

Abstract
Toll-like receptors (TLRs) are a family of cellular structures activated by recognition of pathogen associated molecular sequences. The activation of TLRs triggers a variety of intracellular mechanisms aiming to protect the host from the invading microorganisms. Lipopolysaccharide (LPS) is the main ligand for TLR4. Here we show that resistin, a cystein-rich protein believed to regulate carbohydrate metabolism, competes with LPS for binding to TLR4. Binding of recombinant resistin to human myeloid and epithelial cells was assessed by flow cytometry and its co-precipitation with TLR4 was demonstrated. Antibodies against TLR4 abolished resistin binding to human leucocytes and cytokine production by peripheral blood mononuclear cells in response to resistin stimulation. In contrast, isotype-matched murine IgG or TLR2 antibodies were unable to prevent binding of resistin to the cells. Similarly, TLR4-dependent pattern of resistin binding was observed in epithelial cell line HEK293 (human epithelial kidney cell), where TLR4 transfected, but not myeloid differentiation factor 2/CD14-transfected, TLR2 transfected or HEKnull cells, responded functionally to resistin stimulation. Intracellular signalling of resistin was assessed using inhibitors of transcription factors mitogen activated protein kinases, nuclear factor-κB, phosphoinositide 3-kinase and siRNA targeting TLR4 and human myeloid differentiation factor 88. Results demonstrate that TLR4 serves as a receptor for the pro-inflammatory effects of resistin in human cells. This may partly explain the multifunctional role of resistin in chronic inflammation, atherosclerosis and insulin resistance.
Keywords: toll-like receptor 4, resistin, inflammation, lipopolysaccharide
Introduction
Toll-like receptors (TLRs) are a family of widely distributed cellular structures that are activated by the recognition of pathogen-associated molecular sequences/patterns. The primary role of TLRs is attributed to the protection of the host from invading microorganisms [1]. The activation of TLRs triggers a variety of intracellular mechanisms and transcription factors depending on the receptor and cell type. TLR4 belongs to the TLRs for lipase-based pathogen-associated molecular sequences/patterns having lipopolysaccharide (LPS, a glycolipid component of the outer membrane of gram-negative bacteria) as the main ligand [2]. Intracellular signalling following ligation of the TLR4/myeloid differentiation factor 2 (MD2) complex is achieved through human myeloid differentiation factor 88 (MyD88) resulting in nuclear migration of nuclear factor-κB (NF-κB) transcription factor [3, 4]. Translocation of this transcription factor inevitably leads to a burst of pro-inflammatory cytokines, proteolytic enzymes and adhesion molecules. LPS is known as the main ligand for TLR4. Recently, the presence of endogenous ligands potently activating TLR4 has been reported including hyaluronan, fibronectin and heat shock proteins (HSP60, HSP70). TLR4 mediated effects have been also demonstrated for HMGB-1 [5], certain fatty acids [6], minimally modified LDL and oxidized-LDL [7]. In the present study we show a new endogenous ligand for TLR4.
Resistin is a 12.5-kD protein, a member of a cysteine-rich protein family [8]. Resistin gene is located on chromosome 8 in mice and on chromosome 19 in human beings. It is secreted as a 108aa polypeptide consisting of homodimers linked by disulphide bonds between the cysteine residues. Resistin may also form heterodimeric structures with other members of resistin-like molecules (RELMα and RELMβ) [9]. Human resistin exhibits only 59% amino acid homology to its murine counterpart, predominantly in the cysteine-rich regions [10, 11]. Resistin was originally described as an adipocyte-derived polypeptide modulating the insulin-signalling pathway leading to insulin resistance in obese mice [8, 12]. Indeed, in mice resistin is almost exclusively expressed in white adipose tissue, while in man resistin is not expressed in adipocytes [13, 14] and has no direct relation to obesity or insulin sensitivity. In contrast, high levels of human resistin are expressed by mononuclear leucocytes, spleen and bone marrow cells [15] followed by lung tissue, resting endothelial cells and placenta [16].
Resistin has been described as a protein ‘found in the inflammatory zone’ (FIZZ). Increased expression of resistin has been reported in chronic liver diseases [17, 18], lung diseases [12], in atherosclerotic plaques [19], inside inflamed joints [20, 21] and in inflammatory kidney disease [22, 23]. Resistin has recently attracted attention as a predictive factor for atherosclerosis progression [24], vascular re-occlusion and kidney function in its inflammatory diseases [25]. In the above mentioned conditions, resistin levels showed a strong correlation to other inflammatory markers such as C-reactive protein [17, 24, 25], tumour necrosing factor (TNF)-α[17], interleukine (IL)-6 [20] and soluble vascular adhesion molecules [25]. Active participation of resistin in the inflammatory process is supported by its ability to induce and respond to inflammatory stimuli including cytokines, mitogens and endotoxins. Moreover, the regulatory effect of resistin regarding vascular endothelium has been thoroughly evaluated. It is shown that resistin is only minimally expressed in umbilical endothelial cells and peripheral vein samples while arterial walls from atherosclerotic aorta, aorta aneurysms and occlusive arterial disease displayed increased content of resisting [19, 26]. Resistin exhibits mitogenic properties towards human endothelial [27, 28] and smooth muscle cells [29] by reducing eNOS expression, potentiating expression of adhesion molecules (ICAM, VCAM, ET-1, PAI-1). It up-regulates a cascade of pro-inflammatory cytokines, including TNF-α[18, 20], regulates matrix remodelling in human trophoblast by up-regulating matrix metalloproteinases and down-regulating TIMPs [30] and induces joint inflammation when injected intra-articularly [20]. Participation of resistin in lipid utilization and bone remodelling [15] has also been recently demonstrated. It remains to be elucidated whether all the above properties of resistin are related to its pro-inflammatory effects and how they are mediated at the cellular level since the cellular receptor of resistin is still unknown. Here we demonstrate that TLR4 is a cell surface structure mediating pro-inflammatory effects of resistin and may partly explain the multifunctional role of resistin in such varying processes as chronic inflammation, atherosclerosis and insulin resistance.
Experimental procedures
Cells and culture conditions
Monocytic cell line THP1 (American Type Culture Collection, Manassas, VA, USA) was cultured in RPMI1649 medium supplemented with 10% heat-inactivated foetal calf serum (FCS), 1% sodium pyruvate, gentamycin and 1% L-glutamine. Human epithelial kidney cells (HEK)293-null and HEK293 transfected with MD2/CD14, TLR2 and TLR4 were obtained from InVitroGen Europe (Toulouse, France) and cultured in D-MEM GlutaMAX (Gibco, Invitrogen, Grand Island, NY, USA) medium supplemented with 10% FCS, Normocin (100 μg/ml) and blasticidin (10 μg/ml). For FACS analyses, leucocytes were prepared by lysing erythrocytes with ammonium chloride 0.83% (pH 7.3). Peripheral blood mononuclear cells (PBMC) were separated on Lymphoprep density gradient from heparinized blood, and resuspended in Iscoves medium containing 1% L-glutamine, 5 × 10−5 M β-mercaptoethanol, 50 μg/ml of gentamycin sulphate and 10% FCS. All cells were cultured in a humidified atmosphere of 5% CO2 at 37°C.
Cell stimulation
Recombinant human resistin (PeproTech, London, UK; endotoxin concentration <0.1 ng/μg) was introduced to cell cultures at the final concentrations of 10–1000 ng/ml. Synthetic peptides corresponding to various amino acid sequences of human resistin, assigned as aa23–42, aa43–63, were purchased from Phoenix Biochemicals (Milpitas, CA, USA), and the peptides assigned aa64–88, and aa51–108, were purchased from Immunodiagnostik AG (Bensheim, Germany). The peptide aa43–63 forms a loop by means of Cys51-Cys63 bond. Known ligands for TLR4 (LPS, 10 ng/ml) and for TLR2 (PamCys, 2 ng/ml) were used as positive controls in all the experiments. For extracellular release of cytokines and resistin, supernatants were collected following 48 hrs of stimulation. Total mRNA was prepared at 0, 0.5, 3 and 24 hrs of stimulation. To assess resistin binding to the cell surface, leucocytes were fixed with 1% formaldehyde and washed with 1% FCS-PBS. Following centrifugation, cells were stained with biotinylated anti-human resistin antibodies (clone 184320, R&D Systems, Minneapolis, MN, USA), TLR4 antibodies (phycoerytrin conjugated, Serotec, Oxford, UK) or appropriate mouse IgG isotype controls (BD Diagnostics, Franklin Lakes, NJ, USA) and subjected to FACS analyses. Anti-resistin antibodies (clone 8184335, R&D Systems) were used in the inhibitory experiments. Mouse anti-TLR2 (clone TL2.1) were from Alexis Biochemicals (Lausen, Switzerland) whereas anti-TLR4 (clone HTA125), anti-CD14 (clone UCHM1) antibodies and mouse IgG2a isotype were from Serotec. All the antibodies used had endotoxin concentration <0.01 EU/μg. The inhibitors used in the study were parthenolide (25 μM, Sigma, St Louis, MO, USA) for NF-κB pathway, LY294002 (50 μM) for phosphoinositide 3 (PI3)-kinase, SB203580 (50 μM) for MAP-kinases p38 and PD98059 (50 μM) for p44/42 (MEK1) that were obtained from Biosource (Nivelles, Belgium).
Immunoprecipitation and Western blot assay
THP1 cells (1 × 107) were fixed in 4% paraformaldehyde, and incubated with recombinant resistin (500 ng/ml) for 1 hr at 4°C. Following incubation, the membrane fraction of the cells was prepared using CelLytic™ MEM Protein Extraction kit (Sigma Aldrich, Inc., St. Louis, MO, USA) including protease inhibitor cocktail. Lysates were incubated for 1 hr at 4°C with mouse anti-humanTLR4 (0.5 mg/ml), or mouse anti-human resistin (0.25 mg/ml) or non-specific mouse IgG (0.5 mg/ml). After incubation with the antibody, the lysates were incubated with equal amount of protein A/protein G sepharose beads (Amersham Biosciences, Uppsala, Sweden) for 1 hr at 4°C. After washing the beads were resuspended in sample buffer (50 mM Tris, 1% SDS, 100 μM DTT, pH 7.4) and heated for 3min at 95°C to dissociate captured antigen from beads. Beads were removed by centrifugation at 12,000 ×g. Immunoprecipitates (1μg/sample) from supernatants were separated on a 10% SDS-PAGE Tris-Glycine gel and transferred to a PVDF membrane in a Mini-Trans-Blot electrophoretic unit (Bio-Rad Laboratories, Sundbyberg, Sweden) using Tris-glycine buffer (pH 8.3) containing 20% methanol. The PVDF membrane was washed and blocked with 2% fat-free milk. Following washing, the membrane was incubated with mouse anti-human TLR4 or with biotinylated anti-resistin antibodies (at 5 μg/ml). Proteins were visualized using appropriate HRP-labelled secondary antibody, and detection was performed with H2O2/amino-ethylcarbazole substrate in sodium acetate buffer (pH 5.5). The size of protein complexes was defined according to pre-stained protein weight marker (Invitrogen).
Flow cytometric analysis
Human leucocytes were stimulated as above, washed and incubated with optimal concentrations of mouse monoclonal antibodies and the isotype IgG matched controls. Following staining, cells were analysed with a FACS Calibur equipped with CellQuest software. The results are presented as mean fluorescence intensity (MFI).
siRNA silencing
A pool of four duplex siRNA targeting human TLR4 gene (siGENOME SMARTpool, M-008088–01), as well as MyD88 gene (siGENOME SMARTpool, D-004769) and non-targeting siRNA (siControl) were purchased from Dharmacon, Inc. (Chicago, IL, USA). Transfection of targeting and control siRNA into HEK-TLR4 cells (2.5 × 105/ml) was performed following overnight incubation in serum and antibiotica-free D-MEM GlutaMAX medium without pyruvate (Gibco) using DharmaPHECT 2 transfection reagent. Following 4 hrs incubation at 37°C, the transfection was stopped by introducing FCS to the final concentration 10% and test substances. Down-regulation of the MyD88 protein synthesis following siRNA transfection was proved by Western blot analysis of cell lysates immunoprecipitated using goat anti-human MyD88 antibodies (N-19, Santa Cruz Biotechnologies, CA, USA) as described above. Down-regulation of TLR4 was proved by Western blot analysis of cell membranes precipitated with mouse anti-human TLR4 antibodies (Alexis Biochemicals). Viability of cells was assessed by annexin V/propidium iodide staining and FACS analysis.
RNA isolation and RT-PCR analysis
Total RNA from harvested cells was extracted by an RNeasy kit (Qiagen Nordic, Solna, Sweden) and concentration was assessed spectrophotometrically at 260 nm. The gene expression was measured with TaqMan real-time PCR (Applied Biosystems, Foster City, CA, USA) as previously described [20]. Primers and probes designed by Applied Biosystems were used for the amplification of the samples. Relative quantification was made using the ABI Prism 7700 Sequencing Detection System (Applied Biosystems).
Determination of cytokine levels
The level of IL-6 in supernatants was determined by a bioassay which measures the effect of test samples on proliferation of the IL-6 dependent cell line B13.29 [31]. Levels of IL-8 were measured by a sandwich ELISA using a pair of mouse anti-IL8 antibodies (clone 3IL8-H10, Endogene, Rockford, IL, USA). Levels of TNF-α and IL-1β were measured by a sandwich ELISA using pair of mouse antibodies (R&D Systems). Optical density of the tested samples was related to serial dilution of human recombinant IL-6, IL-8, TNF-α or IL-1β, respectively (R&D Systems), and expressed in pg/ml.
Statistical analysis
The level of continuous variables was expressed as mean ± S.E.M. Difference between the groups was calculated separately employing the Mann-Whitney U-test. Interrelation between parameters studied was calculated employing Spearman’s correlation coefficient. For all the statistical evaluation of the results, P-values below 0.05 were considered significant.
Results
Resistin binding to the surface of human leucocytes requires the presence of TLR4
Human peripheral blood leucocytes exhibited low levels of surface expression of TLR4 in the resting setting (lymphocytes 1.7 ± 2.1%, monocytes 12.4 ± 3.3%). Human leucocytes were prepared from the peripheral blood of healthy individuals (n= 9) as described in the experimental procedures and incubated with recombinant resistin (500 ng/ml) versus buffer. The flow cytometric analysis shows binding of recombinant resistin to the surface of lymphocytes (MFI: 45.6 ± 17.2 versus 26.5 ± 11.3, P= 0.02) and monocytes (MFI: 46.5 ± 5.5 versus 38.5 ± 3.4, P= 0.04). In contrast, human neutrophils expressed resistin on their surface prior to exposure to exogenous resistin and only a minimal de novo binding was observed (MFI: 79.3 ± 2.1 versus 74.3 ± 3.9, not significant, Fig. 1A).
Fig 1.
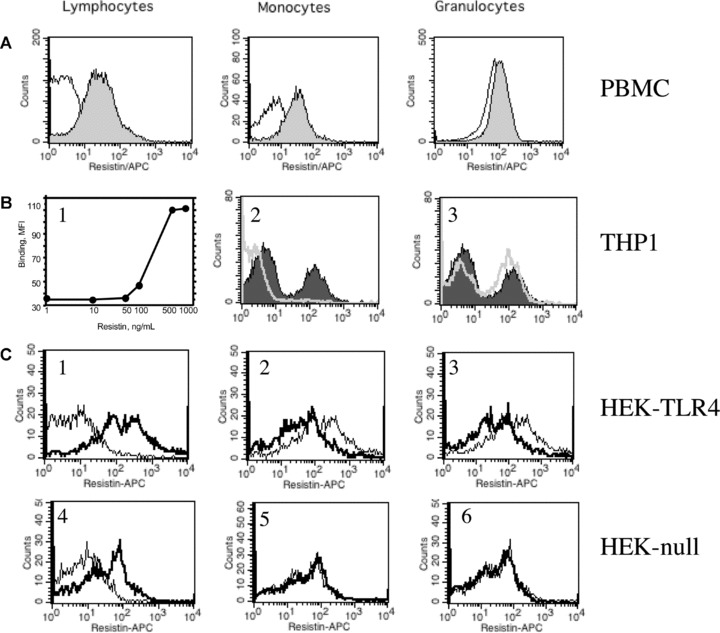
Resistin binding to human leucocytes and epithelial cells. (A) Human leucocytes obtained from peripheral blood were incubated for 30 min with human recombinant resistin (500 ng/ml), washed and cell bound resistin was visualized by anti-resistin antibodies using flow cytometry. Pronounced binding of resistin to lymphocyte and monocyte cell population was observed. (filled area). In contrast, neutrophils display significant expression of resistin on their cell surface prior to incubation with exogenous resistin. (B) THP1 monocytic cells bind exogenous resistin in a dose-dependent manner (B1). This resistin binding (filled area, B2 and B3) was abolished by pre-incubation of THP1 cells with TLR4 antibodies (5 μg/ml) (white line, B2), but not with isotype-matched mouse IgG (white line, B3). (C) HEK293 epithelial cells transfected with TLR4 display significantly higher binding capacity for exogenous resistin (bold, C1) as compared to HEK-null cells transfected with empty vector (bold, C4). Binding of resistin to HEK-TLR4 cells is decreased by pre-treatment of cells with antibodies against TLR4 (C3) and CD14 (C2) prior to exposure to resistin. This is not observed in HEK-null cells treated with the same antibodies (C5, C6).
Analogously, human monocytic THP1 cells bound recombinant resistin in a dose-dependent manner (Fig. 1B1). To evaluate the cellular membrane protein required for resistin binding, THP1 cells were exposed to TLR-4-specific antibodies and mouse isotype-matched Ig. The presence of TLR4 antibodies (Fig. 1B2) but not of isotype matched mouse IgG (Fig. 1B3) totally abrogated binding of resistin to the cell surface indicating the importance of TLR4 for the interaction between resistin and human cell membrane.
To further elaborate this finding, recombinant resistin (500 ng/ml) or buffer alone were added to the cultures of HEK293 cells differing in their expression of TLRs. We observed that resistin binding was significantly higher in the TLR4 expressing HEK293 cells exposed to resistin as compared to buffer (MFI: 28.4 ± 4.6 versus 11.3 ± 3.4, P= 0.027, Fig. 1C1). The binding of resistin to HEK-TLR4 but not HEK-null cells was significantly diminished by introduction of anti-TLR4 (5 μg/ml) (MFI: 28.4 ± 4.6 versus 17.6 ± 2.2, P= 0.038, Fig. 1C2), or anti-CD14 antibodies (5 μg/ml) (MFI: 28.4 ± 4.6 versus 19.4 ± 3.1, P= 0.048, Fig. 1C3) to the cell culture prior to incubation with resistin. In contrast, no differences regarding resistin binding were seen in HEK-null cells (MFI: 12.9 ± 3.1 versus 11.7 ± 2.2, not significant (Fig. 1C4) following addition of TLR4 and CD14 antibodies influenced resistin binding to HEK-null cells (Fig. 1C5,6).
Resistin forms a complex with TLR4 on the cell surface
THP1 cells (1 × 107) expressing TLR4 were incubated with human recombinant resistin (500 ng/ml), lysed and the membrane fraction of the cells was prepared as described in the experimental procedures. The cell membrane-bound resistin was detected by Western blot following immunoprecipitation using anti-resistin antibodies or anti-TLR4 antibodies. Immunoprecipitation of THP1 lysates of non-stimulated cells was used as a control. Following separation on acrylamide gel, the precipitated protein complexes expressed TLR4 (Fig. 2A) and resistin (Fig. 2B) upon development with respective monoclonal antibodies. In contrast, a membrane fraction of THP1 cells incubated with resistin and precipitated with non-specific mouse IgG did not contain any resistin band (Fig. 2B, last lane). Immunoprecipitate of non-stimulated cells revealed TLR4, but no resistin band (not shown). Thus, these results support complex formation between the extracellular resistin and TLR4 molecule exposed on the surface of THP1 cells.
Fig 2.
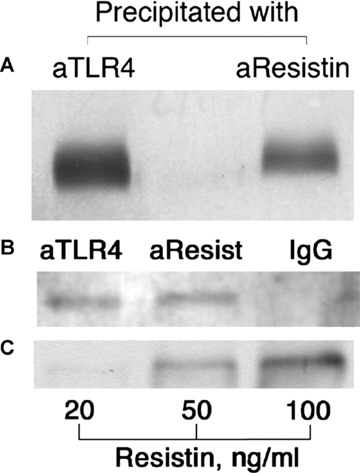
Western blot analysis of the membrane fraction of THP1 cells (1 × 107) incubated with resistin (500 ng/ml). The equal protein amounts of membrane extracts were precipitated using mouse anti-resistin antibodies (7.5 μg), anti-TLR4 antibodies (10 μg) or non-specific mouse IgG (10 μg), followed by incubation with protein A/protein G sepharose beads. Immunoprecipitates obtained by precipitation with either TLR4 ab or resistin ab were subjected to electrophoresis and immunoblotting using TLR4 ab (A). Immunoprecipitates obtained by precipitation with either TLR4 ab, resistin ab or non-specific control ab were subjected to electrophoresis and immunoblotting using resistin antibodies (B). Co-precipitation of TLR4 in the resistin bound membrane fraction is clearly visible. Sensitivity of the system for resistin was tested using various concentrations of human recombinant resistin (C).
Functional properties of resistin are dependent on its interaction with TLR4
To assess the functional role of resistin-TLR4 interaction on the induction of pro-inflammatory responses, PBMC were treated with TLR4 antibodies (dose range 0–1-5–10 μg/ml), and stimulated with recombinant resistin (250 and 1000 ng/ml) and assessed for expression of IL-6 (Fig. 3A) and IL-8 (Fig. 3B). The incubation of PBMC with resistin resulted in a significant up-regulation of IL-6 and IL-8 secretion readily detected in the supernatants. The dose-dependent reduction of IL-6 and IL-8 levels was observed in supernatants of resistin stimulated PBMC pre-treated with TLR4 antibodies. Treatment of PBMC with TLR2 antibodies or with isotype-matched mouse IgG did not prevent resistin-induced secretion of cytokines supporting the specificity of the inhibitory effect of TLR4 interaction (Fig. 3A, B). A prototypic TLR4 ligand, LPS, was used as a positive control. Specificity of resistin to trigger cytokine secretion in the above experimental setting was proved by an abrogation of resistin- but not LPS-induced effects by the addition of anti-resistin antibodies (Fig. 3B).
Fig 3.
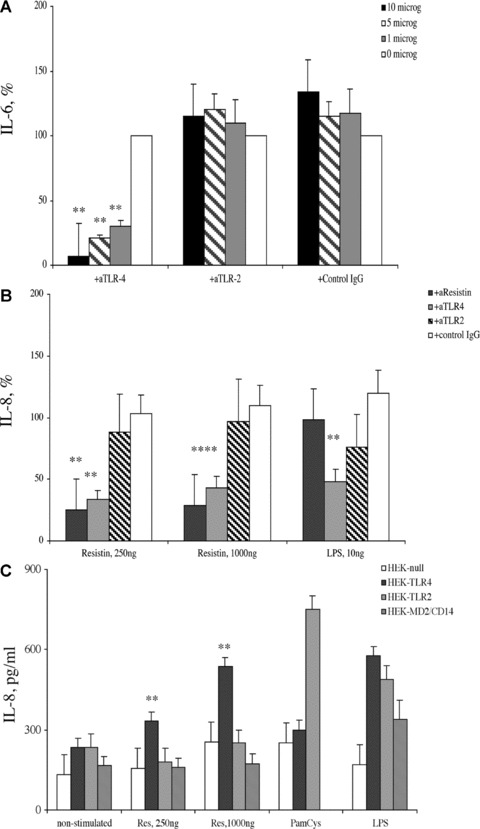
Resistin-induced production of cytokines in human PBMC is dependent on TLR4 expression. Secretion of (A) IL-6 and (B) IL-8 in human PBMC (1 × 106/ml, n= 9) following resistin stimulation was significantly decreased in the PBMC cultures pre-treated with TLR4 antibodies (*P < 0.001). This decrease of cytokine expression was not observed in the PBMC cultures pre-treated with TLR2 antibodies or isotype matched mouse IgG. (C) HEK293 cells transfected with TLR4 display significantly higher secretion of IL-8 following stimulation with resistin (**P < 0.01) as compared to HEK293 cells transfected with TLR2 or those transfected with an empty vector, HEK-null cells. Ligands specific for TLR2 (PamCys, 2 ng/ml) and TLR4 (LPS, 100 ng/ml) were used as positive controls.
Figure 3(C) confirms the requirement of TLR4 for resistin induced effects when HEK293 cells transfected with TLR4 (HEK-TLR4), TLR2 (HEK-TLR2) and empty vector (HEK-null) are stimulated with recombinant resistin. Resistin induced a significant expression of IL-8 in the TLR4 transfected cells that was not observed in the TLR2 transfected cells or in HEK-null cells transfected with an empty vector (Fig. 3C). The ligands of TLR2 (synthetic peptidoglycan, PamCys) and of TLR-4 (LPS) were used as positive controls. Pre-incubation of resistin with anti-resistin antibodies but not with isotype matched mouse IgG resulted in a reduction of IL-8 secretion in HEK-TLR4 cells. Collectively, these results prove that TLR4 is an important receptor and mediator of resistin-induced production of cytokines.
Resistin competes with LPS for binding to TLR4
LPS is the main ligand and has by far the highest affinity for TLR4. To evaluate a role of resistin in the LPS-induced production of pro-inflammatory cytokines, a competitive stimulation of cell cultures was performed. Human PBMC (n= 3) were treated with increasing amounts of resistin for 1 hr followed by stimulation with LPS. The measurement of IL-6 in the supernatants showed that resistin significantly diminished the LPS-induced synthesis of IL-6 (Fig. 4A). Analogous results were obtained when HEK-TLR4 cells were treated with LPS and stimulated with resistin (Fig. 4B). Thus, resistin serves as an endogenous inhibitor of LPS-induced effects, probably by competing for binding to TLR4. A contamination of TLR4 ligands with LPS is an important issue in our study and was, therefore, carefully addressed. Neutralization of resistin using specific monoclonal antibodies almost totally abrogated its cytokine releasing properties, indicating that potential low-grade LPS contamination was not responsible for the effects studied.
Fig 4.
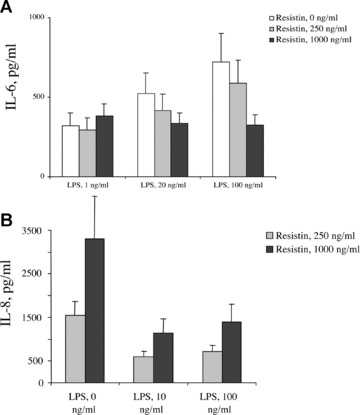
Resistin competes with LPS for binding to TLR4. (A) Treatment of human PBMC (n= 3) with resistin (250 and 1000 ng/ml) significantly diminished secretion of IL-6 in response to LPS. (B) Analogous reduction of LPS-induced production of IL-8 was observed in HEK-TLR4 cells cultured in the presence of resistin.
The requirement of TLR pathways for resistin-induced intracellular signalling
To additionally prove the existence of TLR4 on the cell surface for the pro-inflammatory effects of resistin, HEK-TLR4 cells were transfected with siRNA targeting TLR4 (20 and 100 nM) and non-targeting siRNA sequence (100 nM) (Fig. 5A). The post-transfectional inhibition of TLR-4 was proved by flow cytometry and by Western blot analysis. The HEK-TLR4 cells that underwent inhibition of TLR-4 lost its ability to respond stimulation with resistin (1000 ng/ml) and LPS (10 ng/ml). Similar results were achieved when HEK-TLR4 cells were transfected with siRNA targeting TLRs adaptor molecule, MyD88. Inhibition of MyD88 synthesis following siRNA transfection was proved by Western blot (Fig. 5B). A reduction of IL-8 secretion in response to resistin stimulation was observed in cells exposed to siRNA specific for TLR-4 (50%versus control, P= 0.051) as well as for MyD88 as compared to those transfected with non-targeting siRNA or treated with transfection reagent only (39%versus 106% in the control, P= 0.043, Fig. 5A). Similar pattern was observed in the siRNA transfected cells treated with LPS (36%versus control; P= 0.035, Fig. 5A).
Fig 5.
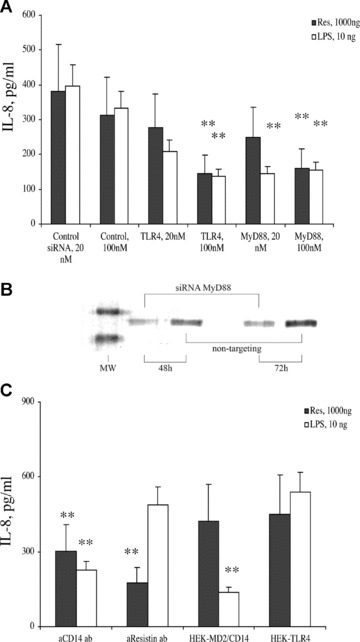
TLR4 is required for the induction of IL-8 expression in HEK293 cells in response to stimulation with resistin. (A) Down-regulation of TLR4 and MyD88 synthesis was achieved by transfecting HEK-TLR4 cells with siRNA sequences specific for these genes. This resulted with a decrease of IL-8 production in response to resistin as well as LPS stimulation in the siRNA transfected cell cultures (*P < 0.05). (B) Down-regulation of MyD88 synthesis in HEK-TLR4 cells transfected with MyD88-specific siRNA and not non-targeting siRNA sequence is shown following 48 and 72 hrs incubation time. (C) Resistin-induced secretion of IL-8 in HEK-TLR4 cells is significantly reduced following incubation with anti-CD14 and anti-resistin antibodies. In contrast, transfection of HEK293 cells with MD2/CD14 in the absence of TLR4 did not lead to resistin-induced activation of HEK293 cells.
For LPS recognition, human cells require a complex consisting of TLR4 in assembly with CD14 and MD2 molecules (4). Interaction between TLR4 and resistin was dependent on the presence of CD14 since resistin binding to HEK-TLR4 cells (Fig. 1C2) and resistin-induced secretion of IL-8 was diminished in the presence of CD14-specific antibodies (Fig. 5C). In contrast, HEK293 transfected with MD2/CD14 genes did not respond to resistin stimulation with IL-8 secretion (Fig. 5C) indicating that the presence of these surface proteins was not sufficient for mediation of resistin effects.
Intracellular pathways mediating resistin effects
We have recently shown that resistin induces activation of the NF-κB pathway [20]. To further evaluate the role of intracellular signalling pathways in the resistin-mediated effects, the inhibitors specific for regulation of NF-κB (parthenolide), MAP-kinases (p44/p42, PD98059 and p38, SB203580) and PI3-kinase (LY294002) were introduced to the PBMC cultures 1 hr prior to stimulation with resistin (Fig. S1). Evaluation of cytokine expression showed that the inhibitors of NF-κB and MAP-kinases efficiently inhibited resistin-induced expression of IL-6, TNF-α and IL-1β on the mRNA (Fig. 6A–C) and protein levels (Fig. 6D–F) in human PBMC (n= 5). The observed inhibitory effects had a dose-dependent pattern (not shown). In contrast, the inhibition of PI3-kinase increased the effect of resistin leading to potentiation of the cytokine expression (Fig. 6). The PI3-kinase has been previously shown as an intracellular inhibitor of TLR2 and TLR4 mediated effects [32, 33].
Fig 6.
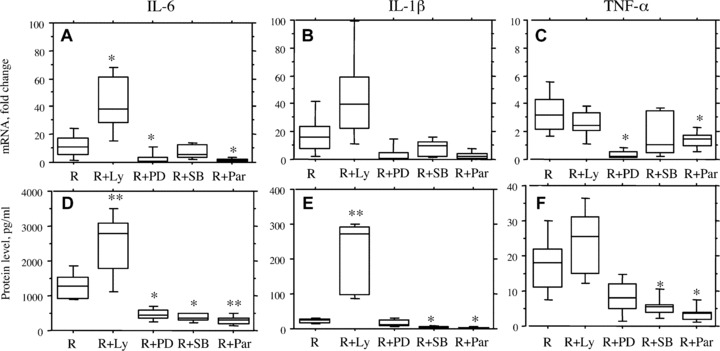
Intracellular regulation of resistin-mediated effects. Human PBMC (1 × 106/ml) were treated with specific inhibitors of signalling pathways including NF-κB (Parthenolide, 25 μM), p38 MAP-kinase (SB203580, 50 μM), p44/p42 MAP-kinase (PD98059, 50 μM) and PI3-kinase (LY294002, 50 μM) prior to stimualtion with resistin (500 ng/ml). Cell cultures were evaluated for the expression of IL-6, TNF-α, and IL-1β on mRNA level (A–C) and on protein level (D–F). We observed that inhibitors of NF-κB and MAP-kinases significantly diminished resistin-induced expression of cytokines, while the inhibitor of PI3-kinase increased resistin stimulation. Differences in cytokine expression as compared to cultures stimulated with resistin are indicated by asterisk (*P < 0.05, and **P < 0.01). Abbreviations: R, resistin; Par, parthrnolide; Ly, LY294002; SB, SB203580; PD, PD98059.
Structural requirements of resistin-TLR-4 interaction
HEK-TLR4, HEK-TLR2 and HEKnull cells were stimulated with equimolar concentrations of synthetic resistin peptides (50 nM), assigned as aa23–42, aa43–64, aa51–108, aa64–88, corresponding to 1000 ng/ml of the whole resistin molecule. All the peptides were recognized by the anti-resistin antibodies in an ELISA-based assay. Functional properties of the peptides were compared to the whole resistin molecule regarding their pro-inflammatory effects, receptor requirement and neutralizing effects of anti-resistin antibodies. Pro-inflammatory effects of resistin-derived peptides were assessed by secretion of IL-8. All the peptides tested were able to significantly induce secretion of IL-8 in HEK-TLR4 as compared to HEK-null cells (Fig. 7A). The level of IL-8 secretion induced by aa43–64, aa51–108 and aa64–88 in HEK-TLR4 were similar to those induced by resistin. The peptides aa43–64, aa51–108 were significantly more efficient in IL-8 induction in TLR-4 expressing cells as compared to those expressing TLR-2. The secretion of IL-8 in HEK-TLR2 cells differed between the peptides. The peptides assigned aa23–42 and aa51–108 induced a significant secretion of IL-8 in HEK-TLR2 cells while aa43–64 and aa64–88 did not. Pre-incubation of the peptides with resistin-specific antibodies significantly diminished IL-8 secretion in HEK-TLR4 cells stimulated with aa23–42 and aa43–64 (Fig. 7B). No significant change in IL-8 secretion was observed following pre-incubation of aa51–108 and aa64–88 peptides with resistin-specific antibodies. The requirement of CD14 for the peptide-induced IL-8 secretion was evaluated using CD14-specific antibodies. The introduction of CD14 antibodies to HEK-TLR4 cells significantly reduced IL-8 secretion triggered by the whole resistin molecule and aa23–42 and aa43–64 resistin-derived peptides (Fig. 7B). Functional properties of resistin-derived peptides in relation to the whole resistin molecule are summarized in Table 1. The peptides aa43–64 and aa51–108 efficiently and TLR-4 dependently induced IL-8 production, thus mimicking functional properties of the whole resistin molecule. Neutralization of IL-8 production in HEK-TLR4 cells using anti-resistin antibodies was achieved using aa43–64 and not aa51–108 peptide. These findings allow to suggest that the peptide sequence aa43–64 is similar in its properties to the whole resistin molecule, and that the interaction between TLR4 complex and resistin occurs at this site.
Fig 7.
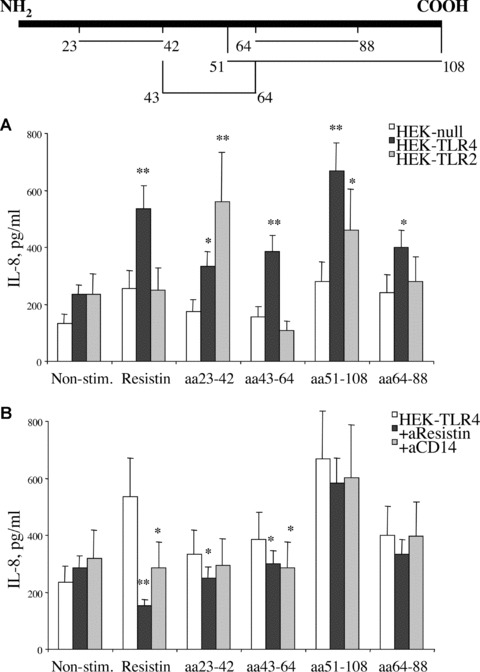
Structural requirements of resistin-TLR4 interaction. HEK293 cells transfected with TLR4, TLR2 or empty vector were stimulated with equimolar concentrations of resistin and resistin peptides, assigned as aa23–42, aa43–64, aa51–108, aa64–88 (50 nM) (A). Localization of the peptides in the whole resistin sequence is schematically shown above the graphs. Differences of IL-8 production in comparison to HEK-null cells are indicated by asterisk (*P < 0.05 and **P < 0.01). Inhibition of IL-8 production by antibodies against resistin and CD14 are evaluated in HEK-TLR4 cells (B). Differences of IL-8 production in HEK-TLR4 cells in the presence of antibodies are indicated by asterisk (*P < 0.05 and **P < 0.01).
Table 1.
Structural requirements for the pro-inflammatory effects (IL-8 secretion) of resistin on HEK293 cells transfected with TLR-4 or TLR-2
| HEK-TLR4 | HEK-TLR2 | |||
|---|---|---|---|---|
| + Anti-resistin ab (resistin dependent) | + Anti-CD14 ab (CD14 dependent) | Compared to HEKnull cells (TLR4 dependent) | Compared to HEKnull cells (TLR-2 dependent) | |
| Resistin, 50 nM | +++ | ++(+) | +++ | − |
| aa 23–42, 50 nM | ++ | + | ++ | +++ |
| aa 43–64, 50 nM | ++ | ++ | +++ | − |
| aa 51–108, 50 nM | + | + | +++ | ++ |
| aa 64–88, 50 nM | + | − | ++ | − |
HEK-TLR4, HEK-TLR2 and HEKnull cells were stimulated with equimolar concentrations of synthetic resistin peptides (50 nM), assigned as aa23–42, aa43–64, aa51–108, aa64–88, corresponding to 1000 ng/ml of the whole resistin molecule. The peptides were compared to the whole resistin molecule regarding their ability to induce IL-8 in relation to TLR4 expression, and neutralizing effects of anti-resistin antibodies. Semi-quantitative scale was used for evaluation of the results, where ‘+++’ corresponded to major effect.
Discussion
This report is the first to demonstrate a cell surface receptor mediating the pro-inflammatory effects of resistin. We show that resistin binds to human leucocytes, monocytic cell line THP1, and epithelial cells (HEK293) transfected with TLR4 inducing the production of cytokines through an interaction with TLR4. Several steps of evidence support the specificity of this interaction. The requirement of TLR4 for the effects induced by resistin was assessed with respect to the cell surface binding, activation of intracellular pathways, and expression of inflammatory cytokines. In all these situations, the effects of resistin were abolished or markedly abrogated by anti-TLR4 antibodies, and by resistin-specific antibodies. In contrast, neither TLR2 antibodies nor isotype-matched mouse IgG were able to inhibit the resistin effects. Additionally, wild-type HEK293 cells, not sensitive to stimulation with resistin, gained sensitivity to resistin following transfection with TLR4 expressing vector. Importantly, HEK293 cells transfected with MD2/CD14 and with TLR2 retained the insensitivity to resistin. Inhibition of the intracellular adaptor protein for TLRs, MyD88, significantly reduced resistin-mediated secretion of IL-8. Taken together these observations support that TLR4 is a receptor for and a mediator of resistin-dependent effects.
TLR4 is characterized by recognition of exogenous bacterial and viral structures and mediating protective inflammatory reactions of the host. TLR4 requires its coreceptor MD-2 for a specific recognition of LPS. CD14 is another essential accessory protein for LPS recognition. LPS is known as the main ligand for TLR4. Recent insights in structure of TLR4 presented a horse shoe-like shape architecture of the extracellular domain common for all TLRs. Recognition sites and binding of LPS to the TLR4-MD-2 complex are now identified [34]. LPS interacts with a hydrophobic pocket in MD-2 and forms a direct bridge to the convex area of the neighbouring TLR4-MD-2 complex resulting in dimerization of TLR4 and recruitment of intracellular adaptor protein MyD88 and IRAK activation [35]. The competitive stimulation of human leucocytes with resistin and LPS showed an inhibitory effect of resistin on LPS-induced production of pro-inflammatory cytokines. These findings suggest resistin to be an endogenous inhibitor of LPS interaction with TLR4 rather than being a co-factor of such an interaction. Several structural variants of TLRs-ligand interaction are described [35]. Analogously, resistin binding to the TLR4 may occur directly at the convex area preventing LPS interaction. Resistin may interact through the part of TLR4 convex occupied with MD-2 or from the MD-2 free side of the convex potentially facilitating dimerization of TLR4-MD-2 complex. The 3D structure proposed for murine resistin [9] reveals a symmetric covalent dimer and/or a trimer of dimer linked through N-terminal cystein residues. A dimer/hexamer of resistin would stretch between the two TLR4-MD-2 complexes (Fig. 8). In such a case, interaction with TLR4 would occur through the loops in the C-terminal part of resistin. Studies with synthetic peptides spanning the whole length of resistin molecule were employed in an attempt to identify amino acid sequence participating in TLR4 binding. The peptide sequence closely mimicking the properties of the whole molecule regarding TLR4-mediation of cytokine induction was located at aa43–64 suggesting that the interaction between TLR4 complex and resistin occurs at this site. Using 3D structure of the molecule [9] the aa43–64 peptide is placed at the globular domain of resistin thereby not participating in the formation of resistin multimers and is potentially available for interaction with TLR4 receptor. Additionally, this is the only of tested peptides possessing a loop structure formed by a cystein 51-cystein 63 bond and potentially exposes epitopes comparable to those exhibited in a properly folded protein. Our results provide experimental evidence suggesting resistin to be an additional, previously unrecognized TLR4 ligand of major importance in the inflammatory cascade in man. Physiological consequences of resistin interaction with TLR4 may explain the spectrum of resistin effects beyond inflammation. Recent reports demonstrated that the activation of TLR4 led to insulin resistance in adipocytes [6, 36] and loss of TLR4 function prevented diet-induced obesity and insulin resistance [37, 38].
Fig 8.
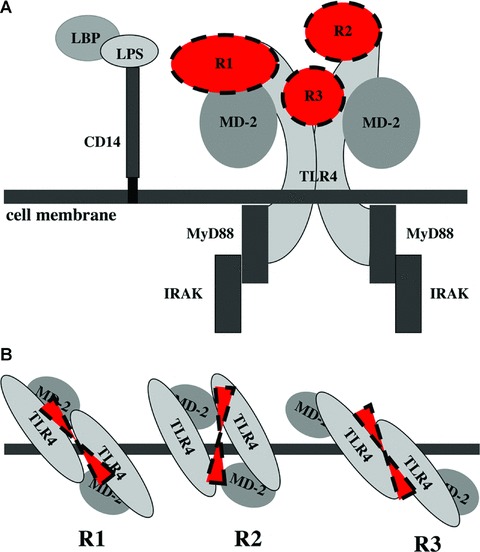
Overall structure of TLR4-resistin complex. Side view of the complex (A) on the cell surface TLR4 is associated with MD-2 and adopts the horseshoe-like shape forming an interface for LPS binding. CD14 delivers LPS molecules to the TLR4-MD-2 complex. Dimerization of TLR4-MD-2 complex is followed by recruitment of adaptor proteins MyD88 and IRAK initiating intracellular signalling. We show that interaction between resistin and TLR4 reduces LPS effects, potentially by interfering with its binding to TLR4-MD-2 complex. Top view (B) shows potential binding sites of resistin. R1, resistin occupies LPS binding site in the convex of TLR4 associated to MD-2; R2, resistin binds to the upper central part of the TLR4 convex facilitating dimerization and recruitment of adaptor proteins; R3, resistin stretches through the central part of TLR4 forming several interactions by identical C-terminal loops of its hexamer.
The employment of specific inhibitors of intracellular transduction pathways showed that intracellular signals evoked by resistin are mediated through NF-κB and MAP-kinase signalling mechanisms and were efficiently abrogated by specific inhibitors of these proteins and by the presence of TLR4 antibodies. The introduction of a PI3-kinase inhibitor to the cell culture prior to stimulation with resistin resulted in a significant up-regulation of the resistin effects including secretion of IL-6 and IL-8. These findings are in agreement with recent reports about PI3-kinase being a negative regulator of inflammatory effects triggered through TLR4 and TLR2 [32, 33]. PI3-kinase is known as a regulator of intracellular signalling originating from insulin and insulin-like receptors [38, 39]. Resistin has been shown to suppress PI3-kinase signalling by up-regulating the PI3-kinase inhibitor PTEN [40]. This may be one of explanations for a decreased sensitivity to insulin in the presence of resistin.
Acknowledgments
The work has been supported by Göteborg Medical Society, Swedish Association against Rheumatism, King Gustaf V Foundation, Swedish Medical Research Council, Professor Nanna Svartz’ Foundation, Rune and Ulla Amlövs Foundation, the Pharmacist Hedberg Foundation, Åke Wiberg Foundation, Börje Dahlin’s Foundation, National Inflammation Network, the Swedish Foundation for Strategic Research, the Swedish Governmental Agency for Innovation Systems and the University of Göteborg.
Supporting Information
Fig. S1 A dose-dependent suppression of resistin-inducedTNF-a production by inhibitors of NF-κB (A) and p38mitogen activated protein kinase (B) signalling.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting material supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–95. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- 2.Medzhitov R, Preston-Hurlburt P, Janeway CA. A human homologue of the Drosophila toll protein signals activation of adaptive immunity. Nature. 1997;388:394–7. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 3.Akashi-Takamura S, Miyake K. Toll-like receptors (TLRs) and immune disorders. J Infect Chemother. 2006;12:233–40. doi: 10.1007/s10156-006-0477-4. [DOI] [PubMed] [Google Scholar]
- 4.Rallabhandi P, Bell J, Boukhvalova MS, et al. Analysis of TLR4 polymorphic variants: new insights into TLR4/MD-2/CD14 stoichiometry, structure, and signaling. J Immunol. 2006;177:322–32. doi: 10.4049/jimmunol.177.1.322. [DOI] [PubMed] [Google Scholar]
- 5.Yu M, Wang H, Ding A, et al. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock. 2006;26:174–9. doi: 10.1097/01.shk.0000225404.51320.82. [DOI] [PubMed] [Google Scholar]
- 6.Shi H, Kokoeva MV, Inouye K, et al. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116:3015–25. doi: 10.1172/JCI28898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walton KA, Hsieh X, Gharavi N, et al. Receptors involved in the oxidized 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphorylcholine-mediated synthesis of interleukin-8. A role for toll-like receptor 4 and a glycosylphosphatidylinositol-anchored protein. J Biol Chem. 2003;278:29661–6. doi: 10.1074/jbc.M300738200. [DOI] [PubMed] [Google Scholar]
- 8.Steppan CM, Brown EJ, Wright CM, et al. A family of tissue-specific resistin-like molecules. Proc Natl Acad Sci USA. 2001;98:502–6. doi: 10.1073/pnas.98.2.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel SD, Rajala MW, Rossetti L, et al. Disulfide-dependent multimeric assembly of resistin family hormones. Science. 2004;304:1154–8. doi: 10.1126/science.1093466. [DOI] [PubMed] [Google Scholar]
- 10.Yang RZ, Huang Q, Xu A, et al. Comparative studies of resistin expression and phylogenomics in human and mouse. Biochem Biophys Res Commun. 2003;310:927–35. doi: 10.1016/j.bbrc.2003.09.093. [DOI] [PubMed] [Google Scholar]
- 11.Ghosh S, Singh AK, Aruna B, et al. The genomic organization of mouse resistin reveals major differences from the human resistin: functional implications. Gene. 2003;305:27–34. doi: 10.1016/s0378-1119(02)01213-1. [DOI] [PubMed] [Google Scholar]
- 12.Holcomb IN, Kabakoff RC, Chan B, et al. FIZZ1, a novel cysteine-rich secreted protein associated with pulmonary inflammation, defines a new gene family. EMBO J. 2000;19:4046–55. doi: 10.1093/emboj/19.15.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nagaev I, Smith U. Insulin resistance and type 2 diabetes are not related to resistin expression in human fat cells or skeletal muscle. Biochem Biophys Res Commun. 2001;285:561–4. doi: 10.1006/bbrc.2001.5173. [DOI] [PubMed] [Google Scholar]
- 14.Patel L, Buckels AC, Kinghorn IJ, et al. Resistin is expressed in human macrophages and directly regulated by PPAR gamma activators. Biochem Biophys Res Commun. 2003;300:472–6. doi: 10.1016/s0006-291x(02)02841-3. [DOI] [PubMed] [Google Scholar]
- 15.Thommesen L, Stunes AK, Monjo M, et al. Expression and regulation of resistin in osteoblasts and osteoclasts indicate a role in bone metabolism. J Cell Biochem. 2006;99:824–34. doi: 10.1002/jcb.20915. [DOI] [PubMed] [Google Scholar]
- 16.Yura S, Sagawa N, Itoh H, et al. Resistin is expressed in the human placenta. J Clin Endocrinol Metab. 2003;88:1394–7. doi: 10.1210/jc.2002-011926. [DOI] [PubMed] [Google Scholar]
- 17.Yagmur E, Trautwein C, Gressner AM, et al. Resistin serum levels are associated with insulin resistance, disease severity, clinical complications, and prognosis in patients with chronic liver diseases. Am J Gastroenterol. 2006;101:1244–52. doi: 10.1111/j.1572-0241.2006.00543.x. [DOI] [PubMed] [Google Scholar]
- 18.Bertolani C, Sancho-Bru P, Failli P, et al. Resistin as an intrahepatic cytokine: overexpression during chronic injury and induction of proinflammatory actions in hepatic stellate cells. Am J Pathol. 2006;169:2042–53. doi: 10.2353/ajpath.2006.060081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jung HS, Park KH, Cho YM, et al. Resistin is secreted from macrophages in atheromas and promotes atherosclerosis. Cardiovasc Res. 2006;69:76–85. doi: 10.1016/j.cardiores.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 20.Bokarewa M, Nagaev I, Dahlberg L, et al. Resistin, an adipokine with potent proinflammatory properties. J Immunol. 2005;174:5789–95. doi: 10.4049/jimmunol.174.9.5789. [DOI] [PubMed] [Google Scholar]
- 21.Senolt L, Housa D, Vernerová Z, et al. Resistin in rheumatoid arthritis synovial tissue, synovial fluid and serum. Ann Rheum Dis. 2007;66:458–63. doi: 10.1136/ard.2006.054734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nusken KD, Kratzsch J, Wienholz V, et al. Circulating resistin concentrations in children depend on renal function. Nephrol Dial Transplant. 2006;21:107–12. doi: 10.1093/ndt/gfi084. [DOI] [PubMed] [Google Scholar]
- 23.Axelsson J, Bergsten A, Qureshi AR, et al. Elevated resistin levels in chronic kidney disease are associated with decreased glomerular filtration rate and inflammation, but not with insulin resistance. Kidney Int. 2006;69:596–604. doi: 10.1038/sj.ki.5000089. [DOI] [PubMed] [Google Scholar]
- 24.Efstathiou SP, Tsiakou AG, Tsioulos DI, et al. Prognostic significance of plasma resistin levels in patients with atherothrombotic ischemic stroke. Clin Chim Acta. 2007;378:78–85. doi: 10.1016/j.cca.2006.10.023. [DOI] [PubMed] [Google Scholar]
- 25.Pilz S, Weihrauch G, Seelhorst U, et al. Implications of resistin plasma levels in subjects undergoing coronary angiography. Clin Endocrinol. 2007;66:380–6. doi: 10.1111/j.1365-2265.2007.02743.x. [DOI] [PubMed] [Google Scholar]
- 26.Burnett MS, Lee CW, Kinnaird TD, et al. The potential role of resistin in atherogenesis. Atherosclerosis. 2005;182:241–8. doi: 10.1016/j.atherosclerosis.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 27.Verma S, Li SH, Wang CH, et al. Resistin promotes endothelial cell activation: further evidence of adipokine-endothelial interaction. Circulation. 2003;108:736–40. doi: 10.1161/01.CIR.0000084503.91330.49. [DOI] [PubMed] [Google Scholar]
- 28.Kougias P, Chai H, Lin Ph, et al. Effects of adipocyte-derived cytokines on endothelial functions: implication of vascular disease. J Surg Res. 2005;126:121–9. doi: 10.1016/j.jss.2004.12.023. [DOI] [PubMed] [Google Scholar]
- 29.Calabro P, Samudio I, Willerson JT, et al. Resistin promotes smooth muscle cell proliferation through activation of extracellular signal-regulated kinase 1/2 and phosphatidylinositol 3-kinase pathways. Circulation. 2004;110:3335–40. doi: 10.1161/01.CIR.0000147825.97879.E7. [DOI] [PubMed] [Google Scholar]
- 30.Di Simone N, Di Nicuolo F, Sanguinetti M, et al. Resistin regulates human choriocarcinoma cell invasive behaviour and endothelial cell angiogenic processes. J Endocrinol. 2006;189:691–9. doi: 10.1677/joe.1.06610. [DOI] [PubMed] [Google Scholar]
- 31.Helle M, Boeije L, De Groot E, et al. Sensitive ELISA for interleukin-6. Detection of IL-6 in biological fluids: synovial fluids and sera. J Immunol Methods. 1991;138:47–56. doi: 10.1016/0022-1759(91)90063-l. [DOI] [PubMed] [Google Scholar]
- 32.Fukao T, Koyasu S. PI3K and negative regulation of TLR signaling. Trends Immunol. 2003;24:358–63. doi: 10.1016/s1471-4906(03)00139-x. [DOI] [PubMed] [Google Scholar]
- 33.Williams DL, Ozment-Skelton T, Li C. Modulation of the phosphoinositide 3-kinase signaling pathway alters host response to sepsis, inflammation, and ischemia/reperfusion injury. Shock. 2006;25:432–9. doi: 10.1097/01.shk.0000209542.76305.55. [DOI] [PubMed] [Google Scholar]
- 34.Park BS, Song DH, Kim HM, et al. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–5. doi: 10.1038/nature07830. [DOI] [PubMed] [Google Scholar]
- 35.Jin MS, Lee JO. Structures of the toll-like receptor family and its ligand complexes. Immunity. 2008;29:182–91. doi: 10.1016/j.immuni.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 36.Song MJ, Kim KH, Yoon JM, et al. Activation of toll-like receptor 4 is associated with insulin resistance in adipocytes. Biochem Biophys Res Commun. 2006;346:739–45. doi: 10.1016/j.bbrc.2006.05.170. [DOI] [PubMed] [Google Scholar]
- 37.Tsukumo DM, Carvalho-Filho MA, Carvalheira JB, et al. Loss-of-function mutation in TLR4 prevents diet-induced obesity and insulin resistance. Diabetes. 2007;56:1986–98. doi: 10.2337/db06-1595. [DOI] [PubMed] [Google Scholar]
- 38.Poggi M, Bastelica D, Gual P, et al. C3H/HeJ mice carrying a toll-like receptor 4 mutation are protected against the development of insulin resistance in white adipose tissue in response to a high-fat diet. Diabetologia. 2007;50:1267–76. doi: 10.1007/s00125-007-0654-8. [DOI] [PubMed] [Google Scholar]
- 39.Taniguchi CM, Tran TT, Kondo T, et al. Phosphoinositide 3-kinase regulatory subunit p85alpha suppresses insulin action via positive regulation of PTEN. Proc Natl Acad Sci USA. 2006;103:12093–7. doi: 10.1073/pnas.0604628103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shen YH, Zhang L, Gan Y, et al. Up-regulation of PTEN (phosphatase and tensin homolog deleted on chromosome ten) mediates p38 MAPK stress signal-induced inhibition of insulin signaling. A cross-talk between stress signaling and insulin signaling in resistin-treated human endothelial cells. J Biol Chem. 2006;281:7727–36. doi: 10.1074/jbc.M511105200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 A dose-dependent suppression of resistin-inducedTNF-a production by inhibitors of NF-κB (A) and p38mitogen activated protein kinase (B) signalling.