

Abstract
Recently, synthesis and secretion of connective tissue growth factor (CTGF)/CYR61/CTGF/NOV-family member 2 (CCN2) in cultures of hepatocytes were shown, which are sensitively up-regulated by exogenous TGF-β. In this study TGF-β-dependent CTGF/CCN2 expression in hepatocytes cultured under completely TGF-β-free conditions was analysed by Western-blots, metabolic labelling, and CTGF-reporter gene assays. In alkaline phosphatase monoclonal anti-alkaline phosphatase complex (APAAP)-staining of cultured hepatocytes it was demonstrated that latent TGF-β within the hepatocytes becomes rapidly detectable during culture indicating an intracellular demasking of the mature TGF-β antigen. Subsequent signaling to theCTGF/CCN2 promoter occurs via p-Smad2, whereas p-Smad3 does not seem to be involved. Cycloheximide did not abolish the rapid immunocytochemical appearance of mature TGF-β, but calpain inhibitors partially suppressed intracellular TGF-β activation and subsequently CTGF up-regulation. Calpain treatment had the reverse effect. None of the inhibitors of extracellular TGF-β signalling was effective in the reduction of spontaneous CTGF synthesis, but intracellularly acting Alk 4-/Alk 5-specific inhibitor SB-431542 was able to diminish CTGF expression. The assumption that latent intracellular TGF-β is activated by calpains during culture-induced stress or injurious conditions in the liver in vivo was further validated by a direct effect of calpains on the activation of recombinant latent TGF-β. In conclusion, these data are the first to suggest the possibility of intracrine TGF-β signalling due to calpain-dependent intracellular proteolytic activation leading to transcriptional activation of CTGF/CCN2 as a TGF-β-sensitive reporter gene. This mechanism might be deleterious for keeping long-term hepatocyte cultures due to TGF-β-induced apoptosis and, further, might be of relevance for induction of apoptosis or epithelial-mesenchymal transition of hepatocytes in injured liver.
Keywords: TGF-β, TGF-β latency; intracellular activation; intracrine signalling; hepatocytes; CTGF/CCN2
Introduction
There is profound evidence for a significant (patho-)physiologic role of the multifunctional polypeptide growth factor transforming growth factor (TGF)-β in the liver [1, 2]. It is an important regulator of cell proliferation and liver regeneration [3, 4], a causative agent of liver cell apoptosis [5, 6], a strong immunosuppressant and viability factor for T-lymphocytes [7], and an important modulator of primary and secondary (liver) tumors [3, 8, 9]. One of the major roles of TGF-β, however, concerns its profibrogenic action on hepatic stellate cells (HSC) [1, 10, 11]. This precursor cell type of myofibroblasts (MFB) is activated by TGF-β to trans-differentiate to MFB [10], and TGF-β stimulates the expression of collagens [12, 13], proteoglycans [14], and hyaluronan [15, 16], enhances the expression of tissue inhibitors of metallopro-teinases (TIMPs). It furthermore down-regulates matrix metallo-proteinases (MMPs) [17, 18], and thus, affects matrix remodeling. In addition, newly discovered fibrogenic pathways in liver and other organs as well, such as epithelial-mesenchymal transition (EMT) [19–21], activation of portal fibroblasts to myofibroblasts [22], and homing of circulating fibrocytes in liver and their transition to (myo-)fibroblasts are critically dependent on the action of TGF-β[23].
The biologic activity of this pluripotent cytokine is mainly regulated by extracellular mechanisms leading to the conversion of the secreted, latent form into the active ligand capable of receptor binding [24, 25] and initiation of the phosphorylation of the intracellular cascade of Smad proteins [26]. But even after activation of the latent complex, the extracellular activity of TGF-β is positively or negatively influenced by several enhancer or trapping proteins, which modulate the receptor activity of mature TGF-β[27]. Among them, CTGF/CCN2, a 36–38 kD cysteine-rich, heparin-binding, and secreted protein belonging to the CCN-gene family [28, 29], plays an important role. It is suggested asa downstream modulator protein of TGF-β action, which facilitates its binding to the signal-generating receptors [30], and which enhances the TGF-β/Smad signalling pathway [31]. Taken together, CTGF/CCN2 is thought to amplify the action of TGF-β[32].
We recently showed that hepatocytes (PC) express CTGF/CCN2, which is strongly upregulated by TGF-β[33]. In comparison to CTGF synthesis in HSC/MFB, which is dependent on endothelin-1, but only moderately regulated by TGF-β, production and secretion of CTGF/CCN2 in diseased liver might be much more attributed to PC than to HSC/MFB and biliary epithelial cells [34], because of the prevalent number of parenchymal liver cells in relation to HSC (hepatic stellate cell index: 109 HSC/1000 PC; [35]) and the sensitive up-regulation by TGF-β in these cells. Therefore, we focused the present study on detailed aspects of TGF-β-dependent regulation of CTGF/CCN2 expression in PC. Based on previously published data showing the presence of several components of the TGF-β-system such as latency associated peptide (LAP) [36], latent TGF-β-binding protein (LTBP) [37], and TGF-β itself within the cultured PC [38], we analysed and characterized the spontaneous CTGF/CCN2 expression in these cells maintained under culture conditions completely free of exogenously added TGF-β. We found that culture-induced stress leads to activation of intracellular latent TGF-β (subtype 1) and signals entirely intracellularly resulting in strong up-regulation of CTGF/CCN2. The data suggest this intracrine mechanism as a very early response of PC to liver injury, which rapidly leads to the secretion of this increasingly important, paracrine acting TGF-β downstream modulator protein in the local tissue or even systemically in the circulation.
Materials and methods
Materials
Recombinant soluble TGF-β type II receptor (rsTβRII) was constructed as previously described [39]; anti hTβRII (AF-241-NA), rhTGFβ-1 (240-B), rhLAP [TGF-β] (246-LP), recombinant latent TGF-β1, neutralizing anti TGF-β1 antibody (AF-101-NA) and mouse monoclonal anti-TGF-β1/-β2/-β3 (MAB1835) were from R&D Systems (Minneapolis, MN); calpain inhibitor III (#208722), calpain inhibitor IV (#208724), furin inhibitor II (#344931), human calpain I (#208713) and rat calpain II (#208718) were from Calbiochem (Darmstadt, Germany); Alk4/5 inhibitor (SB-431542) was from Tocris Bioscience (Ellisville, MO); actinomycin D (A1410), aphidicolin (A0781) and cycloheximide (#01810) were from Sigma Aldrich (St. Louis, MO); rabbit anti-Smad3 (ab28379) was from Abcam (Cambridge, UK); goat anti-CTGF/CCN2 (L-20, sc-14939) was from Santa Cruz (Santa Cruz, CA); rabbit anti-pSmad3 (Ser423/425)/pSmad1 (Ser463/465) (#9514), rabbit anti-pSmad2 (Ser465/467) (#3101) and rabbit anti-Smad2 (#3102) were from Cell Signaling/New England Biolabs (Ipswich, MA); and mouse anti-β-actin was from Cymbus Bioscience (Southampton, UK).
Cell culture and preparation of cells
All animals received human care in compliance with the German Tierschutzgesetz, which is in accordance with the National Research Council's criteria. PC were isolated from male Sprague-Dawley rats (180–320 g body weight) by the two-step collagenase method of Seglen [40] modified as described before [41]. As indicated, cells were isolated and cultured in the complete absence of (TGF-β containing) fetal calf serum (FCS). The viability of the final parenchymal cell suspension, checked by trypan blue exclusion, was around 85%, and cell recovery was 20–50 × 107 cells/liver. Contamination with other nonparenchymal cells was less than 2% and proven by RT-PCR for decorin (data not shown).
Cells were seeded in 2 ml GIBCO™ HepatoZYME-SFM (Invitrogen, Carlsbad, CA) without FCS on type I collagen (BD Bioscience, Erembodegem, Belgium) coated plastic dishes (Becton Dickinson, Franklin Lakes, NJ) with a density of 5.4 × 104 cells/cm2. The presence of mature or latent TGF-β in HepatoZYME-SFM after acid activation was excluded by ELISA (EIA-1864, DRG Diagnostics, Marburg, Germany) [36, 38], which could not detect the respective antigen. Obviously, TGF-β was either absent in the culture medium or below the detection limit of the assay (1.9 pg/mL). PC were cultured at 37°C in a humidified atmosphere of 5% CO2 and 95% air in entirely serum-free (TGF-β-free) medium supplemented with 4 mmol/L L-glutamine, penicillin (100 IU/mL), and streptomycin (100 mg/mL) (all from Cambrex, East Rutherford, NJ). The first change of the medium was 1 h after seeding and unattached PC were washed off with Dulbecco's Modified Eagle's Medium (DMEM) (Cambrex). PC were then cultured for various times in serum-free GIBCO™ HepatoZYME-SFM, supplemented with L-glutamine and penicillin/streptomycin as described above.
HSC isolation
HSC were isolated by the pronase-collagenase perfusion technique, purified by single-step density gradient centrifugation with Nycodenz (Nyegaard, Oslo, Norway) as described [42] and characterized by their typical light microscopic appearance, immunostaining for desmin and vimentin, and vitamin A-specific autofluorescence. Cell viability checked by the trypan blue exclusion test was normally higher than 95%, and the mean purity was 90 ± 5%.
Sodium Dodecyl Sulfate-Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Western Blotting
Preparations of cell extracts, determination of protein concentrations, and Western blot analysis were performed as previously described [36].
Primary antibodies of various species were visualized using horseradish peroxidase- or alkaline phosphatase-conjugated anti-mouse, -goat or -rabbit IgG (Santa Cruz) and the Supersignal West Dura Extended chemiluminescent (Pierce Biotechnology, Rockford, IL) or NBT/BCIP substrate (Perbio Science, Rockford, IL).
Metabolic labelling
The methods of metabolic labelling of PC cultures and immunoprecipita-tion of radioactively labelled proteins were described previously [37].
Briefly, PC cultured in 28 cm2 culture dishes for 24 hrs were labelled with the Pro-Mix L-[35S]in vitro cell labelling mix (Amersham Biosciences, Little Chalfont, UK) 3 hrs before the selected time points in the presence or absence of calpain inhibitor III, cycloheximide and Alk4/5 inhibitor, respectively. Thereafter, the culture medium was discarded, cells were washed and scraped off with lysis buffer (RIPA + Complete™[a mixture of protease-inhibitors; Roche]+ phosphatase inhibitor cocktail II [Sigma-Aldrich]). After a preclearing step with non-immune IgG, the cell lysate was incubated with the CTGF/CCN2 antibody followed by precipitation with protein-G agarose (Santa Cruz) and several washings.
The immunocomplexes were resolved in lysis buffer, LDS (lithium dodecyl sulfate; Invitrogen) and DTT (dithiotreitol; Sigma-Aldrich). The radioactivity incorporated into the CTGF protein was determined using a β-counter (Packard, Downer Grove, IL) and referred to total DNA.
For autoradiography, the beads were resuspended in NuPAGE 2 × LDS sample buffer (Invitrogen) including DTT, heated for 10 min at 70°C and subjected to a 4–12% gel gradient. The gel was fixed, soaked in Amplify (Amersham), dried, and exposed to a BIOMAX MR film (Kodak, Stuttgart, Germany).
Immunocytochemistry
Immunocytochemistry was carried out with the alkaline phosphatase anti-alkaline phosphatase (APAAP) method essentially as previously described [38, 43]. Briefly, PC cultured for 24 hrs were fixed with 95% (v/v) ethanol/5% (v/v) acetic acid at 4°C for 24 hrs. After fixation cells were washed in Tris-buffered saline, and unspecific binding sites were blocked with 1% bovine serum albumin, 0.1% fish gelatin, 0.1% TritonX-100 and 0.05% Tween 20. Cells were then incubated for 1 hr with either goat anti-CTGF/CCN2 (diluted 1:300) or mouse anti-TGF-β1/-β2/-β3 (diluted 1:50) in Tris-buffered saline plus 0.1% bovine serum albumin, followed, when necessary, by additions of the appropriate link antibodies (mouse anti-goat IgG, #31107, Pierce), and finally, by a biotinylated rabbit-anti mouse secondary antibody (Z0259; Dako, Glostrup, Denmark), the mouse monoclonal APAAP complex (Dako), and the Fast Red chromogenic substrate system (Dako). According to the manufacturer, the used mouse monoclonal anti-TGF-β1/-β2/-β3 antibody specifically detects the biologically active, mature peptide. The slides were counterstained with hematoxylin and mounted in glycergel (Dako). Negative controls were performed similarly, but with unspecific mouse or goat immunoglobulin G (IgG) instead of the specific primary antibodies.
For 0 hr stainings (cytospin slides), an aliquot of freshly isolated cell suspension was diluted in GIBCO™ HepatoZYME-SFM in order to obtain a concentration of 105 PC/ml. A total of 200 μl of the suspension were added to cytospin pots (Thermo Scientific, Rockford, IL), and centrifuged for 3 min at 500 rpm. The slides were removed and allowed to dry at room temperature.
Immunohistochemical stainings
Liver specimens of untreated rats or rats subjected to intraperitoneal injection of CCl4 (25% solution in mineral oil [2 ml/kg body weight]; Merck) or D-galactosamine-HCl (D-GalN; 500 mg/kg body weight; Merck, Darmstadt, Germany), respectively, were fixed in 4% paraformaldehyde solution for 4 hrs and embedded in paraffin. After conventional processing as described previously [38, 44], tissue sections (2 μm) were prepared, mounted on glass slides and dried at 56°C for 1 hr. After deparaffinization, endogenous peroxidase blocking as described [38, 44] and incubation with normal serum for 30 min, sections were incubated with the primary monoclonal antibody against goat anti-TGF-β1/-β2/-β3 (dilution, 1:50) at 37°C for 1 hr or overnight at 4°C, respectively, followed by APAAP staining as described above.
RT-PCR for TGF-β1
Total cellular RNA was extracted from PC and HSC, respectively, with the Qiagen RNeasy purification kit (Qiagen, Hilden, Germany). cDNA was reverse-transcribed using the First-Strand cDNA synthesis kit (Invitrogen). RT-PCR was performed using the Biometra T3000 Thermocycler PCR System (Biometra, Göttingen, Germany) and the following primers: rat TGF-β1 (forward: 5′-CTC TCC ACC TGC AAG AC-3′; reverse: 5′-GGA CTC TCC ACC TGC AAG AC-3′) and rat decorin (forward: 5′-CTC TGG CAT AAT CCC TTA CGA-3′; reverse: 5′-GGT ATG CAA GTC CTT CAG GTT-3′). Primers were purchased from MWG Biotech (Ebersberg, Germany). The size of the amplified TGF-β1 fragment was 450 bp and the size of the amplified decorin fragment was 214kb.
In general, 1 μl cDNA in 14.5 μL of RNAse free water (Roche Diagnostics, Mannheim, Germany) and 5 μl 5 × Taq Master PCR Enhancer (Eppendorf, Hamburg, Germany) was mixed with 0.5 μl dNTPs (Qiagen), 2.5 μl of 10 × PCR Buffer/15 mM MgCl2 (Qiagen), 0.5 μl of 10 μM forward primer, 0.5 μl of 10 μM reverse primer and 0.5 μl of Taq polymerase (1U/μl; Roche Diagnostics). The following PCR conditions were used: 95°C/4 min, followed by 40 cycles at 94°C/30 sec, 54°C/30 sec and 72°C/1.5 min, then 72°C/10 min, and stopped at 4°C. PCR products were separated on 4% agarose gels and visualized under UV-Light (MWG Biotech).
hCTGF/CCN2-Promotor-luciferase gene reporter assay
The vector pGL3-hCTGF-Luc was cloned as previously described [33]. Briefly, hCTGF/CCN2 promoter sequences were amplified from human genomic DNA in a standard PCR using primer CTGFu 5′-d(GAG CTC CTG CTG TTT GCC TCT TCA GCT AC)-3′ and CTGFr 5′-d(AAG CTT GTC GTC TCG GGG CTG TCG GA)-3′ containing additional cloning sites for SacI and HindIII (underlined). The amplified fragment containing 508-bp of the annotated CTGF/CCN2 promoter (nt 33708 to 33201, GenBank Accession no. AL354866) was first cloned into pGEM-T-Easy vector, verified by sequencing, and the SacI/HindIII fragment was then subcloned into vector pGL3-Basic (Promega, Madison, WI).
PC were simultaneously transfected with 200 ng of hCTGF/CCN2-luc and 20 pg of promoterless Renilla luciferase as internal standard using FuGENE 6™ according to the manufacturer's instructions (Roche). At specific time points, hCTGF/CCN2 luciferase activities were determined and normalized to Renilla luciferase. Results are presented as luciferase activity relative to Renilla-luciferase activity.
TGF-β1 enzyme linked immunosorbent assay (ELISA)
Freshly isolated PC (107/ml) were lysed on ice by ultrasound 3 × 30 sec (0.5 W) in GIBCO™-HepatoZYME-SFM. After centrifugation (10,000 g, 4°C, 3 hrs), the supernatant was divided into 2 ml aliquots. Some aliquots were incubated each with 0.5 U/ml calpain 1 and calpain 2 for 1 hr, respectively, other aliquots were kept at 0°C and 37°C for 1 hr, respectively, without addition of calpain. Thereafter, the active fraction of TGF-β was measured with an ELISA, which is specific for mature (active) TGF-β1 (EIA-1864, DRG Diagnostics) [36, 38].
Results
Spontaneous and TGF-β-induced expression of CTGF/CCN2 in cultured rat PC
Freshly cultured PC express only trace amounts of CTGF, but during culture under serum-free conditions a gradual increase of CTGF expression can be recorded (Fig. 1A). The expression is rapidly stimulated by TGF-β1. Furthermore, PC transfected with the CTGF-luciferase reporter gene under serum-reduced conditions show significantly more luciferase activity after 24 hrs than at the onset of culture. The 24 hrs expression level is enhanced 5.5-fold by TGF-β (Fig. 1B). In PC-cultures maintained under serum-rich conditions (10% FCS), the expression of CTGF is accelerated reaching a maximum level between 12 and 16 hrs of culture (Fig. 1C). But also under completely serum-free conditions, a gradual, even though less pronounced increase of CTGF in Western blot analysis is noted (Fig. 1C). CTGF expression in PC cultures is due to active de novo synthesis, as both, the transcriptional inhibitor actinomycin D (Fig. 1D) and the translational inhibitor cycloheximide (Fig. 1E), completely suppress the appearance of immuno-blotted CTGF (Fig. 1D) and the biosynthetic labelling of CTGF with [35S] methionine/cysteine (Fig. 1E). The latter one measures the current biosynthetic activity during a pulse labelling of each 3 hrs at the indicated time points. This is in contrast to the immune-blot analyses where total protein after a certain culture time is detected. The incorporation of the radioactive label into immunoprecipitated CTGF increased rapidly during early culture reaching maximum levels about 15 hrs after initiation of the primary culture (Fig. 1E). As expected, the DNA-polymerase inhibitor aphidicolin was without any inhibitory effect on CTGF expression (Fig. 1D).
Fig. 1.
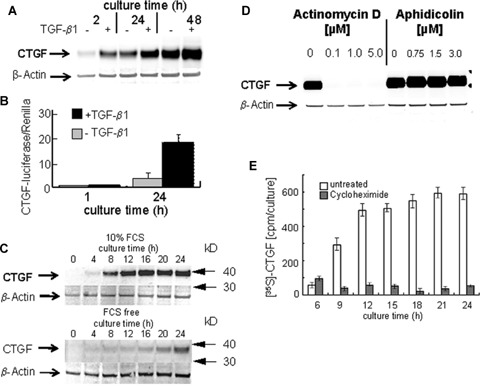
TGF-β-induced and spontaneous expression of CTGF in cultured rat PC (A) Western blot of CTGF of PC cultured for various times under serum-free conditions with or without addition of 5 ng/ml rh TGF-β1. Lysate was separated on 4–12% gel gradient in SDS (25μg protein/lane) under non-reducing conditions and probed for CTGF/CCN2 using a polyclonal anti CTGF-antibody. β-Actin always served as loading control. A representative blot of three independent experiments is shown. (B) CTGF/CCN2 reporter gene activation. PC were transfected simultaneously with 200 ng of reporter plasmid phCTGF-luciferase and 20 pg of promoterless Renilla-luciferase as internal control 1 hr after isolation and cultured under serum-free conditions with or without addition of 5ng/ml rh TGF-β1. Luciferase activity relative to Renilla-luciferase was measured 1 hr and 24 hrs after seeding. Mean values +/- SD of three independent experiments are given. (C) Western blot of CTGF of PC cultured for various times under serum-rich (10% FCS, top) and serum-free (bottom) conditions, respectively. Two representative blots of four independent experiments are shown. (D) Western blot of CTGF of PC cultured under serum-free conditions with or without addition of actinomycin D and aphidicolin, respectively. Representative blots of three independent experiments are shown. (E) Immunoprecipitation of metabolically labeled CTGF. PC were cultured in serum-, cysteine- and methionine-free HepatoZYME-SFM for 24 hrs in the absence or presence of cycloheximide (5 μM) and pulse-labeled with the PRO-MIX L-[35S] methionine/cysteine in vitro cell labeling kit each for 3 hrs before indicated time points. Culture medium was discarded, cells were washed, and cellular protein extracts were prepared, in which CTGF/CCN2 was immunoprecipitated with polyclonal anti-CTGF antibody and protein-G agarose beads. Immunocomplexes were resolved by RIPA+Complete, LDS and DTT. The radioactivity incorporated into CTGF was determined using a β-counter.
Effect of internal and external inhibitors of TGF-β signalling on CTGF/CCN2 expression in PC cultured under TGF-β-free conditions
The data described above suggested that the spontaneous CTGF expression found even under serum- and consequently TGF-β-free culture conditions might as well be regulated by intracellularly deposited TGF-β in PC. To evaluate its potential role, serum-free cultures were exposed to a specific, intracellularly acting Alk4/Alk5-serine/threonine kinase inhibitor (SB-431542) [45], which clearly shows a strong, but not complete inhibition of spontaneous as well as TGF-β-induced CTGF expression (Fig. 2A), indicating a TGF-β/Alk5 dependency of spontaneous CTGF expression. Accordingly, pulsed biosynthetic labelling of CTGF, each for a period of 3 hrs before various time points of PC culture, exhibits a reduction of immuno-precipitated [35S]-labelled CTGF at each time point (Fig. 2B). The consistent reduction of CTGF de novo synthesis eventually results in a strong reduction of overall availability of CTGF within the cell, as seen in Western blot analysis (Fig. 2A). Amazingly, if TGF-β inhibitors are used, which inhibit the extracellular activity of the ligand such as a soluble receptor type II TGF-β (sTβRII), LAP, and neutralizing TGF-β1 antibody (βTGF-β1), no inhibitory effect on spontaneous CTGF expression is noticed (Fig. 2C). In addition, a blocking antibody against type II TGF-β receptor (βTβRII) did not reduce CTGF-luciferase expression of PC transfected with the respective reporter gene construct (Fig. 2D). Functionality of the inhibitors regarding exogenously added TGF-ß was proven in separate experiments (data not shown) and also published previously [39, 46].
Fig. 2.
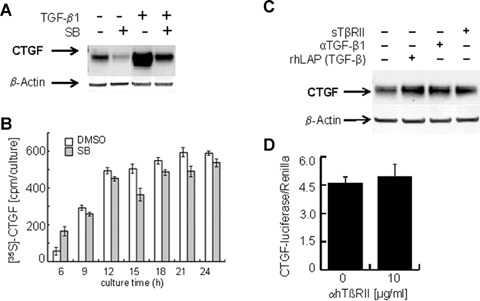
Effect of external and internal inhibitors of TGF-β signalling on CTGF expression in PC cultured under TGF-β-free conditions (A) Western blot of CTGF of PC cultured for 24 hrs under completely serum-free conditions with or without addition of 5ng/ml rh TGF-β1. Some cultures received the Alk4/5 inhibitor (SB-431542, 5 μM), lysates were probed for CTGF/CCN2 and β-actin expression. A representative blot of four independent experiments is shown. (B) Immunoprecipitation of metabolically labeled CTGF. PC were cultured in serum-, cysteine- and methionine-free DMEM for 24 hrs in the absence or presence of Alk4/5 inhibitor (SB-431542, 5μM) and labeled with the PRO-MIX L-[35S] methionine/cysteine in vitro cell labelling kit each for 3 hrs at indicated time points and processed as described in Fig. 1E. (C) Western blot of CTGF of PC cultured for 24 hrs under complete serum-free conditions. Cultures received various inhibitors of extra- and intracellular TGF-β1 signalling like rsTβRII (1 μg/ml), neutralizing anti TGF-β1 antibody (4 μg/ml) and rhLAP [TGF-β] (1 μg/ml). Lysates were probed for CTGF/CCN2 and β-actin expression. A representative blot of three independent experiments is shown. (D) CTGF/CCN2 reporter gene activation. PC were prepared as described in Fig. 1B, but received a blocking anti hTβRII antibody (10 μg/ml). They were cultured for 24 hrs under completely serum-free conditions. CTGF-luciferase activities are shown relative to Renilla luciferase activity. Mean values ± SD of four experiments are shown.
We then analysed the activation status of Smad2 and Smad3 in serum-(i.e. TGF-β-) free cultures of PC, 1 and 2 days after seeding. The expression level of phosphorylated Smad2 could be inhibited only by the intracellularly acting Alk4/Alk5-inhibitor, but not by extracellularly acting TGF-β antagonists or scavengers described above (Fig. 3A). Interestingly, p-Smad3 was not detectable at all (as shown by the missing lower band), but the unphosphorylated form of Smad3 was present at almost the same amounts in cells cultured for 24 hrs (Fig. 3A). Instead, there was a clear expression of phosphorylated Smad1, which, however, could not be reduced by the Alk4/Alk5-inhibitor as expected (Fig. 3A). We further demonstrate in control experiments that TGF-β1 added to PC cultures clearly induced the expected strong phosphorylation of Smad3 (and cross-reacting Smad1) (Fig. 3B). Thus, endogenous TGF-β-dependent CTGF expression in PC involves the phosphorylation of Smad2 but not of Smad3, which obviously is different to the signalling of exogenously acting TGF-β.
Fig. 3.
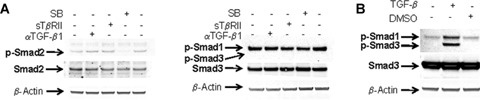
Effect of Alk4-/Alk5-inhibition on Smad-phosphorylation under TGF-β-free conditions (A) Western blot of p-Smad2, 1, 3 of PC cultured for 24 hrs under complete serum-free conditions and subjected to Alk4/5 Inhibitor (SB-431542, 5 μM), rsTβRII (1 μg/ml) and neutralizing anti TGF-β1 antibody (4 μg/ml), respectively. Lysates were probed for p-Smad2, p-Smad1, and p-Smad3, total Smad2, total Smad3, and β-actin, respectively. Representative blots of four independent experiments are shown. (B) Western blot of p-Smad3 (cross-reacting with p-Smad1) of PC cultured for 24 hrs under serum-free conditions with or without addition of rhTGF-β1 (5ng/ml). The blot is representative for three independent experiments.
Demasking of endogenous TGF-β in cultured hepatocytes and in injured liver
The observations described above suggest a CTGF synthesis in cultured PC, which is regulated by intracellular signalling of TGF-β. We favored the hypothesis that the concentration of hepa-tocellular TGF-β might increase during the stress of isolation and cell culture, because freshly isolated PC express only trace amounts of CTGF as described above (Fig. 1A and B) making a steady level of TGF-β within the PC unlikely. To test this assumption, freshly isolated and serum-free PC-cultures (24 hrs) were stained for TGF-β using the APAAP-procedure. As shown in figure 4, cytospins of freshly isolated PC were only marginally stained at the sub-membranous periphery, but 24 hrs later an intense staining of the entire cytosol was observed (Fig. 4A) which confirms previous findings [43]. TGF-β staining was insensitive to the treatment of PC with cycloheximide (Fig. 4B), which rules out de novo synthesis as a cause of increasing TGF-β staining. Instead, we propose a ‘stress-induced’ demasking process of intracellular latent TGF-β during cell culture. To investigate this finding in situ, TGF-β staining of normal, CCl4- and D-galactosamine-injured livers, respectively, was performed. PC of normal livers were completely TGF-β-negative, but 1 week and, more accentuated, 4 weeks of CCl4-treatment resulted in TGF-β positivity of PC spotted in the tissue (Fig. 5). Similarly, D-galactosamine-treated rats contained numerous TGF-β-positive PC one day after onset of liver injury (Fig. 5). Of note, in both models of toxic liver injury, TGF-β positive PC were predominantly located in the periportal fields.
Fig. 4.
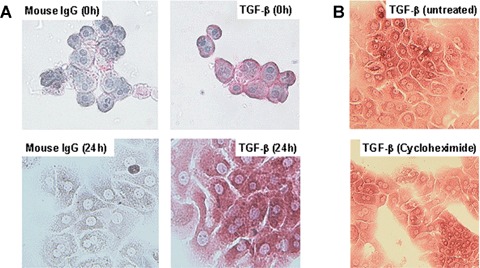
Time course of APAAP immunostainings of TGF-β1, 2,3 in cultured PC (A) APAAP immunostainings of TGF-β in freshly isolated PC (0 hr) and in PC cultured for 24 hrs under serum-free conditions. A monoclonal antibody against the three isoforms of TGF-β was used. According to the manufacturer, the used mouse monoclonal anti-TGF-β1/-β2/-β3 antibody specifically detects the biologically active, mature peptide. Controls were performed with non-specific mouse immunoglobulin G (IgG) instead of TGF-β specific first antibody (Original magnification 40×). (B) APAAP immunostainings of TGF-β in PC cultured for 24 hrs under serum-free conditions but in the presence or absence of cycloheximide (5 μM) (Original magnification 40×).
Fig. 5.
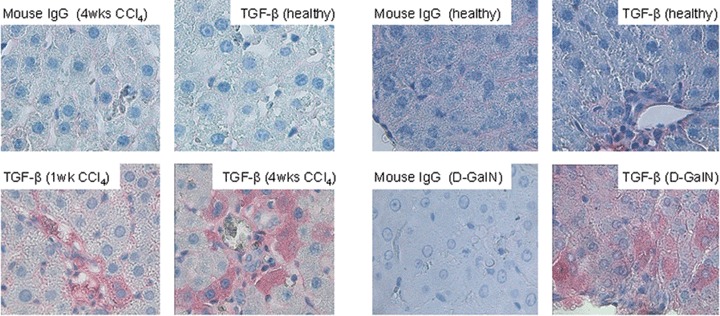
APAAP immunostainings of TGF-β1, 2, 3 in livers of CCl4- and D-galactosamine-injured rats Paraffin-embedded liver tissue of healthy rats and of rats treated intraperitoneally with CCl4 (1 week and 4 weeks) and D-galactosamine-HCl (D-GalN) (24 hrs), respectively were stained as described above (Original magnification 40β). Staining controls received a non-specific IgG instead of TGF-β specific first antibody.
Modulation of TGF-β demasking and CTGF increase in cultured PC by calpains and calpain inhibitors
The following experiments were set up to analyze potential mechanisms of TGF-β demasking in PC stressed by culture conditions and liver injury, respectively. Based on previous results [47], we suspected a role of calpains in this process. Consequently, serum-free PC cultures were treated with calpains 1 and 2 and calpain inhibitors, respectively and TGF-β staining intensity was semi-quantitatively tested against untreated control PC cultures. Calpain inhibitor III clearly reduced the staining intensity of TGF-β compared to control PC and PC treated with calpain 1 and calpain 2, respectively (Fig. 6). Obviously, the latter ones were more intensely stained than the untreated cells. Furthermore, PC treated with calpain 1 and calpain 2, respectively, developed a markably impaired morphology compared to controls (data not shown). These treatments also affected CTGF-immunostaining in a similar way. Compared to untreated PC cultures, addition of both calpain 1 and calpain 2 generated a clearly more intense CTGF-staining, whereas calpain inhibitor reduced the staining intensity (Fig. 7). Here, different time points (12 hrs, 24 hrs) were chosen to make differences in staining intensity more conspicuous.
Fig. 6.
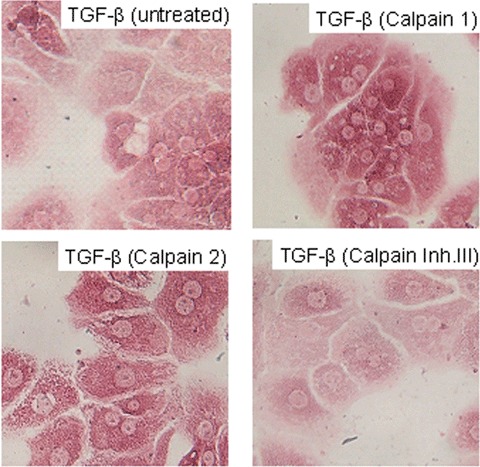
Modulation of TGF-β staining in cultured PC by calpain inhibitors and calpain APAAP immunostainings of TGF-β of PC were performed as described in Fig. 4. Cultured PC were treated with calpain 1 (0.5 U/ml), calpain 2 (0.5 U/ml), and cal-pain inhibitor III (20 μM), respectively for 24 hrs under serum-free conditions (Original magnification 40β).
Fig. 7.
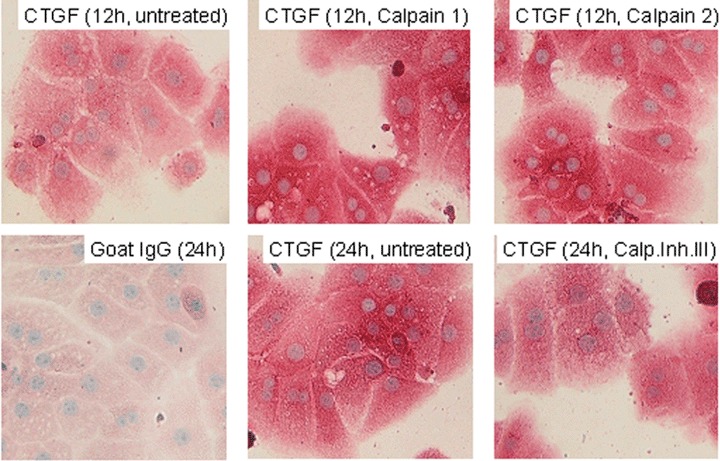
Modulation of CTGF-staining in cultured PC by calpain inhibitors and calpain APAAP immunostainings of CTGF in PC that were treated with calpain 1 (0.5 U/ml), calpain 2 (0.5 U/ml), and calpain inhibitor III (20 μM), respectively and cultured for 12 hrs and 24 hrs under serum-free conditions. Control PC cultures were untreated. Staining was performed with a goat polyclonal antibody against CTGF. The staining control was performed with unspecific goat IgG instead of the specific primary antibody (Original magnification 40β).
Effects of calpain inhibitors on CTGF-expression and of calpains on the activation of latent TGF-β
Next we tested whether calpain inhibition also affects CTGF expression in PC cultures treated with calpain III and calpain IV inhibitors, respectively, in a quantitatively relevant amount. A clear reduction of CTGF protein was observed when calpain inhibitors were added to the cultures (Fig. 8A), but the stimulatory effect of exogenous TGF-β on CTGF remained completely unaffected by both calpain inhibitors (Fig. 8B). The inhibitor of furin, a subtilisin-like proprotein convertase needed for processing of pro-TGF-β[48] had no effect on endogenous CTGF-expression supporting the fact that de novo synthesis of TGF-β only seems to play a minor role, if at all. Analogous to these findings, calpain inhibitors also reduced basal Smad2-phosphorylation while no effect was detectable on Smad3 phosphorylation caused by exogenous TGF-β stimulation (Fig. 8C).
Fig. 8.
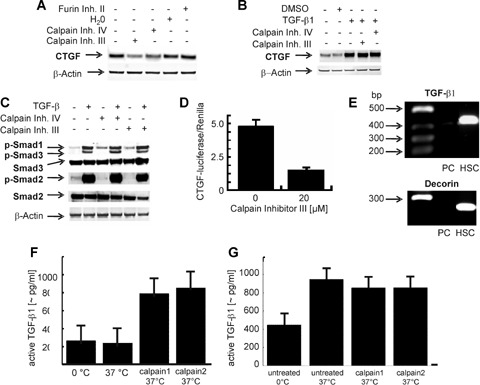
Effects of calpain (inhibitors) on transcriptional activation of the CTGF-gene and subsequent CTGF protein expression (A) Western blot of PC cultured for 24 hrs under serum-free conditions in the presence or absence of calpain inhibitor III (10 μM), calpain inhibitor IV (1 μM) and furin inhibitor (20 μM), respectively. Analysis was as described in Fig. 1A. A representative blot of four independent experiments is shown. (B) Western blot of PC cultured for 24 hrs under serum-free conditions in the presence or absence of calpain inhibitor III (10 μM), calpain inhibitor IV (1 μM) with or without addition of rh TGF-β1 (5 ng/ml). The blot is typical for three independent experiments. (C) Western blot of PC cultured for 24 hrs under serum-free conditions in the presence or absence of calpain inhibitor III (20 μM), calpain inhibitor IV (1 μM) with or without addition of rh TGF-β1 (5 ng/ml). One representative blot out of four independent experiments is shown. (D) CTGF/CCN2 reporter gene activation. Cultured PC were simultaneously transfected with hCTGF/CCN2-luciferase and promoterless Renilla-luciferase as internal control, subjected to calpain inhibitor III (20 μM) and cultured for 24 hrs under serum-free conditions. CTGF-luciferase activity relative to Renilla-luciferase is shown. Mean values ± SD of five experiments are shown. (E) RT-PCR for TGF-β1 and decorin in hepatic stellate cells (HSC) and PC cultured for 24 hrs. RT-PCR was performed using primers for TGF-β1 and decorin as described in Materials and Methods. Shown are the fragments for TGF-β1 and decorin after 32 cycles. The size of the amplified TGF-β1 fragment was 450 bp and the size of the amplified decorin fragment was 214kb. One representative result out of five is shown. (F) Activation of recombinant latent TGF-β (10 μg/ml) by calpain 1 and calpain 2 (each 0.5 U/ml) at 37°C. Thereafter, the concentration of immunologically detectable mature TGF-β was determined with an ELISA. Mean values ± SD of three experiments are given. (G) Activation of latent TGF-β in PC lysate at 37°C. Lysate of freshly isolated PC was kept untreated for 1 hr in ice or at 37°C. Other aliquots of the lysate received calpain 1 (0.5 U/ml) and calpain 2 (0.5 U/ml) prior to incubation. The concentration of mature TGF-β was quantified by an ELISA. Mean values ± SD of three experiments are given.
Supporting these results, we found that calpain inhibitor III reduced spontaneous CTGF-luciferase reporter gene expression of transfected PC by about 75% compared to untreated cultures (Fig. 8D).
In this context it should be noted that no transcript of TGF-β1 could be detected in PC cultured for 24 hrs. Hepatic stellate cells, which are known to express TGF-β1, are strongly positive as expected (Fig. 8E). Furthermore, the absence of transcripts for the small proteoglycan decorin in PC, in contrast to hepatic stellate cells, proves the high purity of the PC cultures used in these experiments (Fig. 8E).
We then tested a direct effect of calpain 1 and 2 on the activation of recombinant latent TGF-β. Both calpains converted the latent TGF-β to the active, immunologically detectable antigen (Fig. 8F). Furthermore, latent TGF-β in the lysate of freshly isolated PC was activated significantly if incubated for 2 hrs at 37°C (Fig. 8G). However, the additions of calpains 1 and 2 did not further enhance the amount of immunologically detectable TGF-β (Fig. 8G) in the cell lysate suspension.
Discussion
Data of this study extend the understanding of the regulation of the newly discovered synthesis of CTGF/CCN2 in PC. As shown by us previously [33] and by a very recent report [49], CTGF expression in PC is efficiently stimulated by exogenous TGF-β using the activin receptor-like kinase (Alk5)-Smad3 pathway. Furthermore, overex-pression of Smad7 in cultured PC and transgenic mice reduced TGF-β-dependent CTGF/CCN2 expression [49]. Thus, signal transduction of exogenous TGF-β to the CTGF promoter follows the common pathway of intracellular signalling, in which phosphorylation of Smad3 plays an important role for CTGF/CCN2 expression [50].
Here we elucidate for the first time the mechanisms that lead to CTGF/CCN2 expression in PC, which were isolated and cultured in an entirely TGF-β-free environment (Fig. 9). We demonstrate that even under these conditions CTGF/CCN2 is regulated by TGF-β, which is present inside PC. However, about 95% of total intracellular TGF-β is functionally inactive and immunocytochemically not detectable in freshly isolated cells [36]. The intracellular existence of LAP, LTBP, mature TGF-β and their subcellular location in cultured PC has been shown previously, but an intracellular function, in particular an intracellular interaction with the mature TGF-β peptide, was not yet reported [36, 37]. Especially the functional meaning of the preferred localization of latent TGF-β in the mitochondrial fraction of PC [36] and in mitochondria of murine heart and liver cells [51] has never been elucidated, although obviously of great importance. The origin of hepatocellular TGF-β (components) might be dual, i.e. receptor-dependent endocytosis of circulating TGF-β[52] and some synthesis, which is absent in normal PC, but up-regulatedduring culture. Transcripts of TGF-β1 were not detectable in freshly isolated and early cultures of PC using RT-PCR, but appeared gradually, even though scarcely, during extended culture time [43].
Fig. 9.
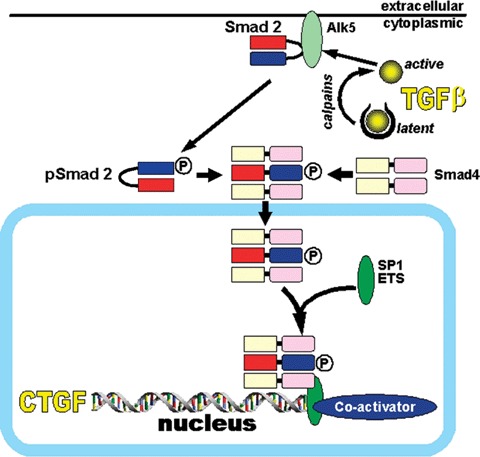
A simplified and schematic overview of the proposed activation process of latent TGF-β within PC and of its intracellular signaling triggering CTGF gene expression under consideration of current literature Latent TGF-β, which is deposited within the hepatocyte is activated by proteases such as calpain within the first 24 hrs following culture-induced stress or in vivo injury of the liver parenchyma. Via intracellular activation of the kinase domain of the Alk5-receptor it triggers Smad2-but not Smad3-phosphorylation leading to nuclear translocation with the common mediator Smad 4 and subsequent CTGF expression. SARA, Smad anchor for receptor activation; SP1, transcription factor; ETS, transcription factor.
Our data suggest deposited and not de novo synthesized latent TGF-β as the (major) source for activation because the immunocytochemically monitored demasking process was insensitive to cycloheximide (which in contrast inhibits CTGF synthesis) and an inhibitor of furin, a subtilisin-like proprotein convertase needed for TGF-β synthesis and processing [48] was not inhibitory to endogenous CTGF/CCN2 expression.
Based on the data presented in this study, we show that (i) conditions of culture-induced stress and liver damage lead to a rapid activation of intracellularly deposited latent TGF-β, which is immuno-cytochemically demonstrated by ‘demasking’ of the mature TGF-β antigen, beginning at the plasma membrane, (ii) the activated (demasked) TGF-β is functionally active, which is seen by up-regulation of CTGF/CCN2 and stimulation of the respective activity of the promoter reporter, (iii) the signalling pathway is exclusively intracellular, because four extracellularly acting inhibitors of the ligand and of type II TGF-β receptor (TβRII) were without any inhibitory effect, whereas the intracellularly acting Alk5 inhibitor SB-431542 [45] strongly reduced CTGF/CCN2 expression, and (iv) the pathway of intracellularly activated TGF-β uses the phosphorylation of Smad2 (p-Smad2) instead of Smad3, which, however, is phosphorylated by exogenous TGF-β as shown. Taken together, these observations support the hypothesis of an intracrine TGF-β signalling, which might occur very rapidly also in situ, e.g. in response to injury as shown here by positive APAAP stainings of PC distributed in a patch-like pattern in CCl4- and D-galactosamine-injured livers, respectively.
The findings pose two important questions, i.e. (i) how might intracrine TGF-β signalling precede and (ii) which processes convert intracellularly latent TGF-β to the functionally active, signalling molecule.
Not in the focus of the presented study is the question whether latent TGF-ß is deposited within the PC through endocytosis from the extracellular space or as a result of de novo synthesis. Both processes are possible [37, 38, 43], but need further investigation.
Exogenous TGF-β transduces signals via heteromeric complexes of type I (TβRI) and type II (TβRII) serine/threonine kinase receptors [26]. Ligand binding to TβRII recruits and phosphorylates TβRI, which then transduces signals through receptor-dependent phosphorylation of Smads2 and 3. The latter ones are complexed with Smad4 to be translocated to the nucleus, where they bind to Smadbinding elements (SBE) of TGF-β responsive genes [53]. It is not known how these components might assemble in intracrine signalling pathways. Histochemical evidence suggested previously an intracellular activation of TGF-β also in B cells and plasma cells of autoimmune mice [54]. Subsequently, a novel ligand-dependent mechanism of intracellular active TGF-β was proposed for plasma cells [55]. Following this report, the formation of active TGF-β within the plasma cell promotes direct intracellular interaction of ligand with TβRII, which effectively traps the receptor and prevents it from trafficking to the plasma membrane [55]. These authors, however, could not demonstrate signalling of the intracellular ligand-receptor complex through Smad2. The evidence presented for the lack of sig-nalling, however, was not firmly established. We show that inhibition of the demasking process by calpain inhibitors reduces the phosphorylation of Smad2 and the expression of CTGF very clearly, whereas the effects of exogenous TGF-β remained completely unaffected by calpain inhibitors. Furthermore, the strong discrepancy between the inhibitory effect of cell permeable, intracellular Alk5 inhibitor and the lacking effect of non-permeable, extracellular inhibitors of TGF-β, such as receptor blocker and ligand scavengers are highly suggestive for entirely intracellular TGF-β signalling. Molecular details of this pathway need to be clarified in following studies.
Based on previous data from this laboratory [44, 47], we have focused our investigation on calpains as potential intracellularly acting proteases responsible for demasking and activation of latent TGF-β. Calpains consist of many types of calcium-dependent cyto-plasmic proteases, which differ in their requirement for free Ca2+[56]. Predominantly, they are in an inactive form, but several cellular events can increase intracellular calpain activity such as rise of cytosolic free Ca2+, dissociation from the membrane, and decreases in the level of calpastatin, the endogenous inhibitor [56]. We propose that some of these cellular events take place in isolated and cultured PC and in injured livers as well leading finally to PC apoptosis and necrosis [57, 58]. This assumption is supported by our earlier findings that inactivation of calpains attenuates the spontaneous apoptosis of PC [47]. Also of interest, calpains were shown to be involved in integrin-mediated signal transduction [59], a process, which is greatly disturbed during isolation, i.e. dissociation between PC and surrounding matrix and culture of PC and in damaged liver as well. Previously, we have shown that the composition of culture matrix determines the staining intensity of intracellular TGF-β, which correlates inversely with the PC-phenotype [44]. These data suggest integrin-linked signals modulating intracellular TGF-β activation and the survival of PC [60]. Several reports point to a significant role of calpain activation for the progression of liver injury, e.g. by hepatotoxins [58]. Even the release of calpains from dying PC is suggested as a mechanism, which mediates progression of acute liver injury [57]. Their proteolytic activity on a plethora of substrates like several cytoskeletal and membrane proteins, metabolic enzymes and transcription factors underlines the patho-physiological significance of calpains. Our data further suggest intracellular latent TGF-β as a further substrate for calpains. Immunochemical and biochemical findings of TGF-β demasking and CTGF expression in response to calpain and calpain-inhibitors argue in favor of calpain-dependent TGF-β activation within PC. The finding of direct activation of recombinant latent TGF-β by calpains confirms a previous report showing cell-associated activation of latent TGF-β by calpain [61]. Furthermore, the faint staining of TGF-β beneath the membrane of PC about 1 hr after isolation (Fig. 4) coincides with a calpain fraction bound to the cell membrane [61]. Another, albeit circumstantial relation might exist between an increase of intracellular calpain activity and loss of mitochondrial membrane potential with increased membrane permeability [62]. The link might be activation of latent TGF-β that is preferentially located in the mitochondrial subcellular fraction of PC [36, 51]. Thus, calpain activation might lead to the induction of PC apoptosis via conversion of intracellular latent to mature TGF-β, as suggested previously from our group [47].
CTGF/CCN2 expression in PC served in this study as a reporter of TGF-β signalling, but the observations described here might be relevant for several or selected TGF-β target genes synthesized by PC. Even more, stress- or injury-dependent calpain activation and subsequent TGF-β activation could be of great relevance for survival or apoptosis of PC [63]. Its preferred location in the mitochondria, key players in the apoptotic cell death in the liver [64], was already mentioned [36, 51]. Finally, intracellular active TGF-β could be an important executor of epithelial-mesenchymal transition (EMT) of PC, which is claimed to be an increasingly important profibrogenic mechanism in liver injury [19, 20]. Indeed, both fractions of TGF-β, i.e. external and endogenous, might be significant for the decision of TGF-β whether to induce apoptosis or EMT [63]. The interrelation of the signalling pathways of TGF-β becomes even more complex if the potential crosstalk of the Alk5/Smad2/3 and the Alk1/Smad1/5/8 cascades is considered [65]. In hepatic stellate cells the significance of the latter TGF-β pathway for the expression of the inhibitor of differentiation 1 (Id1) was shown recently, which is a critical mediator of stellate cell transdifferentiation to myofibroblasts [65]. Furthermore, the modulating effects of BMPs on TGF-β-induced target gene expression needs consideration in future studies. If the interpretation of our data is adequate then a self-limitation of PC cultures should be diminished and prolongation of viable PC in culture could be reached by down-regulation of TGF-β activation potentially by use of calpain inhibitors. The latter ones have already been suggested as a therapeutic drug for liver injury [56–58].
In conclusion, intrinsic activation of latent TGF-β in PC and subsequent signaling to TGF-β-responsive genesis suggested as an immediately, if not the first occurring response of PC to cultural and injurious stress. It should be minimized in improved culture conditions and for the prevention of liver injury, which finally leads to fibrosis.
Acknowledgments
The financial support of this study by grant # 463/14–1 from the Deutsche Forschungsgemeinschaft (DFG) is gratefully acknowledged. We furthermore thank Ms. Sibille Sauer-Lehnen for perfect technical assistance.
References
- 1.Bissell DM, Roulot D, George J. Transforming growth factor β and the liver. Hepatology. 2001;34:859–67. doi: 10.1053/jhep.2001.28457. [DOI] [PubMed] [Google Scholar]
- 2.Gressner AM, Dooley S, Weiskirchen R. TGF-beta and the Smad pathway in liver fibrogenesis. In: Dufour J-F, Clavien P-A, Trautwein C, Graf R, editors. Signalling Pathways in Liver Diseases. Heidelberg. Germany: Springer Verlag; 2005. pp. 139–50. [Google Scholar]
- 3.Rossmanith W, Schulte-Hermann R. Biology of transforming growth factor beta in hepatocarcinogenesis. Microsc Res Tech. 2001;52:430–6. doi: 10.1002/1097-0029(20010215)52:4<430::AID-JEMT1028>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 4.Huang SS, Huang JS. TGF-beta control of cell proliferation. J Cell Biochem. 2005;96:447–62. doi: 10.1002/jcb.20558. [DOI] [PubMed] [Google Scholar]
- 5.Gressner AM, Lahme B, Mannherz HG, Polzar B. TGF-beta-mediated hepatocellu-lar apoptosis by rat and human hepatoma cells and primary rat hepatocytes. J Hepatol. 1997;26:1079–92. doi: 10.1016/s0168-8278(97)80117-1. [DOI] [PubMed] [Google Scholar]
- 6.Oberhammer F, Bursch W, Parzefall W, Breit P, Erber E, Stadler M, Schulte-Hermann R. Effect of transforming growth factor beta on cell death of cultured rat hepa-tocytes. Cancer Res. 1991;51:2478–85. [PubMed] [Google Scholar]
- 7.Cerwenka A, Swain SL. TGF-beta1: immunosuppressant and viability factor for T lymphocytes. Microbes Infect. 1999;1:1291–6. doi: 10.1016/s1286-4579(99)00255-5. [DOI] [PubMed] [Google Scholar]
- 8.Bierie B, Moses HL. TGF-beta and cancer. Cytokine Growth Factor Rev. 2006;17:29–40. doi: 10.1016/j.cytogfr.2005.09.006. [DOI] [PubMed] [Google Scholar]
- 9.Elliott RL, Blobe GC. Role of transforming growth factor beta in human cancer. J Clin Oncol. 2005;23:2078–93. doi: 10.1200/JCO.2005.02.047. [DOI] [PubMed] [Google Scholar]
- 10.Gressner AM, Weiskirchen R, Breitkopf K, Dooley S. Roles of TGF-β in hepatic fibrosis. Front Biosci. 2002;7:D793–807. doi: 10.2741/A812. [DOI] [PubMed] [Google Scholar]
- 11.Gressner AM, Weiskirchen R. Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-beta as major players and therapeutic targets. J Cell Mol Med. 2006;10:76–99. doi: 10.1111/j.1582-4934.2006.tb00292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brenner DA, Rippe RA, Rhodes K, Trotter JF, Breindl M. Fibrogenesis and type I collagen gene regulation. J Lab Clin Med. 1994;124:755–60. [PubMed] [Google Scholar]
- 13.Inagaki Y, Mamura M, Kanamaru Y, Greenwel P, Nemoto T, Takehara K, ten Dijke P, Nakao A. Constitutive phosphorylation and nuclear localization of Smad3 are correlated with increased collagen gene transcription in activated hepatic stellate cells. J Cell Physiol. 2001;187:117–23. doi: 10.1002/1097-4652(2001)9999:9999<00::AID-JCP1059>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 14.Gressner AM, Krull N, Bachem MG. Regulation of proteoglycan expression in fibrotic liver and cultured fat-storing cells. Pathol Res Pract. 1994;190:864–82. doi: 10.1016/S0344-0338(11)80990-8. [DOI] [PubMed] [Google Scholar]
- 15.Gressner AM, Haarmann R. Hyaluronic acid synthesis and secretion by rat liver fat storing cells (perisinusoidal lipocytes) in culture. Biochem Biophys Res Commun. 1988;151:222–9. doi: 10.1016/0006-291x(88)90582-7. [DOI] [PubMed] [Google Scholar]
- 16.Gressner AM, Haarmann R. Regulation of hyaluronate synthesis in rat liver fat storing cell cultures by Kupffer cells. J Hepatol. 1988;7:310–18. doi: 10.1016/s0168-8278(88)80003-5. [DOI] [PubMed] [Google Scholar]
- 17.Iredale JP. Tissue inhibitors of metalloproteinases in liver fibrosis. Int J Biochem Cell Biol. 1997;29:43–54. doi: 10.1016/s1357-2725(96)00118-5. [DOI] [PubMed] [Google Scholar]
- 18.Hemmann S, Graf J, Roderfeld M, Roeb E. Expression of MMPs and TIMPs in liver fibrosis-a systematic review with special emphasis on anti-fibrotic strategies. J Hepatol. 2007;46:955–75. doi: 10.1016/j.jhep.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 19.Kaimori A, Potter J, Kaimori J, Wang C, Mezey E, Koteish A. TGF-beta 1 induces an epithelial-to-mesenchymal transition state in mouse hepatocytes in-vitro. J Biol Chem. 2007;282:22089–101. doi: 10.1074/jbc.M700998200. [DOI] [PubMed] [Google Scholar]
- 20.Zeisberg M, Yang C, Martino M, Duncan M, Rieder F, Tanjore H, Kalluri R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J Biol Chem. 2007;282:23337–47. doi: 10.1074/jbc.M700194200. [DOI] [PubMed] [Google Scholar]
- 21.Zavadil J, Böttinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005;24:5764–74. doi: 10.1038/sj.onc.1208927. [DOI] [PubMed] [Google Scholar]
- 22.Kinnman N, Housset C. Peribiliary myofibroblasts in biliary type liver fibrosis. Front Biosci. 2002;7:d496–d503. doi: 10.2741/A790. [DOI] [PubMed] [Google Scholar]
- 23.Quan TE, Cowper S, Wu SP, Bockenstedt LK, Bucala R. Circulating fibrocytes: collagen-secreting cells of the peripheral blood. Int J Biochem Cell Biol. 2004;36:598–606. doi: 10.1016/j.biocel.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 24.Koli K, Saharinen J, Hyytiainen M, Penttinen C, Keski-Oja J. Latency, activation, and binding proteins of TGF-beta. Microsc Res Technique. 2001;52:354–62. doi: 10.1002/1097-0029(20010215)52:4<354::AID-JEMT1020>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 25.Khalil N. TGF-beta: from latent to active. Microbes Infect. 1999;1:1255–63. doi: 10.1016/s1286-4579(99)00259-2. [DOI] [PubMed] [Google Scholar]
- 26.Inagaki Y, Okazaki I. Emerging insights into Transforming growth factor {beta} Smad signal in hepatic fibrogenesis. Gut. 2007;56:284–92. doi: 10.1136/gut.2005.088690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Neilson EG. Setting a trap for tissue fibrosis. Nat Med. 2005;11:373–4. doi: 10.1038/nm0405-373. [DOI] [PubMed] [Google Scholar]
- 28.Hewitson TD, Martic M, Darby IA, Kelynack KJ, Bisucci T, Tait MG, Becker GJ. Intracellular cyclic nucleotide analogues inhibit in vitro mitogenesis and activation of fibroblasts derived from obstructed rat kidneys. Nephron Exp Nephrol. 2004;96:E59–66. doi: 10.1159/000076405. [DOI] [PubMed] [Google Scholar]
- 29.Moussad EEDA, Brigstock DR. Connective tissue growth factor: What's in a name? Mol Genet Metab. 2000;71:276–92. doi: 10.1006/mgme.2000.3059. [DOI] [PubMed] [Google Scholar]
- 30.Abreu JG, Ketpura NI, Reversade B, De Robertis EM. Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-beta. Nat Cell Biol. 2002;4:599–604. doi: 10.1038/ncb826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wahab NA, Weston BS, Mason RM. Modulation of the TGF beta/Smad signaling pathway in mesangial cells by CTGF/CCN2. Exp Cell Res. 2005;307:305–14. doi: 10.1016/j.yexcr.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 32.Leask A, Abraham DJ. All in the CCN family: essential matricellular signaling modulators emerge from the bunker. J Cell Sci. 2006;119:4803–10. doi: 10.1242/jcs.03270. [DOI] [PubMed] [Google Scholar]
- 33.Gressner O, Lahme B, Demirci I, Gressner AM, Weiskirchen R. Differential effects of TGF-beta on connective tissue growth factor (CTGF/CCN2) expression in hepatic stellate cells and hepatocytes. J Hepatol. 2007;47:699–710. doi: 10.1016/j.jhep.2007.05.015. [DOI] [PubMed] [Google Scholar]
- 34.Sedlaczek N, Jia JD, Bauer M, Herbst H, Ruehl M, Hahn EG, Schuppan D. Proliferating bile duct epithelial cells are a major source of connective tissue growth factor in rat biliary fibrosis. Am J Pathol. 2001;158:1239–44. doi: 10.1016/S0002-9440(10)64074-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marcos R, Rocha E, Henrique RMF, Monteiro RAF. A new approach to an unbiased estimate of the hepatic stellate cell index in the rat liver: An example in healthy conditions. J Histochem Cytochem. 2003;51:1101–4. doi: 10.1177/002215540305100814. [DOI] [PubMed] [Google Scholar]
- 36.Roth-Eichhorn S, Kühl K, Gressner AM. Subcellular localization of (latent) transforming growth factor β and the latent TGF-β binding protein in rat hepatocytes and hepatic stellate cells. Hepatology. 1998;28:1588–96. doi: 10.1002/hep.510280619. [DOI] [PubMed] [Google Scholar]
- 37.Roth S, Schurek J, Gressner AM. Expression and release of the latent TGF-beta binding protein (LTBP) by hepatocytes from rat liver. Hepatology. 1997;25:1398–405. doi: 10.1002/hep.510250616. [DOI] [PubMed] [Google Scholar]
- 38.Roth S, Michel K, Gressner AM. (Latent) transforming growth factor-beta in liver parenchymal cells, its injury-dependent release and paracrine effects on hepatic stellate cells. Hepatology. 1998;27:1003–12. doi: 10.1002/hep.510270416. [DOI] [PubMed] [Google Scholar]
- 39.Borkham-Kamphorst E, Herrmann J, Stoll D, Treptau J, Gressner AM, Weiskirchen R. Dominant-negative soluble PDGF-beta receptor inhibits hepatic stellate cell activation and attenuates liver fibrosis. Lab Invest. 2004;84:766–77. doi: 10.1038/labinvest.3700094. [DOI] [PubMed] [Google Scholar]
- 40.Seglen PO. Preparation of isolated rat liver cells. In: Prescott DM, editor. Methods in cell biology. Vol. 8. New York: Academic Press; 1987. pp. 29–83. [DOI] [PubMed] [Google Scholar]
- 41.Gressner AM, Pfeiffer T. Preventive effects of acute inflammation on liver cell necrosis and inhibition of heparan sulfate synthesis in hepatocytes. J Clin Chem Clin Biochem. 1986;24:821–9. doi: 10.1515/cclm.1986.24.11.821. [DOI] [PubMed] [Google Scholar]
- 42.Schäfer S, Zerbe O, Gressner AM. The synthesis of proteoglycans in fat storing cells of rat liver. Hepatology. 1987;7:680–7. doi: 10.1002/hep.1840070411. [DOI] [PubMed] [Google Scholar]
- 43.Gao CF, Gressner G, Zoremba M, Gressner AM. Transforming growth factor-beta (TGF-beta) expression in isolated and cultured rat hepatocytes. J Cell Physiol. 1996;167:394–405. doi: 10.1002/(SICI)1097-4652(199606)167:3<394::AID-JCP3>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 44.Gressner AM, Wulbrand U. Variation of immunocytochemical expression of transforming growth factor (TGF)-beta hepato-cytes in culture and liver slices. Cell Tissue Res. 1997;287:143–52. doi: 10.1007/s004410050740. [DOI] [PubMed] [Google Scholar]
- 45.Inman GJ, Nicolás FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, Laping NJ, Hill CS. SB-431542 is a potent and specific inhibitor of transforming growth factor-b superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2002;62:65–74. doi: 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- 46.Gressner AM, Polzar B, Lahme B, Mannherz HG. Induction of rat liver parenchymal cell apoptosis by hepatic myofibroblasts via transforming growth factor-beta. Hepatology. 1996;23:571–81. doi: 10.1002/hep.510230324. [DOI] [PubMed] [Google Scholar]
- 47.Gressner AM, Lahme B, Roth S. Attenuation of TGF-beta-induced apoptosis in primary cultures of hepatocytes by cal-pain inhibitors. Biochem Biophys Res Commun. 1997;231:457–62. doi: 10.1006/bbrc.1996.5777. [DOI] [PubMed] [Google Scholar]
- 48.Leitlein J, Aulwurm S, Waltereit R, Naumann U, Wagenknecht B, Garten W, Weller M, Platten M. Processing of immunosuppressive pro-TGF-β1,2 by human glioblastoma cells involves cytoplasmic and secreted furin-like proteases. J Immunol. 2001;166:7238–43. doi: 10.4049/jimmunol.166.12.7238. [DOI] [PubMed] [Google Scholar]
- 49.Weng HL, Ciuclan L, Liu Y, Hamzavi J, Godoy P, Gaitantzi H, Kanzler S, Heuchel R, Üeberham U, Gebhardt R, Breitkopf K, Dooley S. Profibrogenic transforming growth factor-beta/actin receptor-like kinase 5 signaling via connective tissue growth factor expression in hepatocytes. Hepatology. 2007;46:1257–70. doi: 10.1002/hep.21806. [DOI] [PubMed] [Google Scholar]
- 50.Leivonen SK, Hakkinen L, Liu D, Kahari VM. Smad3 and extracellular signal-regulated kinase 1/2 coordinately mediate transforming growth factor-beta-induced expression of connective tissue growth factor in human fibroblasts. J Invest Dermatol. 2005;124:1162–9. doi: 10.1111/j.0022-202X.2005.23750.x. [DOI] [PubMed] [Google Scholar]
- 51.Heine UI, Burmester JK, Flanders KC, Danielpour D, Munoz EF, Roberts AB, Sporn MB. Localization of transforming growth factor-beta1 in mitochondria of murine heart and liver. Cell Regul. 1991;2:467–77. doi: 10.1091/mbc.2.6.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.LaMarre J, Hayes MA, Wollenberg GK, Hussaini I, Hall SW, Gonias SL. An alpha2-macroglobulin receptor-dependent mechanism for the plasma clearance of TGF-beta1 in mice. J Clin Invest. 1991;87:39–44. doi: 10.1172/JCI114998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.ten Dijke P, Hill CS. New insights into TGF-beta-Smad signalling. Trends Biochem Sci. 2004;29:265–73. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 54.Caver TE, O'Sullivan FX, Gold LI, Gresham HD. Intracellular demonstration of active TGF-beta1 in B cells and plasma cells of autoimmune mice. J Clin Invest. 1996;98:2496–506. doi: 10.1172/JCI119068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fernandez T, Amoroso S, Sharpe S, Jones GM, Bliskovski V, Kovalchuk A, Wakefield LM, Kim SJ, Potter M, Letterio JJ. Disruption of transforming growth factor b signaling by a novel ligand-dependent mechanism. J Exp Med. 2002;195:1247–55. doi: 10.1084/jem.20011521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mehendale HM, Limaye PB. Calpain: a death protein that mediates progression of liver injury. Trends Pharmacol Sci. 2005;26:232–6. doi: 10.1016/j.tips.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 57.Limaye PB, Apte UM, Shankar K, Bucci TJ, Warbritton A, Mehendale HM. Calpain released from dying hepatocytes mediates progression of acute liver injury induced by model hepatotoxicants. Toxicol Appl Pharm. 2003;191:211–26. doi: 10.1016/s0041-008x(03)00250-3. [DOI] [PubMed] [Google Scholar]
- 58.Arora AS, de Groen P, Emori Y, Gores GJ. A cascade of degradative hydrolase activity contributes to hepatocyte necrosis during anoxia. Am J Physiol. 1996;270:G238–45. doi: 10.1152/ajpgi.1996.270.2.G238. [DOI] [PubMed] [Google Scholar]
- 59.Inomata M, Hayashi M, Ohnoiwashita Y, Tsubuki S, Saido TC, Kawashima S. Involvement of calpain in integrin-mediated signal transduction. Arch Biochem Biophys. 1996;328:129–34. doi: 10.1006/abbi.1996.0152. [DOI] [PubMed] [Google Scholar]
- 60.Gkretsi V, Mars WM, Bowen WC, Barua L, Yang Y, Guo L, St-Arnaud R, Dedhar S, Wu C, Michalopoulos GK. Loss of inte-grin linked kinase from mouse hepatocytes in vivo and in vitro results in apoptosis and hepatitis. Hepatology. 2007;45:1025–34. doi: 10.1002/hep.21540. [DOI] [PubMed] [Google Scholar]
- 61.Abe M, Oda N, Sato Y. Cell-associated activation of latent transforming growth factor-beta by calpain. J Cell Physiol. 1998;174:186–93. doi: 10.1002/(SICI)1097-4652(199802)174:2<186::AID-JCP6>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 62.Aguilar HI, Botla R, Arora AS, Bronk SF, Gores GJ. Induction of the mitochondrial permeability transition by protease activity in rats: A mechanism of hepatocyte necrosis. Gastroenterology. 1996;110:558–66. doi: 10.1053/gast.1996.v110.pm8566604. [DOI] [PubMed] [Google Scholar]
- 63.Song J. EMT or apoptosis: a decision for TGF-[beta] Cell Res. 2007;17:289–90. doi: 10.1038/cr.2007.25. [DOI] [PubMed] [Google Scholar]
- 64.Gressner O, Schilling T, Lorenz K, Schulze Schleithoff E, Koch A, Schulze-Bergkamen H, Lena AM, Candi E, Terrinoni A, Catani MV, Oren M, Melino G, Krammer PH, Stremmel W, Müller M. TAp63a induces apoptosis by activating signaling via death receptors and mitochondria. EMBO J. 2005;24:2458–71. doi: 10.1038/sj.emboj.7600708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wiercinska E, Wickert L, Denecke B, Said HM, Hamzavi J, Gressner AM, Thorikay M, ten Dijke P, Mertens PR, Breitkopf K, Dooley S. Id1 is a critical mediator in TGF-beta-induced transdiffer-entiation of rat hepatic stellate cells. Hepatology. 2006;43:1032–41. doi: 10.1002/hep.21135. [DOI] [PubMed] [Google Scholar]