

Abstract
Human tumour cells are characterized by their ability to avoid the normal regulatory mechanisms of cell growth, division and death. The classical chemotherapy aims to kill tumour cells by causing DNA damage-induced apoptosis. However, as many tumour cells posses mutations in intracellular apoptosis-sensing molecules like p53, they are not capable of inducing apoptosis on their own and are therefore resistant to chemotherapy. With the discovery of the death receptors the opportunity arose to directly trigger apoptosis from the outside of tumour cells, thereby circumventing chemotherapeutic resistance. Death receptors belong to the tumour necrosis factor receptor superfamily, with tumour necrosis factor (TNF) receptor-1, CD95 and TNF-related apoptosis-inducing ligand-R1 and -R2 being the most prominent members. This review covers the current knowledge about these four death receptors, summarizes pre-clinical approaches engaging these death receptors in anti-cancer therapy and also gives an overview about their application in clinical trials conducted to date.
Keywords: death receptors, trail, TNF, CD95, cancer therapy
Introduction
Death receptor signalling
TNF-R1 signalling
CD95 signalling
TRAIL-R signalling
Targeting death receptors in cancer therapy
-
Targeting TNF-R1
- Isolated limb perfusion (ILP)
- Cell surface antigen-restricted activation of TNF-based fusion proteins
- Use of PEGylation to improve the specific anti-tumour effect of TNF
- Tumour targeted gene delivery of TNF
-
Targeting CD95
- Tissue-specific features of agonistic CD95 antibodies
- Cell surface antigen-restricted activation of CD95L-based fusion proteins
- CD95L-based gene therapy
-
TRAIL as a target for anti-cancer therapy
- TRAIL-receptor agonists and their toxicities
- Responsiveness of primary tumour cells to TRAIL-mediated apoptosis
- Sensitization to TRAIL
- Clinical application of TRAIL-R agonists
Future perspectives
Introduction
Apoptosis is a highly ordered form of programmed cell death crucial for tissue remodelling, homeostasis and the development of multicellular organisms [reviewed in 1, 2–4]. In contrast to necrosis, a form of cell death which is associated with inflammation, apoptosis selectively eliminates abnormal, infected or aged cells without affecting neighbouring tissue.
There is a constant balance between cell death and cell renewal in healthy tissues. However, once the balance is shifted towards the latter and apoptosis is carried out inadequately, cancer may develop. Tumour cells are hallmarked by their ability to escape apoptosis and replicate in an uncontrolled fashion even though their DNA is damaged. Consequently, these malignantly transformed cells carry the potential to proliferate in an unlimited and unregulated fashion.
Apoptosis can be induced by the intrinsic pathway characterized by the involvement of mitochondrial dysfunction and the extrinsic pathway associated with death receptor stimulation on the cell surface. These death receptors belong to the tumour necrosis factor receptor (TNFR) superfamily, with TNF-R1, CD95 (APO-1, Fas), TNF-related apoptosis-inducing ligand (TRAIL)-receptor 1 (TRAIL-R1, DR4) and TRAIL-R2 (APO-2, DR5, KILLER, TRICK2) being the most prominent members of this subfamily. Since the discovery of this class of receptors new strategies in cancer therapy aim to induce apoptosis of tumour cells by the activation of death receptors. Although stimulation of TNF-R1 and CD95 have been shown to be highly efficient in the killing of tumour cells, systemic treatment with TNF or CD95L or agonistic antibodies to their receptors entailed severe side effects due to induction of severe inflammatory conditions in the case of TNF and of apoptosis in normal cells in the case of CD95L [5–10]. New approaches targeting TNF-R1 and CD95 specifically on tumour cells have recently been devised; yet, such cell-specific receptor-targeting therapeutic strategies, albeit potentially very powerful, still await clinical proof of concept.
With the discovery of TRAIL (Apo2L) [11, 12] and its two apoptosis-inducing receptors TRAIL-R1 [13] and TRAIL-R2 [14–18] a new potential opportunity for death receptor targeting arose. For reasons that are still not understood, cancer cells are more susceptible than normal cells to apoptosis induction by TRAIL. Consequently, TRAIL-receptor triggering selectively eliminates cancer cells without life-threatening toxicity in vivo[19]. Further studies demonstrated that a combination of TRAIL with a wide range of chemotherapeutics or irradiation even acts syner-gistically in the killing of tumour cells. Importantly, this can be achieved in the absence of any overt additional side effects [20]. Thus, the efficiency and safety of several TRAIL-receptor agonists for the treatment of a variety of human tumours with and without chemotherapy are currently being investigated in phase I and II clinical trials.
In this review, we will describe different approaches of death receptor-targeting therapies, the side effects associated with them and the current state of clinical trials with a variety of death receptor agonists.
Death receptor signalling
Within the TNFR superfamily, six death receptors are currently known in human beings: CD95 (Fas, APO-1), TNFR-1 (p55, CD120a), TRAMP (APO-3, DR3, WSL-1, LARD), TRAIL-R1 (DR4), TRAIL-R2 (DR5, APO-2, KILLER, TRICK2) and DR6 (TR-7). All these receptors are cell surface cytokine receptors that can induce apoptosis after being crosslinked by their respective ligands or receptor-specific agonistic antibodies (see Fig. 1). Death receptors are type-I transmembrane proteins with an N-terminal extracellular domain responsible for binding to the cognate death ligand and a C-terminal intracellular portion that serves to transmit the death signal to the cell's interior. Two to four characteristic cysteine-rich repeats in the extracellular domain confer significant homology to all death receptors. The presence of these cysteine-rich repeats accounts for their TNFR superfamily membership. In addition, death receptors share a highly conserved death domain (DD), a 60 to 80 amino acid long sequence in the cytoplasmic region which is instrumental in triggering the intracellular death machinery [21].
Fig. 1.
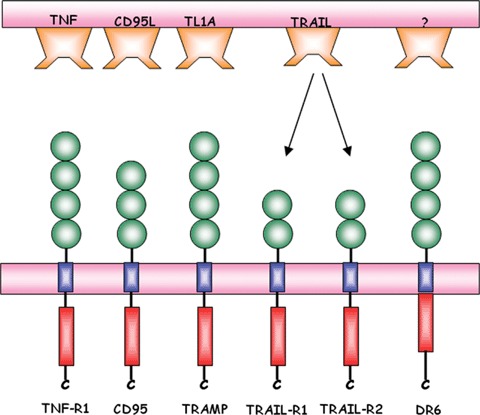
The six currently known death receptors and their respective ligands. All receptors contain several cytokine-rich domains (green) that are responsible for binding to the respective ligands. Following the transmembrane domain, the cytoplasmic region of each receptor pocesses a death domain (red) that is responsible for apoptosis induction.
Death receptors are activated upon binding of their respective ligands. These ligands are type II transmembrane proteins that form a subfamily of the TNF superfamily of cytokines referred to as death ligands. They either occur in a membrane-embedded form or as soluble ligands after being cleaved by metalloproteases [22]. Based on protein crystallographic experiments, it is widely assumed that ligand trimers bind to pre-assembled di- or trimeric receptor complexes that are not yet capable of transmitting a death signal, but are already connected via their pre-ligand assembly domain [23]. As measured by fluorescence resonance energy transfer, ligand binding induces a conformational change in the pre-assembled receptor complex [24] to facilitate downstream signal transduction. Upon receptor oligomerization, the intracellu-lar DDs are juxtaposed. The structural changes associated with this create a structure that allows adaptor proteins to bind via their DD to the death receptor, a prominent example being the Fas-associated protein with DD (FADD, MORT-1). Most of the adaptor proteins do not possess any enzymatic activity themselves but rather serve as linkers to recruit caspases (e.g. caspase-8), the main executioners of apoptosis. This recruitment occurs via the death-effector domain (DED), which is present in both, FADD and caspases 8 and 10. Recruitment of caspases to the receptor-associated protein complex results in the formation of the death-inducing signalling complex (DISC), first described for the CD95 receptor complex in 1995 [25]. The DISC contains active caspases which subsequently trigger a caspase cascade by activation of effector caspases, e.g. caspase-3, caspase-6 and caspase-7 [26]. Bona fide DISC-associated initiator caspases are caspase-8 and caspase-10, respectively.
During the process of apoptosis signalling, the receptor complex is internalized but it is still heavily contested whether this internalization step is an essential prerequisite for successful transmission or whether it may serve to attenuate the death-promoting signal [27].
Besides this extrinsic apoptotic pathway triggered by receptor stimulation, cell death signalling can also be induced or further potentiated by mitochondrial changes. Accordingly, the mitochon-drial pathway is activated by various stimuli, including DNA damage, growth factor withdrawal or cytokine deprivation [28]. DNA damage for instance activates p53, which subsequently induces the expression of the BH3-only proteins Puma and Noxa. Puma and Noxa then inhibit the anti-apoptotic proteins Bcl-2 and Bcl-XL, thus allowing the pro-apoptotic molecules Bax and Bak to multi-merize and to insert into the mitochondrial membrane. Although the exact mechanism is not elucidated yet, Bax/Bak multimeriza-tion triggers the release of several proteins from the intermem-brane space into the cytoplasm, including cytochrome c, Smac/DIABLO and HtrA2/Omi [29]. Smac/DIABLO and HtrA2/Omi block the activity of different inhibitor of apoptosis proteins (IAPs) [30]. In the cytosol, cytochrome cbinds to the apoptotic protease-activating factor-1 (Apaf-1), a cytosolic protein that contains a caspase-recruitment domain and a nucleotide-binding site. Apaf-1/cytochrome c interaction increases the affinity of Apaf-1 for dATP. Binding of dATP triggers a conformational change in Apaf-1 as the caspase-recruitment domain becomes exposed, which subsequently results in the recruitment of procaspase-9 and the formation of a protein complex referred to as the apoptosome [31]. Recruitment of caspase-9 to the apoptosome activates this protease, which is then also processed by autocatalytical cleavage. Subsequently, caspase-9 cleaves and thereby activates downstream effector caspases, among them caspase-3 and caspase-7 which then cleave various substrates leading to the characteristic morphological changes associated with apoptosis [32].
The signalling events downstream of the DISC are dependent on the cellular context. In type-I cells, the DISC is able to introduce strong caspase-8 activation that is followed by rapid activation of caspase-3. This process cannot be blocked by overexpression of the anti-apoptotic proteins Bcl-2 and Bcl-XL, suggesting that in type-I cells the activation of executioner caspases is independent of apoptotic events at the mitochondria. In contrast, overexpres-sion of Bcl-2 and Bcl-XL is sufficient to block death receptor-induced apoptosis in type-II cells, indicating that apoptotic events at the mitochondria are essential for death receptor-mediated apoptosis in type-II cells [33]. As DISC formation is strongly reduced in these cells, lower levels of active caspase-8 are generated that are not sufficient to directly activate caspase-3. The interconnection between the extrinsic and intrinsic mitochondrial pathway is provided by caspase-8. Once activated at the DISC, caspase-8 cleaves the BH3-only protein Bid into its truncated form tBid. This 15-kD fragment then exposes its BH3 domain, thereby facilitating an effective interaction with Bax/Bak at the mitochondria to allow for the release of pro-apoptotic proteins.
As unwanted cell death has to be avoided, death receptor signalling is tightly controlled at several levels. An important regulatory protein is the cellular FLICE (caspase-8)-inhibitory protein, cFLIP. The cFLIP protein has two DEDs that facilitate binding to the DED of FADD, thereby inhibiting the activation of caspase-8. Three different splice variants of cFlip exist referred to as cFLIPL, cFLIPS and cFLIPR[34]. cFLIPL comprises two DEDs and a caspase-like domain, therefore, closely resembling caspase-8. However, due to several amino acid exchanges in the domain which corresponds to the active site of caspase-8, cFLIPL lacks catalytic activity. It does not completely block pro-caspase-8 recruitment. The DED of pro-caspase-8 and cFLIPL compete for binding to the DED of FADD. Their ratio, Hsp90-mediated trafficking and other processes affect this competition, which determines how much pro-caspase-8 versus cFLIPL is recruited to FADD. cFLIPL interferes with the full maturation of DISC-recruited caspase-8 as it inhibits the generation of the active 10- and 18-kD fragments, which are responsible for the transmission of the death signal. Interestingly, a heterodimer of caspase-8 and cFLIPL has been shown to exert stronger caspase-8 activity than a homodimer of caspase-8 in vitro. This result indicates that cFLIPL may not only inhibit apoptosis but may also exhibit pro-apoptotic functions under certain conditions [35]. In contrast, cFLIPS and the closely related cFLIPR seem to exclusively inhibit procaspase-8 activation by competitively inhibiting its recruitment to and activation at the DISC [34].
Other proteins that negatively regulate the activity of initiator and effector caspases belong to the IAP family. Amongst these, XIAP is known to inhibit the autocatalytic activation of caspase-3, therefore preventing a further transmission of the death signal and consequently apoptosis. However, release of Smac/DIABLO from the mitochondria is in turn able to bypass the blockade mediated by XIAP and allow the apoptotic signal to continue [36]. In certain type II cell lines, this process may be more important for the execution of apoptosis than the previous reported weaker DISC formation [33].
Taken together, apoptosis is a complex process regulated and controlled at various stages by a complex protein machinery in order to allow targeted cell death of aged, transformed or infected cells without affecting neighbouring tissue. However, recent evidence has also suggested that TRAIL-R and CD95 signalling does not exclusively result in cell death but rather induce non-apoptotic signalling resulting in proliferation and/or migration [reviewed in 37].
TNF-R1 signalling
The TNF was the first member of the TNF superfamily detected more than 30 years ago. Its name originates from its cytotoxic effects on several tumour cell lines and the induction of tumour necrosis in various animal models. TNF is able to bind to the two receptors referred to as TNF-receptor 1 (TNF-R1) and TNF-receptor 2 (TNF-R2), which are also known to interact with LTα and LTβ[38]. As only TNF-R1 possesses a DD, the current view in the field is that TNF triggers apoptosis only upon binding to TNF-R1. However, TNF-R1 engagement does not necessarily cause cell death. In fact, TNF-R1 signalling does not induce apoptosis in most cell types unless protein biosynthesis is blocked. This is due to the predominance of the non-apoptotic signalling arm induced by TNF in cells under normal circumstances. TNF has been shown to induce the production of pro-inflammatory cytokines, cell proliferation and differentiation [39, 40].
TNF binding to TNF-R1 allows for the adaptor protein TRADD to bind through an interaction of the two proteins' respective DDs. This creates a docking station for further signalling molecules, namely RIP and TRAF-2, forming a complex referred to as complex I. Under apoptotic conditions, a second intracellular complex referred to as complex II is formed. Although the exact mechanism is not elucidated so far, Micheau and Tschopp suggested that FADD substitutes TRADD, therefore allowing the complex to dissociate from the membrane [41]. Subsequently, caspase-8 is recruited to complex II, resulting in its activation and transmission of the apoptotic signal.
As stated above, in most cases the outcome of TNF-R1 signalling is not apoptosis but rather the activation of pro-survival pathways. A pivotal role in this pathway is played by TRAF-2. TRAF-2 is recruited to the TNF-R1 complex via RIP and interferes with apoptosis by two interconnected mechanisms [42]. First, TRAF-2 recruits cIAP-1 and cIAP-2 into the signalling complex, in which the cIAPs inhibit caspase-8 processing. Secondly, TRAF-2 triggers the MAP-kinase cascade leading to the activation of JNK, which in turn activates the transcription of TNF-responsive genes. In addition, TRAF-2 can also induce the translocation of NF-KB into the nucleus thereby initiating the transcription of its targets genes [43–45].
CD95 signalling
The binding of CD95 ligand (CD95L) to its cognate receptor CD95 is the best-characterized system leading to apoptosis. In addition, decoy receptor 3 (DcR3) was described to also bind to and neutralize CD95L, thereby preventing apoptosis induction [46]. However, the physiological importance of this interaction is less clear.
CD95 is ubiquitously expressed, though predominantly in the thymus, liver, heart, kidney and in virus-transformed lymphocytes. In contrast, CD95L expression is very restricted. It is primarily expressed by activated T cells, in which it has been shown to be the mediator of activation-induced cell death [47]. Accordingly, deregulation of the CD95 system, as in ALPS (autoimmune lym-phoproliferative syndrome) patients carrying a heterozygous mutation in the CD95 gene, is characterized by the incapability to shut down an immune response. As activated T cells are not eliminated but remain rather active, autoimmune reactions against self-antigens develop [48]. In addition, the CD95L/CD95 system is also capable of killing virus-infected and oncogenically transformed cells.
CD95 and CD95L are mostly expressed as membrane bound proteins, although a soluble form of both the receptor and the lig-and has been reported. The exact role of soluble CD95 and CD95L is still unclear. However, soluble CD95 is thought to counteract CD95-induced apoptosis, whereas soluble CD95L, generated by metallo-protease-mediated cleavage has been shown to either possess killing activity [49] or to act as an inhibitor of membrane-bound CD95L [50]. As described before and illustrated in Fig. 2, CD95 ligation results in DISC formation and subsequent activation of caspase-8 and -10, which then transmit the apoptotic signal. Although both caspases are processed with similar kinetics, cas-pase-10 cannot functionally substitute for caspase-8 [51, 52].
Fig. 2.
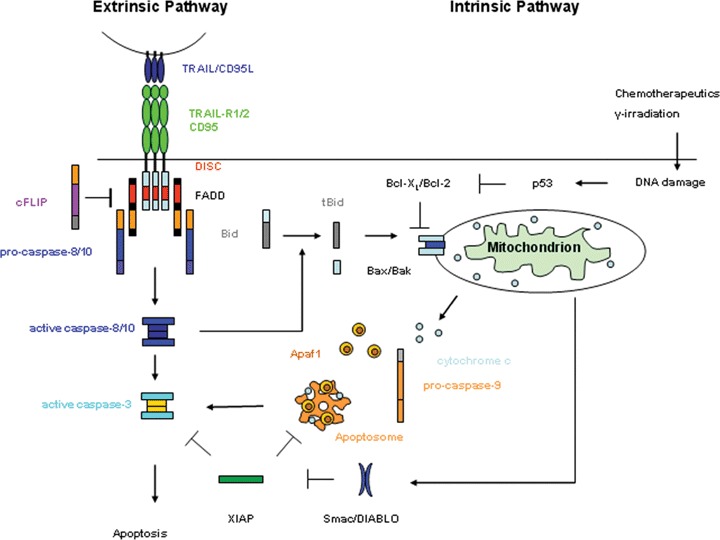
The extrinsic and intrinsic apoptotic pathway.
TRAIL-R signalling
The TRAIL was identified in a screen based on sequence homol-ogy with CD95L [11, 12]. However instead of binding to CD95, TRAIL has been shown to bind to five different receptors [37]. Of these, only TRAIL-R1 and TRAIL-R2 are able to induce apoptosis due to the presence of an intracellular DD (see Fig. 1). Although they share about 51% sequence homology and are similarly expressed on most human tissues as well as in tumour cell lines [53], it is still unclear which different functions are carried out by these two receptors. TRAIL-R3 and TRAIL-R4 are generally referred to as decoy receptors [54, 55]. Their extracellular domains are highly homologous to the ones of TRAIL-R1 and TRAIL-R2, but due to a complete or partial lack of a DD in TRAIL-R3 and TRAIL-R4, respectively, they are unable to transmit a death signal.
It is still a matter of debate whether TRAIL-R3 and TRAIL-R4 act as decoy receptors. So far, a decoy function has almost exclusively been demonstrated in overexpression systems whereas evidence in a more physiological setting is rare. This suggests that even if there is a decoy function of these receptors in some cell lines it is quite unlikely that a decoy function for TRAIL would be a broadly applicable phenomenon and function for TRAIL-R3 and -R4 as has been suggested by some early studies with these receptors.
Osteoprotegerin is the fifth receptor binding to TRAIL, which also binds to the osteoclast differentiation factor [56]. As osteo-protegerin is a soluble molecule, this receptor can only act as a decoy receptor for TRAIL [57]. However, its physiological relevance with respect to TRAIL inhibition remains to be determined whereas its role in bone-morphogenesis by inhibiting the RANKL/RANK interaction is well documented [58].
Despite the vast difference in toxicity between CD95L and TRAIL, the basic apoptosis machinery for TRAIL-R1 and TRAIL-R2 is very similar to the one described earlier for CD95, i.e. DISC formation by recruitment of FADD and subsequent recruitment and activation of pro-caspases 8 and 10. Although TRAIL triggers the extrinsic apoptotic pathway in a way similar to CD95L, systemic application of both death ligands results in opposing effects concerning toxicity. Thus, the mechanisms regulating the basic apoptosis machinery must be quite different between the CD95 and the TRAIL-receptor system.
Although TRAIL-R signalling leads to apoptosis in most cases, a pro-survival function for TRAIL stimulation has only recently been described. Here, NF-κB, JNK and MAPK signalling are supposed to play a major role [reviewed in 37]. However, the exact mechanisms and conditions required to transmit pro-survival signals upon TRAIL-R are just on the way to be investigated.
Targeting death receptors in cancer therapy
To date many of the classical cytotoxic drugs used in chemotherapy kill cells by causing apoptosis induced by, e.g. DNA damage.
Unfortunately, many tumour cells are incapable of inducing apop-tosis on their own due to frequent mutations in intracellular sensor molecules such as p53, which makes them resistant to therapy. In contrast to this, for the targeting of death receptors, the mutant state of these intracellular sensors is less relevant, as apoptosis is directly induced viadeath receptors. Therefore, death receptor targeting represents a feasible strategy to overcome chemotherapeutic resistance.
Targeting TNF-R1
Due to its significant cytotoxicity for normal tissue, TNF treatment has received little attention in recent years. The use of TRAIL, which shows superior tumour killing activity without the toxic side effects associated with TNF treatment, has seemed to be much more feasible.
However, TNF does not only induce apoptosis in tumour cells but also destroys tumour supporting blood vessels and improves the permeability of the vasculature to cytostatic drugs. Therefore, if the toxic side effects linked with TNF treatment could be circumvented, e.g. by means of targeted TNF-activating strategies or locally limited application methods, TNF treatment may prove to be very beneficial.
Isolated limb perfusion (ILP)
Since systemic administration of TNF is highly toxic, Lejeune et al. established a technique referred to as ILP, which facilitates local TNF administration [59]. ILP with TNF in combination with chemotherapeutic drugs led to complete response rates in patients [60] and showed improved penetrance of the cytostatic drugs melphalan and doxorubicin into tumours in animal models [61]. Interestingly, TNF specifically disrupts tumour supporting blood vessels while sparing normal blood vessels.
ILP was approved in Europe in 1998 for unresectable soft tissue sarcoma. It has, however, also been successfully applied in the treatment of various other local tumours.
The induction of apoptosis via TNF-R1 in tumour endothelial cells might not be the only reason for the success of TNF treatment via ILP. In ILP, TNF needs to be applied in very high doses much higher than actually needed for the saturation of TNF-R1. Therefore, signalling of TNF-R2, which has a much lower affinity for soluble TNF than TNF-R1, might also be involved in creating a maximum response of endothelial cells in ILP.
Cell surface antigen-restricted activation of TNF-based fusion proteins
The success of ILP provides proof of principle that TNF might potentially be used as anti-cancer treatment when administered in a locally restricted fashion. Therefore, successful TNF treatment has mainly become a matter of targeted delivery to the tumour site. One way to achieve this is to create fusion proteins in which TNF is fused to antibodies or natural ligands that specifically recognize surface proteins on tumour cells or in the tumour stroma. Soluble TNF is much less effective in inducing apoptosis than membrane bound TNF. Soluble fusion proteins will therefore only be able to efficiently induce apoptosis once they aggregate on the membrane of target cells [62].
The first TNF fusion protein designed by Rosenblum et al. was supposed to target melanoma cells [63]. Here, the anti-melanoma antibody ZME-018 was chemically conjugated to recombinant human TNF. This fusion protein was shown to have improved killing ability against TNF-sensitive tumour cells in culture and, more importantly, also against melanoma cells that were resistant to native TNF alone. As a consequence, the fusion protein turned out to be very effective in a xenograft model of melanoma [64].
Although antibodies have demonstrated great potential in targeted delivery of cytostatic cargos, there are some major drawbacks. Due to their size, treatment with antibodies suffers from rather poor distribution in the tumour and low uptake.
Therefore, the attention shifted towards the development of smaller, genetically engineered, fragments of antibodies, e.g. F(ab)2 fragments or single chain Fv (scFv) antibody fragments linked to TNF. ScFvs only contain one variable heavy chain (VH) and one variable light chain (VL) and are much smaller than whole antibodies. One of the first scFv fusion proteins was developed by Scherf et al.[65]. Here, TNF was fused to B1, an Fv antibody fragment that recognizes the LeY-antigen which is expressed on the surface of tumour cells. This molecule turned out to be highly toxic only for LeY-antigen expressing cancer cells in vitro and induced tumour regression in a xenograft model without inducing severe side effects.
To date, various TNF fusion proteins have been generated, not only using whole antibodies, F(ab)2 fragments or scFv, but also using recognition sites of natural ligands. Curnis et al. coupled TNF to the GNGRC peptide, a ligand of aminopeptidase N, which is a marker for angiogenic vessels [66]. Mice with established xenograft lymphomas could be cured using this approach. Recently, it has also been shown that a combination treatment of TNF-GNGRC and cisplatin, paclitaxel or gemcitabine has synergis-tic anti-tumour activity [67].
Several tumour antigens have been used to direct TNF fusion proteins to the tumour site, among them gp240 [68], EGFR (epidermal growth factor) [69], Her2/Neu [70] and single stranded DNA released from necrotic tumour cells [71].
Antigens targeting the fusion proteins to the tumour sites do not necessarily have to be on the surface of the tumour cell itself, but can rather be proteins of the tumour stroma. Advantages of targeting stroma proteins are that they are genetically stable, abundant and independent of tumour type. A feasible target of tumour stroma seems to be the fibroblast activation protein (FAP), which is a membrane protein of activated stromal fibroblasts present in virtually all solid tumours, but with limited expression in normal tissue [72].
The most advanced approach regarding TNF fusion proteins also uses FAP to target the protein to the tumour site and is referred to as TNF pro-drug. TNF pro-drugs represent an attempt to further increase the safety of systemic TNF treatment. The TNF pro-drug molecule is comprised of a single chain antibody targeting FAP, a trimerization domain, TNF and a TNF-R1 cap separated from TNF by a protease sensitive linker [62]. The strategy is that, to become active at the tumour site, the TNF pro-drug initially needs to be unmasked by the antibody binding to the tumour stroma and, secondly, by processing the linker peptide by tumour associated proteases to remove the TNF-R1 cap. Different recognition sites have been introduced into the linker between TNF and the TNF-R1 cap, e.g.for matrix metalloprotease 2 (MMP-2) which is overexpressed in some tumours [73]. To overcome the heterogeneity of matrix metalloprotease expression, TNF pro-drugs that are sensitive to proteolytic processing by urokinase-type plas-minogen activator were developed [74] as this protease has also been reported to be expressed in various tumour tissues [75].
The TNF pro-drug activity on antigen positive cells is almost 1000-fold higher than on antigen-negative cells and shows no systemic toxicity in mice [76]. It remains to be confirmed in additional animal models whether the individual pro-drugs indeed offer the possibility for systemic TNF treatment with increased biosafety.
Use of PEGylation to improve the specific anti-tumour effect of TNF
One of the most efficient ways to improve the therapeutic potency of a cytokine is by introducing chemical modifications such as polyethylene glycol (PEG)ylation. During the process of PEGylation, the water-soluble polymer PEG is conjugated to the bioactive molecule and serves to increase the molecular size and steric hindrance, thereby reducing renal excretion and increasing the half-life. The functionality of PEGylation has already been verified for PEGylated IFN-α, which has clinically been demonstrated to possess anti-viral activity against hepatitis C [77].
However, the clinical application of most PEGylated proteins has not been successful yet. This is due to the fact that in most cases PEGylations are introduced randomly at lysine residues in the proteins. Some of these lysines may be located near or at the active site of the proteins, thereby reducing its bioactivity. Taking this into consideration, through the use of phage libraries Yamamoto et al. created a PEGylation system in which only the N-terminus of TNF was specifically PEGylated after creation of a lysine-deficient mutant of TNF (mTNF-Lys(-)) [78]. PEGylated mTNF-Lys(-) showed higher bioactivity in vitro and greater anti-tumour therapeutic potency than wtTNF. In their follow-up study the same group showed that the introduction of variations in size and branches of the PEG side chains had a significant impact on the anti-tumour activity of the TNF mutant [79]. Their newest PEGylated mutant molecule mTNF-K90R even possessed an in vivo anti-tumour therapeutic window that is 60-fold wider than that of wt TNF [80]. This property of PEG-mTNF-K90R makes it very attractive, especially when used in an ILP setting, in which very high doses of TNF need to be applied at the moment. However, the higher activity may also result in an increased danger in case of leakage in an ILP setting. In addition, the repeated use of non-native recombinant forms of TNF may induce the formation of a neutralizing antibody response.
Tumour targeted gene delivery of TNF
Targeted gene delivery may be an attractive strategy, which is applicable to highly active yet toxic proteins such as TNF. To date, tumour-restricted non-viral and adenoviral approaches for TNF based gene therapy exist. In the non-viral approach, surface-shielded DNA delivery systems are used that possess virus-like characteristics and target gene expression into distant tumour tissue. TNF-encoding plas-mid DNA is condensed with cationic polyethylenimine (PEI) and displays high transfection efficiency in cell culture [81, 82]. These PEI/DNA polyplexes were conjugated with the cell-binding ligand transferrin to allow for receptor-mediated endocytosis. Systemic application of transferrin-conjugated PEI/DNA polyplexes led to preferential expression of TNF at the tumour site without detectable TNF serum levels [83]. The targeted delivery of TNF was tested in three murine tumour models of different tissue origins and resulted in growth inhibition of the tumour and, in the case of a fibrosarcoma model, even in complete tumour regression. The treatment was well tolerated without any sign of systemic toxicity associated with untargeted treatment by TNF [84].
In further approaches, the adenoviral system TNFerade was generated, which provides the possibility to maximize local anti-tumour activity with concomitant minimization of systemic toxicity. TNFerade is a second-generation replication-deficient adenoviral vector (E1, partial E3 and deleted E4) with a transgene encoding human TNF under control of a radiation-induced promoter, instead of a constitutive promoter. The CArG sequences within the early growth response (EGR-1) gene promoter were shown to be activated by ionizing radiation, thereby making these sequences suitable for ‘genetic radiotherapy’[85, 86]. Upon intratumoral injection of TNFerade and subsequent radiation, the radio-inducible EGR-1 sequences linked to TNF cDNA ensure maximal TNF expression and secretion that is constrained by spatial and temporal parameters of focused ionizing radiation [87, 88]. Thus, TNF location is predominantly restricted to the tumour site with little TNF released into systemic circulation, therefore preventing toxic side effects. A variety of human tumour xenograft models, including prostate [89], glioma [90], laryngeal carcinoma [91] and oesophageal adeno-carcinoma [92] have proven TNFerade to induce high levels of intratumoral TNF and synergistic anti-tumour effects with radiation, even though the tumours were resistant to radiation or TNF alone [93].
Three toxicology studies in mice demonstrated TNFerade to be safe and well tolerated [94]. Currently, TNFerade is tested in differently advanced phase I, II or III studies in locally advanced pancreatic cancer, rectal cancer, melanoma, head and neck cancer, and patients with tissue sarcoma in the extremities. So far, the observed anti-tumour effects were quite promising without any dose-limiting toxicities (DLT) reported. Taken together, the results obtained with TNFerade strongly warrant additional studies in larger controlled prospective trials.
Targeting CD95
Following the discovery of CD95 (Fas, Apo-1) in 1989 [95, 96], hopes were high that agonists of CD95 would become powerful novel agents for the treatment of cancer due to their strong direct apoptosis-inducing capacity. It was thought that they could be used either alternatively to or complementary to irradiation or chemotherapy. However, already in the early 1990s it was demonstrated that systemic administration of agonistic CD95 antibodies and of recombinant CD95L led to severe hepatotoxicity and subsequent death of the treated mice [10, 97]. Thus, although being the most potent physiologically occurring extracellularly triggered apoptosis system, the systemic use of agonistic CD95 antibodies or death ligands seemed to be impeded by severe systemic toxicity associated with unspecific CD95 activation. Hence, considering the use of CD95 agonists as cancer therapeutics, the problem of toxicity needs to be overcome. To date, there are several approaches that try to reinforce CD95 action at the tumour site while at the same time circumventing unwanted side effects. However, these approaches are generally still quite far from being applied in clinical trials.
Tissue-specific features of agonistic CD95 antibodies
The easiest approach in order to circumvent unwanted systemic toxicity of CD95L treatment is to exploit the fact that, for reasons that are not yet understood, some CD95-targeting antibodies seem to induce tissue-specific apoptosis.
For example, the hamster antimouse CD95 antibody RK8 efficiently kills thymocytes, but does not affect hepatocytes. In contrast to this, Jo2, which is also a hamster antimouse CD95 antibody, kills thymocytes with about the same efficiency as reported for RK8, but is additionally toxic for hepatotcytes and confers systemic toxicity [98].
Another antibody showing tissue-specific activity is HFE7A. HFE7A has been derived by the immunization of CD95 deficient mice with human CD95 and has already been tested in many species, among them Macaca fascicularis and marmosets, without showing hepatotoxicity [99].
The most recent anti-CD95 antibody that shows tissue-specific activity is R-125224. This humanized anti-human CD95 monoclonal antibody was shown to selectively induce apoptosis in type I activated lymphocytes but not in type II cells, e.g. Jurkat cells or hepatocytes. In a SCID mouse model, R-125224 reduced the number of activated CD3+ cells in vivo. Taking into consideration that hepatotoxicity is one of the biggest problems of CD95 agonist treatment and that hepatocytes are classified as type II cells, which are not affected by R-125224, this antibody might represent a promising tool in cancer treatment [100].
Cell surface antigen-restricted activation of CD95L-based fusion proteins
The idea to create cell surface-targeted CD95L fusion proteins is based on the fact that soluble CD95L on its own has little bioactivity but becomes bioactive when bound to an extracellular matrix [101]. In light of this, artificial immobilization of soluble inactive CD95L in the tumour area should create bioactive CD95L, which is restricted to the tumour site. Thus, fusion proteins have been generated that contain CD95L connected to an antibody that specifically recognizes tumour cells or tumour stroma.
The first fusion protein was designed by Jung et al.[102]. The study used monomeric CD95 Fab2 fragments hybridized to an anti-CD20 antibody and provided proof of principle of cell-surface-targeted activation of CD95L fusion proteins in vitro. Two years later, the first in vivo study was carried out using a trimeric fusion protein, sc40-FasL, consisting of the extracellular domain of CD95L fused to a single chain antibody that specifically recognizes the tumour stroma marker FAP [103]. The antibody fragment recognizing FAP allows for tumour-specific immobilization of CD95L and, upon binding, converts the inactive protein into a protein with membrane-bound-CD95L-like activity. In line with this, sc40-CD95L solely induced apoptosis in FAP-expressing cells in vitro and intravenous injection into mice did not result in systemic toxicity. Furthermore, it was also able to inhibit the growth of FAP-expressing xenotransplanted tumours.
As FAP is highly expressed in the majority of epithelial cancers including colon cancer [104], the rationale of FAP-fusion to CD95 seems to be promising.
In a similar approach, Bremer et al.recently designed a CD95L fusion protein targeting acute T cell leukaemia [105]. Here, CD95L was fused to a high affinity single-chain fragment of variable regions of an antibody fragment specific for the T cell leukaemia-associated antigen CD7. CD7 expression is present in 10% of patients suffering from acute myeloid leukaemia and in experiments with lymphoblastic leukaemia cell lines and patient-derived lymphoma it could be revealed that application of the fusion protein potently induced apoptosis in CD7-expressing cells.
The most advanced approach concerning CD95L fusion proteins was made by Watermann et al., who also developed the TNF pro-drug described earlier (see Section ‘’) [106]. With the molecular concept of CD95L pro-drugs being basically the same as for TNF pro-drugs, the anti-tumour activity of CD95L pro-drugs in the absence of apparent toxicity is impressive.
A very recent study by Bremer et al.[107] combined soluble CD95L with Rituximab, which is a CD20-specific chimeric monoclonal antibody that activates antibody-dependent cellular cytotoxicity but also induces apoptosis by cross-linking of its target antigen CD20. However, its clinical efficacy is significantly hampered by intrinsic or acquired resistance to therapy. As recent reports indicate that the apoptotic activity of rituximab can be synergized by co-treatment with CD95L, Bremer et al.[107] designed a scFvRit:sCD95L fusion protein which potently induced CD20-restricted apoptosis in various B-cell lines and lacked systemic toxicity in nude mice.
CD95L-based gene therapy
Targeted gene therapy is one possibility to circumvent systemic toxicity of CD95 treatment. The therapeutic potential of a CD95L-transgene expression for cancer treatment has been demonstrated by Arai et al. and Hedlund et al. in 1997 and 1999, respectively [108, 109]. Arai reported tumour regression of mouse epithelial carcinoma and colon carcinoma following the application of a CD95L-containing adenoviral vector. Hedlund demonstrated growth arrest and regression of human prostate cancer cells in vivoupon adenovirus-mediated expression of CD95L. However, in both settings the adenovirus was injected directly into the tumour site, which is not applicable for the majority of tumours in clinical practice. The ideal CD95L-based gene therapy should therefore be inducible and tissue specific.
One way of achieving this is to exploit physical barriers within the body that prevent the distribution of the virus throughout the body. This concept has been applied in a collagen- induced murine arthritis model. The adenovirus encoding for the CD95L was administered directly into the inflamed joint, thereby getting ‘trapped’ in the joint capsule. CD95L successfully induced apoptosis of synovial fibroblasts and significantly ameliorated the disease [110, 111]. However, it has to be made sure that this death-inducing protein or the virus encoding it do not leave the joint capsule in potentially toxic quantities.
Another way to enforce a local expression of adenovirally encoded proteins is the use of tissue-specific promoters which has proven to be successful, e.g.in neurons, glial cells [112] and smooth muscle cells [113].
The next step concerning CD95L-based gene therapy was marked by the development of not only tissue specific but also inducible systems to increase the safety of the application. Rubinchik et al.designed a complex adenoviral vector that can be induced by tetracycline and is under the control of the prostate-specific promoter ARR2P [114]. Additionally, other systems involving, e.g.tamoxifen-inducible constructs have been developed [115].
Adenoviral constructs expressing CD95L have been shown to be much more potent in the induction of apoptosis than anti-CD95 antibodies, such as CH11 or anti-APO-1. It was demonstrated in vitro that the inducible expression of CD95L was able to cause apoptosis in prostate and bladder cancer cells, which were resistant to CH11 treatment [116, 117]. Nevertheless, approaches have been made to amplify the apoptotic response upon adenoviral delivery of CD95L. These approaches comprise the co-delivery of CD95L and TRAIL in a complex adenoviral vector or the combined therapeutic use of adenovirus- encoded CD95L [118] and a small molecule inhibitor of ceramide metabolism [119].
Despite the progress made in the field of CD95-targeted gene therapy approaches, none of them has yet been tested in clinical trials (http://www.wiley.co.uk/genetherapy/clinical).
TRAIL as a target for anti-cancer therapy
In contrast to TNF and CD95L, TRAIL selectively kills a variety of tumour cell lines while sparing the majority of normal cells from apoptosis. This unique feature among the apoptosis-inducing TNF family members makes TRAIL a promising tool for anti-cancer therapy [19, 20]. Yet, when the attention was turned from tumour cell lines to primary tumour cells it became apparent that most primary tumour cells are resistant towards the apoptosis-inducing capacity of TRAIL. However, treatment with chemotherapeutics or irradiation sensitizes many primary tumour cells to TRAIL-mediated apoptosis whereas normal cells remain mainly unaffected by these combinations [120–122]. In the next chapter, we will introduce different TRAIL-receptor agonists currently being developed and summarize the available data about their influence on primary tumour cells in vitro and their effects observed in clinical studies so far.
TRAIL-receptor agonists and their toxicities
In order to use TRAIL-receptor agonists as anti-cancer drugs in the clinics, they require high anti-tumour activity with low toxicity for normal cells. Soluble recombinant TRAIL as well as agonistic antibodies targeting either TRAIL-R1 or TRAIL-R2 can be applied to activate the extrinsic TRAIL-mediated pathway in tumour cells.
A variety of recombinant forms of human TRAIL have been designed, each of them encoding the extracellular domain of human TRAIL. In addition, some preparations were generated that have been amino-terminally fused to distinct tags, including a poly-histidine tag [11], the FLAG epitope (which can be further cross-linked by anti-FLAG antibodies [123]) and the leucine zipper [19] or isoleucine zipper trimerization domain [124]. In vitrotoxicity studies revealed that the ability of recombinant TRAIL to form higher-order complexes or aggregates potentiates its anti-tumour efficiency, but at least in some cases also increased the toxicity to normal cells [125, 126]. Depending on the in vitro culture system and recombinant preparation used, killing of primary human hepatocytes, keratinocytes and astrocytes has been described [reviewed in 127]. However, until recently it has still been a matter of debate whether recombinant TRAIL is indeed toxic for hepatocytes in vivo. Ganten et al. demonstrated that freshly isolated primary human hepatocytes (PHH) at day 1 of in vitro culture were killed by highly aggregated recombinant forms of TRAIL (e.g. His-TRAIL, FLAG-TRAIL), whereas PHH at day 4 of in vitro culture, which resemble normal human liver tissue [128, 129], were TRAIL resistant [124]. Furthermore, whenever TRAIL was applied in vivowith or without sensitizing chemotherapeutic agents, no toxic effects on normal tissues could be monitored in mice, cynomolgus monkeys or chimpanzees [20]. These data imply that the in vitro toxicities observed for some recombinant forms of TRAIL are more likely to represent either cell culture artefacts or are related to the specific recombinant proteins used in these specific studies.
Since non-tagged TRAIL exhibits lower anti-tumour activity compared to tagged versions, it thereby also features the lowest potential for toxicity to normal cells [124]. This property may have been a factor in deciding to develop this form of TRAIL for clinical use. Leucine zipper- and isoleucine zipper-TRAIL represent an intermediate state as they on the one hand possess comparable anti-tumour activity to FLAG-TRAIL and His-TRAIL in vitro, but are on the other hand far less toxic than the latter, highly aggregated preparations.
In order to circumvent TRAIL-mediated killing of normal cells, new approaches similar to TNF-R1- and CD95-targeting therapies aim to selectively target TRAIL to tumour tissue by fusing it to tumour antigen-specific antibodies [130, 131].
While soluble recombinant TRAIL may bind to all TRAIL-receptors, including TRAIL-R3 and -R4, antibodies targeting TRAIL-R1 or -R2 only bind to their respective apoptosis-inducing receptor. Thus, if tumour cells were overcoming TRAIL sensitivity by expression of TRAIL-R3 and -R4 or by signalling events mediated by any of the other TRAIL-receptors which are not triggered by the respective antibody, such a TRAIL-R1 or TRAIL-R2-specific agonist would still be able to transmit the death signal despite the expression of other receptors [132]. However, normal cells may also no longer be safeguarded by these apoptosis-inhibitory signals and may therefore also be more sensitive to anti-TRAIL-receptor-mediated apop-tosis. Thus, on a theoretical basis, although antibodies may be associated with increased antitumoral activity, there could also be an additional toxicity, which is not conferred by soluble ligands. In vivo studies with several anti-TRAIL-receptor antibodies have shown that they can be used to specifically kill tumour cells. TRA-8, as an example of an agonistic anti-TRAIL-R2 antibody, was able to efficiently kill a variety of human tumour cells in mice (including astocytoma, leukaemia and engrafted breast cancer cells) without affecting normal human astrocytes, primary human hepatocytes, B cells or T cells [133, 134].
Takeda et al. suggested antibodies to be highly active due to cross-linking of the antibodies by Fc-receptor-expressing immune cells. In doing so, antibody cross-linking does not only enhance the anti-tumour efficiency due to formation of higher-order structures, it also contributes to the recruitment and activation of innate immune cells [135]. Accordingly, Uno et al.showed that the combination of anti-TRAIL-receptor antibodies with the two immuno-stimulatory antibodies anti-CD40 and anti-4–1BB efficiently eradicated syngenic tumours in vivo without any obvious toxicity to normal murine tissue [136]. Most probably, anti-TRAIL-receptor antibodies killed TRAIL-sensitive tumour cells. Subsequently, apoptotic tumour cells were engulfed by antigen-presenting cells, tumour antigens processed and presented to cytotoxic lymphocytes. Due to additional activation of CD40 on antigen-presenting cells and 4–1BB on T cells, an effective tumour-specific immune response was elicited that eliminated the remaining, TRAIL-resistant tumour burden. Thus, recruitment and activation of the immune system might be an advantage of an antibody-therapy over ligand-induced apoptosis.
In contrast to the short plasma half-life of recombinant ligands (˜30 min. in non-human primates), antibodies are characterized by an improved localization within the tumour site due to their significantly increased half-life (˜14–21 days). Although antibody-mediated tumour cell killing might be prolonged in patients, the risk of toxic side effects is increased at the same time. Studies directly comparing the anti-tumour efficiency and toxicity of TRAIL-R1- and -R2-specific antibodies to soluble recombinant TRAIL are still lacking today. Due to significant tumour penetration, recombinant Apo2L/TRAIL possesses high anti-tumour activity in vivo[137], indicating a possible advantage of the soluble cytokine compared to TRAIL-receptor antibodies. Moreover, despite the potential positive effects mentioned above, death receptor-targeting antibody therapies carry the risk of inducing unwanted and uncontrollable autoimmune responses due to binding of the Fc-part of the antibody to appropriate Fc-receptors on antigen-presenting cells and their subsequent activation.
To decrease possible side effects, new approaches aim to specify the activity of recombinant soluble TRAIL versions for tumour cells. Therefore, fusion proteins have been generated in which the extracellular domain of TRAIL is genetically linked to an scFv. This antibody fragment does not only induce a restricted accumulation of TRAIL at the tumour site, it at the same time contributes to a prolonged binding of TRAIL to tumour cells. A variety of scFv:TRAIL fusion proteins were generated recently, including scFvCD7:sTRAIL [105], scFvCD19:sTRAIL [138] and scFv425:sTRAIL [139]. All these proteins were able to efficiently kill malignantly transformed cells, while sparing normal tissue.
Taken together, these data point towards a conceivable application of distinct TRAIL-receptor agonists, including soluble TRAIL and TRAIL-receptor-targeting antibodies, as anti-cancer therapeutics in the clinic. However, one must take into account that normal cells may be sensitized to TRAIL-induced apoptosis by several patho-physiological mechanisms including inflammation and viral infections [140].
Responsiveness of primary tumour cells to TRAIL-mediated apoptosis
While the effect of TRAIL on several tumour cell lines has been extensively investigated [reviewed in 76, 141], far less is known about the responsiveness of primary tumour cells to TRAIL-mediated apoptosis. Pre-clinical studies revealed that TRAIL induces cell death in otherwise chemotherapy-resistant freshly isolated human multiple myeloma cells [142, 143]. However, such an effect could not be observed upon treatment of primary B cell acute or chronic leukaemia [144, 145], acute lymphoblastic leukaemia, acute myelogenous leukaemia, acute promyelocytic leukaemia or chronic lymphocytic leukaemia cells [146]. For acute myeloid leukaemia, Riccioni et al.reported a correlation between decoy-receptor expression and TRAIL resistance [147]. In addition, only primary glioblastoma cells expressing the wild-type tumour suppressor gene PTEN (Phosphatase and Tensin homolog deleted on chromosome TEN) and low levels of cFLIPS were TRAIL-sensitive [148], whereas those exhibiting a mutation in PTEN produced high levels of cFLIPS and were TRAIL resistant. In contrast, all (oligo-)astrocytoma specimens of all four WHO grades of malignancy as well as isolated tumour cells from medulloblastoma, meningeoma, esthesioneuroblas-toma [120] and soft tissue sarcoma [149] were TRAIL resistant, regardless of cFLIPS expression levels. In line with that, a variety of primary tumour cells from human breast, lung and colon tumour specimen were mostly TRAIL resistant [150], therefore, questioning the use of TRAIL as monotherapeutic anti-cancer agent. Even more strikingly, the migration and metastatic spread of TRAIL-resistant pancreatic cancer and cholangiocarcinoma cells was even enhanced in vitro and in vivo when treated with TRAIL [151, 152].
A major drawback and therefore limitation of TRAIL-based anti-cancer therapies is the need for huge amounts of recombinant, soluble TRAIL. A possible alternative to overcome this demand includes the generation of a replication-deficient adenovirus encoding human TRAIL (Ad5-TRAIL). Administration of this adenovirus at the site of tumour implantation dramatically inhibited the outgrowth of human prostate tumour xenografts in SCID mice [153]. While treatment with soluble, recombinant TRAIL requires consecutive applications due to its short half-life of about 30 min. [19], a single Ad5-TRAIL localized therapy leads to regionally high, sustained TRAIL concentrations. Although only minimal toxicity of adenoviral injections into the prostate were observed so far [154], more studies are required to demonstrate the safety of adenoviral systems in anti-cancer therapy.
Taken together, these results indicate that, unlike most tumour cell lines, many isolates from primary tumours, especially those from solid tumour entities are TRAIL resistant and that in some cases TRAIL treatment might even be counter-productive as it enforces malignancy. These data suggest that the therapeutic potential of TRAIL as a mono-therapeutic agent for the treatment of many types of cancer may be quite narrow. It is therefore essential to characterize each tumour specimen individually before therapy in order to categorize patients for a given TRAIL therapy based on their sensitivity profile. For this purpose, it will be important to develop biomarkers and appropriate sensitivity assays that help to identify those patients potentially benefiting from TRAIL-receptor-targeting therapies in the future [155–157].
Sensitization to TRAIL
Although TRAIL-receptor agonists alone are unable to induce apoptosis in most primary tumour cells, combinatorial treatments with a variety of chemotherapeutic agents or ionizing radiation sensitize otherwise resistant tumour cells to TRAIL-mediated cell death [122, 124, 158–160]. TRAIL engages an extrinsic, p53-independent pathway which can bypass a Bcl-2 or Bcl-2 protein-like imposed block of mitochondrial apoptosis by directly activating a caspase cascade, whereas chemotherapeutic agents and radiation preferentially induce cell death via the p53-dependent intrinsic mitochondrial pathway. The synergy between chemotherapy/ radiation and TRAIL-receptor agonists does not only rely on the stimulation of different targets/pathways, but is moreover due to transcriptional changes of proteins involved in TRAIL-receptor signalling [reviewed in 161, 162]. In this respect, sensitizing agents often shift the threshold of tumour cells for apoptosis by enhancing the cell's capability of forming the DISC by up-regulation of pro-apoptotic molecules including death receptors, caspase-8, FADD, Bax and/or Bak and by down-modulation of the expression of anti-apoptotic molecules like cFLIPL, IAPs, Bcl-XL Mcl-1 and/or Bcl-2 [137, 159, 163]. Tables 1 and 2 summarize the data obtained in pre-clinical studies for TRAIL in combination with different chemotherapeutics and irradiation, the proposed mechanisms responsible for sensitization to TRAIL-induced cell death and possible related toxicities. Although many studies reported up-regulation of TRAIL-R1 and/or TRAIL-R2 following co-treatment with sensitizing agents for a variety of primary tumour cells, this increase does not necessarily have to be the cause for the sensitization. In our own studies, we observed a correlation of TRAIL-R1 and TRAIL-R2 up-regulation and the sensitization by 5-FU and proteasome inhibition to TRAIL-induced cell death. However, this up-regulation turned out not to be responsible for the observed sensitization [120].
Table 1.
Combinational TRAIL treatments- Tumor cells
| Primary Tumour | TRAIL in combination with | Proposed mechanism | Reference |
|---|---|---|---|
| ALL | Vincristine (microtubule inhibitor) | [105] | |
| AML | HDAC inhibitor | [168] | |
| B-CLL | Cycloheximide | Downregulation of cFlipL | [171] |
| CLL | HDAC inhibitor | Signal via TRAIL-R1 | [166,167] |
| Colon cancer | Irinotecan, 5-FU | Upregulation of TRAIL-R2 | [172] |
| Erytholeukemic cells | Irradiation | Upregulation of TRAIL-R1 | [173] |
| Multiple myeloma | NF-κB inhibitor SN50 | Downregulation of Bcl-2, Bfl-1, IAPs, upregulalion of Bax | [163] |
| (Oligo-) astrocytoma | Bortezomib | Upregulation of TRAIL-R1/R2, Bax/Bak Downregulation of cFlipL | [174] |
| Pancreatic cancer | Gemtabicine, Doxorubicin, Cisplatin, Etoposide, Methotrexate | [175] | |
| Soft tissue sarcoma | Cyclophosphamide | [149] |
Table 2.
Combinational TRAIL treatments- Normal cells
| Cultured normal cell | TRAIL In combination with | Toxicity | Reference |
|---|---|---|---|
| Erythoblasts | Irradiation | No | [173] |
| Hepatocytes | 5-FU, Gemtabicine, Irinotecan, Oxaliplatin, Bortezomib (low dose) | No | [122, 124] |
| Hepatocytes | Cisplatin (high dose: 240 μM), Bortezomib (high dose: 3 μM) | Yes | [120, 124] |
| Hepatocytes | HDAC inhibitor | No | [169] |
| Keratinocytes | MG115 (Proteasome inhibitor) | Yes | [121] |
| Myeloid Progenitors | HDAC inhibitor | No | [168] |
| Osteoblasts | Etoposide, Cisplatin; Doxorubicin, Methotrexat, Cyclophosphamide | No | [176, 177] |
| Osteoblasts | Etoposide | No | [178] |
| Osteoblasts | Cisplatin, Doxorubicin | Yes | [178] |
| PBMC | HDAC inhibitor | No | [166, 170] |
| Prostate stromal cells | Doxorubicin | No | [179] |
For a combinatorial therapy to be successful, it is essential that tumour cells are preferentially sensitized to TRAIL-mediated apoptosis, while normal tissue is left relatively unharmed. Although most TRAIL co-treatments did not exert any toxicity on primary human hepatocytes at day 4 of in vitro culture, the combination of TRAIL with high doses of the frequently used chemotherapeutic cisplatin or the proteasome inhibitor bortezomib sensitized these hepatocytes for TRAIL-induced apoptosis [122]. However, the bortezomib concentration necessary to sensitize hepatoma cells is about 40-fold lower than the dose required for primary human hepatocyte sensitization [120, 122]. Thus, a therapeutic window is opened allowing for co-treatment of tumour patients with TRAIL and bortezomib without severe toxicity. In contrast, normal primary human keratinocytes are sensitized to TRAIL-induced cell death even at low concentrations of the proteasome inhibitor MG115 [121]. Most probably, proteasome inhibition enhanced the activation of the mitochondrial pathway in these cells. Due to subsequent release of cytochrome cand Smac/DIABLO, the function of XIAP, which is highly expressed in primary but not in transformed keratinocytes, was antagonized and caspase-3 maturation therefore supported [121]. However, interestingly, the combination of a TRAIL-R-specific monoclonal antibody with bortezomib in vivo was not toxic and did not induce any alterations in the skin [164].
Histone deacetylase inhibitors represent a further class of sensitizing agents that are able to sensitize TRAIL resistant hepatoma cell lines [165], primary acute myeloid leukemia (ALL) and chronic lymphocytic leukaemia (CLL) cells [166–168] to cell death. Again, this co-treatment was not toxic to primary human hepatocytes [169], to normal peripheral blood mononuclear cells [166, 170] and also not to myeloid progenitors [168]. The exact mechanism for histone deacetylase inhibitors-mediated sensitization to TRAIL-induced apoptosis has not yet been elucidated.
Taken together, selective anti-tumour activity was induced by combinatorial TRAIL-based treatment protocols in vitro and in vivo. However, it remains to be analysed why only tumour cells are sensitized to TRAIL-induced apoptosis, with the majority of normal cells not being affected. Understanding the principles underlying the difference between transformed and healthy tissue will allow for further refinement of TRAIL-based cancer treatment strategies.
Clinical application of TRAIL-R agonists
Due to the promising pre-clinical results of TRAIL as an anti-cancer agent presented before, several companies are developing TRAIL-receptor-targeting therapeutics. These TRAIL receptor agonists (TRAs) are currently being tested in phase I and II clinical trials. To date, one recombinant ligand, one anti-TRAIL-R1, five anti-TRAIL-R2 antibodies alone or in combination with distinct chemotherapeutics and Ad5-TRAIL are being evaluated (see Table 3).
Table 3.
TRAIL-receptor agonists
| Company | Treatment | Developmental stage |
|---|---|---|
| HGS | HGS-ETR1 (anti-TRAIL-Rl mAb) | Phase II completed: NHL, colorectal cancer, NSCLC |
| HGS-ETR1 + Paclitaxel + Carboplatin | Phase Ib: advanced solid tumors | |
| HGS-ETR1 + Gemcitabine + Cisplatin | Phase Ib: advanced solid tumors | |
| HGS-ETR1 + Bortezomib | Phase II: advanced multiple myeloma | |
| HGS-ETR2 (anti-TRAIL-R2 mAb) | Phase I: advanced solid tumors | |
| HGS-ETR2 + Chemotherapy | Phase Ib: advanced solid tumors | |
| HGS-TR2J (anti-TRAIL-R2 mAb) | Phase I: advanced solid tumors | |
| Daiichi Sankyo Inc. | TRA-8 (anti-TRAIL-R2) | Phase I: advanced solid tumors and lymphomas |
| CS-1008 (humanized version of TRA-8) | Phase I | |
| Novartis | LBY135 (anti-TRAIL-R2) | Phase I/II: advanced solid tumors |
| LBY135 + Capecitabine | Phase I/II: advanced solid tumors (recruiting since 2006) | |
| Genentech | Apomab (anti-TRAIL-R2) | Phase I/II: advanced solid tumors |
| Apomab + Avastin | Phase II: advanced solid tumors (initiated in 2007) | |
| Amgen | AMG655 (anti-TRAIL-R2) | Phase I: NSCLC, colorectal cancer (initiated in 2005) |
| Genentech/Amgen | Apo2L/TRAlL (soluble) | Phase Ib |
| Apo2L/TRAIL + Rituximab | Phase Ib/II: NHL (recruiting since 2006) | |
| Ad5-TRAIL | Phase Ia: organ-confined prostate cancer |
Human Genome Science (HGS, Rockville MD, USA) was the first company to test a TRA in clinical trials and is currently being investigating the activity and toxicity of two fully humanized monoclonal antibodies activating TRAIL-R1 (HGS-ETR1; Mapatumumab) and TRAIL-R2 (HGS-ETR2; Lexatumumab), respectively. In pre-clinical models, both antibodies rapidly increased the activation of cell death cascades and potently induced apoptosis across a wide range of human tumour cell lines and primary cells from both solid and haematological malignancies.
So far, the activity of HGS-ETR1 could be validated in three phase II clinical studies with patients suffering from non-Hodgkin's lymphoma (NHL), colorectal cancer and non-small cell lung cancer (NSCLC). Around 30% of the patients enrolled in the NSCLC or CRC studies exhibited stable disease after treatment with HGS-ETR1 as monotherapy. Clinical response or stable disease was observed in 14/17 patients with NHL diagnosed with follicular lymphomas. At the same time, no DLT, even at the highest doses applied (10 mg/kg), could be noticed in these clinical trials.
In addition, phase Ib clinical studies were initiated investigating the anti-tumour effect of HGS-ETR1 in combination with different panels of chemotherapeutics. From 32 patients with advanced tumours treated in combination with gemcitabine and cisplatin, partial response was observed in 9 and stable disease in 14 patients. The observed adverse effects may be attributable to the toxicity profile associated with the concomitantly applied chemotherapeutics. Accordingly, the combination of HGS-ETR1 with paclitaxel and carboplatin induced partial response in 4/20 and stable disease in 10/20 patients analysed. DLT has been neutropenic fever (attributed to chemotherapy) and hypersensitivity (attributed to HGS-ETR1) [180].
The efficiency and safety of HGS-ETR1 in combination with the proteasome inhibitor bortezomib in patients with advanced multiple myeloma is currently under investigation in phase II clinical studies (study number: HGS1012-C1055).
HGS-ETR2, the TRAIL-R2-specific antibody developed by HGS, is currently evaluated in phase Ib clinical trials in combination with a variety of chemotherapeutics. In particular, the combination with FOLFIRI and doxorubicin was well tolerated and associated with the induction of tumour shrinkage and partial responses in a wide range of cancer types. However, several adverse effects including anaemia, fatigue and dehydration were related to HGS-ETR2 treatment. Nevertheless, HGS-ETR2 could be safely administered, making further evaluations in combination with chemotherapy warranted. Interestingly, pre-clinical evaluation demonstrated a complete regression of different tumour cell line xenografts in vivo upon combinatorial treatment with HGS-ETR2 and the Smac mimetic SM-164 (Ascenta Therapeutics, Malvern, PA, USA).
Daiichi Sankyo, Tokyo, Japan is another company developing a TRAIL-receptor agonist for the treatment of advanced solid tumours and lymphomas. TRA-8, a humanized anti-TRAIL-R2 antibody, exhibited high anti-tumour activity against astrocytoma and leukaemia cells in vitro and engrafted breast cancer cells in vivo[133, 134].
The chimeric TRAIL-R2-targeting antibody LBY135 from Novartis induces apoptosis in 50% of a panel of 40 human colon cancer cell lines with an IC50 of 10 nM and less. Since the in vivo anti-tumour activity of LBY135 has been verified in human colorectal xenograft models in mice [181], Novartis is recruiting patients for an open-label, multi-centre, two-arm phase I/II trial of LBY135 alone and in combination with capecitabine in advanced solid tumours (Nevada Cancer Institute, Las Vegas, NV, USA).
The fully humanized monoclonal anti-TRAIL-R2 antibody Apomab generated by Genentech (South California, CA, USA) is currently investigated in phase I and II clinical trials in solid advanced tumours. First pharmacokinetic studies have proven it to be generally well tolerated even at doses up to 20 mg/kg, yet two disparate DLTs, e.g.one asymptomatic transaminitis and one pulmonary embolism developed among eleven patients treated with 10 mg/kg. However, a 28% shrinkage of the target lesions and symptomatic improvement could be shown in at least 1/6 patients receiving at least four cycles of Apomab [182]. Accordingly, a phase II study with Apomab as monotherapy for sarcoma or in combination with Avastatin against NSCLC was initiated in 2007 and a further treatment of NHL in combination with rituximab, the CD20-targeting antibody is underway.
AMG655, another fully human antibody targeting TRAIL-R2 which is developed by Amgen (Thousand Oaks, CA, USA) is currently enrolled in phase Ib clinical studies. The anti-tumour activity of AMG655 was confirmed by partial response in NSCLC and a metabolic partial response in colorectal cancer. Although no DLTs or severe side effects have been reported so far even when administered at doses of up to 20 mg/kg every two weeks, adverse effects including fever, fatigue and hypomagnesaemia were observed in nine patients enrolled in the study. A combination of AMG655 with chemotherapy might further enhance the anti-tumour responses, making the analysis of such treatment protocols warranted.
Furthermore, Genentech and Amgen have joined forces in the development of a recombinant untagged version of human TRAIL (Apo2L or AMG951), which is currently the only ligand being evaluated in clinical trials. Phase Ib/II pharmacokinetic and safety studies in patients suffering from low-grade NHL have already proven Apo2L/TRAIL to be active and safe both, alone or in combination with rituximab, without any incidence of DLT or severe side effects. In combination with rituxibam, 10 NHL patients have been assessed with 8 responding to varying degrees [183].
Future perspectives
Systemic TNF-R1 and CD95-targeting therapies are associated with severe side effects impeding their clinical application. However, new approaches discussed in this review aim to selectively target these death receptors expressed on tumour cells or cells important for tumour maintenance, thereby minimizing damage to healthy tissue. Although several pre-clinical studies have highlighted the ongoing progress of these approaches, most of them are still quite far from clinical applicability. In contrast, a variety of TRAIL-receptor agonists is currently being investigated in clinical trials and all of them have yielded promising results so far. Currently, the application of these agonists is restricted to tumours which are either primarily TRAIL sensitive or to tumours which can be sensitized to TRAIL-induced apoptosis by the application of currently available cancer drugs. To overcome resistance and the current limitations in the treatment of cancer it seems unavoidable to better understand the mechanisms of resistance in tumour cells.
References
- 1.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–19. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 3.Steller H. Mechanisms and genes of cellular suicide. Science. 1995;267:1445–9. doi: 10.1126/science.7878463. [DOI] [PubMed] [Google Scholar]
- 4.Thompson CB. Apoptosis in the pathogenesis and treatment of disease. Science. 1995;267:1456–62. doi: 10.1126/science.7878464. [DOI] [PubMed] [Google Scholar]
- 5.Creagan ET, Kovach JS, Moertel CG, Frytak S, Kvols LK. A phase I clinical trial of recombinant human tumor necrosis factor. Cancer. 1988;62:2467–71. doi: 10.1002/1097-0142(19881215)62:12<2467::aid-cncr2820621202>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 6.Creaven PJ, Plager JE, Dupere S, Huben RP, Takita H, Mittelman A, Proefrock A. Phase I clinical trial of recombinant human tumor necrosis factor. Cancer Chemother Pharmacol. 1987;20:137–44. doi: 10.1007/BF00253968. [DOI] [PubMed] [Google Scholar]
- 7.Daniel PT, Wieder T, Sturm I, Schulze-Osthoff K. The kiss of death: promises and failures of death receptors and ligands in cancer therapy. Leukemia. 2001;15:1022–32. doi: 10.1038/sj.leu.2402169. [DOI] [PubMed] [Google Scholar]
- 8.Galle PR, Hofmann WJ, Walczak H, Schaller H, Otto G, Stremmel W, Krammer PH, Runkel L. Involvement of the CD95 (APO-1/Fas) receptor and ligand in liver damage. J Exp Med. 1995;182:1223–30. doi: 10.1084/jem.182.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hersh EM, Metch BS, Muggia FM, Brown TD, Whitehead RP, Budd GT, Rinehart JJ, Crawford ED, Bonnet JD, Behrens BC. Phase II studies of recombi-nant human tumor necrosis factor alpha in patients with malignant disease: a summary of the Southwest Oncology Group experience. J Immunother. 1991;10:426–31. doi: 10.1097/00002371-199112000-00006. [DOI] [PubMed] [Google Scholar]
- 10.Ogasawara J, Watanabe-Fukunaga R, Adachi M, Matsuzawa A, Kasugai T, Kitamura Y, Itoh N, Suda T, Nagata S. Lethal effect of the anti-Fas antibody in mice. Nature. 1993;364:806–9. doi: 10.1038/364806a0. [DOI] [PubMed] [Google Scholar]
- 11.Pitti RM, Marsters SA, Ruppert S, Donahue CJ, Moore A, Ashkenazi A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J Biol Chem. 1996;271:12687–90. doi: 10.1074/jbc.271.22.12687. [DOI] [PubMed] [Google Scholar]
- 12.Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK, Sutherland GR, Smith TD, Rauch C, Smith CA, Goodwin RG. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity. 1995;3:673–82. doi: 10.1016/1074-7613(95)90057-8. [DOI] [PubMed] [Google Scholar]
- 13.Pan G, O'Rourke K, Chinnaiyan AM, Gentz R, Ebner R, Ni J, Dixit VM. The receptor for the cytotoxic ligand TRAIL. Science. 1997;276:111–3. doi: 10.1126/science.276.5309.111. [DOI] [PubMed] [Google Scholar]
- 14.Pan G, Ni J, Wei YF, Yu G, Gentz R, Dixit VM. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science. 1997;277:815–8. doi: 10.1126/science.277.5327.815. [DOI] [PubMed] [Google Scholar]
- 15.Screaton GR, Mongkolsapaya J, Xu XN, Cowper AE, McMichael AJ, Bell JI. TRICK2, a new alternatively spliced receptor that transduces the cytotoxic signal from TRAIL. Curr Biol. 1997;7:693–6. doi: 10.1016/s0960-9822(06)00297-1. [DOI] [PubMed] [Google Scholar]
- 16.Sheridan JP, Marsters SA, Pitti RM, Gurney A, Skubatch M, Baldwin D, Ramakrishnan L, Gray CL, Baker K, Wood WI, Goddard AD, Godowski P, Ashkenazi A. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science. 1997;277:818–21. doi: 10.1126/science.277.5327.818. [DOI] [PubMed] [Google Scholar]
- 17.Walczak H, Degli-Esposti MA, Johnson RS, Smolak PJ, Waugh JY, Boiani N, Timour MS, Gerhart MJ, Schooley KA, Smith CA, Goodwin RG, Rauch CT. TRAIL-R2: a novel apoptosis-mediating receptor for TRAIL. EMBO J. 1997;16:5386–97. doi: 10.1093/emboj/16.17.5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu GS, Burns TF, McDonald ER, 3rd, Jiang W, Meng R, Krantz ID, Kao G, Gan DD, Zhou JY, Muschel R, Hamilton SR, Spinner NB, Markowitz S, Wu G, el-Deiry WS. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat Genet. 1997;17:141–3. doi: 10.1038/ng1097-141. [DOI] [PubMed] [Google Scholar]
- 19.Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M, Chin W, Jones J, Woodward A, Le T, Smith C, Smolak P, Goodwin RG, Rauch CT, Schuh JC, Lynch DH. Tumoricidal activity of tumor necrosis factor-related apoptosis-induc-ing ligand in vivo. Nat Med. 1999;5:157–63. doi: 10.1038/5517. [DOI] [PubMed] [Google Scholar]
- 20.Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA, Blackie C, Chang L, McMurtrey AE, Hebert A, DeForge L, Koumenis IL, Lewis D, Harris L, Bussiere J, Koeppen H, Shahrokh Z, Schwall RH. Safety and antitumor activity of recombinant soluble Apo2 ligand. J Clin Invest. 1999;104:155–62. doi: 10.1172/JCI6926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–8. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 22.Zhou T, Mountz JD, Kimberly RP. Immunobiology of tumor necrosis factor receptor superfamily. Immunol Res. 2002;26:323–36. doi: 10.1385/IR:26:1-3:323. [DOI] [PubMed] [Google Scholar]
- 23.Chan FK, Chun HJ, Zheng L, Siegel RM, Bui KL, Lenardo MJ. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science. 2000;288:2351–4. doi: 10.1126/science.288.5475.2351. [DOI] [PubMed] [Google Scholar]
- 24.Chan FK, Siegel RM, Zacharias D, Swofford R, Holmes KL, Tsien RY, Lenardo MJ. Fluorescence resonance energy transfer analysis of cell surface receptor interactions and signaling using spectral variants of the green fluorescent protein. Cytometry. 2001;44:361–8. doi: 10.1002/1097-0320(20010801)44:4<361::aid-cyto1128>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 25.Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, Peter ME. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995;14:5579–88. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Slee EA, Adrain C, Martin SJ. Executioner caspase-3, -6, and -7 perform distinct, non-redundant roles during the demolition phase of apoptosis. J Biol Chem. 2001;276:7320–6. doi: 10.1074/jbc.M008363200. [DOI] [PubMed] [Google Scholar]
- 27.Lee KH, Feig C, Tchikov V, Schickel R, Hallas C, Schutze S, Peter ME, Chan AC. The role of receptor internalization in CD95 signaling. EMBO J. 2006;25:1009–23. doi: 10.1038/sj.emboj.7601016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roos WP, Kaina B. DNA damage-induced cell death by apoptosis. Trends Mol Med. 2006;12:440–50. doi: 10.1016/j.molmed.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 29.Willis SN, Fletcher JI, Kaufmann T, van Delft MF, Chen L, Czabotar PE, Ierino H, Lee EF, Fairlie WD, Bouillet P, Strasser A, Kluck RM, Adams JM, Huang DC. Apoptosis initiated when BH3 ligands engage multiple Bcl-2 homologs, not Bax or Bak. Science. 2007;315:856–9. doi: 10.1126/science.1133289. [DOI] [PubMed] [Google Scholar]
- 30.Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 31.Acehan D, Jiang X, Morgan DG, Heuser JE, Wang X, Akey CW. Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol Cell. 2002;9:423–32. doi: 10.1016/s1097-2765(02)00442-2. [DOI] [PubMed] [Google Scholar]
- 32.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–89. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 33.Barnhart BC, Alappat EC, Peter ME. The CD95 type I/type II model. Semin Immunol. 2003;15:185–93. doi: 10.1016/s1044-5323(03)00031-9. [DOI] [PubMed] [Google Scholar]
- 34.Golks A, Brenner D, Fritsch C, Krammer PH, Lavrik IN. c-FLIPR, a new regulator of death receptor-induced apoptosis. J Biol Chem. 2005;280:14507–13. doi: 10.1074/jbc.M414425200. [DOI] [PubMed] [Google Scholar]
- 35.Micheau O, Thome M, Schneider P, Holler N, Tschopp J, Nicholson DW, Briand C, Grutter MG. The long form of FLIP is an activator of caspase-8 at the Fas death-inducing signaling complex. J Biol Chem. 2002;277:45162–71. doi: 10.1074/jbc.M206882200. [DOI] [PubMed] [Google Scholar]
- 36.Srinivasula SM, Hegde R, Saleh A, Datta P, Shiozaki E, Chai J, Lee RA, Robbins PD, Fernandes-Alnemri T, Shi Y, Alnemri ES. A conserved XIAP-interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature. 2001;410:112–6. doi: 10.1038/35065125. [DOI] [PubMed] [Google Scholar]
- 37.Falschlehner C, Emmerich CH, Gerlach B, Walczak H. TRAIL signalling: decisions between life and death. Int J Biochem Cell Biol. 2007;39:1462–75. doi: 10.1016/j.biocel.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 38.Bossen C, Ingold K, Tardivel A, Bodmer JL, Gaide O, Hertig S, Ambrose C, Tschopp J, Schneider P. Interactions of tumor necrosis factor (TNF) and TNF receptor family members in the mouse and human. J Biol Chem. 2006;281:13964–71. doi: 10.1074/jbc.M601553200. [DOI] [PubMed] [Google Scholar]
- 39.Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science. 2002;296:1634–5. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- 40.Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 41.Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114:181–90. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- 42.Wajant H, Scheurich P. Tumor necrosis factor receptor-associated factor (TRAF) 2 and its role in TNF signaling. Int J Biochem Cell Biol. 2001;33:19–32. doi: 10.1016/s1357-2725(00)00064-9. [DOI] [PubMed] [Google Scholar]
- 43.Devin A, Cook A, Lin Y, Rodriguez Y, Kelliher M, Liu Z. The distinct roles of TRAF2 and RIP in IKK activation by TNF-R1: TRAF2 recruits IKK to TNF-R1 while RIP mediates IKK activation. Immunity. 2000;12:419–29. doi: 10.1016/s1074-7613(00)80194-6. [DOI] [PubMed] [Google Scholar]
- 44.Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity. 1998;8:297–303. doi: 10.1016/s1074-7613(00)80535-x. [DOI] [PubMed] [Google Scholar]
- 45.Yeh WC, Shahinian A, Speiser D, Kraunus J, Billia F, Wakeham A, de la Pompa JL, Ferrick D, Hum B, Iscove N, Ohashi P, Rothe M, Goeddel DV, Mak TW. Early lethality, functional NF-kappaB activation, and increased sensitivity to TNF-induced cell death in TRAF2-deficient mice. Immunity. 1997;7:715–25. doi: 10.1016/s1074-7613(00)80391-x. [DOI] [PubMed] [Google Scholar]
- 46.Wroblewski VJ, Witcher DR, Becker GW, Davis KA, Dou S, Micanovic R, Newton CM, Noblitt TW, Richardson JM, Song HY, Hale JE. Decoy receptor 3 (DcR3) is proteolytically processed to a metabolic fragment having differential activities against Fas ligand and LIGHT. Biochem Pharmacol. 2003;65:657–67. doi: 10.1016/s0006-2952(02)01612-x. [DOI] [PubMed] [Google Scholar]
- 47.Miethke T, Vabulas R, Bittlingmaier R, Heeg K, Wagner H. Mechanisms of peripheral T cell deletion: anergized T cells are Fas resistant but undergo proliferation-associated apoptosis. Eur J Immunol. 1996;26:1459–67. doi: 10.1002/eji.1830260709. [DOI] [PubMed] [Google Scholar]
- 48.Rieux-Laucat F, Le Deist F, Hivroz C, Roberts IA, Debatin KM, Fischer A, de Villartay JP. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995;268:1347–9. doi: 10.1126/science.7539157. [DOI] [PubMed] [Google Scholar]
- 49.Schneider P, Holler N, Bodmer JL, Hahne M, Frei K, Fontana A, Tschopp J. Conversion of membrane-bound Fas(CD95) ligand to its soluble form is associated with downregulation of its proapoptotic activity and loss of liver toxicity. J Exp Med. 1998;187:1205–13. doi: 10.1084/jem.187.8.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Knox PG, Milner AE, Green NK, Eliopoulos AG, Young LS. Inhibition of metalloproteinase cleavage enhances the cytotoxicity of Fas ligand. J Immunol. 2003;170:677–85. doi: 10.4049/jimmunol.170.2.677. [DOI] [PubMed] [Google Scholar]
- 51.Kischkel FC, Lawrence DA, Tinel A, LeBlanc H, Virmani A, Schow P, Gazdar A, Blenis J, Arnott D, Ashkenazi A. Death receptor recruitment of endogenous caspase-10 and apoptosis initiation in the absence of caspase-8. J Biol Chem. 2001;276:46639–46. doi: 10.1074/jbc.M105102200. [DOI] [PubMed] [Google Scholar]
- 52.Sprick MR, Rieser E, Stahl H, Grosse-Wilde A, Weigand MA, Walczak H. Caspase-10 is recruited to and activated at the native TRAIL and CD95 death-inducing signalling complexes in a FADD-dependent manner but can not functionally substitute caspase-8. EMBO J. 2002;21:4520–30. doi: 10.1093/emboj/cdf441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schneider P, Bodmer JL, Thome M, Hofmann K, Holler N, Tschopp J. Characterization of two receptors for TRAIL. FEBS Lett. 1997;416:329–34. doi: 10.1016/s0014-5793(97)01231-3. [DOI] [PubMed] [Google Scholar]
- 54.Degli-Esposti MA, Smolak PJ, Walczak H, Waugh J, Huang CP, DuBose RF, Goodwin RG, Smith CA. Cloning and characterization of TRAIL-R3, a novel member of the emerging TRAIL receptor family. J Exp Med. 1997;186:1165–70. doi: 10.1084/jem.186.7.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Degli-Esposti MA, Dougall WC, Smolak PJ, Waugh JY, Smith CA, Goodwin RG. The novel receptor TRAIL-R4 induces NF-kappaB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity. 1997;7:813–20. doi: 10.1016/s1074-7613(00)80399-4. [DOI] [PubMed] [Google Scholar]
- 56.Emery JG, McDonnell P, Burke MB, Deen KC, Lyn S, Silverman C, Dul E, Appelbaum ER, Eichman C, DiPrinzio R, Dodds RA, James IE, Rosenberg M, Lee JC, Young PR. Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J Biol Chem. 1998;273:14363–7. doi: 10.1074/jbc.273.23.14363. [DOI] [PubMed] [Google Scholar]
- 57.Truneh A, Sharma S, Silverman C, Khandekar S, Reddy MP, Deen KC, McLaughlin MM, Srinivasula SM, Livi GP, Marshall LA, Alnemri ES, Williams WV, Doyle ML. Temperature-sensitive differential affinity of TRAIL for its receptors. DR5 is the highest affinity receptor. J Biol Chem. 2000;275:23319–25. doi: 10.1074/jbc.M910438199. [DOI] [PubMed] [Google Scholar]
- 58.Boyce BF, Xing L. Biology of RANK, RANKL, and osteoprotegerin. Arthritis Res Ther. 2007;9:S1. doi: 10.1186/ar2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lejeune F, Lienard D, Eggermont A, Schraffordt Koops H, Rosenkaimer F, Gerain J, Klaase J, Kroon B, Vanderveken J, Schmitz P. Administration of high-dose tumor necrosis factor alpha by isolation perfusion of the limbs. Rationale and results. J Infus Chemother. 1995;5:73–81. [PubMed] [Google Scholar]
- 60.Eggermont AM, ten Hagen TL. Isolated limb perfusion for extremity soft-tissue sarcomas, in-transit metastases, and other unresectable tumors: credits, debits, and future perspectives. Curr Oncol Rep. 2001;3:359–67. doi: 10.1007/s11912-001-0090-8. [DOI] [PubMed] [Google Scholar]
- 61.Brunstein F, Santos ID, Ferreira LM, van Tiel ST, Eggermont AM, Ten Hagen TL. Histamine combined with melphalan in isolated limb perfusion for the treatment of locally advanced soft tissue sarcomas: preclinical studies in rats. Acta Cir Bras. 2005;20:275–9. doi: 10.1590/s0102-86502005000400003. [DOI] [PubMed] [Google Scholar]
- 62.Wuest T, Gerlach E, Banerjee D, Gerspach J, Moosmayer D, Pfizenmaier K. TNF-Selectokine: a novel prodrug generated for tumor targeting and site-specific activation of tumor necrosis factor. Oncogene. 2002;21:4257–65. doi: 10.1038/sj.onc.1205193. [DOI] [PubMed] [Google Scholar]
- 63.Rosenblum MG, Cheung L, Murray JL, Bartholomew R. Antibody-mediated delivery of tumor necrosis factor (TNF-alpha): improvement of cytotoxicity and reduction of cellular resistance. Cancer Commun. 1991;3:21–7. [PubMed] [Google Scholar]
- 64.Rosenblum MG, Cheung L, Mujoo K, Murray JL. An antimelanoma immunotoxin containing recombinant human tumor necrosis factor: tissue disposition, pharmacokinetic, and therapeutic studies in xenograft models. Cancer Immunol Immunother. 1995;40:322–8. doi: 10.1007/BF01519633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Scherf U, Benhar I, Webber KO, Pastan I, Brinkmann U. Cytotoxic and antitumor activity of a recombinant tumor necrosis factor-B1(Fv) fusion protein on LeY antigen-expressing human cancer cells. Clin Cancer Res. 1996;2:1523–31. [PubMed] [Google Scholar]
- 66.Curnis F, Sacchi A, Borgna L, Magni F, Gasparri A, Corti A. Enhancement of tumor necrosis factor alpha antitumor immunotherapeutic properties by targeted delivery to aminopeptidase N (CD13) Nat Biotechnol. 2000;18:1185–90. doi: 10.1038/81183. [DOI] [PubMed] [Google Scholar]
- 67.Sacchi A, Gasparri A, Gallo-Stampino C, Toma S, Curnis F, Corti A. Synergistic antitumor activity of cisplatin, paclitaxel, and gemcitabine with tumor vasculature-targeted tumor necrosis factor-alpha. Clin Cancer Res. 2006;12:175–82. doi: 10.1158/1078-0432.CCR-05-1147. [DOI] [PubMed] [Google Scholar]
- 68.Liu Y, Zhang W, Cheung LH, Niu T, Wu Q, Li C, Van Pelt CS, Rosenblum MG. The antimelanoma immunocytokine scFvMEL/ TNF shows reduced toxicity and potent antitumor activity against human tumor xenografts. Neoplasia. 2006;8:384–93. doi: 10.1593/neo.06121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Christ O, Matzku S, Burger C, Zoller M. Interleukin 2-antibody and tumor necrosis factor-antibody fusion proteins induce different antitumor immune responses in vivo. Clin Cancer Res. 2001;7:1385–97. [PubMed] [Google Scholar]
- 70.Rosenblum MG, Horn SA, Cheung LH. A novel recombinant fusion toxin targeting HER-2/NEU-over-expressing cells and containing human tumor necrosis factor. Int J Cancer. 2000;88:267–73. doi: 10.1002/1097-0215(20001015)88:2<267::aid-ijc19>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 71.Sharifi J, Khawli LA, Hu P, Li J, Epstein AL. Generation of human interferon gamma and tumor Necrosis factor alpha chimeric TNT-3 fusion proteins. Hybrid Hybridomics. 2002;21:421–32. doi: 10.1089/153685902321043954. [DOI] [PubMed] [Google Scholar]
- 72.Scanlan MJ, Raj BK, Calvo B, Garin-Chesa P, Sanz-Moncasi MP, Healey JH, Old LJ, Rettig WJ. Molecular cloning of fibroblast activation protein alpha, a member of the serine protease family selectively expressed in stromal fibroblasts of epithelial cancers. Proc Natl Acad Sci USA. 1994;91:5657–61. doi: 10.1073/pnas.91.12.5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gerspach J, Muller D, Munkel S, Selchow O, Nemeth J, Noack M, Petrul H, Menrad A, Wajant H, Pfizenmaier K. Restoration of membrane TNF-like activity by cell surface targeting and matrix metalloproteinase-mediated processing of a TNF prodrug. Cell Death Differ. 2006;13:273–84. doi: 10.1038/sj.cdd.4401735. [DOI] [PubMed] [Google Scholar]
- 74.Gerspach J, Nemeth J, Munkel S, Wajant H, Pfizenmaier K. Target-selective activation of a TNF prodrug by urokinase-type plasminogen activator (uPA) mediated proteolytic processing at the cell surface. Cancer Immunol Immunother. 2006;55:1590–600. doi: 10.1007/s00262-006-0162-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Andreasen PA, Kjoller L, Christensen L, Duffy MJ. The urokinase-type plasminogen activator system in cancer metastasis: a review. Int J Cancer. 1997;72:1–22. doi: 10.1002/(sici)1097-0215(19970703)72:1<1::aid-ijc1>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 76.Wajant H, Gerspach J, Pfizenmaier K. Tumor therapeutics by design: targeting and activation of death receptors. Cytokine Growth Factor Rev. 2005;16:55–76. doi: 10.1016/j.cytogfr.2004.12.001. [DOI] [PubMed] [Google Scholar]
- 77.Poynard T, McHutchison J, Manns M, Trepo C, Lindsay K, Goodman Z, Ling MH, Albrecht J. Impact of pegylated inter-feron alfa-2b and ribavirin on liver fibrosis in patients with chronic hepatitis C. Gastroenterology. 2002;122:1303–13. doi: 10.1053/gast.2002.33023. [DOI] [PubMed] [Google Scholar]
- 78.Yamamoto Y, Tsutsumi Y, Yoshioka Y, Nishibata T, Kobayashi K, Okamoto T, Mukai Y, Shimizu T, Nakagawa S, Nagata S, Mayumi T. Site-specific PEGylation of a lysine-deficient TNF-alpha with full bioactivity. Nat Biotechnol. 2003;21:546–52. doi: 10.1038/nbt812. [DOI] [PubMed] [Google Scholar]
- 79.Yoshioka Y, Tsutsumi Y, Ikemizu S, Yamamoto Y, Shibata H, Nishibata T, Mukai Y, Okamoto T, Taniai M, Kawamura M, Abe Y, Nakagawa S, Nagata S, Yamagata Y, Mayumi T. Optimal site-specific PEGylation of mutant TNF-alpha improves its antitumor potency. Biochem Biophys Res Commun. 2004;315:808–14. doi: 10.1016/j.bbrc.2004.01.125. [DOI] [PubMed] [Google Scholar]
- 80.Shibata H, Yoshioka Y, Ikemizu S, Kobayashi K, Yamamoto Y, Mukai Y, Okamoto T, Taniai M, Kawamura M, Abe Y, Nakagawa S, Hayakawa T, Nagata S, Yamagata Y, Mayumi T, Kamada H, Tsutsumi Y. Functionalization of tumor necrosis factor-alpha using phage display technique and PEGylation improves its antitumor therapeutic window. Clin Cancer Res. 2004;10:8293–300. doi: 10.1158/1078-0432.CCR-04-0770. [DOI] [PubMed] [Google Scholar]
- 81.Kircheis R, Kichler A, Wallner G, Kursa M, Ogris M, Felzmann T, Buchberger M, Wagner E. Coupling of cell-binding ligands to polyethylenimine for targeted gene delivery. Gene Ther. 1997;4:409–18. doi: 10.1038/sj.gt.3300418. [DOI] [PubMed] [Google Scholar]
- 82.Zou SM, Erbacher P, Remy JS, Behr JP. Systemic linear polyethylenimine (L-PEI)-mediated gene delivery in the mouse. J Gene Med. 2000;2:128–34. doi: 10.1002/(SICI)1521-2254(200003/04)2:2<128::AID-JGM95>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 83.Kircheis R, Wightman L, Kursa M, Ostermann E, Wagner E. Tumor-targeted gene delivery: an attractive strategy to use highly active effector molecules in cancer treatment. Gene Ther. 2002;9:731–5. doi: 10.1038/sj.gt.3301748. [DOI] [PubMed] [Google Scholar]
- 84.Ogris M, Walker G, Blessing T, Kircheis R, Wolschek M, Wagner E. Tumor-targeted gene therapy: strategies for the preparation of ligand-polyethylene glycol-polyethylenimine/DNA complexes. J Control Release. 2003;91:173–81. doi: 10.1016/s0168-3659(03)00230-x. [DOI] [PubMed] [Google Scholar]
- 85.Datta R, Rubin E, Sukhatme V, Qureshi S, Hallahan D, Weichselbaum RR, Kufe DW. Ionizing radiation activates transcription of the EGR1 gene via CArG elements. Proc Natl Acad Sci USA. 1992;89:10149–53. doi: 10.1073/pnas.89.21.10149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Weichselbaum RR, Kufe DW, Hellman S, Rasmussen HS, King CR, Fischer PH, Mauceri HJ. Radiation-induced tumour necrosis factor-alpha expression: clinical application of transcriptional and physical targeting of gene therapy. Lancet Oncol. 2002;3:665–71. doi: 10.1016/s1470-2045(02)00900-2. [DOI] [PubMed] [Google Scholar]
- 87.Hallahan DE, Mauceri HJ, Seung LP, Dunphy EJ, Wayne JD, Hanna NN, Toledano A, Hellman S, Kufe DW, Weichselbaum RR. Spatial and temporal control of gene therapy using ionizing radiation. Nat Med. 1995;1:786–91. doi: 10.1038/nm0895-786. [DOI] [PubMed] [Google Scholar]
- 88.Hallahan DE, Vokes EE, Rubin SJ, O'Brien S, Samuels B, Vijaykumar S, Kufe DW, Phillips R, Weichselbaum RR. Phase I dose-escalation study of tumor necrosis factor-alpha and concomitant radiation therapy. Cancer J Sci Am. 1995;1:204–9. [PubMed] [Google Scholar]
- 89.Chung TD, Mauceri HJ, Hallahan DE, Yu JJ, Chung S, Grdina WL, Yajnik S, Kufe DW, Weichselbaum RR. Tumor necrosis factor-alpha-based gene therapy enhances radiation cytotoxicity in human prostate cancer. Cancer Gene Ther. 1998;5:344–9. [PubMed] [Google Scholar]
- 90.Staba MJ, Mauceri HJ, Kufe DW, Hallahan DE, Weichselbaum RR. Adenoviral TNF-alpha gene therapy and radiation damage tumor vasculature in a human malignant glioma xenograft. Gene Ther. 1998;5:293–300. doi: 10.1038/sj.gt.3300594. [DOI] [PubMed] [Google Scholar]
- 91.Hanna NN, Mauceri HJ, Wayne JD, Hallahan DE, Kufe DW, Weichselbaum RR. Virally directed cytosine deaminase/5-fluorocytosine gene therapy enhances radiation response in human cancer xenografts. Cancer Res. 1997;57:4205–9. [PubMed] [Google Scholar]
- 92.Gupta VK, Park JO, Jaskowiak NT, Mauceri HJ, Seetharam S, Weichselbaum RR, Posner MC. Combined gene therapy and ionizing radiation is a novel approach to treat human esophageal adenocarcinoma. Ann Surg Oncol. 2002;9:500–4. doi: 10.1007/BF02557275. [DOI] [PubMed] [Google Scholar]
- 93.Mauceri HJ, Hanna NN, Wayne JD, Hallahan DE, Hellman S, Weichselbaum RR. Tumor necrosis factor alpha (TNF-alpha) gene therapy targeted by ionizing radiation selectively damages tumor vasculature. Cancer Res. 1996;56:4311–4. [PubMed] [Google Scholar]
- 94.Rasmussen H, Rasmussen C, Lempicki M, Durham R, Brough D, King CR, Weichselbaum R. TNFerade Biologic: preclinical toxicology of a novel adenovec-tor with a radiation-inducible promoter, carrying the human tumor necrosis factor alpha gene. Cancer Gene Ther. 2002;9:951–7. doi: 10.1038/sj.cgt.7700518. [DOI] [PubMed] [Google Scholar]
- 95.Yonehara S, Ishii A, Yonehara M. A cell-killing monoclonal antibody (anti-Fas) to a cell surface antigen co-downregulated with the receptor of tumor necrosis factor. J Exp Med. 1989;169:1747–56. doi: 10.1084/jem.169.5.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Trauth BC, Klas C, Peters AM, Matzku S, Moller P, Falk W, Debatin KM, Krammer PH. Monoclonal antibody-mediated tumor regression by induction of apoptosis. Science. 1989;245:301–5. doi: 10.1126/science.2787530. [DOI] [PubMed] [Google Scholar]
- 97.Kondo T, Suda T, Fukuyama H, Adachi M, Nagata S. Essential roles of the Fas ligand in the development of hepatitis. Nat Med. 1997;3:409–13. doi: 10.1038/nm0497-409. [DOI] [PubMed] [Google Scholar]
- 98.Nishimura Y, Hirabayashi Y, Matsuzaki Y, Musette P, Ishii A, Nakauchi H, Inoue T, Yonehara S. In vivo analysis of Fas antigen-mediated apoptosis: effects of agonistic anti-mouse Fas mAb on thymus, spleen and liver. Int Immunol. 1997;9:307–16. doi: 10.1093/intimm/9.2.307. [DOI] [PubMed] [Google Scholar]
- 99.Ichikawa K, Yoshida-Kato H, Ohtsuki M, Ohsumi J, Yamaguchi J, Takahashi S, Tani Y, Watanabe M, Shiraishi A, Nishioka K, Yonehara S, Serizawa N. A novel murine anti-human Fas mAb which mitigates lymphadenopathy without hepatotoxicity. Int Immunol. 2000;12:555–62. doi: 10.1093/intimm/12.4.555. [DOI] [PubMed] [Google Scholar]
- 100.Nakayama J, Ogawa Y, Yoshigae Y, Onozawa Y, Yonemura A, Saito M, Ichikawa K, Yamoto T, Komai T, Tatsuta T, Ohtsuki M. A humanized anti-human Fas antibody, R-125224, induces apoptosis in type I activated lymphocytes but not in type II cells. Int Immunol. 2006;18:113–24. doi: 10.1093/intimm/dxh353. [DOI] [PubMed] [Google Scholar]
- 101.Aoki K, Kurooka M, Chen JJ, Petryniak J, Nabel EG, Nabel GJ. Extracellular matrix interacts with soluble CD95L: retention and enhancement of cytotoxicity. Nat Immunol. 2001;2:333–7. doi: 10.1038/86336. [DOI] [PubMed] [Google Scholar]
- 102.Jung G, Grosse-Hovest L, Krammer PH, Rammensee HG. Target cell-restricted triggering of the CD95 (APO-1/Fas) death receptor with bispecific antibody fragments. Cancer Res. 2001;61:1846–8. [PubMed] [Google Scholar]
- 103.Samel D, Muller D, Gerspach J, Assohou-Luty C, Sass G, Tiegs G, Pfizenmaier K, Wajant H. Generation of a FasL-based proapoptotic fusion protein devoid of systemic toxicity due to cell-surface antigen-restricted Activation. J Biol Chem. 2003;278:32077–82. doi: 10.1074/jbc.M304866200. [DOI] [PubMed] [Google Scholar]
- 104.Henry LR, Lee HO, Lee JS, Klein-Szanto A, Watts P, Ross EA, Chen WT, Cheng JD. Clinical implications of fibroblast activation protein in patients with colon cancer. Clin Cancer Res. 2007;13:1736–41. doi: 10.1158/1078-0432.CCR-06-1746. [DOI] [PubMed] [Google Scholar]
- 105.Bremer E, Samplonius DF, Peipp M, van Genne L, Kroesen BJ, Fey GH, Gramatzki M, de Leij LF, Helfrich W. Target cell-restricted apoptosis induction of acute leukemic T cells by a recombinant tumor necrosis factor-related apoptosis-inducing ligand fusion protein with specificity for human CD7. Cancer Res. 2005;65:3380–8. doi: 10.1158/0008-5472.CAN-04-2756. [DOI] [PubMed] [Google Scholar]
- 106.Watermann I, Gerspach J, Lehne M, Seufert J, Schneider B, Pfizenmaier K, Wajant H. Activation of CD95L fusion protein prodrugs by tumor-associated proteases. Cell Death Differ. 2007;14:765–74. doi: 10.1038/sj.cdd.4402051. [DOI] [PubMed] [Google Scholar]
- 107.Bremer E, ten Cate B, Samplonius DF, Mueller N, Wajant H, Stel AJ, Chamuleau M, van de Loosdrecht AA, Stieglmaier J, Fey GH, Helfrich W. Superior activity of fusion protein scFvRit:sFasL over cotreatment with rituximab and Fas agonists. Cancer Res. 2008;68:597–604. doi: 10.1158/0008-5472.CAN-07-5171. [DOI] [PubMed] [Google Scholar]
- 108.Arai H, Gordon D, Nabel EG, Nabel GJ. Gene transfer of Fas ligand induces tumor regression in vivo. Proc Natl Acad Sci USA. 1997;94:13862–7. doi: 10.1073/pnas.94.25.13862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hedlund TE, Meech SJ, Srikanth S, Kraft AS, Miller GJ, Schaack JB, Duke RC. Adenovirus-mediated expression of Fas ligand induces apoptosis of human prostate cancer cells. Cell Death Differ. 1999;6:175–82. doi: 10.1038/sj.cdd.4400477. [DOI] [PubMed] [Google Scholar]
- 110.Zhang H, Gao G, Clayburne G, Schumacher HR. Elimination of rheumatoid synovium in situ using a Fas ligand ‘gene scalpel’. Arthritis Res Ther. 2005;7:R1235–43. doi: 10.1186/ar1811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Kim SH, Kim S, Oligino TJ, Robbins PD. Effective treatment of established mouse collagen-induced arthritis by systemic administration of dendritic cells genetically modified to express FasL. Mol Ther. 2002;6:584–90. [PubMed] [Google Scholar]
- 112.Morelli AE, Larregina AT, Smith-Arica J, Dewey RA, Southgate TD, Ambar B, Fontana A, Castro MG, Lowenstein PR. Neuronal and glial cell type-specific promoters within adenovirus recombinants restrict the expression of the apoptosis-inducing molecule Fas ligand to predetermined brain cell types, and abolish peripheral liver toxicity. J Gen Virol. 1999;80:571–83. doi: 10.1099/0022-1317-80-3-571. [DOI] [PubMed] [Google Scholar]
- 113.Aoki K, Akyurek LM, San H, Leung K, Parmacek MS, Nabel EG, Nabel GJ. Restricted expression of an adenoviral vector encoding Fas ligand (CD95L) enhances safety for cancer gene therapy. Mol Ther. 2000;1:555–65. doi: 10.1006/mthe.2000.0076. [DOI] [PubMed] [Google Scholar]
- 114.Rubinchik S, Wang D, Yu H, Fan F, Luo M, Norris JS, Dong JY. A complex adenovirus vector that delivers FASL-GFP with combined prostate-specific and tetracy-cline-regulated expression. Mol Ther. 2001;4:416–26. doi: 10.1006/mthe.2001.0478. [DOI] [PubMed] [Google Scholar]
- 115.Kodaira H, Kume A, Ogasawara Y, Urabe M, Kitano K, Kakizuka A, Ozawa K. Fas and mutant estrogen receptor chimeric gene: a novel suicide vector for tamoxifen-inducible apoptosis. Jpn J Cancer Res. 1998;89:741–7. doi: 10.1111/j.1349-7006.1998.tb03279.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hyer ML, Voelkel-Johnson C, Rubinchik S, Dong J, Norris JS. Intracellular Fas lig-and expression causes Fas-mediated apoptosis in human prostate cancer cells resistant to monoclonal antibody-induced apoptosis. Mol Ther. 2000;2:348–58. doi: 10.1006/mthe.2000.0139. [DOI] [PubMed] [Google Scholar]
- 117.Sudarshan S, Holman DH, Hyer ML, Voelkel-Johnson C, Dong JY, Norris JS. In vitro efficacy of Fas ligand gene therapy for the treatment of bladder cancer. Cancer Gene Ther. 2005;12:12–8. doi: 10.1038/sj.cgt.7700746. [DOI] [PubMed] [Google Scholar]
- 118.Rubinchik S, Yu H, Woraratanadharm J, Voelkel-Johnson C, Norris JS, Dong JY. Enhanced apoptosis of glioma cell lines is achieved by co-delivering FasL-GFP and TRAIL with a complex Ad5 vector. Cancer Gene Ther. 2003;10:814–22. doi: 10.1038/sj.cgt.7700651. [DOI] [PubMed] [Google Scholar]
- 119.Norris JS, Bielawska A, Day T, El-Zawahri A, ElOjeimy S, Hannun Y, Holman D, Hyer M, Landon C, Lowe S, Dong JY, McKillop J, Norris K, Obeid L, Rubinchik S, Tavassoli M, Tomlinson S, Voelkel-Johnson C, Liu X. Combined therapeutic use of AdGFPFasL and small molecule inhibitors of ceramide metabolism in prostate and head and neck cancers: a status report. Cancer Gene Ther. 2006;13:1045–51. doi: 10.1038/sj.cgt.7700965. [DOI] [PubMed] [Google Scholar]
- 120.Koschny R, Ganten TM, Sykora J, Haas TL, Sprick MR, Kolb A, Stremmel W, Walczak H. TRAIL/bortezomib cotreatment is potentially hepatotoxic but induces cancer-specific apoptosis within a therapeutic window. Hepatology. 2007;45:649–58. doi: 10.1002/hep.21555. [DOI] [PubMed] [Google Scholar]
- 121.Leverkus M, Sprick MR, Wachter T, Mengling T, Baumann B, Serfling E, Brocker EB, Goebeler M, Neumann M, Walczak H. Proteasome inhibition results in TRAIL sensitization of primary keratinocytes by removing the resistance-mediating block of effector caspase maturation. Mol Cell Biol. 2003;23:777–90. doi: 10.1128/MCB.23.3.777-790.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ganten TM, Koschny R, Haas TL, Sykora J, Li-Weber M, Herzer K, Walczak H. Proteasome inhibition sensitizes hepato-cellular carcinoma cells, but not human hepatocytes, to TRAIL. Hepatology. 2005;42:588–97. doi: 10.1002/hep.20807. [DOI] [PubMed] [Google Scholar]
- 123.Schneider P. Production of recombinant TRAIL and TRAIL receptor: Fc chimeric proteins. Methods Enzymol. 2000;322:325–45. doi: 10.1016/s0076-6879(00)22031-4. [DOI] [PubMed] [Google Scholar]
- 124.Ganten TM, Koschny R, Sykora J, Schulze-Bergkamen H, Buchler P, Haas TL, Schader MB, Untergasser A, Stremmel W, Walczak H. Preclinical differentiation between apparently safe and potentially hepatotoxic applications of TRAIL either alone or in combination with chemotherapeutic drugs. Clin Cancer Res. 2006;12:2640–6. doi: 10.1158/1078-0432.CCR-05-2635. [DOI] [PubMed] [Google Scholar]
- 125.Gores GJ, Kaufmann SH. Is TRAIL hepatotoxic? Hepatology. 2001;34:3–6. doi: 10.1053/jhep.2001.25173. [DOI] [PubMed] [Google Scholar]
- 126.Lawrence D, Shahrokh Z, Marsters S, Achilles K, Shih D, Mounho B, Hillan K, Totpal K, DeForge L, Schow P, Hooley J, Sherwood S, Pai R, Leung S, Khan L, Gliniak B, Bussiere J, Smith CA, Strom SS, Kelley S, Fox JA, Thomas D, Ashkenazi A. Differential hepatocyte toxicity of recombinant Apo2L/TRAIL versions. Nat Med. 2001;7:383–5. doi: 10.1038/86397. [DOI] [PubMed] [Google Scholar]
- 127.Koschny R, Walczak H, Ganten TM. The promise of TRAIL–potential and risks of a novel anticancer therapy. J Mol Med. 2007;85:923–35. doi: 10.1007/s00109-007-0194-1. [DOI] [PubMed] [Google Scholar]
- 128.Ballet F, Bouma ME, Wang SR, Amit N, Marais J, Infante R. Isolation, culture and characterization of adult human hepato-cytes from surgical liver biopsies. Hepatology. 1984;4:849–54. doi: 10.1002/hep.1840040509. [DOI] [PubMed] [Google Scholar]
- 129.Dunn JC, Tompkins RG, Yarmush ML. Hepatocytes in collagen sandwich: evidence for transcriptional and translational regulation. J Cell Biol. 1992;116:1043–53. doi: 10.1083/jcb.116.4.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bremer E, Kuijlen J, Samplonius D, Walczak H, de Leij L, Helfrich W. Target cell-restricted and -enhanced apoptosis induction by a scFv:sTRAIL fusion protein with specificity for the pancarcinoma-associated antigen EGP2. Int J Cancer. 2004;109:281–90. doi: 10.1002/ijc.11702. [DOI] [PubMed] [Google Scholar]
- 131.Bremer E, Samplonius D, Kroesen BJ, van Genne L, de Leij L, Helfrich W. Exceptionally potent anti-tumor bystander activity of an scFv:sTRAIL fusion protein with specificity for EGP2 toward target antigen-negative tumor cells. Neoplasia. 2004;6:636–45. doi: 10.1593/neo.04229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Buchsbaum DJ, Zhou T, Lobuglio AF. TRAIL receptor-targeted therapy. Future Oncol. 2006;2:493–508. doi: 10.2217/14796694.2.4.493. [DOI] [PubMed] [Google Scholar]
- 133.Buchsbaum DJ, Zhou T, Grizzle WE, Oliver PG, Hammond CJ, Zhang S, Carpenter M, LoBuglio AF. Antitumor efficacy of TRA-8 anti-DR5 monoclonal antibody alone or in combination with chemotherapy and/or radiation therapy in a human breast cancer model. Clin Cancer Res. 2003;9:3731–41. [PubMed] [Google Scholar]
- 134.Ichikawa K, Liu W, Zhao L, Wang Z, Liu D, Ohtsuka T, Zhang H, Mountz JD, Koopman WJ, Kimberly RP, Zhou T. Tumoricidal activity of a novel anti-human DR5 monoclonal antibody without hepatocyte cytotoxicity. Nat Med. 2001;7:954–60. doi: 10.1038/91000. [DOI] [PubMed] [Google Scholar]
- 135.Takeda K, Yamaguchi N, Akiba H, Kojima Y, Hayakawa Y, Tanner JE, Sayers TJ, Seki N, Okumura K, Yagita H, Smyth MJ. Induction of tumor-specific T cell immunity by anti-DR5 antibody therapy. J Exp Med. 2004;199:437–48. doi: 10.1084/jem.20031457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Uno T, Takeda K, Kojima Y, Yoshizawa H, Akiba H, Mittler RS, Gejyo F, Okumura K, Yagita H, Smyth MJ. Eradication of established tumors in mice by a combination antibody-based therapy. Nat Med. 2006;12:693–8. doi: 10.1038/nm1405. [DOI] [PubMed] [Google Scholar]
- 137.Kelley SK, Ashkenazi A. Targeting death receptors in cancer with Apo2L/TRAIL. Curr Opin Pharmacol. 2004;4:333–9. doi: 10.1016/j.coph.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 138.Stieglmaier J, Bremer E, Kellner C, Liebig TM, ten Cate B, Peipp M, Schulze-Koops H, Pfeiffer M, Buhring HJ, Greil J, Oduncu F, Emmerich B, Fey GH, Helfrich W. Selective induction of apoptosis in leukemic B-lymphoid cells by a CD19-specific TRAIL fusion protein. Cancer Immunol Immunother. 2008;57:233–46. doi: 10.1007/s00262-007-0370-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bremer E, Samplonius DF, van Genne L, Dijkstra MH, Kroesen BJ, de Leij LF, Helfrich W. Simultaneous inhibition of epidermal growth factor receptor (EGFR) signaling and enhanced activation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor-mediated apoptosis induction by an scFv:sTRAIL fusion protein with specificity for human EGFR. J Biol Chem. 2005;280:10025–33. doi: 10.1074/jbc.M413673200. [DOI] [PubMed] [Google Scholar]
- 140.Volkmann X, Fischer U, Bahr MJ, Ott M, Lehner F, Macfarlane M, Cohen GM, Manns MP, Schulze-Osthoff K, Bantel H. Increased hepatotoxicity of tumor necrosis factor-related apoptosis-inducing ligand in diseased human liver. Hepatology. 2007;46:1498–508. doi: 10.1002/hep.21846. [DOI] [PubMed] [Google Scholar]
- 141.Wu XX, Ogawa O, Kakehi Y. TRAIL and chemotherapeutic drugs in cancer therapy. Vitam Horm. 2004;67:365–83. doi: 10.1016/S0083-6729(04)67019-1. [DOI] [PubMed] [Google Scholar]
- 142.Gazitt Y. TRAIL is a potent inducer of apoptosis in myeloma cells derived from multiple myeloma patients and is not cytotoxic to hematopoietic stem cells. Leukemia. 1999;13:1817–24. doi: 10.1038/sj.leu.2401501. [DOI] [PubMed] [Google Scholar]
- 143.Mitsiades CS, Treon SP, Mitsiades N, Shima Y, Richardson P, Schlossman R, Hideshima T, Anderson KC. TRAIL/Apo2L ligand selectively induces apoptosis and overcomes drug resistance in multiple myeloma: therapeutic applications. Blood. 2001;98:795–804. doi: 10.1182/blood.v98.3.795. [DOI] [PubMed] [Google Scholar]
- 144.Clodi K, Wimmer D, Li Y, Goodwin R, Jaeger U, Mann G, Gadner H, Younes A. Expression of tumour necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) receptors and sensitivity to TRAIL-induced apoptosis in primary B-cell acute lymphoblastic leukaemia cells. Br J Haematol. 2000;111:580–6. doi: 10.1046/j.1365-2141.2000.02404.x. [DOI] [PubMed] [Google Scholar]
- 145.MacFarlane M, Harper N, Snowden RT, Dyer MJ, Barnett GA, Pringle JH, Cohen GM. Mechanisms of resistance to TRAIL-induced apoptosis in primary B cell chronic lymphocytic leukaemia. Oncogene. 2002;21:6809–18. doi: 10.1038/sj.onc.1205853. [DOI] [PubMed] [Google Scholar]
- 146.Snell V, Clodi K, Zhao S, Goodwin R, Thomas EK, Morris SW, Kadin ME, Cabanillas F, Andreeff M, Younes A. Activity of TNF-related apoptosis-inducing ligand (TRAIL) in haematological malignancies. Br J Haematol. 1997;99:618–24. doi: 10.1046/j.1365-2141.1997.4393250.x. [DOI] [PubMed] [Google Scholar]
- 147.Riccioni R, Pasquini L, Mariani G, Saulle E, Rossini A, Diverio D, Pelosi E, Vitale A, Chierichini A, Cedrone M, Foa R, Lo Coco F, Peschle C, Testa U. TRAIL decoy receptors mediate resistance of acute myeloid leukemia cells to TRAIL. Haematologica. 2005;90:612–24. [PubMed] [Google Scholar]
- 148.Panner A, James CD, Berger MS, Pieper RO. mTOR controls FLIPS translation and TRAIL sensitivity in glioblastoma multi-forme cells. Mol Cell Biol. 2005;25:8809–23. doi: 10.1128/MCB.25.20.8809-8823.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Clayer M, Bouralexis S, Evdokiou A, Hay S, Atkins GJ, Findlay DM. Enhanced apoptosis of soft tissue sarcoma cells with chemotherapy: a potential new approach using TRAIL. J Orthop Surg. 2001;9:19–22. doi: 10.1177/230949900100900205. [DOI] [PubMed] [Google Scholar]
- 150.Todaro M, Lombardo Y, Francipane MG, Alea MP, Cammareri P, Iovino F, Di Stefano AB, Di Bernardo C, Agrusa A, Condorelli G, Walczak H, Stassi G. Apoptosis resistance in epithelial tumors is mediated by tumor-cell-derived inter-leukin-4. Cell Death Differ. 2008;15:762–72. doi: 10.1038/sj.cdd.4402305. [DOI] [PubMed] [Google Scholar]
- 151.Ishimura N, Isomoto H, Bronk SF, Gores GJ. Trail induces cell migration and invasion in apoptosis-resistant cholangiocarci-noma cells. Am J Physiol Gastrointest Liver Physiol. 2006;290:G129–36. doi: 10.1152/ajpgi.00242.2005. [DOI] [PubMed] [Google Scholar]
- 152.Trauzold A, Siegmund D, Schniewind B, Sipos B, Egberts J, Zorenkov D, Emme D, Roder C, Kalthoff H, Wajant H. TRAIL promotes metastasis of human pancreatic ductal adenocarcinoma. Oncogene. 2006;25:7434–9. doi: 10.1038/sj.onc.1209719. [DOI] [PubMed] [Google Scholar]
- 153.Griffith TS, Broghammer EL. Suppression of tumor growth following intralesional therapy with TRAIL recombinant adenovirus. Mol Ther. 2001;4:257–66. doi: 10.1006/mthe.2001.0439. [DOI] [PubMed] [Google Scholar]
- 154.Herman JR, Adler HL, Aguilar-Cordova E, Rojas-Martinez A, Woo S, Timme TL, Wheeler TM, Thompson TC, Scardino PT. In situ gene therapy for adenocarcinoma of the prostate: a phase I clinical trial. Hum Gene Ther. 1999;10:1239–49. doi: 10.1089/10430349950018229. [DOI] [PubMed] [Google Scholar]
- 155.Jonsson G, Paulie S, Grandien A. High level of cFLIP correlates with resistance to death receptor-induced apoptosis in bladder carcinoma cells. Anticancer Res. 2003;23:1213–8. [PubMed] [Google Scholar]
- 156.McCarthy MM, Sznol M, DiVito KA, Camp RL, Rimm DL, Kluger HM. Evaluating the expression and prognostic value of TRAIL-R1 and TRAIL-R2 in breast cancer. Clin Cancer Res. 2005;11:5188–94. doi: 10.1158/1078-0432.CCR-05-0158. [DOI] [PubMed] [Google Scholar]
- 157.Strater J, Hinz U, Walczak H, Mechtersheimer G, Koretz K, Herfarth C, Moller P, Lehnert T. Expression of TRAIL and TRAIL receptors in colon carcinoma: TRAIL-R1 is an independent prognostic parameter. Clin Cancer Res. 2002;8:3734–40. [PubMed] [Google Scholar]
- 158.Anan A, Gores GJ. A new TRAIL to therapy of hepatocellular carcinoma: blocking the proteasome. Hepatology. 2005;42:527–9. doi: 10.1002/hep.20869. [DOI] [PubMed] [Google Scholar]
- 159.Held J, Schulze-Osthoff K. Potential and caveats of TRAIL in cancer therapy. Drug Resist Updat. 2001;4:243–52. doi: 10.1054/drup.2001.0208. [DOI] [PubMed] [Google Scholar]
- 160.Wissink EH, Verbrugge I, Vink SR, Schader MB, Schaefer U, Walczak H, Borst J, Verheij M. TRAIL enhances efficacy of radiotherapy in a p53 mutant, Bcl-2 overexpressing lymphoid malignancy. Radiother Oncol. 2006;80:214–22. doi: 10.1016/j.radonc.2006.07.030. [DOI] [PubMed] [Google Scholar]
- 161.Cretney E, Shanker A, Yagita H, Smyth MJ, Sayers TJ. TNF-related apoptosis-inducing ligand as a therapeutic agent in autoimmunity and cancer. Immunol Cell Biol. 2006;84:87–98. doi: 10.1111/j.1440-1711.2005.01413.x. [DOI] [PubMed] [Google Scholar]
- 162.Wajant H, Pfizenmaier K, Scheurich P. TNF-related apoptosis inducing ligand (TRAIL) and its receptors in tumor surveillance and cancer therapy. Apoptosis. 2002;7:449–59. doi: 10.1023/a:1020039225764. [DOI] [PubMed] [Google Scholar]
- 163.Mitsiades N, Mitsiades CS, Poulaki V, Chauhan D, Richardson PG, Hideshima T, Munshi N, Treon SP, Anderson KC. Biologic sequelae of nuclear factor-kappaB blockade in multiple myeloma: therapeutic applications. Blood. 2002;99:4079–86. doi: 10.1182/blood.v99.11.4079. [DOI] [PubMed] [Google Scholar]
- 164.Shanker A, Brooks AD, Tristan CA, Wine JW, Elliott PJ, Yagita H, Takeda K, Smyth MJ, Murphy WJ, Sayers TJ. Treating metastatic solid tumors with bortezomib and a tumor necrosis factor-related apoptosis-inducing ligand receptor agonist antibody. J Natl Cancer Inst. 2008;100:649–62. doi: 10.1093/jnci/djn113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Schuchmann M, Schulze-Bergkamen H, Fleischer B, Schattenberg JM, Siebler J, Weinmann A, Teufel A, Worns M, Fischer T, Strand S, Lohse AW, Galle PR. Histone deacetylase inhibition by valproic acid down-regulates c-FLIP/CASH and sensitizes hepatoma cells towards CD95- and TRAIL receptor-mediated apoptosis and chemotherapy. Oncol Rep. 2006;15:227–30. doi: 10.3892/or.15.1.227. [DOI] [PubMed] [Google Scholar]
- 166.Inoue S, MacFarlane M, Harper N, Wheat LM, Dyer MJ, Cohen GM. Histone deacetylase inhibitors potentiate TNF-related apoptosis-inducing ligand (TRAIL)-induced apoptosis in lymphoid malignancies. Cell Death Differ. 2004;11:S193–206. doi: 10.1038/sj.cdd.4401535. [DOI] [PubMed] [Google Scholar]
- 167.MacFarlane M, Inoue S, Kohlhaas SL, Majid A, Harper N, Kennedy DB, Dyer MJ, Cohen GM. Chronic lymphocytic leukemic cells exhibit apoptotic signaling via TRAIL-R1. Cell Death Differ. 2005;12:773–82. doi: 10.1038/sj.cdd.4401649. [DOI] [PubMed] [Google Scholar]
- 168.Nebbioso A, Clarke N, Voltz E, Germain E, Ambrosino C, Bontempo P, Alvarez R, Schiavone EM, Ferrara F, Bresciani F, Weisz A, de Lera AR, Gronemeyer H, Altucci L. Tumor-selective action of HDAC inhibitors involves TRAIL induction in acute myeloid leukemia cells. Nat Med. 2005;11:77–84. doi: 10.1038/nm1161. [DOI] [PubMed] [Google Scholar]
- 169.Pathil A, Armeanu S, Venturelli S, Mascagni P, Weiss TS, Gregor M, Lauer UM, Bitzer M. HDAC inhibitor treatment of hepatoma cells induces both TRAIL-independent apoptosis and restoration of sensitivity to TRAIL. Hepatology. 2006;43:425–34. doi: 10.1002/hep.21054. [DOI] [PubMed] [Google Scholar]
- 170.Nakata S, Yoshida T, Horinaka M, Shiraishi T, Wakada M, Sakai T. Histone deacetylase inhibitors upregulate death receptor 5/TRAIL-R2 and sensitize apoptosis induced by TRAIL/APO2-L in human malignant tumor cells. Oncogene. 2004;23:6261–71. doi: 10.1038/sj.onc.1207830. [DOI] [PubMed] [Google Scholar]
- 171.Olsson A, Diaz T, Aguilar-Santelises M, Osterborg A, Celsing F, Jondal M, Osorio LM. Sensitization to TRAIL-induced apop-tosis and modulation of FLICE-inhibitory protein in B chronic lymphocytic leukemia by actinomycin D. Leukemia. 2001;15:1868–77. doi: 10.1038/sj.leu.2402287. [DOI] [PubMed] [Google Scholar]
- 172.Naka T, Sugamura K, Hylander BL, Widmer MB, Rustum YM, Repasky EA. Effects of tumor necrosis factor-related apoptosis-inducing ligand alone and in combination with chemotherapeutic agents on patients' colon tumors grown in SCID mice. Cancer Res. 2002;62:5800–6. [PubMed] [Google Scholar]
- 173.Di Pietro R, Secchiero P, Rana R, Gibellini D, Visani G, Bemis K, Zamai L, Miscia S, Zauli G. Ionizing radiation sensitizes erythroleukemic cells but not normal erythroblasts to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)–mediated cytotoxicity by selective up-regulation of TRAIL-R1. Blood. 2001;97:2596–603. doi: 10.1182/blood.v97.9.2596. [DOI] [PubMed] [Google Scholar]
- 174.Koschny R, Holland H, Sykora J, Haas TL, Sprick MR, Ganten TM, Krupp W, Bauer M, Ahnert P, Meixensberger J, Walczak H. Bortezomib sensitizes primary human astrocytoma cells of WHO grades I to IV for tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. Clin Cancer Res. 2007;13:3403–12. doi: 10.1158/1078-0432.CCR-07-0251. [DOI] [PubMed] [Google Scholar]
- 175.Hylander BL, Pitoniak R, Penetrante RB, Gibbs JF, Oktay D, Cheng J, Repasky EA. The anti-tumor effect of Apo2L/TRAIL on patient pancreatic adenocarcinomas grown as xenografts in SCID mice. J Transl Med. 2005;3:22. doi: 10.1186/1479-5876-3-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Atkins GJ, Bouralexis S, Evdokiou A, Hay S, Labrinidis A, Zannettino AC, Haynes DR, Findlay DM. Human osteoblasts are resistant to Apo2L/TRAIL-mediated apoptosis. Bone. 2002;31:448–56. doi: 10.1016/s8756-3282(02)00858-x. [DOI] [PubMed] [Google Scholar]
- 177.Evdokiou A, Bouralexis S, Atkins GJ, Chai F, Hay S, Clayer M, Findlay DM. Chemotherapeutic agents sensitize osteogenic sarcoma cells, but not normal human bone cells, to Apo2L/TRAIL-induced apoptosis. Int J Cancer. 2002;99:491–504. doi: 10.1002/ijc.10376. [DOI] [PubMed] [Google Scholar]
- 178.Van Valen F, Fulda S, Schafer KL, Truckenbrod B, Hotfilder M, Poremba C, Debatin KM, Winkelmann W. Selective and nonselective toxicity of TRAIL/Apo2L combined with chemotherapy in human bone tumour cells vs. normal human cells. Int J Cancer. 2003;107:929–40. doi: 10.1002/ijc.11503. [DOI] [PubMed] [Google Scholar]
- 179.Wu XX, Kakehi Y, Mizutani Y, Kamoto T, Kinoshita H, Isogawa Y, Terachi T, Ogawa O. Doxorubicin enhances TRAIL-induced apoptosis in prostate cancer. Int J Oncol. 2002;20:949–54. [PubMed] [Google Scholar]
- 180.Chow LQ, Eckhardt SG, Gustafson DL, O'Bryant C, Hariharan S, Diab S, Fox NL, Corey A, Padavic K, Brown M, Cohen RB. HGS-ETR1, an antibody targeting TRAIL-R1, in combination with paclitaxel and carboplatin in patients with advanced solid malignancies: results of a phase 1 and PK study. J Clin Oncol 2006 ASCO Annu Meet Proc I. 2006;24:2515. [Google Scholar]
- 181.Natoni A, MacFarlane M, Inoue S, Walewska R, Majid A, Knee D, Stover DR, Dyer MJ, Cohen GM. TRAIL signals to apoptosis in chronic lymphocytic leukaemia cells primarily through TRAIL-R1 whereas cross-linked agonistic TRAIL-R2 antibodies facilitate signalling via TRAIL-R2. Br J Haematol. 2007;139:568–77. doi: 10.1111/j.1365-2141.2007.06852.x. [DOI] [PubMed] [Google Scholar]
- 182.Camidge DHR, Gordon M, Eckhardt S, Kurzroc R, Durbin B, Ing J, Ling J, Sager J, Mendelsohn D. ASCO annual Meeting Proceedings Part I. Journal of Clinical Oncology. 2007;25:3582. [Google Scholar]
- 183.Yee L, Fanale M, Dimick K, Calvert S, Robins C, Ing J, Ling J, Novotny W, Ashkenazi A, Burris H. A phase IB safety and pharmacokinetic (PK) study of recom-binant human Apo2L/TRAIL in combination with rituximab in patients with low-grade non-Hodgkin lymphoma. J Clin Oncol 2007 ASCO Annu Meet Proc I. 2007;25:8078. [Google Scholar]