

Summary
In order to combat oxidative damage, eukaryotic cells have evolved with numerous anti-oxidant factors that are often distributed between cytosolic and mitochondrial pools. Glutathione reductase, which regenerates the reduced form of glutathione, represents one such anti-oxidant factor, yet nothing is known regarding the partitioning of this enzyme within the cell. Using the bakers’ yeast Saccharomyces cerevisiae as a model, we provide evidence that a single gene, namely GLR1, encodes both the mitochondrial and cytosolic forms of glutathione reductase. A deletion in GLR1 drastically increases levels of oxidized glutathione in these two subcellular compartments. The GLR1 gene has two in-frame start codons that are both used as translation initiation sites. Translation from the 1st codon generates the mitochondrial form that includes a mitochondrial targeting signal, while translation from the 2nd codon produces the cytosolic form that lacks this sequence. Our results indicate that the sequence context of the two AUG codons influences the efficiency of translation initiation at each site, which in turn affects the relative levels of cytosolic and mitochondrial Glr1p. This method of subcellular distribution of glutathione reductase may be conserved in mammalian cells as well.
Introduction
The abundant tripeptide glutathione (γ-glutamylcysteinylglycine) plays a pivotal role in the oxidative stress defense systems of the cell. Glutathione is a ubiquitous thiol that maintains the intracellular redox state by reducing cellular disulfide bonds and detoxifying damaging molecules such as xenobiotics and heavy metals (1). Glutathione also serves as a reductive cofactor for anti-oxidant enzymes such as glutaredoxins and glutathione peroxidases (1,2). The anti-oxidant function of glutathione depends upon its redox-active thiol group that becomes oxidized when glutathione reduces target molecules; two molecules of reduced glutathione (GSH) are thereby converted to glutathione disulfide (GSSG). The enzyme glutathione reductase catalyzes the reduction of GSSG back to GSH, therefore it plays a critical role in the cellular defense against oxidative damage as well. Glutathione reductase (GR) is a flavin-containing oxidoreductase similar in sequence and structure to thioredoxin reductase (TR). It contains a redox-active disulfide in its active site and requires NADPH for its catalytic activity. GR is found in many types of organisms including bacteria, plants, and yeast, as well as higher eukaryotes such as mice and humans. However, the intracellular localization of GR is not completely understood.
Since mitochondria are an important source and target of oxidative damage, it would seem critical to have anti-oxidant factors such as GR housed in this organelle, along with other cellular locations. In eukaryotes, anti-oxidant factors typically have both mitochondrial and cytosolic versions that are often encoded by different genes. For example, the matrix of the mitochondria harbors a manganese-containing superoxide dismutase (SOD2), whereas the cytosol contains a Cu/Zn superoxide dismutase (SOD1) (3). Both yeast and mammalian cells express two different TRs as well as numerous thioredoxins and glutaredoxins that are specifically targeted to the mitochondria or the cytosol (4–9). There are also different mechanisms for the generation of cytosolic and mitochondrial NADPH, an essential cofactor for both TR and GR. In the cytosol, NADPH is primarily supplied by the pentose phosphate pathway (10,11), whereas in the mitochondria, NADPH is either provided by an NADH kinase (e.g., S. cerevisiae POS5) (12) or by NADP+-dependent isocitrate dehydrogenase as in mammalian cells (13). Another method by which anti-oxidant enzymes may be distributed between the cytosol and mitochondria is via dual targeting of a single gene product. For example, the Cu/Zn SOD1 polypeptide is targeted to both the cytosol and the intermembrane space of mitochondria (14).
As with other anti-oxidant factors, GR is clearly expected to reside in the mitochondria and cytosol. Reduced GSH is synthesized in the cytosol and can be transported into the mitochondria (15), but the GSSG formed in the matrix is unable to exit this compartment (16). Therefore the GR-mediated regeneration of GSH must take place inside the mitochondria as well as in the cytosol. In mammals and yeast, only a single GR-encoding gene has been identified, yet GR activity has been detected in both the cytosol and mitochondria of mammalian cells (17). The question remains as to how GR is localized to both compartments.
Our objective was to use the S. cerevisiae as a model system to study the intracellular compartmentalization of GR. Previous researchers have shown that the GLR1 gene, encoding yeast GR, is required to maintain a high intracellular GSH to GSSG ratio, however the localization of this protein has not been determined (18,19). We show here that the GLR1 gene of S. cerevisiae encodes both mitochondrial and cytosolic forms of the enzyme, and that the protein accumulates to approximately equal concentrations in these compartments. The mechanism for cytosolic/mitochondrial distribution of Glr1p appears to involve two in-frame start codons; translation from the 1st start codon generates the mitochondrial form, while translation from the 2nd start codon generates the cytosolic form. Furthermore, our experiments indicate that the relative levels of Glr1p in the cytosol and mitochondria are determined by the sequence context of each start codon. A preliminary examination of mammalian GR sequences suggests that this mechanism may be conserved in higher eukaryotes as well.
Experimental Procedures
Strains and Plasmids
S. cerevisiae strains used in this study were BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) and BY4741 glr1Δ::kanMX4 obtained from Research Genetics. Strains were maintained at 30 °C on either enriched yeast extract-peptone-based medium supplemented with 2% glucose (YPD) or minimal synthetic defined medium (SD) supplemented with the appropriate amino acids (20). Yeast transformations were performed by the lithium acetate procedure (21).
The GLR1 CEN plasmid pCO113 was originally constructed as a GFP fusion with one copy of green fluorescent protein (GFP) fused to the C-terminus of Glr1p. The GLR1 gene was amplified from −1011 (with respect to the +1 start codon) to the stop codon with primers that introduced a HindIII site at −1002 and a NotI site at +1450. This PCR fragment and the plasmid pAA1 (LEU2 CEN) were digested with HindIII and NotI and ligated together, producing an in-frame fusion of Glr1p with the GFP sequence already present in pAA1 (22). The GFP tag was later removed by site-directed mutagenesis (QuikChange kit, Stratagene) of the NotI site to recreate the GLR1 stop codon at +1449 and introduce a SacII site at +1454. After mutagenesis, the plasmid was digested with SacII and religated to remove the GFP tag located between the two SacII sites. pCO114 (M17L), pCO115 (Δ1–16), pCO116 (M1L), pCO117 (FS1), pCO118 (FS2), and pCO119 (U−3A(M1)) were constructed by site-directed mutagenesis of pCO113 using the following primers and their anti-parallel complements (mutated sequences underlined) : M17L, 5’-CTACAGATAAGAACTTTGTCCACGAACACCAAGC-3’; Δ1–16, 5’-CATATTAGTTTACAGAACTTTATGTCCACGAACACCAAGCATTAC-3’; M1L, 5’-GTTTACAGAACTTTTTGCTTTCTGCAACCAAAC-3’; FS1, 5’-CAACCAAACAAACATTCTAGAAGTCTACAGATAAG-3’; FS2, 5’-GAAGTCTACAGATAACGAACTATGTCCACGAAC-3’; U−3A(M1), 5’-GTTTACAGAACATTATGCTTTCTGCAACC-3’. pCO121 (U−3A(M1)/M17L) and pCO122 (M1L/M17L) were created by site-directed mutagenesis of pCO114 (M17L) using the U−3A(M1) primers or the M1L primers, respectively. The sequence integrity of all plasmids was confirmed by double-stranded DNA sequencing (DNA Analysis Facility, Johns Hopkins University School of Medicine).
Subcellular Fractionation
For subcellular fractionation experiments, yeast cells were grown aerobically to mid-log phase in selecting SD medium with 2% glucose. Mitochondrial and post-mitochondrial supernatant (PMS) fractions were obtained as described previously by converting cells to spheroplasts followed by gentle lysis by Dounce homogenization and differential centrifugation (23,24). For intermembrane space (IMS) and matrix fractionation, mitochondria were prepared from cells grown in SD with 2% galactose so that cytochrome b2, which is glucose-repressed, could be used as an IMS marker.
GR Activity Assays
For GR activity assays, yeast cells were grown aerobically to mid-log phase in glucose SD medium, then fractionated into PMS and mitochondrial fractions. GR activity was measured using the Glutathione Reductase Assay kit from Sigma, which is a colorimetric assay monitoring the reduction of 5,5’-dithiobis(2-nitrobenzoic acid) (DTNB). 140–300 µg of PMS extract was used directly in the assay. For mitochondrial fractions, 30–70 µg of extract was diluted to 200 µL in assay buffer with 1% Tween-20 and vortexed for 1 min prior to analysis.
GSH/GSSG Assays
Total glutathione and oxidized glutathione were measured by the DTNB-GR recycling assay (25,26). Cells were grown to mid-log phase in glucose SD medium and fractionated into mitochondria and PMS fractions as described above. Freshly-made extracts were deproteinated by the addition of 10% 5-sulfosalicylic acid to a final concentration of 1% and incubated on ice for 30 minutes. Precipitated protein was then removed by centrifugation at 13,000 × g for 5 min. Measurement of total GSH and GSSG in acidified extracts was conducted by spectrophotometric analysis of DTNB reduction as described previously (26).
Immunoblotting Techniques
Yeast extracts were subjected to electrophoresis on a 12% SDS-polyacrylamide gel and analyzed by Western blotting using an anti-Glrlp antibody diluted to 1:10,000 and a secondary anti-rabbit IgG (Amersham) diluted to 1:12,500. For Glr1p antibody production, S. cerevisiae Glr1p was purchased from Sigma and used to prepare rabbit-generated anti-Glr1p antibodies (Cocalico Biologicals, Reamstown, PA). PMS fractions were monitored by anti-3-phosphoglycerate kinase (PGK) antibodies diluted to 50 ng/mL (Molecular Probes). Mitochondrial fractions were monitored by using antibodies directed against cytochrome b2 (diluted to 1:10,000) in the intermembrane space (IMS), and Mas2 (diluted to 1:25,000) in the matrix (kind gifts of R. Jensen). Detection employed the ECL kit (Amersham) used according to the manufacturer’s specifications. Protein concentrations were determined using the Bradford method (Bio-Rad) with bovine serum albumin as the calibration standard.
Results
Glr1p Subcellular Localization
S. cerevisiae is reported to contain a single GR-encoding gene, namely GLR1 (27). A single GR-encoding gene has also been identified in mammalian species such as humans and mice (28,29). A comparison of the amino acid sequences of GR from human, mouse, and yeast to that of E. coli GR reveals that the eukaryotic homologues all have a 19–60 amino acid extension at the N-terminus of the protein that precedes the homology region to E. coli GR (Fig. 1). For the mammalian sequences, this extension includes two in-frame start codons with an abundance of arginine residues in between. Previous researchers have proposed that these mammalian N-termini encode a putative mitochondrial targeting signal (MTS) (28,29). The N-terminal sequence in the yeast Glr1p protein also includes two in-frame start codons and exhibits the hallmarks of a MTS, including an abundance of positively-charged and hydroxylated amino acids with no negatively-charged residues (30,31). The putative cleavage site for mitochondrial processing of yeast Glr1p is between Met17 and Ser18, as determined by TargetP v1.0 (32). The presence of this putative MTS in S. cerevisiae Glr1p would suggest that the protein is exclusively targeted to the mitochondria.
Fig. 1. Sequence alignment of human, mouse, S. cerevisiae, and E. coli GR.

The amino acid sequences of the N-termini of human, mouse, yeast and E. coli GR were aligned using ClustalW. Identical residues are highlighted in black, and similar residues are outlined in gray. For the eukaryotic sequences, the two methionines encoded by the two in-frame start codons are shown in bold. The predicted cleavage site for the MTS in yeast Glr1p is designated by an arrow (↑).
To address the localization of Glr1p in yeast, we assayed for GR activity in cell lysates that were resolved into crude mitochondria and largely cytosolic (PMS) fractions. As seen in Fig. 2A, GR activity is found in both the mitochondria and cytosolic fractions of wild-type (WT) cells. Furthermore, both compartments have similar concentrations of GR activity, although mitochondrial levels are somewhat higher with ~35% more activity per mg of protein compared to the PMS fraction. As seen in Fig. 2A, deletion of GLR1 results in loss of GR activity in both compartments. When the GLR1 gene is re-introduced on a CEN plasmid in glr1Δ strains, activity in both compartments is restored to wild-type levels. These results indicate that GLR1 is responsible for both the cytosolic and mitochondrial forms of GR.
Fig. 2. GLR1 encodes cystosolic and mitochondrial forms of GR.

A, GR activity assays of post-mitochondrial supernatant (PMS) and mitochondrial (Mito) extracts from the indicated strains. Strains utilized: wild-type (WT), BY4741; glr1Δ, BY4741 glr1Δ; glr1Δ + vector, glr1Δ transformed with pAA1; glr1Δ + GLR1, glr1Δ transformed with pCO113. Activity is reported as units per mg of protein in the extract. One unit of enzyme activity = 1.0 µmol DTNB reduced/min at 25 °C at pH 7.5. B, Total glutathione (GSH + GSS) for PMS and mitochondrial fractions from WT and glr1Δ strains was assayed as described in Experimental Procedures. GSS = 2 * GSSG. C, The % oxidized glutathione (% GSS/(GSH + GSS) was calculated from total GSH and GSSG levels in each extract. For A–C, the reported values are the mean of three independent experiments; error bars are standard deviations.
The levels of total glutathione as well as the ratio of oxidized (GSSG) to reduced (GSH) glutathione were measured in WT and glr1Δ strains. As shown in Fig. 2B, the amount of total glutathione (GSH + GSSG) in both the mitochondria and cytosol are similar for WT and glr1Δ strains. When the ratio of oxidized to reduced was examined, the mitochondria of WT strains exhibited a higher percentage of oxidized glutathione (~9%) compared to the cytosol (~0.4%) (Fig. 2C). A similar situation has been reported for the cytosol and mitochondria of mammalian cells (9,33). This result is expected since mitochondria are a major source of reactive oxygen species in the cell, which would lead to a higher proportion of GSSG within this compartment. Upon comparison of the glr1Δ mutant to WT, we found that glr1Δ has a much greater percentage of oxidized glutathione in both the cytosol (~57%) and the mitochondria (~41%) (Fig. 2C). Since the consequence of loss of GLR1 is a vast increase in oxidized glutathione in both compartments, this provides further evidence that GLR1 encodes both the mitochondrial and cytosolic isoforms of GR in yeast.
In order to confirm that Glr1p is localized to both the cytosol and mitochondria, we conducted a Western blot analysis of the PMS (largely cytosolic) and mitochondrial fractions of WT and glr1Δ strains. Bakers’ yeast GR was purchased from Sigma and used to prepare anti-Glr1p antibodies. This antibody recognizes a major species of ~50 kDa that corresponds to Glr1p since it is seen in WT cells, but not in glr1Δ mutants (Fig. 3A, top panel, lanes 1–2). This antibody also occasionally recognizes a minor contaminant that migrates slightly faster than Glr1p, but this does not reflect Glr1p since it also observed in glr1Δ strains. In Fig. 3A, lanes 3–6, identical cell equivalents of PMS and crude mitochondria were analyzed to approximate the proportion of Glr1p protein that localizes to the different cellular compartments. These results indicate that Glr1p is predominantly found in the cytoplasm, but a small fraction of total Glr1p is also seen in mitochondria. A titration of the PMS fraction (lanes 7–9) reveals that the total amount of Glr1p present in the mitochondria represents approximately 5–10% of cytosolic Glr1p. Given that mitochondria constitute ~3% of the total cell volume under these growth conditions (34), it appears that the concentration of Glr1p is roughly equivalent in the two compartments. This result is in accordance with the activity assays in Fig. 2A, which demonstrate that GR activity levels are similar in the mitochondria and cytosol.
Fig. 3. Glr1p is located in the PMS and mitochondrial matrix.

Wild type (BY4741) and glr1Δ (BY4741 glr1Δ) strains were grown to mid-log phase in a synthetic medium containing either glucose (A) or galactose (B). Cells were lysed and fractionated, and fractions analyzed by SDS-PAGE and immunoblotting using antibodies directed against Glr1p, Pgk1p (PMS marker), Cyt b2 (mitochondrial IMS marker), or Mas2p (mitochondrial matrix marker). A, 75 µg total cell protein (Total) was fractionated into PMS and crude mitochondria (Mito) and the entire amount of each fraction (lanes 1–6) or the indicated percentage of the PMS fraction (lanes 7–9) was analyzed. B, Mitochondria (15 µg protein) were further fractionated into intermembrane space (IMS) and matrix components and the entire amount of each fraction analyzed. The anti-Glr1p antibody often recognizes non-specific band(s) of slightly faster mobility that are present in both WT and glr1Δ mutants.
To identify the mitochondrial compartment that houses Glr1p, we fractionated the mitochondria into intermembrane space (IMS) and matrix components. The results shown in Fig. 3B indicate that Glr1p is specifically localized to the matrix as determined by co-localization with the mitochondrial processing protease, Mas2p.
Glr1p Distribution in Translation Initiation Mutants
As shown in Table 1, the GLR1 gene has two in-frame start codons corresponding to Met1 and Met17. Translation from the first AUG codon would produce a 483 amino acid (aa), 53.4 kDa protein including the putative MTS. Translation from the second AUG codon would produce a 467 aa, 51.6 kDa protein that lacks the MTS. Through site-directed mutagenesis studies, we tested whether these potential translation start sites had any role in determining Glr1p subcellular distribution.
Table 1.
Predicted mRNA and protein sequences of GLR1 mutant constructs.
 |
Mutations in the RNA and protein sequences are shown in bold letters and start codons are highlighted in gray. The predicted protein sequences translating from the first (AUG1) and second start codons (AUG17) are shown in italics above and below the RNA sequence, respectively. RNA sequence insertions are shown with a carat (^).
Several different mutants were designed with variations in the translation initiation sequences (Table 1). First, the putative MTS was deleted by removing amino acids 1–16 (Δ1–16). We also mutated the two potential start sites, creating M1L and M17L single mutants and an M1L/M17L double mutant. As shown in the Western blot in Fig. 4A, lanes 1–6, the Δ1–16 and M1L mutants both have no detectable mitochondrial Glr1p, although cytosolic Glr1p is still present. Together, these results suggest that translation from the 1st AUG codon produces the mitochondrial Glr1p isoform and that amino acids 1–16 are important for mitochondrial targeting. Furthermore, the abundant expression of cytosolic Glr1p in the M1L mutant indicates that in the absence of AUG1, AUG17 is translated efficiently.
Fig. 4. Glr1p mutants have altered Glr1p expression and distribution.

PMS (P) and mitochondrial (M) fractions were prepared from the glr1Δ mutant strain transformed with the indicated plasmids, and fractions were analyzed by immunoblot as in Fig. 3. In all panels except C (bottom), the entire fraction derived from 75 µg total cell protein was analyzed such that the same cell equivalents of PMS and mitochondria are examined, as in Fig. 3. In C (bottom), PMS and mitochondria extracts are compared on the basis of equal protein (20 µg protein analyzed in each case). Unprocessed (u) and processed (p) forms of Glr1p are indicated. Glr1p-expressing plasmids utilized include: pCO113 (WT), pCO115 (Δ1–16), pCO116 (M1L), pCO114 (M17L), pCO117 (FS1), pCO118 (FS2), pCO119 (U−3A(M1)), and pCO121 (U−3A(M1)/M17L).
Conversely, mutation of the second AUG codon (M17L) generates the opposite result. This mutant has very low levels of cytosolic Glr1p accompanying an increase in mitochondrial Glr1p (Fig. 4A, lanes 7–8). In fact, the cytosolic isoform of M17L Glr1p runs higher on the Western blot than the mitochondrial protein. We believe this band actually corresponds to the long, mitochondrial form that has not been processed and is trapped in the cytoplasm. This phenomenon may occur because the mitochondrial import process for Glr1p is overwhelmed by the increased levels of the mitochondrial isoform in this mutant, creating a backlog of Glr1p proteins waiting to be imported. These results indicate that in the absence of AUG17, translation from AUG1 exclusively generates the long form of the protein that is either imported into the mitochondria and processed, or trapped in the cytosol as the unprocessed form. AUG1 and AUG17 are indeed the only possible start sites for Glr1p because a double mutant (M1L/M17L) lacking both start sites results in loss of GR activity in both the PMS and the mitochondria (Fig. 5).
Fig. 5. GR activity in mitochondrial and PMS extracts from Glr1p mutants.

GR activity assays on mitochondrial and PMS extracts from glr1Δ strains transformed with pAA1 (vector), pCO113 (WT), pCO115 (Δ1–16), pCO116 (M1L), pCO114 (M17L), pCO122 (M1L/M17L), pCO117 (FS1), pCO118 (FS2), pCO119 (U−3A(M1)), or pCO121 (U−3A(M1)/M17L). Enzyme units, reported values, and error bars are the same as in Fig. 2A.
Glr1p Distribution in Frameshift Mutants
Based on our results with the M1L and M17L mutants, it appears that either of these two start codons can be used to initiate translation of Glr1p. However, even though the 2nd AUG codon is translated in the M1L mutant, this does not necessarily mean that the ribosome naturally initiates at this site in the wild-type GLR1 mRNA. Typically, translational initiation of eukaryotic mRNAs proceeds via the “scanning model” in which the ribosome binds to the 5’ end of the mRNA and migrates linearly, initiating translation at the 1st AUG codon it encounters (35). Therefore, in the case of the M1L mutant, the ribosome might initiate at AUG17 only because it is the first available initiation site. Results with the M1L mutant did not exclude the possibility that both cytosolic and mitochondrial Glr1p normally result from initiation at AUG1, with the shorter, cytosolic Glr1p produced from processing of the longer form, as has been described for yeast fumarase (36–38).
To more definitively determine whether the 2nd start codon of GLR1 is utilized when the 1st start codon is still intact, two different frameshift (FS) mutations were introduced between AUG1 and AUG17. For the FS1 and FS2 mutants, translation from AUG1 will produce a nonsense polypeptide that is 9 aa and 52 aa long, respectively (Table 1). If AUG1 is the only start site employed, then Glr1p should be absent from both the cytosol and mitochondria in both mutants. However, as shown in Fig. 4B, lanes 1–6, both frameshift mutants still express Glr1p in the cytosol, but the protein is not detected in the mitochondria. Therefore, translation must be initiating at AUG17. Taken together, the data from the translation initiation and frameshift mutants suggest that both start codons are naturally used as initiation sites in vivo, with translation from AUG1 generating mitochondrial Glr1p, while translation from AUG17 produces the cytosolic isoform of the protein.
As another means of assaying the effects of all these mutations on Glr1p distribution, we monitored GR enzymatic activity in the cytosol and mitochondria. As seen in Fig. 5, GR activity closely correlates with the level of Glr1p protein, i.e., mitochondrial GR activity is increased in the M17L mutant and reduced in Δ1–16, M1L, FS1 and FS2 mutants. Yet there remains some residual mitochondrial activity in the latter mutants that is not detected by Western blots. This may represent GR activity from the PMS or other cellular compartments that contaminate the crude mitochondrial prep.
AUG Codon Context and Translation Initiation Efficiency
Although both start sites are utilized in vivo, it appears that translation initiation at AUG17 is preferred over AUG1 since the cytosolic isoform of G1r1p is the predominant form of the protein in the cell. According to the scanning model for translation, this result is unexpected since the first AUG codon in an mRNA sequence is ordinarily preferred as the ribosome initiation site (35). However, “leaky scanning,” or translation from downstream AUG codons, may occur when the sequence context of the first AUG codon is unfavorable (39). To investigate whether leaky scanning was occurring in Glr1p translation, we examined the sequences surrounding each AUG codon. These flanking sequences, also called the Kozak sequence, have been shown to influence initiation efficiency (40,41). In yeast, the preferred consensus sequence is 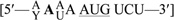 , with the A in position −3 (in bold) being the most highly conserved of the residues surrounding the AUG codon (42). As shown in Table 2, AUG17 more closely matches this consensus sequence, notably having an A in the critical −3 position, whereas AUG1 contains a U at this site. This suggests that the relatively poor utilization of AUG1 may be due to absence of an A in position −3.
, with the A in position −3 (in bold) being the most highly conserved of the residues surrounding the AUG codon (42). As shown in Table 2, AUG17 more closely matches this consensus sequence, notably having an A in the critical −3 position, whereas AUG1 contains a U at this site. This suggests that the relatively poor utilization of AUG1 may be due to absence of an A in position −3.
Table 2.
AUG Sequence Context for Yeast Glr1p Start Sites
| Initiation Codon | AUG Sequence Contexta | −3 Conserved? |
|---|---|---|
| Yeast consensus | Yes | |
| WT AUG1 | No | |
| WT AUG17 | Yes | |
| U−3A(M1) AUG1 | Yes |
The conserved residue at position −3 is shown in bold and the AUG start codon is underlined.
To test this hypothesis, we introduced an A residue at position −3 with respect to AUG1 creating the U−3A(M1) mutant (Table 1,2). This single mutation resulted in a large increase in the levels of mitochondrial Glr1p (Fig. 4C, lane 4), indicative of a dramatic increase in translation initiation at AUG1. Furthermore, this increase in mitochondrial Glr1p is accompanied by a decrease in cytosolic Glr1p in the U−3A(M1) mutant. This result is most obvious when equal protein levels are analyzed for mitochondrial and cytosolic extracts (bottom panel, Fig. 4C, compare lanes 1 and 3). The results obtained from Western blot analysis were also confirmed in enzymatic assays of GR activity (Fig. 5). However, initiation at AUG17 is still evident in the U−3A(M1) mutant because a substantial level of Glr1p still accumulates in the cytosol (Fig. 4C, lane 3, and Fig. 5). The majority of this cytosolic protein indeed results from initiation at AUG17 because a double U−3A(M1)/M17L mutant abolishes production of the major cytosolic species of Glr1p (Fig. 4C, lane 5). A minor species corresponding to the unprocessed mitochondrial isoform (u) is also detected in the cytosol of the U−3A(M1) and U−3A(M1)/M17L mutants (Fig. 4C, top panel, lanes 3 and 5). As is the case with the M17L mutant (Fig. 4C, lane 7), the increased levels of polypeptide initiated at AUG1 may not be efficiently imported into the mitochondria, resulting in accumulation of a portion of the long, unprocessed isoform in the cytosol. Overall, these results suggest that improvement of the AUG1 sequence context reduces but does not completely preclude translation from AUG17 by limiting the occurrence of leaky scanning (see Discussion).
Discussion
In the present study, we provide evidence that yeast Glr1p is localized to both the mitochondria (~5–10%) and the cytosol (~90–95%). Although the cytosolic isoform is predominant in terms of total cellular levels, the concentration of Glr1p is each compartment appears to be similar when the volume of the cytosol vs. the mitochondria is taken into account. When considering the generation and distribution of GSH within the cell, the importance of having a mitochondrial version of Glr1p becomes clear. GSH is exclusively synthesized in the cytosol (15) and then transported into the mitochondria, presumably via organic anion carriers (43). Within the mitochondria, GSH reverses oxidative damage directly, or functions as a cofactor for mitochondrial glutaredoxins and glutathione peroxidases (5,44). Since the mitochondrion is the primary source of reactive oxygen species in the cell, maintaining a store of reduced GSH in this compartment is crucial for combating these damaging molecules. However, once oxidized, glutathione (GSSG) cannot be transported back across the mitochondrial membrane into the cytosol (16). This fact underscores the importance of having a mitochondrial isoform of Glr1p that can regenerate reduced GSH thereby preventing accumulation of GSSG in this compartment.
The mechanism dictating the dual targeting of yeast GR involves two in-frame start codons, AUG1 and AUG17. Our data indicate that translation from AUG1 produces the long, mitochondrial form including the MTS, while translation from AUG17 generates the short, cytosolic form. Upon import, the MTS is removed from the mitochondrial isoform, generating mature Glr1p that is similar in length to the cytosolic version. The sequences flanking each start codon, especially in the −3 position, appear to influence the relative levels of each isoform in the cell. Since AUG17 has a more favorable sequence context based on the Kozak consensus, translation from this start codon is more efficient than AUG1, resulting in a predominance of the cytosolic isoform. A leaky scanning mechanism, in which some ribosomes initiate at AUG1, but most bypass AUG1 and preferentially initiate at AUG17, appears to largely account for this result. When the context of the AUG1 codon is improved, as in the U−3A(M1) mutant, the levels of mitochondrial Glr1p (translated from AUG1) increase with a concomitant decrease in cytosolic levels (translated from AUG17). Presumably, leaky scanning is reduced in this mutant as more ribosomes initiate translation at AUG1 rather than AUG17. Therefore, this result suggests that the long, mitochondrial form and the short, cytosolic form are translated from one mRNA that includes both start codons.
Although leaky scanning appears to be the main factor controlling the dual distribution of yeast GR, we cannot rule out other factors that could contribute to localization of this protein, including slow mitochondrial import and processing. Under certain conditions, we see a small percentage of the unprocessed mitochondrial isoform in the cytosol. This phenomenon is much more pronounced in mutants with increased levels of mitochondrial Glr1p (e.g. M17L, U−3A(M1)), however it is still apparent in WT Glr1p (see Fig. 3A, lane 4 and Fig. 4A–C, lanes 1). It is possible that the mitochondrial import of Glr1p may be somewhat inefficient such that a small fraction of the polypeptide folds before import and remains trapped in the cytosol. This type of mechanism has been described for the localization of yeast fumarase (Fum1p). A portion of the FUM1 translation product is processed and fully imported into the mitochondria, while the majority is released back into the cytosol after processing, presumably due to rapid folding of the polypeptide into an import-incompetent state (37,38). It is possible that a similar mechanism contributes to incomplete mitochondrial uptake of Glr1p, although our results indicate that leaky scanning translation is the driving force for Glr1p distribution.
Can the mechanism for dual targeting of GR described here be extrapolated to other organisms as well? An examination of the gene sequences of mammalian GRs provides some clues to answer this question. As shown in Figure 1, human and mouse GRs also exhibit two potential in-frame start codons at their N-termini (28,29). Translation from the 1st AUG codon is predicted to encode the mitochondrial isoform since this sequence includes the putative MTS, while translation from the 2nd AUG codon could encode the shorter, cytosolic isoform. The preferred AUG context sequence for mammalian genes 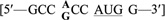 is somewhat different from that of S. cerevisiae, although the purine at position −3 (in bold), is still the most critical in determining translation efficiency (39,41). As shown in Table 3, the AUG1 sequences for both human and mouse GRs lack this critical residue. In contrast, the 2nd AUG codons for both mammalian GRs (AUG44 in human GR and AUG27 in mouse GR) have the appropriate residue (an A or G) at this important position. Based on this analysis alone, the second AUG is predicted to be the preferential start site for translation of mammalian GR, just as we have demonstrated here for yeast Glr1p. Therefore, the leaky scanning mechanism for translation may be the conserved driving force across eukaryotes that ensures the dual targeting of GR to both the mitochondria and cytoplasm.
is somewhat different from that of S. cerevisiae, although the purine at position −3 (in bold), is still the most critical in determining translation efficiency (39,41). As shown in Table 3, the AUG1 sequences for both human and mouse GRs lack this critical residue. In contrast, the 2nd AUG codons for both mammalian GRs (AUG44 in human GR and AUG27 in mouse GR) have the appropriate residue (an A or G) at this important position. Based on this analysis alone, the second AUG is predicted to be the preferential start site for translation of mammalian GR, just as we have demonstrated here for yeast Glr1p. Therefore, the leaky scanning mechanism for translation may be the conserved driving force across eukaryotes that ensures the dual targeting of GR to both the mitochondria and cytoplasm.
Table 3.
AUG Sequence Context for Human and Mouse GR Putative Start Sites
| Initiation Codon | AUG Sequence Contexta | −3 Conserved? |
|---|---|---|
| Mammalian consensus | Yes | |
| Human AUG1 | No | |
| Human AUG44 | Yes | |
| Mouse AUG1 | No | |
| Mouse AUG27 | Yes |
The conserved residue at position −3 is shown in bold and the AUG start codon is underlined.
Acknowledgements
We would like to thank R. Jensen for the cytochrome b2 and Mas2 antibodies and plasmid pAA1, and F.W. Outten for critical reading of the manuscript. This work was supported by the JHU NIEHS center and by the NIH grant GM 50016 to V.C.C. C.E.O. was supported by the NIH post-doctoral fellowship GM 66594.
The abbreviations used are
- GSH
reduced glutathione
- GSSG
oxidized glutathione
- GR
glutathione reductase
- TR
thioredoxin reductase
- GFP
green fluorescent protein
- PMS
post-mitochondrial supernatant
- IMS
intermembrane space
- DTNB
5,5’-dithiobis(2-nitrobenzoic acid)
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- PGK
3-phosphoglycerate kinase
- MTS
mitochondrial targeting signal
- aa
amino acid
References
- 1.Jamieson DJ. Yeast. 1998;14:1511–1527. doi: 10.1002/(SICI)1097-0061(199812)14:16<1511::AID-YEA356>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 2.Carmel-Harel O, Storz G. Annu. Rev. Microbiol. 2000;54:439–461. doi: 10.1146/annurev.micro.54.1.439. [DOI] [PubMed] [Google Scholar]
- 3.Fridovich I. J. Biol. Chem. 1989;264:7761–7764. [PubMed] [Google Scholar]
- 4.Pedrajas JR, Kosmidou E, Miranda-Vizuete A, Gustafsson JA, Wright AP, Spyrou G. J. Biol. Chem. 1999;274:6366–6373. doi: 10.1074/jbc.274.10.6366. [DOI] [PubMed] [Google Scholar]
- 5.Rodriguez-Manzaneque MT, Tamarit J, Belli G, Ros J, Herrero E. Mol. Biol. Cell. 2002;13:1109–1121. doi: 10.1091/mbc.01-10-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pedrajas JR, Porras P, Martinez-Galisteo E, Padilla CA, Miranda-Vizuete A, Barcena JA. Biochem. J. 2002;364:617–623. doi: 10.1042/BJ20020570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miranda-Vizuete A, Damdimopoulos AE, Spyrou G. Antioxid. Redox Signal. 2000;2:801–810. doi: 10.1089/ars.2000.2.4-801. [DOI] [PubMed] [Google Scholar]
- 8.Lundberg M, Johansson C, Chandra J, Enoksson M, Jacobsson G, Ljung J, Johansson M, Holmgren A. J. Biol. Chem. 2001;276:26269–26275. doi: 10.1074/jbc.M011605200. [DOI] [PubMed] [Google Scholar]
- 9.Gladyshev VN, Liu A, Novoselov SV, Krysan K, Sun QA, Kryukov VM, Kryukov GV, Lou MF. J. Biol. Chem. 2001;276:30374–30380. doi: 10.1074/jbc.M100020200. [DOI] [PubMed] [Google Scholar]
- 10.Juhnke H, Krems B, Kotter P, Entian KD. Mol. Gen. Genet. 1996;252:456–464. doi: 10.1007/BF02173011. [DOI] [PubMed] [Google Scholar]
- 11.Pandolfi PP, Sonati F, Rivi R, Mason P, Grosveld F, Luzzatto L. EMBO J. 1995;14:5209–5215. doi: 10.1002/j.1460-2075.1995.tb00205.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Outten CE, Culotta VC. EMBO J. 2003;22:2015–2024. doi: 10.1093/emboj/cdg211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jo SH, Son MK, Koh HJ, Lee SM, Song IH, Kim YO, Lee YS, Jeong KS, Kim WB, Park JW, Song BJ, Huh TL, Huhe TL. J. Biol. Chem. 2001;276:16168–16176. doi: 10.1074/jbc.M010120200. [DOI] [PubMed] [Google Scholar]
- 14.Sturtz LA, Diekert K, Jensen LT, Lill R, Culotta VC. J. Biol. Chem. 2001;276:38084–38089. doi: 10.1074/jbc.M105296200. [DOI] [PubMed] [Google Scholar]
- 15.Griffith OW, Meister A. Proc. Natl. Acad. Sci. USA. 1985;82:4668–4672. doi: 10.1073/pnas.82.14.4668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olafsdottir K, Reed DJ. Biochim. Biophys. Acta. 1988;964:377–382. doi: 10.1016/0304-4165(88)90038-4. [DOI] [PubMed] [Google Scholar]
- 17.Mbemba F, Houbion A, Raes M, Remacle J. Biochim. Biophys. Acta. 1985;838:211–220. doi: 10.1016/0304-4165(85)90081-9. [DOI] [PubMed] [Google Scholar]
- 18.Muller EG. Mol. Biol. Cell. 1996;7:1805–1813. doi: 10.1091/mbc.7.11.1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trotter EW, Grant CM. EMBO Rep. 2003;4:184–188. doi: 10.1038/sj.embor.embor729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sherman F, Fink GR, Lawrence CW. Methods in Yeast Genetics. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory Press; 1978. [Google Scholar]
- 21.Gietz RD, Schiestl RH. Yeast. 1991;7:253–263. doi: 10.1002/yea.320070307. [DOI] [PubMed] [Google Scholar]
- 22.Hobbs AE, Srinivasan M, McCaffery JM, Jensen RE. J. Cell Biol. 2001;152:401–410. doi: 10.1083/jcb.152.2.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Daum G, Bohni PC, Schatz G. J. Biol. Chem. 1982;257:13028–13033. [PubMed] [Google Scholar]
- 24.Jensen LT, Culotta VC. Mol. Cell. Biol. 2000;20:3918–3927. doi: 10.1128/mcb.20.11.3918-3927.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Anderson ME. Methods Enzymol. 1985;113:548–555. doi: 10.1016/s0076-6879(85)13073-9. [DOI] [PubMed] [Google Scholar]
- 26.Cuozzo JW, Kaiser CA. Nat. Cell Biol. 1999;1:130–135. doi: 10.1038/11047. [DOI] [PubMed] [Google Scholar]
- 27.Collinson LP, Dawes IW. Gene. 1995;156:123–127. doi: 10.1016/0378-1119(95)00026-3. [DOI] [PubMed] [Google Scholar]
- 28.Kelner MJ, Montoya MA. Biochem. Biophys. Res. Commun. 2000;269:366–368. doi: 10.1006/bbrc.2000.2267. [DOI] [PubMed] [Google Scholar]
- 29.Tamura T, McMicken HW, Smith CV, Hansen TN. Biochem. Biophys. Res. Commun. 1997;237:419–422. doi: 10.1006/bbrc.1997.7153. [DOI] [PubMed] [Google Scholar]
- 30.Neupert W. Annu. Rev. Biochem. 1997;66:863–917. doi: 10.1146/annurev.biochem.66.1.863. [DOI] [PubMed] [Google Scholar]
- 31.Schatz G, Butow RA. Cell. 1983;32:316–318. doi: 10.1016/0092-8674(83)90450-6. [DOI] [PubMed] [Google Scholar]
- 32.Emanuelsson O, Nielsen H, Brunak S, von Heijne G. J. Mol. Biol. 2000;300:1005–1016. doi: 10.1006/jmbi.2000.3903. [DOI] [PubMed] [Google Scholar]
- 33.Lenton KJ, Therriault H, Wagner JR. Anal. Biochem. 1999;274:125–130. doi: 10.1006/abio.1999.4258. [DOI] [PubMed] [Google Scholar]
- 34.Stevens BJ. Biol. Cellulaire. 1977;28:37–56. [Google Scholar]
- 35.Kozak M. Cell. 1978;15:1109–1123. doi: 10.1016/0092-8674(78)90039-9. [DOI] [PubMed] [Google Scholar]
- 36.Stein I, Peleg Y, Even-Ram S, Pines O. Mol. Cell. Biol. 1994;14:4770–4778. doi: 10.1128/mcb.14.7.4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Knox C, Sass E, Neupert W, Pines O. J. Biol. Chem. 1998;273:25587–25593. doi: 10.1074/jbc.273.40.25587. [DOI] [PubMed] [Google Scholar]
- 38.Sass E, Blachinsky E, Karniely S, Pines O. J. Biol. Chem. 2001;276:46111–46117. doi: 10.1074/jbc.M106061200. [DOI] [PubMed] [Google Scholar]
- 39.Kozak M. Gene. 2002;299:1–34. doi: 10.1016/S0378-1119(02)01056-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kozak M. Nature. 1984;308:241–246. doi: 10.1038/308241a0. [DOI] [PubMed] [Google Scholar]
- 41.Kozak M. Cell. 1986;44:283–292. doi: 10.1016/0092-8674(86)90762-2. [DOI] [PubMed] [Google Scholar]
- 42.Cigan AM, Donahue TF. Gene. 1987;59:1–18. doi: 10.1016/0378-1119(87)90261-7. [DOI] [PubMed] [Google Scholar]
- 43.Chen Z, Lash LH. J. Pharmacol. Exp. Ther. 1998;285:608–618. [PubMed] [Google Scholar]
- 44.Green RC, O'Brien PJ. Biochim. Biophys. Acta. 1970;197:31–39. doi: 10.1016/0005-2728(70)90005-8. [DOI] [PubMed] [Google Scholar]