro RC. SSEA-4 identifies mesenchymal stem cells from BM. Blood. 2007;109:1743–51. doi: 10.1182/blood-2005-11-010504. [DOI] [PubMed] [Google Scholar]
- 5.Sacchetti B, Funari A, Michienzi S, Di Cesare S, Piersanti S, Saggio I, Tagliafico E, Ferrari S, Robey PG, Riminucci M, Bianco P. Self-renewing osteoprogenitors in BM sinusoids can organize a hematopoietic microenvironment. Cell. 2007;131:324–36. doi: 10.1016/j.cell.2007.08.025. [DOI] [PubMed] [Google Scholar]
- 6.Digirolamo CM, Stokes D, Colter D, Phinney DG, Class R, Prockop DJ. Propagation and senescence of human marrow stromal cells in culture: a simple colony-forming assay identifies samples with the greatest potential to propagate and differentiate. Br J Haematol. 1999;107:275–81. doi: 10.1046/j.1365-2141.1999.01715.x. [DOI] [PubMed] [Google Scholar]
- 7.Muraglia A, Cancedda R, Quarto R. Clonal mesenchymal progenitors from human BM differentiate in vitro according to a hierarchical model. J Cell Sci. 2000;113:1161–6. doi: 10.1242/jcs.113.7.1161. [DOI] [PubMed] [Google Scholar]
- 8.Siena S, Schiavo R, Pedrazzoli P, Carlo-Stella C. Therapeutic relevance of CD34 cell dose in blood cell transplantation for cancer therapy. J Clin Oncol. 2000;18:1360–77. doi: 10.1200/JCO.2000.18.6.1360. [DOI] [PubMed] [Google Scholar]
- 9.Menendez P, Perez-Simon JA, Mateos MV, Caballero MD, Gonzalez M, San Miguel JF, Orfao A. Influence of the different CD34 + and CD34-cell subsets infused on clinical outcome after non-mye-loablative allogeneic peripheral blood transplantation from human leucocyte antigen-identical sibling donors. Br. J. Haematol. 2002;119:135–43. doi: 10.1046/j.1365-2141.2002.03794.x. [DOI] [PubMed] [Google Scholar]
- 10.Fernández-Miguel G, Bovy T. Production of cell-based medicines for somatic cell therapy. In: Garcia-Olmo D, García-Verdugo JM, Alemany J, Gutiérrez-Funetes JA, editors. Cell therapy. Madrid: McGraw-Hill/Interamericana de España; 2008. pp. 33–41. [Google Scholar]
- 11.Devine SM, Bartholomew AM, Mahmud N, Nelson M, Patil S, Hardy W, Sturgeon C, Hewett T, Chung T, Stock W, Sher D, Weissman S, Ferrer K, Mosca J, Deans R, Moseley A, Hoffman R. Mesenchymal stem cells are capable of homing to the BM of non-human primates following systemic infusion. Exp Hematol. 2001;29:244–55. doi: 10.1016/s0301-472x(00)00635-4. [DOI] [PubMed] [Google Scholar]
- 12.Horwitz EM, Gordon PL, Koo WK, Marx JC, Neel MD, McNall RY, Muul L, Hofmann T. Isolated allogeneic BM-derived mesenchymal cells engraft and stimulate growth in children with osteogenesis imperfecta: implications for cell therapy of bone. Proc Natl Acad Sci USA. 2002;99:8932–7. doi: 10.1073/pnas.132252399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le Blanc K, Rasmusson I, Sundberg B, Gotherstrom C, Hassan M, Uzunel M, Ringden O. Treatment of severe acute graft-versus-host disease with third party haploidentical mesenchymal stem cells. Lancet. 2004;36:1439–41. doi: 10.1016/S0140-6736(04)16104-7. [DOI] [PubMed] [Google Scholar]
- 14.Bernardo ME, Zaffaroni N, Novara F, Cometa AM, Avanzini MA, Moretta A, Montagna D, Maccario R, Villa R, Daidone MG, Zuffardi O, Locatelli F. Human BM derived mesenchymal stem cells do not undergo transformation after long-term in vitro culture and do not exhibit telomere maintenance mechanisms. Cancer Res. 2007;67:9142–9. doi: 10.1158/0008-5472.CAN-06-4690. [DOI] [PubMed] [Google Scholar]
- 15.Miura M, Miura Y, Padilla-Nash HM, Molinolo AA, Fu B, Patel V, Seo BM, Sonoyama W, Zheng JJ, Baker CC, Chen W, Ried T, Shi S. Accumulated chromosomal instability in murine BM mesenchymal stem cells leads to malignant transformation. Stem Cells. 2006;24:1095–103. doi: 10.1634/stemcells.2005-0403. [DOI] [PubMed] [Google Scholar]
- 16.Rubio D, Garcia-Castro J, Martin MC, de la Fuente R, Cigudosa JC, Lloyd AC, Bernad A. Spontaneous human adult stem cell transformation. Cancer Res. 2005;65:3035–9. doi: 10.1158/0008-5472.CAN-04-4194. [DOI] [PubMed] [Google Scholar]
- 17.Cobo F, Navarro JM, Herrera MI, Vivo A, Porcel D, Jurado M, García-Castro J, Menendez P. Electron microscopy reveals the presence of viruses in mouse embryonic fibroblasts but neither in human embryonic fibroblasts nor in human mes-enchymal cells used for hESC maintenance: toward an implementation of microbiological quality assurance program in stem cell banks. Cloning Stem Cells. 2008;10:65–74. doi: 10.1089/clo.2007.0020. [DOI] [PubMed] [Google Scholar]
- 18.Menendez P, Wang Bhatia M. Genetic manipulation of human embryonic stem cells: a system to study early human development and potential therapeutic applications. Curr Gene Ther. 2005;5:375–85. doi: 10.2174/1566523054546198. [DOI] [PubMed] [Google Scholar]
- 19.Kon E, Muraglia A, Corsi A, Bianco P, Marcacci M, Martin I, Boyde A, Ruspantini I, Chistolini P, Rocca M, Giardino R, Cancedda R, Quarto R. Autologous BM stromal cells loaded onto porous hydroxyapatite ceramic accelerate bone repair in critical-size defects of sheep long bones. J Biomed Mater Res. 2000;49:328–37. doi: 10.1002/(sici)1097-4636(20000305)49:3<328::aid-jbm5>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 20.Horwitz EM, Prockop DJ, Fitzpatrick LA, Koo WW, Gordon PL, Neel M, Sussman M, Orchard P, Marx JC, Pyeritz RE, Brenner MK. Transplantability and therapeutic effects of BM-derived mesenchymal cells in children with osteogenesis imper-fecta. Nat Med. 1999;5:309–13. doi: 10.1038/6529. [DOI] [PubMed] [Google Scholar]
- 21.Koc ON, Gerson SL, Cooper BW, Dyhouse SM, Haynesworth SE, Caplan AI, Lazarus HM. Rapid hematopoietic recovery after coinfusion of autologous-blood stem cells and culture-expanded marrow mesenchy-mal stem cells in advanced breast cancer patients receiving high-dose chemotherapy. J Clin Oncol. 2000;18:307–16. doi: 10.1200/JCO.2000.18.2.307. [DOI] [PubMed] [Google Scholar]
- 22.Le Blanc K, Samuelsson H, Gustafsson B, Remberger M, Sundberg B, Arvidson J, Ljungman P, Lonnies H, Nava S, Ringden O. Transplantation of mesenchy-mal stem cells to enhance engraftment of hematopoietic stem cells. Leukemia. 2007;21:1733–8. doi: 10.1038/sj.leu.2404777. [DOI] [PubMed] [Google Scholar]
- 23.Sykes M. Immune tolerance: mechanisms and application in clinical transplantation. J Intern Med. 2007;262:288–310. doi: 10.1111/j.1365-2796.2007.01855.x. [DOI] [PubMed] [Google Scholar]
- 24.Singh NJ, Schwartz RH. Primer: mechanisms of immunologic tolerance. Nat Clin Pract Rheumatol. 2006;2:44–52. doi: 10.1038/ncprheum0049. [DOI] [PubMed] [Google Scholar]
- 25.Sundrud MS, Rao A. New twists of T cell fate: control of T cell activation and tolerance by TGF-beta and NFAT. Curr Opin Immunol. 2007;19:287–93. doi: 10.1016/j.coi.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 26.Sohn SJ, Thompson J, Winoto A. Apoptosis during negative selection of autoreactive thymocytes. Curr Opin Immunol. 2007;19:510–5. doi: 10.1016/j.coi.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 27.Gauld SB, Benschop RJ, Merrell KT, Cambier JC. Maintenance of B cell anergy requires constant antigen receptor occupancy and signaling. Nat Immunol. 2005;6:1160–7. doi: 10.1038/ni1256. [DOI] [PubMed] [Google Scholar]
- 28.Cambier JC, Gauld SB, Merrell KT, Vilen BJ. B-cell anergy: from transgenic models to naturally occurring anergic B cells? Nat Rev Immunol. 2007;7:633–43. doi: 10.1038/nri2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Madrenas J, Chau LA, Smith J, Bluestone JA, Germain RN. The efficiency of CD4 recruitment to ligand-engaged TCR controls the agonist/partial agonist properties of peptide-MHC molecule ligands. J Exp Med. 1997;185:219–29. doi: 10.1084/jem.185.2.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tarazona R, Williams O, Moskophidis D, Smyth LA, Tanaka Y, Murdjeva M, Wack A, Mamalaki C, Kioussis D. Susceptibility and resistance to antigen-induced apopto-sis in the thymus of transgenic mice. J Immunol. 1998;160:5397–403. [PubMed] [Google Scholar]
- 31.Bensinger SJ, Bandeira A, Jordan MS, Caton AJ, Laufer TM. Major histocompat-ibility complex class II-positive cortical epithelium mediates the selection of CD4(+)25(+) immunoregulatory T cells. J Exp Med. 2001;194:427–38. doi: 10.1084/jem.194.4.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang Q, Bluestone JA. The Foxp3 + regulatory T cell: a jack of all trades, master of regulation. Nat Immunol. 2008;9:239–44. doi: 10.1038/ni1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wells AD, Li XC, Li Y, Walsh MC, Zheng XX, Wu Z, Nunez G, Tang A, Sayegh M, Hancock WW, Strom TB, Turka LA. Requirement for T-cell apoptosis in the induction of peripheral transplantation tolerance. Nat Med. 1999;5:1303–7. doi: 10.1038/15260. [DOI] [PubMed] [Google Scholar]
- 34.Chau LA, Rohekar S, Wang JJ, Lian D, Chakrabarti S, Zhang L, Zhong R, Madrenas J. Thymic re-entry of mature activated T cells and increased negative selection in vascularized allograft recipients. Clin Exp Immunol. 2002;127:43–52. doi: 10.1046/j.1365-2249.2002.01717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tian C, Bagley J, Lacomini J. Homeostatic expansion permits T cells to re-enter the thymus and deliver antigen in a tolerogenic fashion. Am J Transplant. 2007;7:1934–41. doi: 10.1111/j.1600-6143.2007.01891.x. [DOI] [PubMed] [Google Scholar]
- 36.Reich-Zeliger S, Bachar-Lustig E, Bar-Man A, Reisner Y. Tolerance induction in presensitized BM recipients by veto CTLs: effective deletion of host anti-donor memory effector cells. J Immunol. 2007;179:6389–94. doi: 10.4049/jimmunol.179.10.6389. [DOI] [PubMed] [Google Scholar]
- 37.Madrenas J, Schwartz RH, Germain RN. Interleukin 2 production, not the pattern of early T-cell antigen receptor-dependent tyrosine phosphorylation, controls anergy induction by both agonists and partial agonists. Proc Natl Acad Sci USA. 1996;93:9736–41. doi: 10.1073/pnas.93.18.9736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Salojin KV, Zhang J, Madrenas J, Delovitch TL. T-cell anergy and altered T-cell receptor signaling: effects on autoimmune disease. Immunol Today. 1998;19:468–73. doi: 10.1016/s0167-5699(98)01326-7. [DOI] [PubMed] [Google Scholar]
- 39.Choi S, Schwartz RH. Molecular mechanisms for adaptive tolerance and other T cell anergy models. Semin Immunol. 2007;19:140–52. doi: 10.1016/j.smim.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perez VL, Van Parijs L, Biuckians A, Zheng XX, Strom TB, Abbas AK. Induction of peripheral T cell tolerance in vivo requires CTLA-4 engagement. Immunity. 1997;6:411–7. doi: 10.1016/s1074-7613(00)80284-8. [DOI] [PubMed] [Google Scholar]
- 41.Greenwald RJ, Boussiotis VA, Lorsbach RB, Abbas AK, Sharpe AH. CTLA-4 regulates induction of anergy in vivo. Immunity. 2001;14:145–55. doi: 10.1016/s1074-7613(01)00097-8. [DOI] [PubMed] [Google Scholar]
- 42.O'Neill HC. Dendritic cell therapy for tolerance induction to stem cell transplants. Curr Stem Cell Res Ther. 2006;1:121–5. doi: 10.2174/157488806775269160. [DOI] [PubMed] [Google Scholar]
- 43.Zha Y, Marks R, Ho AW, Peterson AC, Janardhan S, Brown I, Praveen K, Stang S, Stone JC, Gajewski TF. T cell anergy is reversed by active Ras and is regulated by diacylglycerol kinase-alpha. Nat Immunol. 2006;7:1166–73. doi: 10.1038/ni1394. [DOI] [PubMed] [Google Scholar]
- 44.Zheng Y, Zha Y, Gajewski TF. Molecular regulation of T-cell anergy. EMBO Rep. 2008;9:50–5. doi: 10.1038/sj.embor.7401138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang ZX, Yang L, Young KJ, DuTemple B, Zhang L. Identification of a previously unknown antigen-specific regulatory T cell and its mechanism of suppression. Nat Med. 2000;6:782–9. doi: 10.1038/77513. [DOI] [PubMed] [Google Scholar]
- 46.Zhang ZX, Young K, Zhang L. CD3+CD4-CD8- alphabeta-TCR+ T cell as immune regulatory cell. J Mol Med. 2001;79:419–27. doi: 10.1007/s001090100238. [DOI] [PubMed] [Google Scholar]
- 47.Roncarolo MG, Gregori S, Battaglia M, Bacchetta R, Fleischhauer K, Levings MK. Interleukin-10-secreting type 1 regulatory T cells in rodents and humans. Immunol Rev. 2006;212:28–50. doi: 10.1111/j.0105-2896.2006.00420.x. [DOI] [PubMed] [Google Scholar]
- 48.Starzl TE. Immunosuppressive therapy and tolerance of organ allografts. N Engl J Med. 2008;358:407–11. doi: 10.1056/NEJMe0707578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Roelen D, Brand A, Claas FH. Pretransplant blood transfusions revisited: a role for CD(4+) regulatory T cells? Transplantation. 2004;77:S26–8. doi: 10.1097/01.TP.0000106469.12073.01. [DOI] [PubMed] [Google Scholar]
- 50.Chavers BM, Sullivan EK, Tejani A, Harmon WE. Pre-transplant blood transfusion and renal allograft outcome: a report of the North American Pediatric Renal Transplant Cooperative Study. Pediatr Transplant. 1997;1:22–8. [PubMed] [Google Scholar]
- 51.Fandrich F, Lin X, Chai GX, Schulze M, Ganten D, Bader M, Holle J, Huang DS, Parwaresch R, Zavazava N, Binas B. Preimplantation-stage stem cells induce long-term allogeneic graft acceptance without supplementary host conditioning. Nat Med. 2002;8:171–8. doi: 10.1038/nm0202-171. [DOI] [PubMed] [Google Scholar]
- 52.Drukker M, Katz G, Urbach A, Schuldiner M, Markel G, Itskovitz-Eldor J, Reubinoft B, Mandelboim O, Benvenisty N. Characterization of the expression of MHC proteins in human embryonic stem cells. Proc Natl Acad Sci USA. 2002;99:9864–9. doi: 10.1073/pnas.142298299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Menendez P, Bueno C, Wang L, Bhatia M. Human embryonic stem cells: potential tool for achieving immunotolerance? Stem Cell Rev. 2005;1:151–8. doi: 10.1385/SCR:1:2:151. [DOI] [PubMed] [Google Scholar]
- 54.Le Blanc K, Tammik C, Rosendahl K, Zetterberg E, Ringdén O. HLA expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Exp Hematol. 2003;31:890–6. doi: 10.1016/s0301-472x(03)00110-3. [DOI] [PubMed] [Google Scholar]
- 55.Tuan RS, Boland G, Tuli R. Adult mes-enchymal stem cells and cell-based tissue engineering. Arthritis Res Ther. 2003;5:32–45. doi: 10.1186/ar614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Breitbart AS, Grande DA, Mason JM, Barcia M, James T, Grant RT. Gene-enhanced tissue engineering: applications for bone healing using cultured periosteal cells transduced retrovirally with the BMP-7 gene. Ann Plast Surg. 1999;42:488–95. [PubMed] [Google Scholar]
- 57.Cetrulo CL., Jr Cord-blood mesenchymal stem cells and tissue engineering. Stem Cell Rev. 2006;2:163–8. doi: 10.1007/s12015-006-0023-x. [DOI] [PubMed] [Google Scholar]
- 58.Studeny M, Marini FC, Dembinski JL, Zompetta C, Cabreira-Hansen M, Bekele BN, Champlin RE, Andreeff M. Mesenchymal stem cells: potential precursors for tumor stroma and targeted-deliv-ery vehicles for anticancer agents. J Natl Cancer Inst. 2004;96:1593–603. doi: 10.1093/jnci/djh299. [DOI] [PubMed] [Google Scholar]
- 59.Spaeth E, Klopp A, Dembinski J, Andreeff M, Marini F. Inflammation and tumor microenvironments: defining the migratory itinerary of mesenchymal stem cells. Gene Ther. 2008;15:730–8. doi: 10.1038/gt.2008.39. [DOI] [PubMed] [Google Scholar]
- 60.Koc ON, Day J, Nieder M, Gerson SL, Lazarus HM, Krivit W. Allogeneic mes-enchymal stem cell infusion for treatment of metachromatic leukodystrophy (MLD) and Hurler syndrome (MPSIH) BM Transplant. 2002;30:215–22. doi: 10.1038/sj.bmt.1703650. [DOI] [PubMed] [Google Scholar]
- 61.M̧ller P, Bulnheim U, Diener A, Ļthen F, Teller M, Klinkenberg ED, Neumann HG, Nebe B, Liebold A, Steinhoff G, Rychly J. Calcium phosphate surfaces promote osteogenic differentiation of mesenchymal stem cells. J Cell Mol Med. 2008;12:281–91. doi: 10.1111/j.1582-4934.2007.00103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Marí-Beffa M, Santamaría JA, Murciano C, Santos-Ruiz L, Andrades JA, Guerado E, Becerra J. Zebrafish fins as a model system for skeletal human studies. Sci World J. 2007;7:1114–27. doi: 10.1100/tsw.2007.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Karlsson C, Brantsing C, Svensson T, Brisby H, Asp J, Tallheden T, Lindahl A. Differentiation of human mesenchymal stem cells and articular chondrocytes: analysis of chondrogenic potential and expression pattern of differentiation related transcription factors. J Orthop Res. 2007;25:152–63. doi: 10.1002/jor.20287. [DOI] [PubMed] [Google Scholar]
- 64.Kurth T, Hedbom E, Shintani N, Sugimoto M, Chen FH, Haspl M, Martinovic S, Hunziker EB. Chondrogenic potential of human synovial mesenchymal stem cells in alginate. Osteoarthritis Cartilage. 2007;15:1178–89. doi: 10.1016/j.joca.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 65.Mazhari R, Hare JM. Advances in cell-based therapy for structural heart disease. Prog Cardiovasc Dis. 2007;49:387–95. doi: 10.1016/j.pcad.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 66.Fuster V, Sanz J. Gene therapy and stem cell therapy for cardiovascular diseases today: a model for translational research. Nat Clin Prac Cardiovas Med. 2007;4:S1–8. doi: 10.1038/ncpcardio0737. [DOI] [PubMed] [Google Scholar]
- 67.Zietlow R, Lane EL, Dunnett SB, Rosser AE. Human stem cells for CNS repair. Cell Tissue Res. 2008;331:301–22. doi: 10.1007/s00441-007-0488-1. [DOI] [PubMed] [Google Scholar]
- 68.Chen L, Tredget EE, Wu PY, Wu Y. Paracrine factors of mesenchymal stem cells recruit macrophages and endothelial lineage cells and enhance wound healing. PLoS ONE. 2008;3:e1886–94. doi: 10.1371/journal.pone.0001886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Breitbach M, Bostani T, Roell W, Xia Y, Dewald O, Nygren JM, Fries JW, Tiemann K, Bohlen H, Hescheler J, Welz A, Bloch W, Jacobsen SE, Fleischmann BK. Potential risks of bone marrow cell transplantation into infarcted hearts. Blood. 2007;110:1362–9. doi: 10.1182/blood-2006-12-063412. [DOI] [PubMed] [Google Scholar]
- 70.Darwin J. Prockop, Scott DO. Clinical trials with adult stem/progenitor cells for tissue repair: let's not overlook some essential precautions. Blood. 2007;109:3147–51. doi: 10.1182/blood-2006-03-013433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Duncan AW, Rattis FM, DiMascio L, Congdon KL, Pazianos G, Zhao C, Yoon K, Cook JM, Willert K, Gaiano N, Reya T. Integration of Noch and Wnt signalling in hematopoietic stem cell maintenance. Nat Immunol. 2005;6:314–22. doi: 10.1038/ni1164. [DOI] [PubMed] [Google Scholar]
- 72.Arai F, Hirao A, Ohmura M, Sato H, Matsuoka S, Takubo K, Ito K, Koh GY, Suda T. Tie2/angiopoietic-1 signaling regulates hematopoietic stem cell quiescence in the BM niche. Cell. 2004;118:149–61. doi: 10.1016/j.cell.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 73.Li L, Xie T. Stem cell niche: structure and function. Ann Rev Cell Dev Biol. 2005;21:605–31. doi: 10.1146/annurev.cellbio.21.012704.131525. [DOI] [PubMed] [Google Scholar]
- 74.Zhang J, Niu C, Ye L, Huang H, He X, Tong WG, Ross J, Haug J, Johnson T, Feng JQ, Harris S, Wiedemann LM, Mishina Y, Li L. Identification of the hematopoietic stem cell niche and control of the niche size. Nature. 2003;425:836–41. doi: 10.1038/nature02041. [DOI] [PubMed] [Google Scholar]
- 75.Aggarwal S, Pittenger MF. Human mes-enchymal stem cells modulate allogeneic immune cell responses. Blood. 2005;105:1815–22. doi: 10.1182/blood-2004-04-1559. [DOI] [PubMed] [Google Scholar]
- 76.Glennie S, Soeiro I, Dyson PJ, Lam EW, Dazzi F. BM mesenchymal stem cells induce division arrest anergy of activated T cells. Blood. 2005;105:2821–7. doi: 10.1182/blood-2004-09-3696. [DOI] [PubMed] [Google Scholar]
- 77.Tabera S, Pérez-Simón J, Díez-Campelo M, Sánchez-Abarca L, Blanco B, López A, San Miguel JF. The effect of mesenchymal stem cells on the viability, proliferation and differentiation of B lymphocytes. Haematologica. 2008;93:1301–9. doi: 10.3324/haematol.12857. [DOI] [PubMed] [Google Scholar]
- 78.Beyth S, Borovsky Z, Mevorach D, Liebergall M, Gazit Z, Aslan H, Galun E, Rachmilewitz J. Human mesenchymal stem cells alter antigen-presenting cell maturation and induce T-cell unrespon-siveness. Blood. 2005;105:2214–9. doi: 10.1182/blood-2004-07-2921. [DOI] [PubMed] [Google Scholar]
- 79.Jiang XX, Zhang Y, Liu B, Zhang SX, Wu Y, Yu XD, Mao N. Human mesenchymal stem cells inhibit differentiation and function of monocyte-derived dendritic cells. Blood. 2005;105:4120–6. doi: 10.1182/blood-2004-02-0586. [DOI] [PubMed] [Google Scholar]
- 80.Stanley RR, Frederick RA. Graft-versus-host disease: a surge of developments. PLoS Medicine. 2007;4:e198–205. doi: 10.1371/journal.pmed.0040198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Collins RH, Shpilberg O, Drobyski O, Porter DL, Giralt S, Champlin R, Goodman SA, Wolff SN, Hu W, Verfaillie C, List A, Dalton W, Ognoskie N, Chetrit A, Antin JH, Nemunaitis J. Donor leukocyte infusions in 140 patients relapsed with malignancy after allogeneic BM transplantation. J Clin Oncol. 1997;15:433–44. doi: 10.1200/JCO.1997.15.2.433. [DOI] [PubMed] [Google Scholar]
- 82.Valcárcel D, Martino R, Caballero D, Martin J, Ferra C, Nieto JB, Sampol A, Bernal MT, Piñana JL, Vazquez L, Ribera JM, Besalduch J, Moraleda JM, Carrera D, Brunet MS, Perez-Simón JA, Sierra J. Sustained remissions of high-risk acute myeloid leukemia and myelodysplastic syndrome after reduced-intensity conditioning allogeneic hematopoietic transplantation: chronic graft-versus-host disease is the strongest factor improving survival. J Clin Oncol. 2008;26:577–84. doi: 10.1200/JCO.2007.11.1641. [DOI] [PubMed] [Google Scholar]
- 83.Pérez-Simón J, Sureda A, Fernández-Avilés F, Sampol A, Cabrera JR, Caballero D, Martino R, Petit J, Tomás JF, Moraleda JM, Alegre A, Cañizo C, Brunet S, Rosiñol L, Lahuerta J, Díez-Martín JL, León A, García A, Vazquez L, Sierra J, San Miguel JF. Reduced intensity conditioning allogeneic transplantation is associated with a high incidence of extramedullary relapses in multiple myeloma patients. Leukemia. 2006;20:542–5. doi: 10.1038/sj.leu.2404085. [DOI] [PubMed] [Google Scholar]
- 84.Le Blanc K, Ringden O. Immunomodulation by mesenchymal stem cells and clinical experience. J Intern Med. 2007;262:509–25. doi: 10.1111/j.1365-2796.2007.01844.x. [DOI] [PubMed] [Google Scholar]
- 85.Ringdén O, Uzunel M, Rasmusson I, Remberger M, Sundberg B, Lönnies H, Marschall HU, Dlugosz A, Szakos A, Hassan Z, Omazic B, Aschan J, Barkholt L, Le Blanc K. Mesenchymal stem cells for treatment of therapy-resistant graft-ver-sus-host disease. Transplantation. 2006;81:1390–7. doi: 10.1097/01.tp.0000214462.63943.14. [DOI] [PubMed] [Google Scholar]
- 86.Le Blanc K, Frassoni F, Ball L, Locatelli F, Roelofs H, Lewis I, Lanino E, Sundberg B, Bernardo ME, Remberger M, Dini G, Egeler RM, Bacigalupo A, Fibbe W, Ringdén O. Mesenchymal stem cells for the treatment of steroid resistant, severe, acute graft versus host disease: a phase II study. Lancet. 2008;371:1579–89. doi: 10.1016/S0140-6736(08)60690-X. [DOI] [PubMed] [Google Scholar]
- 87.Yãnez R, Lamana M, García-Castro J, Colmenero I, Ramirez M, Buren JA. Adipose tissue-derived mesenchymal stem cells have in vivo immunosuppressive properties applicable for the control of the graft-versus-host disease. Stem Cells. 2006;24:2582–91. doi: 10.1634/stemcells.2006-0228. [DOI] [PubMed] [Google Scholar]
- 88.Fang B, Liao S, Zhang Y, Zhao R. Favorable response to human adipose tissue-derived mesenchymal stem cells in steroid-refractory acute graft-versus-host disease. Transplant Proceeding. 2007;39:3358–62. doi: 10.1016/j.transproceed.2007.08.103. [DOI] [PubMed] [Google Scholar]
- 89.Ning H, Yang F, Jiang M, Hu L, Feng K, Zhang J, Yu, Li B, Xu C, Li Y, Wang J, Hu J, Lou X, Chen H. The correlation between cotransplantation of MSC and higher recurrence rate in hematologic malignancy patients: outcome of a pilot clinical trial. Leukemia. 2008;22:593–9. doi: 10.1038/sj.leu.2405090. [DOI] [PubMed] [Google Scholar]
- 90.Vianello F, Dazzi F. Mesenchymal stem cells for graft-versus-host disease: a double edge sword? Leukemia. 2008;22:463–5. doi: 10.1038/leu.2008.25. [DOI] [PubMed] [Google Scholar]
- 91.Bueno C, Lopes LF, Menendez P. BM stromal cell-derived Wnt signals as a potential underlying mechanism for cyclin D1 deregulation in multiple myeloma lacking t(11;14)(q13;q32) Blood Cells Mol Dis. 2007;39:366–8. doi: 10.1016/j.bcmd.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 92.Lazarus H, Koc O, Devine S, Curtin P, Maziarz R, Holland H, Shpall EJ, McCarthy P, Atkinson K, Cooper BW, Gerson SL, Laughlin MJ, Loberiza FR, Jr, Moseley AB, Bacigalupo A. Cotransplantation of HLA-identical sibling culture-expanded mes-enchymal stem cells andhematopoietic stem cells in hematologic malignancy patients. Biol Blood Marrow Transplant. 2005;11:389–98. doi: 10.1016/j.bbmt.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 93.Villarón EM, Pérez-Simón JA, San Miguel JF, Del Cañizo C. BM mesenchymal stem cells chimerism after allogeneic hematopoietic transplantation. Exp Hematol. 2006;34:7. doi: 10.1016/j.exphem.2005.09.014. [DOI] [PubMed] [Google Scholar]
- 94.Ball L, Bernardo ME, Roelofs H, Lankester A, Cometa A, Egeler R, Locatelli F, Fibbe WE. Co-transplantation of ex-vivo expanded mesenchymal stem cells accelerates lymphocyte recovery and may reduce the risk of graft failure in haploidentical hematopoietic stem cell transplantation. Blood. 2007;110:2764–7. doi: 10.1182/blood-2007-04-087056. [DOI] [PubMed] [Google Scholar]
- 95.Ringdén O, Uzunel M, Sundberg B, Lönnies L, Nava S, Gustafsson J, Henningsohn L, Le Blanc K. Tissue repair using allogeneic mesenchymal stem cells for hemorrhagic cystitis, pneumomediastinum and perforated colon. Leukemia. 2007;21:2271–6. doi: 10.1038/sj.leu.2404833. [DOI] [PubMed] [Google Scholar]
- 96.Nöth U, Steiner AF, Tuan RS. Technology insight: adult mesenchymal stem cells for osteoarthritis therapy. Nat Clin Pract Rheumatol. 2008;4:371–80. doi: 10.1038/ncprheum0816. [DOI] [PubMed] [Google Scholar]
- 97.Tzaribachev N, Vaegler M, Schaefer J, Reize P, Rudert M, Handgretinger Muller I. Mesenchymal stromal cells: a novel treatment option for steroid-induced avas-cular osteonecrosis. Isr Med Assoc J. 2008;10:232–4. [PubMed] [Google Scholar]
- 98.Richadson S, Hoyland JA. Stem cell regeneration of degenerated intervertebral discs: current status. Curr Pain Headache Rep. 2008;12:83–8. doi: 10.1007/s11916-008-0016-3. [DOI] [PubMed] [Google Scholar]
- 99.Vermeulen L, Sprick MR, Kemper K, Stassi G, Medema JP. Cancer stem cells – old concepts, new insights. Cell Death Differ. 2008;15:947–58. doi: 10.1038/cdd.2008.20. [DOI] [PubMed] [Google Scholar]
- 100.Perez-Mancera PA, Sanchez-Garcia I. Understanding mesenchymal cancer: the liposarcoma-associated FUS-DDIT3 fusion gene as a model. Semin Cancer Biol. 2005;15:206–14. doi: 10.1016/j.semcancer.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 101.Riggi N, Cironi L, Provero P, Suvà ML, Stehle JC, Baumer K, Guillou L, Stamenkovic I. Expression of the FUS-CHOP fusion protein in primary mesenchymal progenitor cells gives rise to a model of myxoid liposarcoma. Cancer Res. 2006;66:7016–23. doi: 10.1158/0008-5472.CAN-05-3979. [DOI] [PubMed] [Google Scholar]
- 102.Castillero-Trejo Y, Eliazer S, Xiang L, Richardson JA, Ilaria RL., Jr Expression of the EWS/FLI-1 oncogene in murine primary bone-derived cells Results in EWS/FLI-1dependent, ewing sarcoma-like tumors. Cancer Res. 2005;65:8698–705. doi: 10.1158/0008-5472.CAN-05-1704. [DOI] [PubMed] [Google Scholar]
- 103.Riggi N, Cironi L, Provero P, Suvà ML, Kaloulis K, Garcia-Echeverria C, Hoffmann F, Trumpp A, Stamenkovic I. Development of Ewing's sarcoma from primary BM-derived mesenchymal progenitor cells. Cancer Res. 2005;65:11459–68. doi: 10.1158/0008-5472.CAN-05-1696. [DOI] [PubMed] [Google Scholar]
- 104.Riggi N, Suva ML, Suva D, Cironi L, Provero P, Tercier S, Joseph JM, Stehle JC, Baumer K, Kindler V, Stamenkovic I. EWS-FLI-1 expression triggers a Ewing's sarcoma initiation program in primary human mesenchymal stem cells. Cancer Res. 2008;68:2176–85. doi: 10.1158/0008-5472.CAN-07-1761. [DOI] [PubMed] [Google Scholar]
- 105.Matushansky I, Hernando E, Socci ND, Mills JE, Matos TA, Edgar MA, Singer S, Maki RG, Cordon-Cardo C. Derivation of sarcomas from mesenchymal stem cells viainactivation of the Wnt pathway. J Clin Invest. 2007;117:3248–57. doi: 10.1172/JCI31377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Matushansky I, Hernando E, Socci ND, Matos T, Mills J, Edgar MA, Schwartz GK, Singer S, Cordon-Cardo C, Maki RG. A developmental model of sarcomagenesis defines a differentiation-based classification for liposarcomas. Am J Pathol. 2008;172:1069–80. doi: 10.2353/ajpath.2008.070284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Serakinci N, Guldberg P, Burns JS, Abdallah B, Schr̄dder H, Jensen T, Kassem M. Adult human mesenchymal stem cell as a target for neoplastic transformation. Oncogene. 2004;23:5095–8. doi: 10.1038/sj.onc.1207651. [DOI] [PubMed] [Google Scholar]
- 108.Shima Y, Okamoto T, Aoyama T, Yasura K, Ishibe T, Nishijo K, Shibata KR, Kohno Y, Fukiage K, Otsuka S, Uejima D, Nakayama T, Nakamura T, Kiyono T, Toguchida J. In vitro transformation of mesenchymal stem cells by oncogenic H-rasVal12. Biochem Biophys Res Commun. 2007;353:60–6. doi: 10.1016/j.bbrc.2006.11.137. [DOI] [PubMed] [Google Scholar]
- 109.Liu C, Chen Z, Chen Z, Zhang T, Lu Y. Multiple tumor types may originate from BM-derived cells. Neoplasia. 2006;8:716–24. doi: 10.1593/neo.06253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Houghton J, Stoicov C, Nomura S, Rogers AB, Carlson J, Li H, Cai X, Fox JG, Goldenring JR, Wang TC. Gastric cancer originating from BM-derived cells. Science. 2004;306:1568–71. doi: 10.1126/science.1099513. [DOI] [PubMed] [Google Scholar]
- 111.Karnoub AE, Dash AB, Vo AP, Sullivan A, Brooks MW, Bell GW, Richardson AL, Polyak K, Tubo R, Weinberg RA. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature. 2007;449:557–63. doi: 10.1038/nature06188. [DOI] [PubMed] [Google Scholar]
- 112.Djouad F, Bony C, Apparailly F, Louis-Plence P, Jorgensen C, Noel D. Earlier onset of syngeneic tumors in the presence of mesenchymal stem cells. Transplantation. 2006;82:1060–6. doi: 10.1097/01.tp.0000236098.13804.0b. [DOI] [PubMed] [Google Scholar]
- 113.Galie M, Konstantinidou G, Peroni D, Scambi I, Marchini C, Lisi V, Krampera M, Magnani P, Merigo F, Montani M, Boschi F, Marzola P, Orrù R, Farace P, Sbarbati A, Amici A. Mesenchymal stem cells share molecular signature with mesenchymal tumor cells and favor early tumor growth in syngeneic mice. Oncogene. 2008;27:2542–51. doi: 10.1038/sj.onc.1210920. [DOI] [PubMed] [Google Scholar]
- 114.Corre J, Mahtouk K, Attal M, Gadelorge M, Huynh A, Fleury-Cappellesso S, Danho C, Laharrague P, Klein B, Rème T, Bourin P. BM mesenchymal stem cells are abnormal in multiple myeloma. Leukemia. 2007;21:1079–88. doi: 10.1038/sj.leu.2404621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Iwamoto S, Mihara K, Downing JR, Pui CH, Campana D. Mesenchymal cells regulate the response of acute lymphoblastic leukemia cells to asparaginase. J Clin Invest. 2007;117:1049–57. doi: 10.1172/JCI30235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mueller LP, Luetzkendorf J, Mueller T, Reichelt K, Simon H, Schmoll HJ. Presence of mesenchymal stem cells in human BM after exposure to chemotherapy: evidence of resistance to apoptosis induction. Stem Cells. 2006;24:2753–65. doi: 10.1634/stemcells.2006-0108. [DOI] [PubMed] [Google Scholar]
- 117.Meads MB, Hazlehurst LA, Dalton WS. The BM microenvironment as a tumor sanctuary and contributor to drug resistance. Clin Cancer Res. 2008;14:2519–26. doi: 10.1158/1078-0432.CCR-07-2223. [DOI] [PubMed] [Google Scholar]
- 118.Rubio D, Garcia S, De la Cueva T, Paz MF, Lloyd AC, Bernad A, Garcia-Castro J. Human mesenchymal stem cell transformation is associated with a mesenchymal-epithelial transition. Exp Cell Res. 2008;314:691–8. doi: 10.1016/j.yexcr.2007.11.017. [DOI] [PubMed] [Google Scholar]
- 119.Wang Y, Huso DL, Harrington J, Kellner J, Jeong DK, Turney J, McNiece IK. Outgrowth of a transformed cell population derived from normal human BM mes-enchymal stem cell culture. Cytotherapy. 2005;7:509–19. doi: 10.1080/14653240500363216. [DOI] [PubMed] [Google Scholar]
- 120.Li H, Fan X, Kovi RC, Moquin B, Konz R, Stoicov C, Kurt-Jones E, Grossman SR, Lyle S, Rogers AB, Montrose M, Houghton J. Spontaneous expression of embryonic factors and p53 point mutations in aged mesenchymal stem cells: a model of age-related tumorigenesis in mice. Cancer Res. 2007;67:10889–98. doi: 10.1158/0008-5472.CAN-07-2665. [DOI] [PubMed] [Google Scholar]
- 121.Tolar J, Nauta AJ, Osborn MJ, Panoskaltsis A, McElmurry RT, Bell S, Xia L, Zhou N, Riddle M, Schroeder TM, Westendorf JJ, McIvor RS, Hogendoorn PC, Szuhai K, Oseth L, Hirsch B, Yant SR, Kay MA, Peister A, Prockop DJ, Fibbe WE, Blazar BR. Sarcoma derived from cultured mesenchym al stem cells. Stem Cells. 2007;25:371–9. doi: 10.1634/stemcells.2005-0620. [DOI] [PubMed] [Google Scholar]
- 122.Izadpanah R, Kaushal D, Kriedt C, Tsien F, Patel B, Dufour J, Bunnell BA. Long-term in vitro expansion alters the biology of adult mesenchymal stem cells. Cancer Res. 2008;68:4229–38. doi: 10.1158/0008-5472.CAN-07-5272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nakamizo A, Marini F, Amano T, Khan A, Studeny M, Gumin J, Chen J, Hentschel S, Vecil G, Dembinski J, Andreeff M, Lang FF. Human BM-derived mesenchy-mal stem cells in the treatment of gliomas. Cancer Res. 2005;65:3307–18. doi: 10.1158/0008-5472.CAN-04-1874. [DOI] [PubMed] [Google Scholar]
- 124.Khakoo AY, Pati S, Anderson SA, Reid W, Elshal MF, Rovira II, Nguyen AT, Malide D, Combs CA, Hall G, Zhang J, Raffeld M, Rogers TB, Stetler-Stevenson W, Frank JA, Reitz M, Finkel T. Human mes-enchymal stem cells exert potent antitu-morigenic effects in a model of Kaposi's sarcoma. J Exp Med. 2006;203:1235–47. doi: 10.1084/jem.20051921. 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lu YR, Yuan Y, Wang XJ, Wei LL, Chen YN, Cong C, Li SF, Long D, Tan WD, Mao YQ, Zhang J, Li YP, Cheng JQ. The growth inhibitory effect of mesenchymal stem cells on tumor cells in vitro and in vivo. Cancer Biol Ther. 2007;7:2–8. doi: 10.4161/cbt.7.2.5296. [DOI] [PubMed] [Google Scholar]
- 126.Qiao L, Xu Z, Zhao T, Zhao Z, Shi M, Zhao RC, Ye L, Zhang X. Suppression of tumorigenesis by human mesenchymal stem cells in a hepatoma model. Cell Res. 2008;18:500–7. doi: 10.1038/cr.2008.40. [DOI] [PubMed] [Google Scholar]
- 127.Hakkarainen T, Sarkioja M, Lehenkari P, Miettinen S, Ylikomi T, Suuronen R, Desmond RA, Kanerva A, Hemminki A. Human mesenchymal stem cells lack tumor tropism but enhance the antitumor activity of oncolytic adenoviruses in ortho-topic lung and breast tumors. Hum Gene Ther. 2007;18:627–41. doi: 10.1089/hum.2007.034. [DOI] [PubMed] [Google Scholar]
- 128.Stoff-Khalili MA, Rivera AA, Mathis JM, DeBenedetti A, Li XL, Odaka Y, Podduturi J, Sibley DA, Siegal GP, Stoff A, Young S, Zhu ZB, Curiel DT, Mathis JM. Mesenchymal stem cells as a vehicle for targeted delivery of CRAds to lung metas-tases of breast carcinoma. Breast Cancer Res Treat. 2007;105:157–67. doi: 10.1007/s10549-006-9449-8. [DOI] [PubMed] [Google Scholar]
- 129.Mohr A, Lyons M, Deedigan L, Harte T, Shaw G, Howard L, Barry F, O'Brien T, Zwacka R. Mesenchymal Stem Cells expressing TRAIL lead to tumour growth inhibition in an experimental lung cancer model. J Cell Mol Med. 2008 doi: 10.1111/j.1582-4934.2008.00317.x. (doi: 10.1111/j.15824934.2008.00317.x) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rubio D, Garcia S, Paz MF, De la Cueva T, Lopez-Fernandez LA, Lloyd AC, Garcia-Castro J, Bernad A. Molecular characterization of spontaneous mes-enchymal stem cell transformation. PLoS ONE. 2008;3:e1398–406. doi: 10.1371/journal.pone.0001398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Fodde R, Brabletz T. Wnt/beta-catenin signaling in cancer stemness and malignant behavior. Curr Opin Cell Biol. 2007;19:150–8. doi: 10.1016/j.ceb.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 132.De Boer J, Wang HJ, Van Blitterswijk C. Effects of Wnt signaling on proliferation and differentiation of human mesenchymal stem cells. Tissue Eng. 2004;10:393–401. doi: 10.1089/107632704323061753. [DOI] [PubMed] [Google Scholar]
- 133.Neth P, Ciccarella M, Egea V, Hoelters J, Jochum M, Ries C. Wnt signaling regulates the invasion capacity of human mes-enchymal stem cells. Stem Cells. 2006;24:1892–903. doi: 10.1634/stemcells.2005-0503. [DOI] [PubMed] [Google Scholar]
- 134.Neth P, Ries C, Karow M, Egea V, Ilmer M, Jochum M. The Wnt signal transduc-tion pathway in stem cells and cancer cells: influence on cellular invasion. Stem Cell Rev. 2007;3:18–29. doi: 10.1007/s12015-007-0001-y. [DOI] [PubMed] [Google Scholar]
- 135.Nauta AJ, Westerhuis G, Kruisselbrink AB, Lurvink EG, Willemze R, Fibbe WE. Donor-derived mesenchymal stem cells are immunogenic in an allogeneic host and stimulate donor graft rejection in a non-myeloablative setting. Blood. 2006;108:2114–20. doi: 10.1182/blood-2005-11-011650. [DOI] [PMC free article] [PubMed] [Google Scholar]