

Abstract
Wound repair requires both recruitment and well co-ordinated actions of many cell types including inflammatory cells, endothelial cells, epithelial cells and importantly fibroblast cells. Urokinase-type plasminogen activator (uPA) system plays a vital role in wound healing phenomenon. We have previously demonstrated that C-phycocyanin (C-pc), a biliprotein from blue-green algae, transcriptionally regulates uPA through cAMP-dependent protein kinase A (PKA) pathway. To date, a role for C-pc in wound-healing scenario is not elucidated. This study was designed to examine the wound-healing property of C-pc in relation to fibroblast proliferation and migration. C-pc increased fibroblast proliferation in a dose-dependent manner. It also enhanced G1 phase of cell cycle and increased the expressions of cyclin-dependent kinases 1 and 2, which facilitate cell cycle progression, in a uPA-independent manner. In vitro wound healing and migration assays revealed the pro-migratory properties of C-pc. Short-interference RNA studies demonstrated that uPA was necessary for C-pc-induced fibroblast migration. C-pc also significantly elevated the expressions of chemokines (MDC, RANTES, Eotaxin, GRO α, ENA78 and TARC) and Rho-GTPases (Cdc 42 and Rac 1) in a uPA-dependent manner. Pre-treatment of C-pc-stimulated cells with pharmacological inhibitor of PI-3K (LY294002) annulled the expression of GTPases implying that Rac 1 and Cdc 42 were induced through PI-3K pathway. C-pc-induced cellular migration towards wounded area was also negatively affected by PI-3K inhibition. In vivo wound-healing experiments in mice validated our finding that C-pc accelerates wound healing. Our data provides conclusive evidence of a novel therapeutic usage for C-pc as a wound-healing agent. C-pc is a food and drug administration (FDA)-approved health supplement. We believe this compound can also be beneficial in healing of internal wounds, such as ulcers.
Keywords: C-phycocyanin, cell migration, cell proliferation, PI-3K pathway, uPA, wound healing
Introduction
Wound healing is a complex biological and physiological process involving interaction of many cell types, cytokines, growth factors and their inhibitors [1, 2]. The first stage of wound healing involves rapid deposition and polymerization of fibrin. Plasma fibronectin binds and covalently cross-links with fibrin to form a fibrous clot structure that promotes migration and attachment of leukocytes and fibroblasts [3]. The effect on fibroblast migration is of particular interest to wound repair in that it could dictate the pace of cell recruitment into the wound bed as well as subsequent activities including contraction of wound extracellular matrix. High level of fibroblast migration and proliferation, collectively termed as fibroplasias, in a dermal substitute is a hallmark phenomenon that leads to improved healing [4]. Matrix remodelling and fibrob-last cell response are integrally linked. Changes in fibroblast migration and proliferation depend upon the extracellular matrix environment in which they are encased. In connection to extracel-lular matrix and tissue remodelling, urokinase-type plasminogen activator (uPA) system has been discussed extensively [5, 6]. The system includes the serine protease uPA, its receptor uPAR and two specific inhibitors, type 1 and type 2 plasminogen activator inhibitors (PAI-1 and PAI-2). Generation of pericellular plasmin by uPA induces matrix proteolysis and is thought to be essential in matrix remodelling, cell adhesion and cell migration. Several studies show tight correlation between expression of uPA system components and wound healing in human cell lines [7–9].
C-phycocyanin (C-pc) is a protein-bound pigment found in some blue-green algae. Recent reports have credited C-pc with many pharmacological properties such as anti-inflammatory [10], antioxidant [10, 11] and anti-tumor activities [12]. We had earlier purified C-pc from Spirulina fusiformis[13] and demonstrated its uPA-enhancing activity in human fibroblasts [14]. We questioned if C-pc could play a role in wound healing, through its effect on uPA.
In this study, we have systematically investigated the effect of C-pc on fibroblast proliferation and migration; and the associated mechanisms by which these phenomena exert their effects on wound healing. We also evaluated the significance of uPA in these phenomena. Additionally, we validated the in vitro observations with in vivo dermal wound healing in mice models. To our knowledge, this is the first report revealing the wound-healing properties of C-pc.
Materials and methods
Cell culture and reagents
Human fibroblast cells TIG 3–20 (HSRBB Cell bank, Osaka, Japan) were cultured in Modified Eagles Medium with 10% heat inactivated foetal bovine serum, 100 U/ml penicillin and 100 mg/ml streptomycin at 37°C, 5% CO2 and 95% humidity. Semi-confluent cells between passages 3 and 5 were used for the studies. Dimethyl sulfoxide (DMSO) was used as the solvent for dissolving all compounds tested. DMSO concentration (0.25%), held constant in all experiments, did not cause cytotoxicity in TIG 3–20 cells.
Transient transfection of uPA siRNA
A lyophilized pool of three target-specific 20–25 nt siRNAs designed to knock-down uPA gene expression (sc-36779) was purchased from Santa Cruz Biotechnology, Inc, USA. Control siRNA consisting of a scrambled sequence that does not degrade any known cellular mRNA (sc-37007, Santa Cruz Biotechnology, Inc, Santa Cruz, CA, USA) served as a negative control for the experiments. Transient transfection of cells with siRNA was performed at 50–70% confluence using siRNA transfection reagent (sc-29528) and medium (sc-36868) from Santa Cruz Biotechnology, Inc, USA, following manufacturer's protocol. Preliminary experiments conducted over a range of concentrations revealed 60% inhibition of uPA expression at a siRNA concentration of 200 nM. Thereafter, all siRNA experiments were performed using 200 nM siRNA. The day after transfection, cells were provided with fresh normal growth medium. All assays were conducted 48 hrs after addition of normal growth medium. Cells transfected with uPA siRNA are henceforth referred to as uPA knock-down (or uPA−) cells and non-transfected cells as uPA+ cells.
Real time PCR
Following transfection, total RNA was isolated and RT-PCR was carried out as per standard procedures using ReverTra Ace enzyme (Toyobo, Tokyo, Japan). For real-time PCR, 5 μl cDNA was amplified in a 20 μl PCR volume containing 2x Power SYBR green PCR master mix (Applied Biosystems, Foster City, CA, USA) and primers specific for uPA or 18S rRNA. Sequences targeting thecoding region of the genes were selected for primer designing. Primer sequences were as follows: uPA (125 bp) forward: 5′-TCACCAC-CAAAATGCTGTGT-3′ and reverse 5′- CCAGCTCACAATTCCAGTCA-3′; 18SrRNA (76bp) forward: 5′-TGCATGGCCGTTCTTAGTTG-3 and reverse 5′- AGTTAGCATGCCAGAGTCTCGTT-3′.
LDH cytotoxicity assay
Cytotoxicity assay was performed to determine the amount of lactate dehydrogenase (LDH) released from C-pc stimulated fibroblast cells using CytoTox 96(r) Non-Radioactive kit (Promega Corp., Madison, USA). Experiments were conducted in both normal as well as uPA knock-down cells.
Cell viability and proliferation assay
Semi-confluent TIG 3–20 cells were incubated with different concentrations (10 μg/ml to 200 μg/ml) of C-pc in minimal essential medium (MEM) for 24 hrs at 37°C. Cells that did not receive C-pc were treated with vehicle (DMSO) and served as control. Cell proliferation and viability tests were determined by trypan blue dye exclusion assay using Automated Cell Viability Analysis Robot (Beckman Coulter Inc., Vi-CELL XR, Fullerton, CA, USA). The percentage of cell death and viability was estimated in three independent experiments.
In vitro wound-healing assay
Fibroblasts were grown in 6-well plates at a density of approximately 4 × 104 and a small linear wound was created in the confluent monolayer by gently scratching with sterile cell scrapper as per standard methods [15]. Cells were extensively rinsed with medium to remove cellular debris before treating with 75 μg/ml of C-pc in fetal bovine serum (FBS)-deprived condition. Twenty-four hours later, images of the cells were obtained using digital camera (Nikon, Tokyo, Japan) connected to the inverted microscope (Nikon TMS-F, Japan) and analysed by image analysis software (Image J 1.32e, National Institutes of Health, Bethesda, MD, USA). Extent of wound healing was determined by the distance traversed by cells migrating into the denuded area. Similar experiments were conducted in uPA knock-down conditions. Representative data is cumulative of three independent experiments.
In vitro cell outgrowth assay
Cell outgrowth assays were performed as per standard procedure [16]. Briefly, sterile glass 0.5 cm diameter cloning cylinders (Sigma) were placed in the centre of wells of a 24-well plate. Fibroblasts were plated into the center of the cloning cylinder at a density of 2 × 104 cells per cylinder. Cells were cultured for 24 hrs to allow them to attach to the plastic in a circular ‘island’. Cloning cylinders were then removed and unattached cells were rinsed off the plastic. The cells were treated with 75 μg/ml C-pc for 24 hrs. Cell outgrowths were fixed in 3.7% formalin for 10 min., washed with phosphate-buffered saline (PBS), stained with 0.2% crystal violet blue for 5 min. and washed several times in distilled water. Stained cells were air dried and scanned. Total area of cell outgrowth was analysed using Image J software (Image J 1.32e, National Institutes of Health, Bethesda, MD, USA). Results of triplicate assays are expressed as mean cell outgrowth in mm. Experiments were repeated in uPA knock-down conditions.
Chemotaxis/ cell migration assay
Cultrex(R) 96-well basement membrane extract (BME) cell invasion assay for cell migration was used as per manufacturers' instructions (Trevigen, Inc., MD, USA). Briefly, fibroblasts were grown to sub confluence in Cultrex(R) 96-well upper plate. A day prior to the assay, cells were starved in serum free medium. C-pc was then added to the bottom chamber along with fresh medium and cells incubated for 24 hrs. Later, cells in both upper and bottom chambers were washed and 100 μl of calcein-AM/ cell dissociation solution was added to bottom chamber. Plates were read at 485 nm excitation and 520 nm emissions with spectrofluorometer (Spectra MAX Gemini XS, Molecular devices, Sunnyvale, CA, USA) to estimate the number of cells that migrated to the bottom chamber. Experiments were repeated in uPA knock-down conditions.
Cell cycle analysis by flow cytometer
uPA+ and uPA− fibroblasts were treated with vehicle or C-pc for 24 hrs and then fixed in 70% ethanol at 4°C. Cells were stored in the fixative at 20°C for 1 hr. Following fixation, cells were centrifuged at 800 ×g for 5 min., re-suspended in phosphate-citrate (PC) buffer at room temperature for 30 min. and again centrifuged at 1000 ×g for 5 min. The cells were re-suspended in 800 l PBS before incubation in 100 μl each of 100 μg/ml RNase A and 0.01% propidium iodide (PI). Flow cytometry analysis was performed using EPICS XL flow cytometer (Coulter, USA). The cells were excited by a 488 nm argon ion laser and analysed at 530 nm (FL1, showing CFSE) and 585 (FL2, showing PI). Signal height, area and width were recorded for the PI channel. Gating was done with traditional gating set on PI-Area versus PI-Width. Cell debris was removed during histogram analysis by modelling the cell debris. Computer-based analysis programs can compensate the debris with build-in algorithms. The algorithm can predict the small amount of debris with high reliability. Data was analysed with Win MDI v2.8 software and Mod Fit LT (Verity Software House Inc.). Gating results were expressed as histograms.
Western blot analysis
Following incubation with C-pc, cells were harvested, washed twice with cold PBS and lysed in a lysis buffer containing protease inhibitors. 10 μg proteins were resolved over 8–12% SDS-PAGE gels and transblotted onto nitrocellulose membrane. After blocking non-specific binding sites using blocking buffer (5% non-fat dry milk, 1% Tween 20 in 20 mM TBS, pH 7.6), blots were incubated overnight with primary antibodies specific for the proteins to be assessed. Antibodies anti-cdK 1 and anti-cdK 2 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA); anti-Cdc 42 from Biomol Research (Plymouth, PA, USA); anti-Rac 1 from R & D Systems (Minneapolis, MN, USA) and anti-β-actin from Sigma Chemical (St. Louis, MO, USA). Blots was washed and incubated with appropriate horseradish peroxidase (HRP)-conjugated secondary antibody. Expression of proteins was detected by chemiluminescence using ECL™ detection system (Amersham Life Science, Inc., USA). To ensure equal protein loading, membranes were stripped and re-probed with anti-β-actin antibodies using the protocol detailed above. Intensities of the bands were measured using digitized scientific software program UN-SCAN-IT (Silk Scientific Corp., Orem, UT, USA). Density values of experimental conditions were normalized to that of control (vehicle-treated; C-pc-). Ratio of density of a given protein to that of -actin of the same sample was represented graphically as the relative density of the protein. Experiments were repeated three times.
Human chemokine array
Endogen SearchLight(R) IR human chemokine multiplex sandwich array ELISA was used as per manufacturer's instructions (Pierce Biotechnology, Inc., CA, USA), for the quantification of 12 human chemokines in the cell culture supernatants. Briefly, culture supernatants from C-pc treated normal as well as uPA knock-down cells were applied to ELISA plates pre-coated with antibodies relevant to the above mentioned array. Plates were incubated at room temperature for 1 hr with shaking, after which 500 μl of biotinylated antibody reagent was added. Plates were further incubated at room temperature for 30 min. Following a brief wash, 50 μl of SA-DyLight™ 800 was added to each well and incubated at room temperature for 30 min. Plates were washed and scanned with LI-COR Aerius infrared imaging system (LI-COR Biosciences, Linclon, NE, USA). Results were quantified for control as well as treated sets in both normal as well as uPA knock-down groups in three independent experiments.
Phosphoinositide-3 kinase (PI-3K) pathway inhibition study
LY294002, a pharmacological inhibitor of PI-3K, was used to analyse the role of PI-3K pathway in the expressions of Cdc 42 and Rac 1, as well as in wound healing. In the case of Cdc 42 and Rac 1, 15 nM LY294002 was added 2 hrs prior to C-pc addition. In wound-healing experiments, 15 nM LY294002 was added 24 hrs prior to scraping of the monolayer. Following LY294002 addition, experiments were performed as described above.
Preparation of C-pc incorporated collagen films
Commercial collagen was purchased from Funakoshi, Japan. Collagen films were prepared as per the procedures of Gopinath et al. [17]. Briefly, collagen concentration was adjusted to 3.5 mg/ml with 0.05 M acetic acid. 75 μg/ml C-pc in absolute ethanol was mixed with 50 ml of collagen solution with constant stirring for 48 hrs at 4°C. The suspension was squeezed through muslin cloth to remove any precipitate formed during the process. The solution was placed in glass trays and vacuum dried under aseptic conditions. Control collagen films without C-pc were similarly prepared.
In vivo dermal wound-healing experiments
Male ICR mice of a mixed race were used as animal models for wound healing. All experiments were performed with 8–12-week-old mice. The mice were kept under specific pathogen-free conditions. Experiments were conducted in accordance with the guidelines of the Ethical Committee on Animal Experimentation of the University of Miyazaki. Skin wounds were performed as per standard protocols. Briefly, mice were anaesthetized by intraperitoneous injection of pentobarbitone. After shaving and cleaning the exposed skin with 70% ethanol, full-thickness wounds were made on the upper side of the dorsal middle line using 8 mm biopsy-punch (Accuderm, Ft. Lauderdale, FL, USA). Collagen films with/ without C-pc were placed over the wounds. Healing was monitored over a period of 1 week and photographs were taken at the indicated time-points. Wound area in mm2 was calculated for each time-point, from three independent experiments.
Statistical analyses
All in vitro and in vivo experiments were conducted in triplicates. Data were expressed as mean standard deviation, and the difference between the groups was analysed by Student's t-test. P < 0.05 was considered as statistically significant.
Results
Effect of C-pc on cell proliferation and cell viability
Initially we addressed the cytotoxic and cell viability profiles of C-pc in TIG 3–20 cells. LDH assay was used to study the cytotoxicity induced by exposure to different doses of C-pc. Lower doses of C-pc did not generate any increase in LDH from cells, implying non-cytotoxicity (Fig. 1). An increase in LDH was noticed at a concentration of 200 μg/ml of C-pc, suggesting that high doses of C-pc can be toxic to cells. Different concentrations of C-pc had no significant inhibitory effect on cell proliferation and viability (Fig. 2). On the other hand, C-pc significantly stimulated cell proliferation compared to controls in a dose-dependent manner (P < 0.05). Peak activities were observed at concentrations of 75 μg/ml C-pc. There was no difference in the patterns of cell viability and proliferation between uPA+ and uPA− cells. The data confirms that C-pc is not cytotoxic and rather helps in cell proliferation. Additionally, it also shows that viability and proliferation of TIG 3–20 cells were uPA-independent.
Fig. 1.
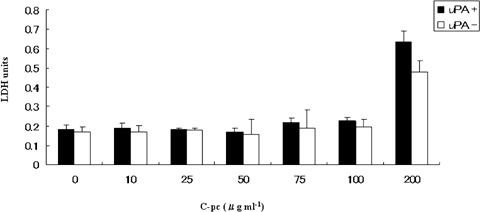
Cytotoxic profile of C-pc. Cytotoxicity was measured by quantifying the amount of LDH released from dead cells. C-pc was non-toxic to TIG 3–20 cells upto 100 μg/ml concentration. The effect was similar in normal (uPA+)as well as uPA silenced (uPA−) cells. Values are mean ±S.D. of three independent experiments.
Fig. 2.
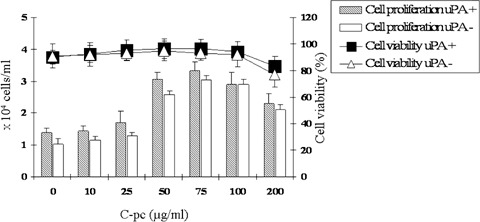
Cell viability and proliferation tests. Cell viability is represented by line diagram. Cell proliferation is represented by bar diagram and is denoted by number of cells (× 104 cells/ml). Viability of TIG 3–20 cells was not affected by C-pc up to a dose of 100 μg/ml. C-pc increased cell proliferation in a dose-dependent manner. Values are mean S.D. of three independent experiments.
C-pc stimulates cell cycle independent of uPA
Stimulation of cell cycle at various junctions is considered a major cause for cell proliferation and multiplication [18]. We performed flow cytometry-aided cell cycle analysis in uPA+ as well as uPA cells stimulated with 75 μg/ml C-pc. Cell cycle distribution analysis showed that C-pc treatment resulted in an increase in G1 phase with no significant changes in G2 or S phases (Fig. 3). Thus, C-pc accelerates cell growth by inducing G1 phase, an early event of cell cycle and helps in cell proliferation. Induction of cell cycle and proliferation by C-pc in TIG 3–20 cells was independent of uPA as there was no difference in the cell cycle patterns in uPA+ and uPA conditions.
Fig. 3.
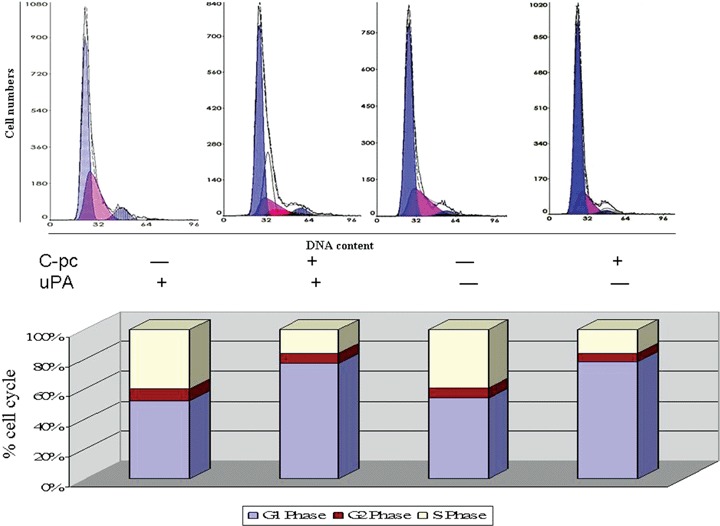
Cell cycle analysis. Different phases of cell cycle were analysed by flow cytometry. Treatment with 75 μg/ml C-pc increased the number of cells in the G1 phase. C-pc increased the G1 phase of cell cycle in uPA-independent manner. Values of different cell cycle stage are adjusted to 100%. Results are mean ± S.D. of three independent experiments.
uPA is not necessary for cdK 1 and cdK 2 expression stimulated by C-pc
It is now clear that uPA is not a key molecule governing proliferation and cell cycle in TIG 3–20 fibroblasts. However, the molecular event associated with C-pc-induced proliferation is not clear.
Cyclin-dependent kinases (cdK 1 and 2)play important roles in the regulation of cell cycle progression [19]. We studied the effect of C-pc on the protein expression of cdK 1 and cdK 2. As shown in Fig. 4, treatment with 75 μg/ml C-pc resulted in a marked increase in cdK 1 and cdK 2 expressions. The effect on the cdKs was not affected by the presence/ absence of uPA. CdKs up-regulated by C-pc help in cell cycle progression and ultimately lead to cell proliferation. CdK inhibitory proteins suppress cell cycle progression by binding to and inhibiting the kinase activity of cdK-cyclin complex [19, 20]. We assessed the effect of C-pc on cdK inhibitory pro-teinsand found that there was no change in the expression patterns of cdKi (data not shown). These results indicate that C-pc induced enhancement of cdK 1 and cdK 2 may play an important role in the cell cycle progression in TIG 3–20 cells, possibly through G1 phase acceleration, leading to cell proliferation. This part of study clearly shows that C-pc is an important factor for cell division and proliferation in TIG 3–20 cells and the mechanism is independent of uPA.
Fig. 4.
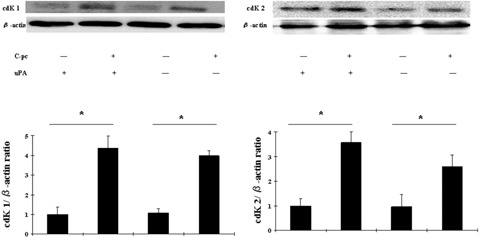
Expression of cyclin dependent kinases upon stimulation by C-pc: Lysates from uPA+ and uPA− cells treated with 75μg/ml C-pc were analyzed by western blot to study the expression of cdK 1 and cdK 2 proteins. Equal loading of proteins was verified using β-actin antibody. Upper panel is a representative image of three independent experiments. The relative density of the proteins was expressed as the ratio of cdK 1/β-actin and cdK 2/β-actin, as shown in lower panel. Values are expressed as mean ± S.D. of three independent experiments. * p < 0.05.
In vitro wound healing and cell migration involves uPA
Fibroblast cell migration to wounded area is a hallmark of wound closure [21]. We used a standard in vitro wound-healing assay [15] to observe the effect of C-pc on cell migration. As depicted in Fig. 5, C-pc-stimulated cells displayed significantly (P < 0.05) higher rate of migration into the wounded area, than non-stimulated cells. However, C-pc addition to uPA knock-down cells failed to increase the migratory rate of the cells. Further experiments were carried out to examine whether C-pc could stimulate random movement of cells in a normal microenvironment. Figure 6 shows the results of in vitro cell outgrowth assay in uPA+ and uPA− cells stimulated with 75 μg/ml C-pc. C-pc significantly increased fibroblast cell motility in uPA condition, but was ineffective in inducing cell motility in the absence of uPA. These results indicate that C-pc accelerated cell migration in an uPA-dependent manner. We also observed that uPA− fibroblasts exposed to C-pc exhibited broad lamellae and revealed long dendritic extensions (data not shown). Elongation of dendrites in fibroblast and migration of border cells is indicative of cell invasiveness [22]. It is reasonable to consider that border cell migration could be evoked upon C-pc stimulation, with a roleplay by uPA attached to the process. We also studied the effect of C-pc on cell chemotaxis using Boyden chamber assay. Over the range of C-pc concentrations studied, significant enhancement of cell migration was observed at 50 μg/ml and higher (Fig. 7). However, no discernable increase in cell migration in uPA knock-down cells was observed when stimulated with the same concentrations of C-pc. These findings point to chemotactic activity of C-pc that is dependent on uPA. Various studies have demonstrated the importance of chemokines in wound healing [23]. To validate this phenomenon, we studied the expression of a panel of chemokines. Chemokines analysed were MCP, MIP1 α, MIP1 β, TARC, MDC, RANTES, Eotaxin, GRO α, IP-10, ENA78, MIG and MPIF1. Among tested chemokines, C-pc (75 μg/ml) treatment led to a significant (P < 0.05) elevation of MDC, RANTES, Eotaxin, GRO α, ENA78 and TARC in uPA cells (Fig. 8). Silencing of uPA affected the expression of chemokines. While expression of RANTES increased, albeit to a lower degree, expression of other chemokines, such as MDC, eotaxin, GRO and ENA-78 was completely blocked in uPA- condition. However, TARC seemed to be unaffected by uPA. A comparison of the two groups of cells (uPA+ and uPA−) reflects the view that uPA differentially regulates the expression of chemokines induced by C-pc.
Fig. 5.
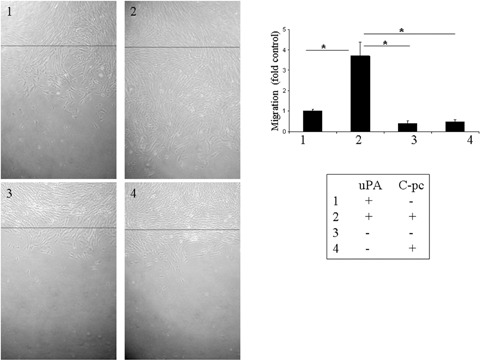
In vitro wound healing assay: Confluent monolayers of uPA and uPA cells were wounded by gently scratching with a cell scraper and treated with 75 μg/ml C-pc. Photographs were taken 24hrs after wounding. Black line denotes the wound edge at the start of experiments. Wound healing was measured by the distance traveled by the cells from the wound edge. C-pc induced cell migration towards the wounded area in an uPA-dependent manner. *p < 0.05.
Fig. 6.
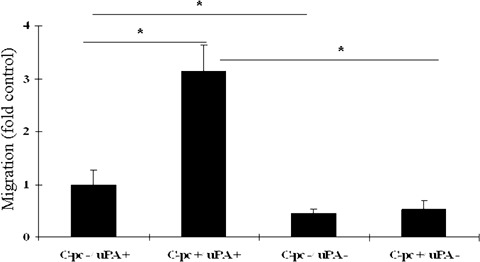
In vitro cell outgrowth assay: Cell outgrowth assay was performed to study the effect of C-pc on random migration of TIG 3–20 cells. uPA+ cells stimulated with 75 μg/ml C-pc were significantly more migratory compared to control. The migratory rate was retarded in uPA cells. Values are expressed as mean ± S.D. of three independent experiments. *p < 0.05.
Fig. 7.
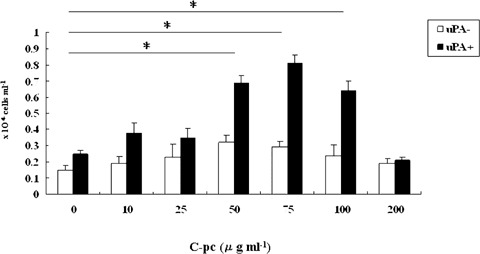
Effect of C-pc on directed chemotactic migration ofTIG 3–20 cells: Cell migration in response to a range of concentrations of C-pc was measured by Boyden chamber assay. uPA cells responded with higher rate of migration in a dose-dependent manner. C-pc had little effect on uPA+ cells. Values are expressed as mean ± S.D. of three independent experiments. *p < 0.05.
Fig. 8.
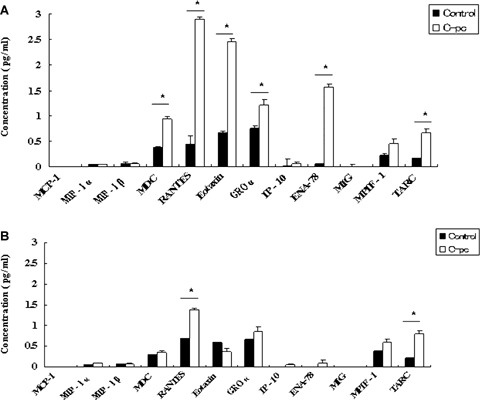
Human chemokine array: After 24 hrs treatment with 75 μg/ml C-pc, supernatants from uPA+ (panel A) and uPA− (panel B) cells were analyzed for expression of various chemokines. C-pc induced the expression of a group of chemokines. Values are expressed as mean ± S.D. of three independent experiments. *p < 0.05.
Cdc 42 and Rac 1 over expression is uPA dependent and mediated through PI-3 kinase pathway
We observed structural modifications, such as broad lamellae and dendritic extensions, in cells treated with C-pc (data not shown). Since Rho-family GTPases including Cdc 42 and Rac 1 regulate fibroblast cell migration and polarization [24], we addressed the question whether Rho-family proteins were stimulated in response to C-pc. Figure 9 shows a representative result of Western blot analyses of Cdc 42 and Rac 1. Immunoblotting with antibodies specific for Cdc 42 and Rac1 showed significant (P < 0.05) overex-pression of the proteins when stimulated with 75 μg/ml C-pc in uPA+ cells, but abrogated in uPA− cells. These results demonstrate that C-pc stimulates the Rho-family GTPases through uPA.
Fig. 9.
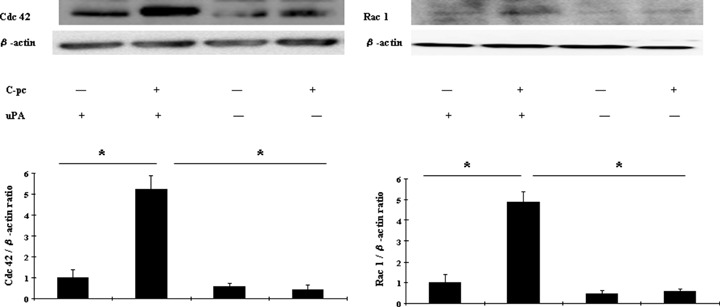
C-pc induced Cdc 42 and Rac1 in uPA-dependent manner: uPA+ and uPA− cells were treated with 75 μg/ml C-pc for 24 hrs and proteins were analyzed by western blotting. -actin was used as control for equal loading. Upper left panel denotes a representative blot of Cdc 42 and upper right panel denotes Rac 1. The relative density of the proteins was expressed as the ratio of Cdc 42/β-actin or Rac 1/β-actin, as shown in lower panel. Data represents the values of three independent values. *p < 0.05
PI-3K plays an important role in signal transduction leading to cellular processes that promote cell migration and survival. We next examined whether stimulation of cell migration by C-pc is associated with PI-3K pathway. uPA cells were pre-treated with 15 nM LY294002 (specific PI-3K inhibitor) for 2 hrs prior to C-pc treatment. Western blot analysis revealed the inhibition of Cdc 42 and Rac 1 when PI-3K pathway was blocked (Fig. 10). C-pc could not induce the proteins in the event of PI-3K inhibition. Inhibition of PI-3K pathway also displayed a dramatic effect on cell migration stimulated by C-pc, as revealed in subsequent wound-healing assay (Fig. 11). Addition of LY294002 totally blocked C-pc-stimulated cell migration. These sets of experiments throw light on the findingthat uPA plays an important role in C-pc-induced fibroblast migration and the migratory signals are facilitated through the Rho-family GTPases via PI-3K pathway.
Fig. 10.
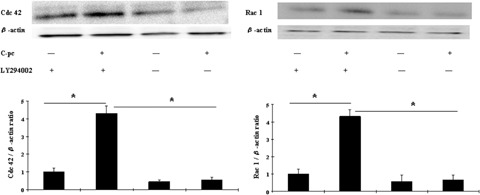
PI-3K pathway is necessary for C-pc induced expression of Cdc 42 and Rac 1. uPA+ cells were pretreated with PI-3K inhibitor, LY294002 (15 nM) for 2 hrs prior to stimulation with 75 μg/ml C-pc. Expression patterns were studied as outlined for Fig. 9. *p < 0.05.
Fig. 11.
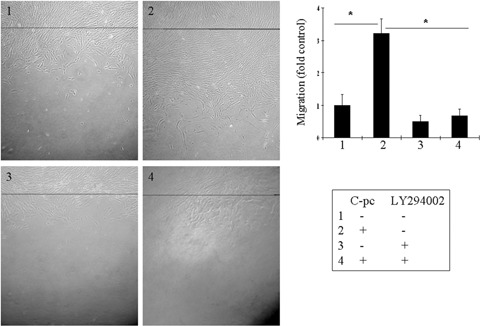
PI-3K pathway mediates C-pc induced fibroblast migration: uPa+ cells were treated with 15 nM LY294002 for 24 h, wounded by gently scratching the monolayer and incubated with 75 μg/ml C-pc for 24 hrs. Black line denotes the wound edge at 0 h. Wound healing was studied as explained in Fig. 5. Data represent mean ± S.D. of three independent experiments. *p < 0.05.
C-pc promotes in vivo wound healing in mouse dermal wound model
Finally, in vivo experiments were performed to evaluate the healing efficiency of C-pc on dermal wounds in mouse models. Collagen films containing C-pc were laid over the dermal wound area and rate of wound healing was monitored over a period of 1 week. In the initial stage, no significant difference was observed between control and C-pc treatment. However, from the fourth day onwards, C-pc-treated groups displayed significant reduction of wound area, demonstrating faster wound healing (Fig. 12). By the end of the trial period, C-pc treatment exhibited 80% closure of wound in contrast to 50% closure in control models. These in vivo experiments positively projected the efficacy of C-pc in wound healing.
Fig. 12.
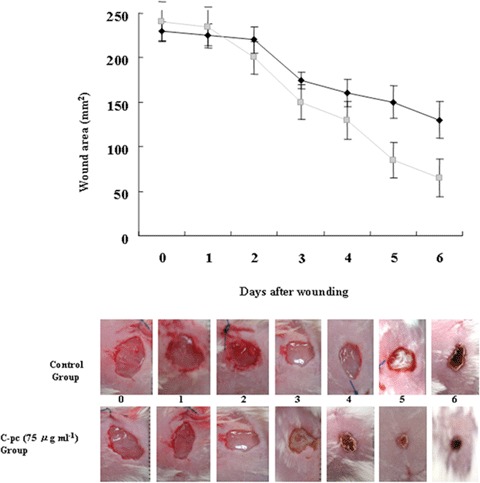
Dermal wound healing in mice models: C-pc incorporated collagen films were placed over dermal wound as explained in Materials and Methods. Wound healing was measured by quantifying the total wound area over a period of one week. Images shown are representative of three independent experiments. Data represent mean ± S.D.
Discussion
Following dermal wound, a cascade of cellular events mediates tissue repair, re-establishment of granulation, synthesis of new connective tissue and ultimately wound closure. The whole repair process calls for the interaction between different cell types. In the mid and late phases of wound closure, cellular and physiological interplay becomes dominated by the proliferation and migration of fibroblasts into the wound environment. Functional importance of fibroblasts in wound healing has been demonstrated [25]. In this study, we demonstrated the dual function of C-pc, an algal protein, in enhancing proliferation and migration of fibroblast during wound healing. Furthermore, using siRNA-mediated blockade, we showed that uPA is a key regulator of fibroblast migration but is not vital for proliferation and cell cycle patterns in TIG 3–20 fibroblasts.
The first phase of the present investigation demonstrated the relationship between C-pc and fibroblast proliferation. Cell cycle is a critical regulator of the process of cell proliferation and growth. In this study, we stimulated human fibroblasts with C-pc and showed that it helped in cell division at G1 phase. It also up-regulated the cyclin-dependent kinases, cdK 1 and cdK 2. CdKs regulate the cell cycle [26] and are particularly active at G1-S phase of cell cycle [27]. It is reasonable to conclude that C-pc promotes cell proliferation through its effect on the cyclin-dependent kinases. Recent studies highlight the importance of uPA in promoting cell growth and proliferation [28–30]. Contrary to these reports, our findings failed to provide evidence of the vitality of uPA in C-pc-induced proliferation of TIG 3–20 fibroblast cells.
Proliferation and migration of fibroblasts into the wound site are essential events in the early phase of wound repair. We demonstrated the efficacy of C-pc in promoting fibroblast migration using three in vitro models; wound-healing assay, cell outgrowth assay and transwell migration assay. These techniques allowed us to analyse cell migration from different viewpoints, namely wound environment (wound-healing assay), normal environment (cell outgrowth assay) and chemotaxis environment (transwell migration assay). All the assays highlighted the efficiency of C-pc in enhancing cell migration in a uPA-dependent manner. The functional link between uPA and cell motility was established over a decade ago [31, 32]. uPA-related migratory responses seem to be highly cell specific, implying some structural specificity and diversity of underlying signalling events. Recent reports underscore the importance of PI-3K signalling pathway in cell migration [33–37] and we reasoned that PI-3K could be a candidate for mediating C-pc-induced fibroblast migration. Our studies using LY294002, a pharmacologic inhibitor of PI-3K, demonstrated the involvement of PI-3K signalling in C-pc-induced cell migration. Rho-family GTPases, such as Rho, Rac1 and Cdc 42 are key regulators of cell migration [36] in fibroblasts by regulating and modifying the microtubules orientation [24, 38]. Rho family GTPases act as molecular switches in signal transduction pathways linking cell surface receptors to the actin cytoskeleton [39].
Studies using fibroblasts and astrocytes demonstrated a pivotal role of Rac1 for protrusion formation and forward motion, whereas Cdc 42 and RhoA regulated the directionality of movement and maintenance of cell adhesion respectively [24, 40]. The increase in cell migration was associated with PI-3K activation of Rac1 and Cdc 42 in intestinal epithelial cell restitution [41]. RhoA and Rac 1, together with Rho kinase are necessary to mediate the uPA/uPAR-directed migration via the Tyk2/PI-3K signalling complex in human vascular smooth muscle cells [42]. Our accumulated data are consistent with earlier reports demonstrating the central role of PI-3K in initiating cell migration [43–45].
Upon appropriate stimulation, fibroblasts are potent producers of a variety of chemokines [23]. Chemokines have been associated with a variety of important functions with relevance to wound healing [46]. Chemokines, such as MCP1, MIP1 α, MIP1 β, RANTES, IL-8 and IP-10 have been shown to cause the in vitro migration of rat brain microglia [47]. C-pc amplified the expression of MDC, RANTES, eotaxin, GROα and ENA-78. This amplification called for the presence of uPA.
Compilation of our data reveals that C-pc promotes wound healing by up-regulating uPA, stimulating the GTPases Cdc 42 and Rac 1 through PI-3K pathway, besides enhancing the expression of chemokines and cyclin-dependent kinases 1 and 2. In vivo wound models authenticated our findings on the therapeutic properties of C-pc as a wound-healing agent. Topical application of C-pc on dermal wounds was effective in accelerating the rate of wound healing. The effect was noticeable from the fourth day of treatment. It has been reported that as an early response to injury, fibroblasts in the wound edges begin to proliferate and by approximately day 4 start to migrate into the provisional matrix of the wound clot, where they lay down a collagen-rich matrix, including collagens, proteoglycans and elastin [48].
Summary
The present study unravelled two important therapeutic properties of C-pc with relevance to wound healing. The traffic and signal transduction pathway of C-pc-induced wound healing is depicted in Fig. 13; first, it induces fibroblast proliferation via the cyclin-dependent kinases, cdK 1 and cdK 2. Second, it also enhances cellular migration towards the wound. On migration aspects, uPA plays a pivotal role which is mediated on one hand through the GTPases, Rac 1 and Cdc 42 via PI-3K pathway, and on the other hand, the differential expression of chemokines.
Fig. 13.
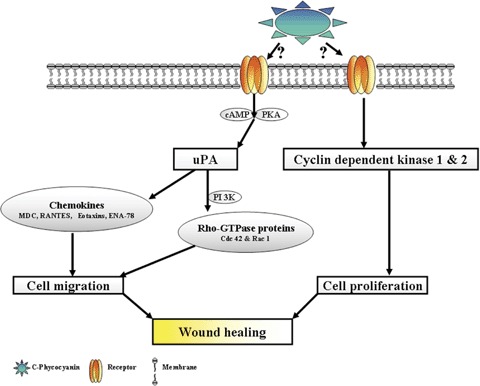
Schematic representation of C-pc induced traffic and signal transduction pathway during wound healing.
References
- 1.Davis GE, Saunders WB. Molecular balance of capillary tube formation versus regression in wound repair: role of matrix metalloproteinases and their inhibitors. J Invest Dermatol. 2006;126:44–56. doi: 10.1038/sj.jidsymp.5650008. [DOI] [PubMed] [Google Scholar]
- 2.Idell S. Coagulation, fibrinolysis, and fibrin deposition in acute lung injury. Crit Care Med. 2003;31:S213–20. doi: 10.1097/01.CCM.0000057846.21303.AB. [DOI] [PubMed] [Google Scholar]
- 3.Maquerlot F, Galiacy S, Malo M, Guignabert C, Lawrence DA, d'Ortho MP, Barlovatz-Meimon G. Dual role for plasminogen activator inhibitor type 1 as soluble and as matricellular regulator of epithelial alveolar cell wound healing. Am J Pathol. 2006;169:1624–32. doi: 10.2353/ajpath.2006.051053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lamme EN, Van Leeuwen RT, Brandsma K, Van Marle J, Middelkoop E. Higher numbers of autologous fibroblasts in an artificial dermal substitute improve tissue regeneration and modulate scar tissue formation. J Pathol. 2000;190:595–603. doi: 10.1002/(SICI)1096-9896(200004)190:5<595::AID-PATH572>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 5.Nicholl SM, Roztocil E, Davies MG. Urokinase-induced smooth muscle cell responses require distinct signaling pathways: a role for the epidermal growth factor receptor. J Vasc Surg. 2005;41:672–81. doi: 10.1016/j.jvs.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 6.Tanski WJ, Fegley AJ, Roztocil E, Davies MG. Domain-dependent action of urokinase on smooth muscle cell responses. J Vasc. Surg. 2004;39:214–22. doi: 10.1016/s0741-5214(03)01031-0. [DOI] [PubMed] [Google Scholar]
- 7.Planus E, Barlovatz-Meimon G, Rogers RA, Bonavaud S, Ingber DE, Wang N. Binding of urokinase to plasminogen activator inhibitor type-1 mediates cell adhesion and spreading. J Cell Sci. 1997;110:1091–8. doi: 10.1242/jcs.110.9.1091. [DOI] [PubMed] [Google Scholar]
- 8.Providence KM, Kutz SM, Staiano-Coico L, Higgins PJ. PAI-1 gene expression is regionally induced in wounded epithelial cell monolayers and required for injury repair. J Cell Physiol. 2000;182:269–80. doi: 10.1002/(SICI)1097-4652(200002)182:2<269::AID-JCP16>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 9.Romer J, Bugge TH, Pyke C, Lund LR, Flick MJ, Degen JL, Dano K. Plasminogen and wound healing. Nat Med. 1996;2:725. doi: 10.1038/nm0796-725a. [DOI] [PubMed] [Google Scholar]
- 10.Romay C, Gonzalez R, Ledon N, Remirez D, Rimbau V. C-phycocyanin: a biliprotein with antioxidant, anti-inflammatory and neuroprotective effects. Curr Protein Pept Sci. 2003;4:207–16. doi: 10.2174/1389203033487216. [DOI] [PubMed] [Google Scholar]
- 11.Ge B, Qin S, Han L, Lin F, Ren Y. Antioxidant properties of recombinant allophycocyanin expressed in Escherichia coli. J Photochem Photobiol B-Biol. 2006;84:175–80. doi: 10.1016/j.jphotobiol.2006.02.008. [DOI] [PubMed] [Google Scholar]
- 12.Li B, Zhang X, Gao M, Chu X. Effects of CD59 on antitumoral activities of phycocyanin from Spirulina platensis. Biomed Pharmacother. 2005;59:551–60. doi: 10.1016/j.biopha.2005.06.012. [DOI] [PubMed] [Google Scholar]
- 13.Madhyastha HK, Radha KS, Sugiki M, Omura S, Maruyama M. Purification of c-phycocyanin from Spirulina fusiformis and its effect on the induction of urokinase-type plasminogen activator from calf pulmonary endothelial cells. Phytomedicine. 2006;13:564–9. doi: 10.1016/j.phymed.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 14.Madhyastha HK, Radha KS, Sugiki M, Omura S, Maruyama M. C-phycocyanin transcriptionally regulates uPA mRNA through cAMP mediated PKA pathway in human fibroblast WI-38 cells. Biochim Biophys Acta. 2006;1760:1624–30. doi: 10.1016/j.bbagen.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 15.Liang CC, Park AY, Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protocol. 2007;2:329–33. doi: 10.1038/nprot.2007.30. [DOI] [PubMed] [Google Scholar]
- 16.Wilson SE, He YG, Weng J, Zieske JD, Jester JV, Schultz GS. Effect of epidermal growth factor, hepatocyte growth factor, and keratinocyte growth factor, on proliferation, motility and differentiation of human corneal epithelial cells. Exp Eye Res. 1994;59:665–78. doi: 10.1006/exer.1994.1152. [DOI] [PubMed] [Google Scholar]
- 17.Gopinath D, Ahmed MR, Gomathi K, Chitra K, Sehgal PK, Jayakumar R. Dermal wound healing processes with curcumin incorporated collagen films. Biomaterials. 2004;25:1911–7. doi: 10.1016/s0142-9612(03)00625-2. [DOI] [PubMed] [Google Scholar]
- 18.Coller HA. What's taking so long? S-phase entry from quiescence versus proliferation. Nat. Rev. 2007;8:667–70. doi: 10.1038/nrm2223. [DOI] [PubMed] [Google Scholar]
- 19.Morgan DO. Principles of CDK regulation. Nature. 1995;374:131–4. doi: 10.1038/374131a0. [DOI] [PubMed] [Google Scholar]
- 20.Hunter T, Pines J. Cyclins and cancer. II: Cyclin D and CDK inhibitors come of age. Cell. 1994;79:573–82. doi: 10.1016/0092-8674(94)90543-6. [DOI] [PubMed] [Google Scholar]
- 21.Singer AJ, Clark RA. Cutaneous wound healing. N Eng J Med. 1999;341:738–46. doi: 10.1056/NEJM199909023411006. [DOI] [PubMed] [Google Scholar]
- 22.Fulga TA, Rorth P. Invasive cell migration is initiated by guided growth of long cellular extensions. Nat Cell Biol. 2002;4:715–9. doi: 10.1038/ncb848. [DOI] [PubMed] [Google Scholar]
- 23.Gillitzer R, Goebeler M. Chemokines in cutaneous wound healing. J Leukoc Biol. 2001;69:513–21. [PubMed] [Google Scholar]
- 24.Nobes CD, Hall A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J Cell. Biol. 1999;144:1235–44. doi: 10.1083/jcb.144.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev. 2002;3:349–63. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 26.Malumbres M, Barbacid M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer. 2001;1:222–31. doi: 10.1038/35106065. [DOI] [PubMed] [Google Scholar]
- 27.Laronga C, Yang HY, Neal C, Lee MH. Association of the cyclin-dependent kinases and 14–3-3 sigma negatively regulates cell cycle progression. J Biol Chem. 2000;275:23106–12. doi: 10.1074/jbc.M905616199. [DOI] [PubMed] [Google Scholar]
- 28.Plekhanova OS, Stepanova VV, Ratner EI, Bobik A, Tkachuk VA, Parfyonova YV. Urokinase plasminogen activator in injured adventitia increases the number of myofi-broblasts and augments early proliferation. J Vas Res. 2006;43:437–46. doi: 10.1159/000094906. [DOI] [PubMed] [Google Scholar]
- 29.Basire A, Sabatier F, Ravet S, Lamy E, Mialhe A, Zabouo G, Paul P, Gurewich V, Sampol J, Dignat-George F. High urokinase expression contributes to the angio-genic properties of endothelial cells derived from circulating progenitors. Thromb Haemost. 2006;95:678–88. [PubMed] [Google Scholar]
- 30.Alfano D, Franco P, Vocca I, Gambi N, Pisa V, Mancini A, Caputi M, Carriero MV, Iaccarino I, Stoppelli MP. The urokinase plasminogen activator and its receptor: role in cell growth and apoptosis. Thromb Haemost. 2005;93:205–11. doi: 10.1160/TH04-09-0592. [DOI] [PubMed] [Google Scholar]
- 31.Gudewicz PW, Gilboa N. Human urokinase-type plasminogen activator stimulates chemotaxis of human neutrophils. Biochem Biophys Res Commun. 1987;147:1176–81. doi: 10.1016/s0006-291x(87)80193-6. [DOI] [PubMed] [Google Scholar]
- 32.Fibbi G, Ziche M, Morbidelli L, Magnelli L, Del Rosso M. Interaction of urokinase with specific receptors stimulates mobilization of bovine adrenal capillary endothelial cells. Exp. Cell Res. 1988;179:385–95. doi: 10.1016/0014-4827(88)90277-7. [DOI] [PubMed] [Google Scholar]
- 33.Adam L, Vadlamudi R, Kondapaka SB, Chernoff J, Mendelsohn J, Kumar R. Heregulin regulates cytoskeletal reorganization and cell migration through the p21-activated kinase-1 via phosphatidylinositol-3 kinase. J Biol Chem. 1998;273:28238–46. doi: 10.1074/jbc.273.43.28238. [DOI] [PubMed] [Google Scholar]
- 34.Reiske HR, Kao SC, Cary LA, Guan JL, Lai JF, Chen HC. Requirement of phosphatidylinositol 3-kinase in focal adhesion kinase-promoted cell migration. J Biol Chem. 1999;274:12361–6. doi: 10.1074/jbc.274.18.12361. [DOI] [PubMed] [Google Scholar]
- 35.Van haesebroeck B, Jones GE, Allen WE, Zicha D, Hooshmand-Rad R, Sawyer C, Wells C, Waterfield MD, Ridley AJ. Distinct PI(3)Ks mediate mitogenic signalling and cell migration in macrophages. Nat Cel Biol. 1999;1:69–71. doi: 10.1038/9045. [DOI] [PubMed] [Google Scholar]
- 36.Fukata M, Nakagawa M, Kaibuchi K. Roles of Rho-family GTPases in cell polarisation and directional migration. Curr Opin Cell Biol. 2003;15:590–7. doi: 10.1016/s0955-0674(03)00097-8. [DOI] [PubMed] [Google Scholar]
- 37.Kusch A, Tkachuk S, Haller H, Dietz R, Gulba DC, Lipp M, Dumler I. Urokinase stimulates human vascular smooth muscle cell migration via a phosphatidylinositol 3-kinase-Tyk2 interaction. J Biol Chem. 2000;275:39466–73. doi: 10.1074/jbc.M003626200. [DOI] [PubMed] [Google Scholar]
- 38.Gundersen GG, Bulinski JC. Selective stabilization of microtubules oriented toward the direction of cell migration. Proc Natl Acad Sci. USA. 1988;85:5946–50. doi: 10.1073/pnas.85.16.5946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–14. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 40.Etienne-Manneville S, Hall A. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell. 2001;106:489–98. doi: 10.1016/s0092-8674(01)00471-8. [DOI] [PubMed] [Google Scholar]
- 41.Babbin BA, Jesaitis AJ, Ivanov AI, Kelly D, Laukoetter M, Nava P, Parkos CA, Nusrat A. Formyl peptide receptor-1 activation enhances intestinal epithelial cell restitution through phosphatidylinositol 3-kinase-dependent activation of Rac1 and Cdc42. J Immunol. 2007;179:8112–21. doi: 10.4049/jimmunol.179.12.8112. [DOI] [PubMed] [Google Scholar]
- 42.Kiian I, Tkachuk N, Haller H, Dumler I. Urokinase-induced migration of human vascular smooth muscle cells requires coupling of the small GTPases RhoA and Rac1 to the Tyk2/PI3-K signalling pathway. Thromb Haemost. 2003;89:904–14. [PubMed] [Google Scholar]
- 43.Vicente-Manzanares M, Rey M, Jones DR, Sancho D, Mellado M, Rodriguez-Frade JM, del Pozo MA, Yanez-Mo M, de Ana AM, Martinez AC, Merida I, Sanchez-Madrid F. Involvement of phosphatidylinositol 3-kinase in stromal cell-derived factor-1 alpha-induced lymphocyte polarization and chemotaxis. J Immunol. 1999;163:4001–12. [PubMed] [Google Scholar]
- 44.Hooshmand-Rad R, Claesson-Welsh L, Wennstrom S, Yokote K, Siegbahn A, Heldin CH. Involvement of phosphatidylinositide 3 -kinase and Rac in platelet-derived growth factor-induced actin reorganization and chemotaxis. Exp Cell Res. 1997;234:434–41. doi: 10.1006/excr.1997.3636. [DOI] [PubMed] [Google Scholar]
- 45.Kunigal S, Gondi CS, Gujrati M, Lakka SS, Dinh DH, Olivero WC, Rao JS. SPARC-induced migration of glioblastoma cell lines via uPA-uPAR signaling and activation of small GTPase RhoA. Int J Oncol. 2006;29:1349–57. [PMC free article] [PubMed] [Google Scholar]
- 46.Fivenson DP, Faria DT, Nickoloff BJ, Poverini PJ, Kunkel S, Burdick M, Strieter RM. Chemokine and inflammatory cytokine changes during chronic wound healing. Wound Repair Regen. 1997;5:310–22. doi: 10.1046/j.1524-475X.1997.50405.x. [DOI] [PubMed] [Google Scholar]
- 47.Cross AK, Woodroofe MN. Chemokine modulation of matrix metalloproteinase and TIMP production in adult rat brain microglia and a human microglial cell line in vitro. Glia. 1999;28:183–9. [PubMed] [Google Scholar]
- 48.Li J, Chen J, Kirsner R. Pathophysiology of acute wound healing. Clin Dermatol. 2007;25:9–18. doi: 10.1016/j.clindermatol.2006.09.007. [DOI] [PubMed] [Google Scholar]