

Abstract
Recent studies suggest that mesenchymal stem cells (MSCs) possess a greater differentiation potential than once thought and that they have the capacity to regenerate damaged tissues/organs. However, the evidence is insufficient, and the mechanism governing the recruitment and homing of MSCs to these injured sites is not well understood. We first examined the MSCs circulating in peripheral blood and then performed chemotaxis, wound healing and tubule-formation assays to investigate the migration capability of mouse bone marrow MSCs (mBM-MSCs) in response to liver-injury signals. In addition, BM-MSCs from donor enhanced green fluorescent protein transgenic male mice were transplanted into liver-injured co-isogenic female recipients, either by intra-bone marrow injection or through the caudal vein, to allow in vivo tracking analysis of the cell fate after transplantation. Donor-derived cells were analysed by in vivo imaging analysis, PCR, flow cytometry and frozen sections. Microarray and real-time PCR were used for chemokine/cytokine and receptor analyses. We successfully isolated circulating MSCs in peripheral blood of liver-injured mice and provided direct evidence that mBM-MSCs could be mobilized into the circulation and recruited into the liver after stimulation of liver injury. CCR9, CXCR4 and c-MET were essential for directing cellular migration towards the injured liver. The recruited mBM-MSCs may play different roles, including hepatic fate specification and down-regulation of the activity of hepatic stellate cells which inhibits over-accumulation of collagen and development of liver fibrosis. Our results provide new insights into liver repair involving endogenous BM-MSCs and add new information for consideration when developing clinical protocols involving the MSCs.
Keywords: mesenchymal stem cells, recruitment, homing, liver, cytokine receptor
Introduction
Mesenchymal stem cells (MSCs) are multipotent non-haematopoietic cells capable of differentiating into mesoderm cell lineages. There is a general consensus that bone marrow MSCs (BM-MSCs) provide a suitable haematopoietic microenvironment and that they can exert both positive and negative regulatory effects on the self-renewal, proliferation and differentiation of haematopoietic stem/progenitor cells. It is well established that MSCs serve as precursors of bone, cartilage and adipocytes [1–3]. Emerging new findings now suggest that MSCs are able to give rise to a broad range of cells in vitro, including hepatocytes, neurons, epithelial cells and keratinocytes [4–8]. This in vitro plasticity has attracted much attention and more studies are now focused on whether MSCs possess the same potential for contributing to different tissue cell-types in vivo, under either metabolical or pathological conditions.
So far, several in vivo studies involving a variety of animal models have shown beneficial effects of MSC-based therapy on tissue structural repair, including that of bone, myocardial tissue, skin, kidney and liver [9–19]. It has also been suggested that MSCs could be widely delivered in minimally injured syngeneic mice, where they acquired multiple tissue specific morphology and antigen expression [20]. These accumulating clues now suggest that endogenous MSCs may ‘naturally’ be involved in wound healing and tissue regeneration [11]. This has encouraged further studies on in vivo MSC biology and the mechanisms underlying their mobilization or migration.
A number of investigators, including ourselves, have suggested that induction of stress in specific tissues may result in the release of various cytokines. These cytokines then facilitate the mobilization of MSCs into the peripheral blood and their homing to sites of wound healing. Furthermore, engrafted MSCs at wound sites are able to trans-differentiate into multiple component cell types, thus directly contributing to wound healing [5, 13, 16, 21, 22]. This process, as the part of the inflammatory response and tissue repair, requires signalling mediated primarily by chemokines. This is followed by the presence of a mobile pool of MSCs that are mobilized to the location of tissue injury, with subsequent homing and engraftment at the injury site. To provide direct evidence in support of this hypothesis, a liver-injury mouse model was employed in our present study. We demonstrated that both injured-liver culture medium and liver-injured serum dramatically facilitated the trans-differentiation of BM-MSCs into functional hepatocyte-like cells in vitro. This suggested that injured livers are able to secrete cytokines that direct hepatic fate specification in mouse BM-MSCs (mBM-MSCs) [4, 23].
In the present study, we characterized peripheral blood MSCs (PB-MSCs) in the circulating blood by expansion in culture, and we showed the migration capability of these cells by in vitro chemotaxis, wound healing and tubule formation assays. The mBM-MSCs from donor enhanced green fluorescent protein (EGFP)-transgenic mice were also transplanted into liver-injured co-isogenic recipients, either by intra-bone marrow (IBM) injection or through caudal vein inoculation, for in vivo tracking analysis of the cell fate after transplantation. The donor-derived cells were examined by in vivo bio-imaging, PCR, flow cytometry and frozen section analyses; while the homing-related chemokines/cytokines and receptors were analysed by microarray and real-time PCR. Under liver-injured conditions, mBM-MSCs could migrate into peripheral blood and home towards the injured liver by chemoattraction mediated by stromal cell-derived factor (SDF)-1, chemokine (C-C motif) ligand (CCL)25, hepatocyte growth factors (HGF) and their receptors chemokine (C-C motif) receptor (CCR)9, chemokine (C-X-C motif) receptor (CXCR)4 and mesenchymal-epithelial transition factor (c-MET). The recruited mBM-MSCs may play different roles in injured liver, including hepatic fate specification and down-regulation of the activity of hepatic stellate cells (HSCs). We hope this finding may contribute to the better understanding of the interaction between stem cells and the environment that leads to homing and tissue integration. This knowledge will not only be beneficial for the improvement of existing cell-based therapeutic approaches, but also will allow interpretation of the possible involvement of MSCs in multiple tissue/organ development and regeneration.
Materials and methods
Experimental animals
Six- to 10-week-old female C57BL/6 and EGFP-transgenic male mice (C57BL/6 background) were obtained from Zhejiang Academy of Medical Sciences (Hangzhou, People’s Republic of China). Eight-week-old C57BL/6 female mice were subjected to acute liver injury by i.p. injection of CCL4 (20 μl/g body weight, 10% solution mixed in mineral oil) twice over a 48-hr period [24]. Experiments analysing hepatic injury were performed before collection of serum (liver-injury mouse serum, LIMS). Animals were housed under specified pathogen-free conditions. All animal experiments were performed in accordance with all legal regulations, which included approval by a local ethical committee.
Isolation and culture of MSCs from bone marrow and peripheral blood
Mouse MSCs were prepared as described previously [4, 13]. Briefly, bone marrow-derived cells were collected by flushing the femurs and tibias from C57BL/6 and EGFP-transgenic mice. These cells were cultured in Iscove’s modified Dulbecco’s medium (IMDM; Sigma, St. Louis, MO, USA) supplemented with 10% foetal bovine serum (FBS; Hyclone, Rockville, MD, USA) and 1% penicillin/streptomycin. After 3 days, non-adherent cells and debris were removed and the adherent cells were continuously cultured. To obtain MSC clones, cells at near confluence were harvested and re-plated into 96-well plates by limited dilution. Individual clones were then picked out and expanded. Cells were used at third to seventh passage.
The PB-MSCs were isolated from adult mice peripheral blood mononuclear cells (PBMCs) by primary culture. Peripheral blood samples were collected by tail bleeding into heparinized tubes. PBMCs were isolated by diluting the heparinized blood 1:2 in Hanks balanced salt solution followed by Ficoll–Hypaque centrifugation (1.092 g/ml). After three washes in Hanks, the isolated PBMCs were adjusted to a concentration of 5 × 106 cells/ml. These cells were cultured in IMDM supplemented and purified as above.
Staining of peripheral blood derived colony-forming unit-fibroblastic (CFU-Fs)
After 14 days’ culture of the PBMCs, the medium was removed and the cells were stained with 0.5% crystal violet in methanol for 5 min. The dishes were washed twice with distilled water and cells were directly visualized using a Zeiss Axiovert 400 microscope (Carl Zeiss, Inc., Jena, Germany).
Flow cytometry
Cells for flow cytometry analyses were incubated with anti-CD11b, CD44, CD45, CD73, CD90, CD105, SCA-1, STRO-1, c-MET (Abcam, Cambridge, UK) and secondary R-PE or FITC-conjugated Abs (MultiScience, Hangzhou, China). They were then analysed by a fluorescence activated cell sorter (FACS, BD Biosciences, San Jose, CA, USA).
Differentiation culture of MSCs for mesenchymal lineage
MSCs were grown to 90% confluence and then specific supplements for mesenchymal lineage differentiation were added. Osteogenic differentiation was induced by basic medium with 10 mM β-glycerophosphate (Sigma), 50 mg/ml ascorbic acid (Sigma) and 10−9 M dexamethasone (Sigma). Adipogenic differentiation was induced by basic medium containing 5 mg/ml insulin (Sigma) and 10−9 M dexamethasone. Chondrogenic differentiation was achieved by basic medium containing 50 μM ascorbic acid, 0.1 μM dexamethasone and 10 ng/ml TGF-β (R&D Systems, Minneapolis, MN, USA). Each specific differentiation medium was changed every 3 days. Confirmation of differentiation of the cells to osteocytes, adipocytes and chondrocytes were performed by staining with alizarin red (for calcium deposits), oil red O (for lipids) and toluidine blue (for proteoglycans), respectively.
Boyden chamber assay
Migration was assayed using inserts with 8-μm-pore membranes (Millpore, Billerica, MA, USA) as described previously [25]. Serum-starved (12 hrs) MSCs were added to the upper chamber at 4 × 105 cells/ml. In the bottom chamber, LIMS or HGF (R&D Systems) was used as a chemoattractant. Serum-free IMDM or IMDM containing healthy mouse serum (HMS) and 10% FBS were added to the bottom chamber as negative and positive controls, respectively. After incubation at 37°C overnight, cells on the upper side of the membrane were wiped off, and the migrated cells were visualized by crystal violet staining. For neutralization studies, cells were incubated with antimouse CCR1, CCR2, CCR9, CXCR4 and/or c-MET Abs (20 μg/ml, Abcam). A total of 1% LIMS in the bottom chamber was used as a chemoattractant. Antimouse IgG (20 μg/ml, Abcam) was used as a negative control. Migration was quantified by counting the cells that passed through the filter. Stained cells from a minimum of five fields of view (200×) for three replicates were counted and the data were expressed as the average number of migrated cells.
Assays for wound healing and in vitro tubule formation
Assays for wound healing and in vitro tubule formation were performed as described previously [25]. Briefly, MSCs were plated in 12-well plates to 70% confluence and serum starved. A ‘wound’ was made by scoring the confluent monolayer of MSCs with a pipette tip across the plate well. Scored wells were washed twice with PBS to remove cell debris followed by the addition of IMDM containing LIMS or HGF. Serum-free IMDM or IMDM containing HMS and 10% FBS served as negative and positive controls, respectively. Invasion or growth into the scored area was observed by phase-contrast microscopy. For in vitro tubule formation assay, culture plates were coated with undiluted Matrigel solution and allowed to solidify overnight in a humidified 37°C CO2 incubator. MSCs were serum starved overnight and then plated at 50,000 cells per test well. IMDM medium containing LIMS or growth factors was added and further incubated overnight. Tubule formation was monitored by Zeiss Axiovert 400 phase-contrast and fluorescence microscopy (Carl Zeiss, Inc.).
Intra-bone marrow injection and detection of donor-derived cells
Male EGFP-MSCs were delivered by IBM injection into each female C57BL/6 mouse as described previously [26]. Briefly, the region from the inguen to the knee joint was shaved of hair and a 5 mm incision was made on the thigh. The knee was flexed to 90° and the proximal side of the tibia was drawn to the anterior. A 26-gauge needle was inserted into the joint surface of the tibia through the patellar tendon and then inserted into the bone marrow cavity. Donor EGFP-MSCs (2 × 106 cells /30 μl) were injected into the bone marrow cavity through the bone hole using a 50 ml microsyringe. A volume of CCL4 (10 μl/g body weight, 10% solution mixed in mineral oil) was subsequently injected twice a week to maintain persistent liver damage. Individual mice were then killed on selected days, and peripheral blood and liver tissues were collected. Genomic DNA was isolated from each tissue in lysis buffer containing 1% SDS and 0.2 mg/ml Proteinase K (Sigma) followed by phenol-chloroform extraction. The genomic DNA was used for detection of EGFP and the Y chromosome sequence (primers are shown in supplement 1). The PCR products were fractionated by 1.2% agarose gel electrophoresis and visualized under UV illumination after staining with ethidium bromide. PBMCs were separated from 1 ml heparinized peripheral blood by gradient centrifugation. Single cell suspensions were analysed for EGFP signals by flow cytometry. A minimum of 1,000,000 cells per sample was analysed. The liver tissues were fixed in 10% neutral buffered formaldehyde, cryoprotected in 30% sucrose overnight and embedded in OCT compound. Sections (5 μm thick) were screened for the presence of EGFP by direct visualization using a fluorescence microscope.
Caudal vein injection and detection of donor-derived cells
EGFP+ mBM-MSCs from liver-injured EGFP-transgenic donors were transplanted into liver-injured recipients through the caudal vein (1 × 106 cells/0.1 ml/mouse) for in vivo tracking analysis of their fate after transplantation. The mice were killed 2 weeks after transplantation. In vivo bio-imaging was conducted under a Nightowl imaging system, using WinLight software (Berthold Technologies). The liver tissues were then fixed, sectioned and visualized as described above.
Immunofluorescence staining
Liver frozen sections were permeabilized with cold methanol and incubated with primary antibodies, including rabbit anti-AFP (α-fetoprotein), rat anti-CK19, -CK18, -CD146 and goat anti-ALB (albumin) (Biodesign, Saco, ME, USA). The secondary antibodies, including TRITC-conjugated rabbit anti-goat IgG, goat anti-rabbit IgG and goat anti-rat IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA), were used according to the manufacturer’s instructions. Samples were photomicrographed under a confocal laser-scanning microscope (LSM 510; Carl Zeiss, Inc.).
Expression analysis of chemokine receptors
Total RNA was isolated from MSCs induced by LIMS and from un-induced control cells. RT-PCR was initially performed for the analysis of chemokine receptor transcripts. The PCR primers are shown in Table 1. The expression levels of the genes of interest were further quantified by real-time PCR using the Mastercycler ep realplex (Eppendorf, Germany) and detection system software. The total amount of mRNA was normalized to endogenous β-actin mRNA.
Table 1.
Primers used for PCR
| Genes | Sequences (forward) | Sequences (reverse) |
|---|---|---|
| Ccr1 | GACCAGCATCTACCTGTTCA | GCAGAAACAAATACACTCAG |
| Ccr 2 | GTTACCTCAGTTCATCCA | CAAGGCTCACCATCATCGTAGTC |
| Ccr 3 | TTGCAGGACTGGCAGCATT | TTCATTCTTAGAGCATGGAAACGTT |
| Ccr 4 | TCATGACTTCCGTGACGCTTT | GTTTTCTTCCTCAGAGCCCTGTT |
| Ccr 5 | GACATCGATTATGGTATG | GAAGAGCAGGTCAGAGATGGC |
| Ccr 6 | TCGTCCAGGCAACCAAATCTTTCC | TGTTGTATGCGTTTATTGGCCAGA |
| Ccr 7 | CACGCTGAGATGCTCACTGG | CCATCTGGGCCACTTGGA |
| Ccr 8 | TGACCGACTACTACCCTGATTTCTT | GCTGCCCCTGAGGAGGAA |
| Ccr 9 | GTCAGCTGTCTTGATCCTGAAG | CATAGAGAACTGGGTTCAGACAA |
| Ccr 10 | AGAGCTCTGTTACAAGGCTGATGTC | CAGGTGGTACTTCCTAGATTCCAGC |
| Cxcr1 | CTGCTATGAAGTCCTGGGTG | TCAATCAAGTGGGCTCCTAA |
| Cxcr 2 | GTGCCGCTGCTCATCATG | AAGGACGACAGCGAAGATGAC |
| Cxcr 3 | TCCCAACCACAAGTGCCAAAG | AGAAAGGCAAAGTCCGAGGC |
| Cxcr 4 | GGCTGTAGAGCGAGTGTTGC | GTAGAGGTTGACAGTGTAGAT |
| Cxcr 5 | AAACGAAGCGGAAACTAGAGCC | GCCCAGCTTGGTCAGAAGCC |
| Cxcr 6 | TACGATGGGCACTACGAGGGAG | GCAAAGAAACCAACAGGGAGAC |
| Xcr1 | ATACCTGTCTGTAGTGAGC | AAAGCACTGGGTTGAAACA |
| Cx3cr1 | TGTCCACCTCCTTCCCTGAA | TCGCCCAAATAACAGGCC |
| Ccl25 | GTGAAGAGGGCGATGAGAAT | TTCAGCAGACTCCTCCCATC |
| Cxcl12 | GACCCGAAATTAAAGTGGAT | GAACACCCAGTTCCTATTCAG |
| Hgf | GTGCCAACAGGTGTATCAG | TGTCACAGACTTCGTAGCG |
| Egfp | CAACAGCCACAACGTCTATATCATG | GAAGTTCTAGGCGGTGTTGTA |
| Ymt2/b | CTGGAGCTCTACAGTGATGA | CAGTTACCAATCAACACATCAC |
| α-Sma | GGGAGTAATGGTTGGAATGG | GGCAGTAGTCACGAAGGAATAG |
| Col(I)α1-f | AACTTTGCTTCCCAGATGTCCT | TCGGTGTCCCTTCATTCCAG |
| β-Actin | TTCCTTCTTGGGTATGGAAT | GAGCAATGATCTTGATCTTC |
Microarray analysis
Global gene expression analysis was performed to identify which genes were responsible for hepatic commitment. Total RNA and complementary DNA were prepared from CCl4-treated and untreated mouse livers. An Illumina MouseWG-6 v2.0 BeadChip was used to generate expression profiles of more than 48,000 transcripts with 500 ng of labelled cDNA for each sample, following manufacturer’s recommended protocols. A randomized design was used to minimize chip effects. Four individuals were replicated in the two batches. Expression intensity measures were obtained from an average of 30 beads for each transcript. The BeadChips were imaged with an Illumina BeadArray Reader. The raw intensities were extracted with the Gene Expression Module in Illumina’s BeadStudio software. Expression intensities were log2 transformed and median-centred by subtracting the mean value of each array from each intensity value.
Statistical analysis
Statistical analysis was performed with SPSS version 16.0, and data were expressed as means ± S.D. Differences between the values were determined by 2-tailed paired t-tests.
Results
Identification of migrated MSCs in the peripheral blood of liver-injured mice
To evaluate whether MSCs could chemotactically migrate from their residential niche into peripheral blood circulation under liver-injured conditions, MSCs in the peripheral blood were carefully examined. The PB-MSCs were isolated from peripheral blood of adult liver-injured mice by primary culture as discrete plastic-adherent colonies consisting of fibroblast-like cells (CFU-Fs), which were similar to the CFU-Fs formed in the BM-MSCs culture. In 4 of the 21 cases, one to five fibroblastic colonies per 107 PBMCs were identified after 10 to 30 days of culture (Fig. 1A-II). The adherent cells grew in a disarrayed fashion and were distinguished by their fibroblastic morphology. After these cultures became confluent, the cells were split into three plates. The ‘stemness’ was defined by their ability to differentiate into osteoblasts, adipocytes and chondrocytes, as well as by the surface marker phenotype of CD11b−CD44+CD45−CD73+CD90+CD105+SCA-1+ STRO-1+. As shown in Fig. 1B, results indicated that these peripheral blood-derived cells were typical MSC and share common characteristics with BM-MSCs. However, these in vitro expanded PB-MSCs grew for up to four passages before senescence in all cases. We were unable to isolate PB CFU-Fs from healthy mice (>30 cases, Fig. 1A-I).
Fig 1.
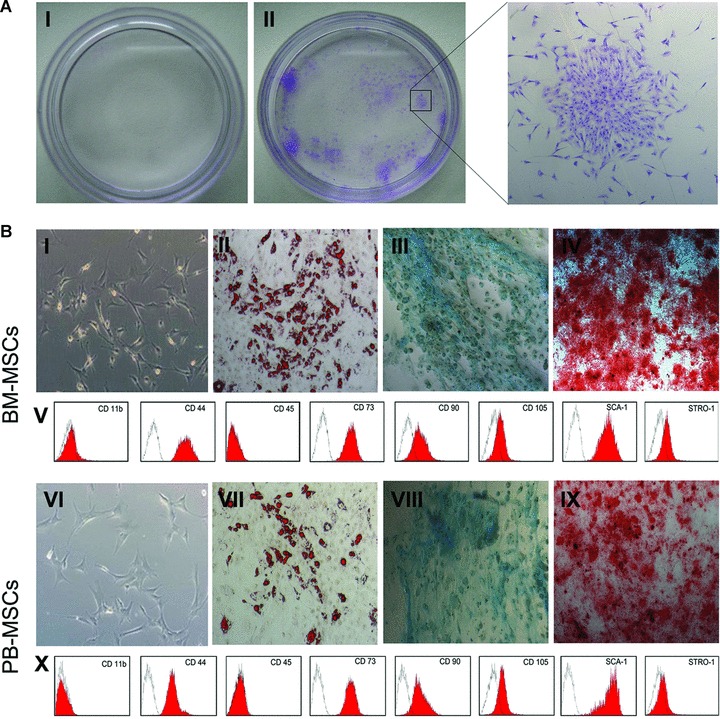
Identification and comparative characterization of MSCs in peripheral blood and bone marrow. (A) CFU-Fs were successfully isolated from peripheral blood of liver-injured mice, although they were extremely low in frequency (II, 4 out of the 21 cases). In comparison, in healthy mice CFU-Fs were rarely found (I). (B) Cultured PB-MSCs are adherent, clonogenic and fibroblast-like but more stretched out in shape, pale in colour, and do not have two pointed ends (VI). Like BM-MSCs (I), PB-MSCs also contain heterogeneous cell populations. Adipogenic differentiation of MSCs: MSCs were cultured for 14 days alternately in adipogenic induction medium. Lipid vacuoles were visualized by oil red O staining (II, VII). Chondrogenic differentiation of MSCs: culture of mMSCs after 28 days in chondrogenic induction medium. Proteoglycans were stained with Toluidine blue (III, VIII). Osteogenic differentiation of MSCs: cells were cultured for 21 days with osteogenic induction medium, and calcium deposits were visualized by alizarin red staining (IV, IX). Cell surface markers of MSCs were assessed using FACS. Similar to BM-MSCs (V), PB-MSCs expressed CD44, CD73, CD90, CD105, SCA-1 and STRO-1, but not CD11b or CD45 (X).
In vitro migration assays
Further evidence that the migration of mBM-MSCs could be triggered by the liver injury signals was obtained by an in vitro migration assay using a Boyden Chamber. After incubation, there was a dramatic increase in migration of mBM-MSCs incubated with medium containing 25 ng/ml HGF, (or) 1% HMS, (or) 1% LIMS or 10% FBS (Fig. 2A). By contrast, few mBM-MSCs were detectable in serum/cytokine-free medium. MSCs migrated towards serum derived from liver-injured mice to a greater extent than towards serum from healthy mice, suggesting that chemokines/cytokines were more plentiful in LIMS. The ‘wound healing’ assay showed similar results to the Boyden Chamber migration assay. After 20 hrs culture with 25 ng/ml HGF or 1% HMS, mBM-MSCs were able to ‘heal’ part of the wound area (Fig. 2B). Cells in 1% LIMS showed higher efficiency in this ‘healing’ process. Positive controls were obtained using mBM-MSCs in 10% FBS, and these cells completely sealed the originally scored area.
Fig 2.
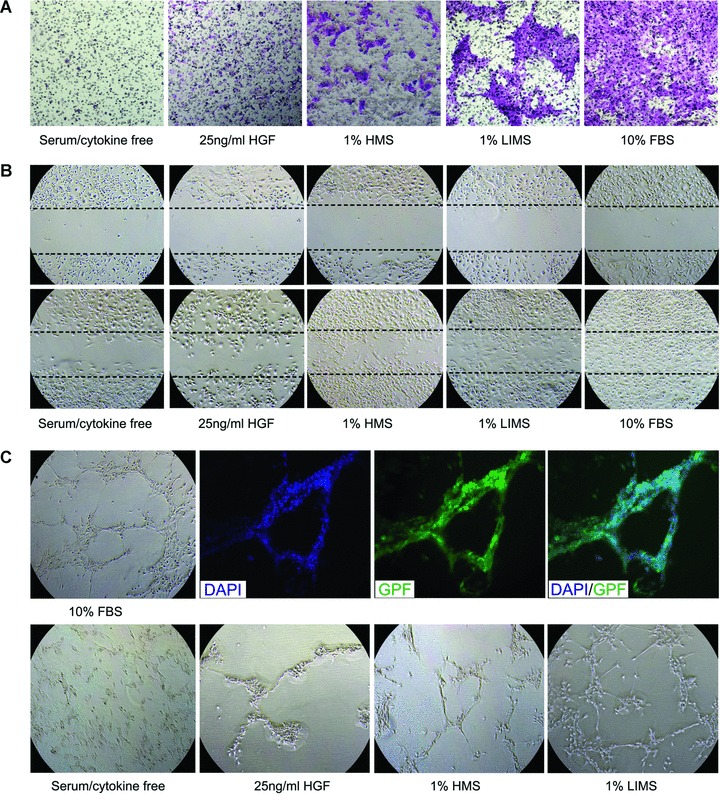
In vitro Cellular migration assays. (A) mBM-MSCs were exposed to HGF, HMS and LIMS in a Boyden microchemotaxis chamber to identify potential chemoattractants. Cells that had migrated towards the source of chemoattractant (lower chamber) by passing through a membrane filter (8 μm) were visualized by crystal violet staining (original magnification, 40×). (B) For ‘wound healing’ assays, confluent cell monolayers of mBM-MSCs were wounded by scratching with a pipette tip. After 20 hrs of stimulation with HGF, HMS, LIMS and FBS in culture medium, wound closure was documented photographically. These figures show one representative result out of five (40×). (C) For in vitro capillary-like tube formation assays, EGFP+ BM-MSCs were plated on reduced growth factor Matrigel-coated plates as indicated. Nuclei were visualized by staining with DAPI. Phase contrast (40×) and fluorescence microscopy (100×) was performed after 20 hrs of incubation. Representative images of tubule formation from six independently experiments are shown. The mBM-MSCs had the most capillary-like tubule formation in 1% LIMS-containing medium, followed by 1% HMS and 25 ng/ml HGF. No tube formation was observed in the serum/cytokine-free culture conditions.
The ability of mBM-MSCs to form tubules is demonstrated in the fluorescence images presented in Fig. 2C. Tubule formation by mBM-MSCs was more effective in 10% FBS-containing medium, followed closely by 1% LIMS, 1% MS, then 25 ng/ml HGF. Serum-free culture conditions resulted in the slowest tube formation.
In vivo migration assay and detection of donor-derived cells
The migration of mBM-MSCs out of bone marrow to the injured liver was evident by the detection of donor-derived signals in bone marrow, peripheral blood and livers of all transplanted mice killed at 72 and 120 hrs after IBM injection (Fig. 3A and B). In separate experiments, livers were isolated and sectioned at days 3, 7 and 30 after IBM injection (Fig. 4). Analysis of liver sections obtained 3 days after IBM injection showed that a small number of EGFP+ cells were localized in close proximity to the blood vessels. EGFP+ cell clusters were readily detected 30 days after IBM injection. No EGFP+ cells were detected in livers of mice that had not been pre-treated with CCL4 before IBM injection, suggesting that injured liver may secrete chemokines or cytokines that recruit BM-MSCs.
Fig 3.
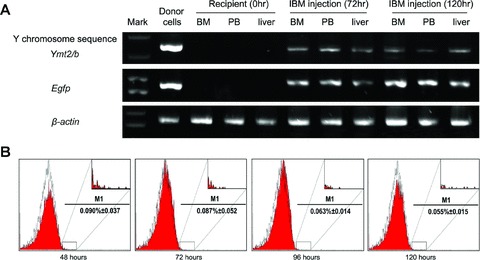
Transplantation of mBM-MSCs and detection of donor-derived cells. EGFP+ mBM-MSCs from male donors were IBM injected into liver-injured female mice. (A) Detection of donor-derived cells in different organs of recipients by PCR. (B) Representative flow cytometric analysis of EGFP signals in PBMCs at different time-points after IBM injection. The inserts denoted the enlargements of positive area. A minimum of 1,000,000 cells per sample were analysed.
Fig 4.
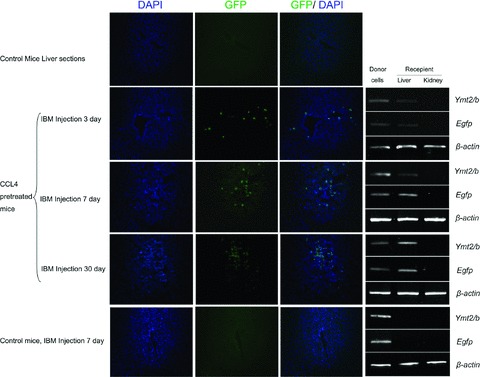
Efficient homing of mBM-MSCs to injured livers. EGFP+ mBM-MSCs from male donors were IBM injected into liver-injured female mice. Frozen sections of livers at different time-points after IBM injection were scanned by Zeiss confocal scanning laser microscopy. Analysis of liver sections obtained 3 days after IBM showed that a small number of EGFP+ cells were localized in the proximity of the blood vessels. A significantly higher number of EGFP+ cells were detected on day 7. Distribution of EGFP+ cell clusters was readily detected 30 days after IBM injection. Y chromosome and EGFP signals in livers were also determined by PCR, kidneys were used as negative controls. EGFP+ cells were not detected in livers of healthy mice (100×).
EGFP-expressing mBM-MSCs were transplanted into liver-injured mice via caudal vein, which allowed in vivo tracking analysis of the cell fate after transplantation. As shown in Fig. 5A, the EGFP signals were detected only in livers but not in hearts or kidneys from the same individuals with acute liver injury, and these results indicated that BM-MSCs homed primarily to the injured tissues. Stronger EGFP signals were detected at the ligation position of a partial hepatectomy, which suggested that more engrafted MSCs had migrated to the wound site and may be involved in the cicatrization. Immunofluorescence staining results showed that some of the engrafted MSCs expressed epithelial markers, including AFP, CK19 and CD146, and the marker of mature hepatocyte CK18, ALB (Fig. 5B). This further suggested that partially recruited BM-MSCs may undergo epithelial or hepatic fate specification when a suitable microenvironment is available. Evaluation of the expressions of smooth muscle α-actin (α-Sma) and collagen I α1 in the recipient livers was also carried out. It has been reported that hepatic injury can induce HSCs to undergo an activation process to form a more myofibroblast-like cell type with expression of α-SMA and an increase in the production of ECM proteins, including type I collagen [27, 28]. As shown in Fig. 6, the expressions of α-Sma and collagen I α1 were significantly decreased in the livers of mice that received MSC transplantation, suggesting that the MSCs may be involved in the down-regulation of the activation of HSCs and thus contribute to the reduction of the over-accumulation of collagen and liver fibrosis.
Fig 5.
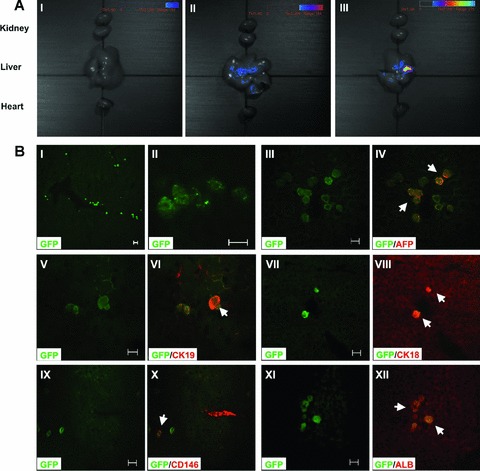
In vivo analysis of EGFP+ cells in recipient livers 14 days after transplantation. EGFP+ mBM-MSCs were transplanted into liver-injured mice through the caudal vein (1 × 106 cells/0.1 ml/mouse). (A) Visible light image superimposed on the optical CCD image. A pseudocolour luminescent image from blue (least intense) to red (most intense), represents the spatial distribution of the detected photons emitted from EGFP+ cell within the liver. An EGFP signal was not detected in the healthy recipients (I). In the experimental models of acute liver injury, including the CCL4-induced acute liver injury group (II) and the partial hepatectomy group (III), an EGFP signal was observed in livers, but not in hearts or kidneys. (B) MSCs differentiated into multiple liver cell types at the wound site. (I, II) EGFP+ MSCs were engrafted in the livers and differentiated into multiple components of the liver. GFP positive cells (green) were colocalized with (III, IV) AFP (red), (V, VI) CK19 (red), (VII, VIII) CK18 (red), (IX, X) CD146 (red) and (XI, XII) ALB (red), respectively. White arrows indicate the positive cells. The results shown are representative of at least five separate experiments. Scale bars represent 20 μm.
Fig 6.
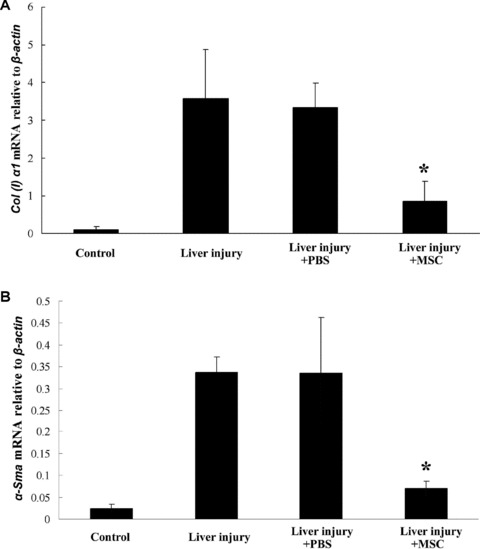
Real-time PCR analysis of α-Sma and collagen I α1 gene expression in mice livers. The expressions of α-Sma and collagen I α1 were significantly decreased in the livers of mice that received MSC transplantation, suggesting that the MSCs regulated the activity of the HSC, and thus contributed to the reduction of collagen accumulation and liver fibrosis. The total amount of mRNA was normalized to endogenous β-Actin mRNA. *:P < 0.05, compared with the data in liver-injured mice without transplantation.
Identification of homing-related chemokine/cytokine receptors
The distribution of EGFP+ cells in recipient mice livers indicated the presence of a specific homing mechanism that was guiding transplanted MSCs from bone marrow to injured livers. We therefore investigated the potential homing-related receptors in mBM-MSCs. As chemokine/cytokine receptors are well-known factors that control multiple cell-trafficking and localization in tissue compartments, we suggested that chemokine/cytokine receptors might also play roles in MSC activation, migration, engraftment and differentiation. RT-PCR, flow cytometry and microarray analysis showed that cultured mBM-MSCs widely expressed Ccr1, Ccr2, Ccr7, Ccr9, Cxcr1, Cxcr3, Cxcr4 and Cxcr6 to different degrees (Fig. 7A and B). In addition, Fgfr1IIIc, Fgfr2IIIc and c-Met were also detectable in cultured mBM-MSCs (Supplement 1).
Fig 7.
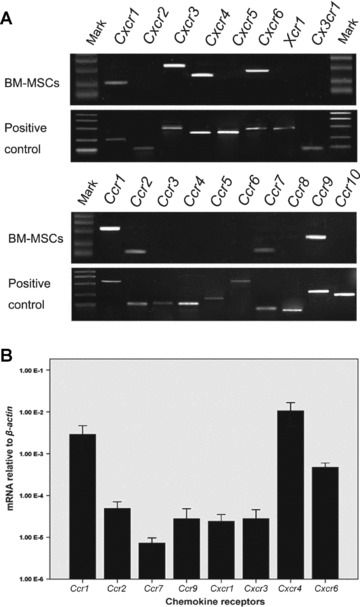
The expression of chemokine receptors in mBM-MSCs. (A) RT-PCR analysis of chemokine receptor mRNA in mBM-MSCs. The mRNA mixture of mouse PBMCs and splenocytes were used as positive controls. mBM-MSCs expressed the transcripts for Ccr1, Ccr2, Ccr7, Ccr9, Cxcr1, Cxcr3, Cxcr4 and Cxcr6. Transcripts for other chemokine receptors were not detected. (B) Real-time PCR was used to analyse mRNA expression of detectable chemokine receptors.
To identify the exact receptors involved in cellular recruitment in response to the liver injury signals, expression of the receptors examined above were further analysed after exposure of the mBM-MSCs to LIMS. Expression of the entire range of receptors examined was up-regulated at the mRNA level to some extent (Fig, 8A and B). Maximum expression of a 2.3-fold increase for CCR9, a 3.1-fold increase for CXCR4, and a 4.8-fold increase for c-MET (P < 0.05) was reached. Similar results were also seen for receptor protein levels analysed by flow cytometry. Short-term (24 hrs) incubation in medium containing LIMS resulted in an increase of surface CCR9 (from 3.29 ± 2.51% to 8.78 ± 1.55%), CXCR4 (from 30.09 ± 2.11% to 45.68 ± 4.34%) and c-MET (from 6.08 ± 1.26% to 24.80 ± 3.35%) proteins, which accompanied increases in mRNA expression (Fig. 8C).
Fig 8.
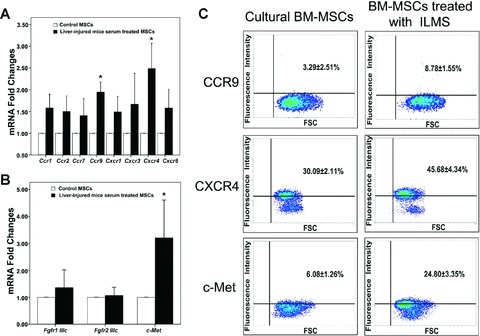
Effect of liver-injured mouse serum on culture of mBM-MSCs. Gene expression profiles of chemokine receptors (A), Fdf receptors and c-Met (B) in MSCs before/after exposure to LIMS for 24 hrs were determined by real-time PCR analyses and compared. *:P < 0.05, compared with the data in control group. (C) Flow cytometry analysis of CCR9, CXCR4 and c-MET by cultured mBM-MSCs. Short-term (24 hrs) incubation in medium containing LIMS resulted in an increase in surface CCR9, CXCR4 and c-MET proteins. These figures show one representative result out of five.
Microarray analysis for homing-related chemokines/cytokines
Microarray analysis showed that numerous chemokine/cytokine transcripts were up-regulated after CCL4-induce acute liver injury. Chemokines related to the receptors that were expressed on mBM-MSCs are listed in Table 2. Coinciding with the results from chemokine receptor analysis, Ccl25 (binding with CCR9) and Sdf-1 (Cxcl12, binding with CXCR4) were significantly up-regulated (2.0- and 1.3-fold, respectively). Other chemokines, including Ccl2, Ccl3, Ccl4, Ccl7, Ccl8, Cxcl11 and Cxcl16, were also significantly up-regulated (1.7∼61.9-fold). However, their corresponding receptors on BM-MSCs did not substantially change after LIMS treatment. Because previous reports have shown that chemokines and their receptors largely contribute to the initiation of inflammation, we supposed that these up-regulated chemokines may have been involved in the homing of leukocytes towards the wound sites, but only slightly contribute to the migration of MSCs. The expression of Hgf, a cytokine that is well known to augment cell migration, scattering and proliferation of many different cell types through HGF/c-MET axis [29, 30], was also significantly up-regulated (2.4-fold). These results indicated that Ccl25, Sdf-1 and Hgf may play essential roles in the recruitment of mBM-MSCs towards injured liver.
Table 2.
GeneChip analysis of changes in chemokine gene expression in CCL4-treated mouse liver
 |
To provide further evidence, the expression levels of these three chemokines/cytokines in injured livers were carefully examined by real-time PCR at different time-points. As shown in Fig. 9A, injured livers increasingly expressed Sdf-1, Ccl25 and Hgf mRNAs in a time-dependent manner. Similar results were also seen at the protein level, as SDF-1, CCL25 and HGF were measurably elevated in the peripheral blood after CCL4 injection or partial hepatectomy (Fig. 9B).
Fig 9.
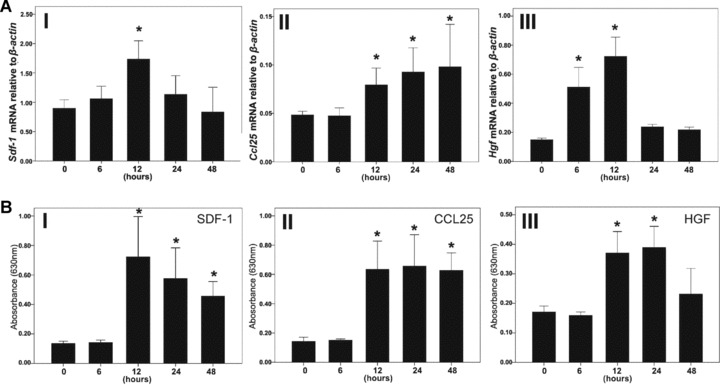
Expression profiles of CCL25, SDF-1 and HGF in injured mouse livers. (A) Ccl25, Sdf-1 and Hgf mRNA in livers at different times after injury was quantified by real-time PCR using the Mastercycler ep realplex (Eppendorf) and Real-Time Detection System software. The total amount of mRNA was normalized to endogenous β-Actin mRNA. (B) The serum levels of CCL25, SDF-1 and HGF protein were measured by an ELISA method. *: P < 0.05, compared with the data of healthy mice (0 hr).
Migration inhibition by blockage of chemokine/cytokine receptors
Migration inhibition assays were performed to confirm whether the chemokine/cytokine receptors were involved in the migration of BM-MSCs. We investigated whether pre-treatment of mBM-MSCs with anti-CCR1, anti-CCR2, anti-CCR9, anti-CXCR4 or anti-c-MET antibodies could abrogate cellular migration in response to LIMS. Quantitative assessments of the number of migrated mBM-MSCs showed only a slight change in anti-CCR1 (21.6 ± 4.6 cells/field) and anti-CCR2 (25.8 ± 5.2 cells/field) groups compared to the control group (24.1 ± 6.7 cells/field). However, much stronger decreases were seen with anti-CCR9 (20.7 ± 2.3 cells/field), anti-c-MET (17.5 ± 1.9 cells/field) and anti-CXCR4 (15.5 ± 1.7 cells/field). On the other hand, treatment of mBM-MSCs with a cocktail of anti-CCR9, anti-CXCR4 and anti-c-MET reduced the cellular migration by 80%, which was statistically significant (P < 0.01) (Fig. 10). Thus, blocking of CCR9, CXCR4 and c-MET strongly inhibited the migration of mBM-MSCs to LIMS, suggesting that CCR9, CXCR4 and c-MET expressed on mBM-MSCs might be instrumental for liver-targeted migration and homing of these cells.
Fig 10.
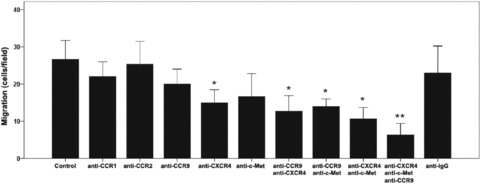
Pre-incubation of BM-MSCs with antibodies against chemokine/cytokine receptors inhibited LIMS-induced target migration. For blockade studies, cells were pre-incubated with antimouse CCR1, CCR2, CCR9, CXCR4 and c-MET polyclonal antibodies. mBM-MSCs were then seeded in the top compartment of a Boyden chamber, and 1% LIMS in the bottom chamber was used as a chemoattractant. After 12 hrs incubation at 37°C, cells on the upper side of the membrane were wiped off, and the migrated cells were visualized by crystal violet staining. Migration was quantified by counting the cells that had passed through the filter. Stained cells from a minimum of five fields of view (200×) for three replicates were counted and the data were expressed as the average number of migrated cells. Antimouse IgG was used as negative control antibody. *: P < 0.05, **: P < 0.01 compared with the data in negative control group.
Discussion
MSCs were initially identified from bone marrow, and later studies indicated the MSC compartment is more widely distributed than was previously thought. Other organs and tissues, including brain, spleen, liver, kidney, lung, muscle, thymus, lymph node, adipose tissue, muscle and exfoliated deciduous teeth, were also shown to be sources of MSCs [31]. However, at the current stage of investigation, bone marrow still is the richest and most reliable reservoir for MSCs. So far, information is still very limited regarding the MSCs in the peripheral blood, as these cells cannot be easily isolated. Compared with BM CFU-Fs, the yield of PB CFU-Fs is extremely poor and it is quite common that none is detected. Moreover, maintaining these cell cultures also seems to be difficult [32, 33]. The origin and physio-pathological function of PB-MSCs are still unclear, and their presence in the adult peripheral blood might relate to some interesting subjects in the field of adult stem cell biology. Recently, research has also indicated that the numbers of PB-MSCs are low per se but may change in response to certain pathological conditions [32, 34] (e.g. the colony-forming efficiency of PB CFU-Fs seems to increase in mice with phenylhydrazine-induced haemolytic anaemia [35] and in the growth factor-mobilized peripheral blood of breast cancer patients [36]).
In this study, we successfully isolated PB CFU-Fs in both CCL4-induced acute liver injury mice and partial hepatectomy mice although in extremely low frequency. By contrast, very few CFU-Fs were found in peripheral blood of healthy mice. We also performed flow cytometric analysis to evaluate the percentage of CD45−SCA-1+ and CD45−STRO-1+ cells in PBMCs from both CCL4-induced acute liver injury mice and partial hepatectomy mice, and the percentages were measurably elevated compared with those of healthy mice (data not shown). The isolated PB-MSCs shared most of the characteristics of mBM-MSCs, including surface marker phenotypes and multi-lineage mesenchymal differentiation potentials. The PB-MSCs cultured were adherent, clonogenic and fibroblast-like but had a more stretched-out shape, were paler in colour, and did not have two sharp ends. These findings led us to investigate whether BM-MSCs or MSCs in other tissue niches naturally migrate into the circulation, and what the fate of these migrated MSCs might be. The in vitro chemotaxis, wound healing, tubule formation assays, as well as in vivo tracking analysis with EGFP-labelling BM-MSCs supported our hypothesis that injured liver may release various cytokines and recruit BM-MSCs to the injured sites. This is one possible interpretation of the involvement of MSCs in tissue regeneration.
The stem cell migration property, termed homing, is thought to be a coordinated multi-step process that involves intracellular-surface antigens and cell adhesion molecules. Chemokines, which are categorized into four groups, CC chemokines, CXC chemokines, C chemokines and CX3C chemokines, have the ability to induce directed chemotaxis in nearby responsive cells. These proteins exert their biological effects by interacting with G protein-linked transmembrane receptors called chemokine receptors, which are selectively found on the target cell surface. The major role of chemokines is to guide the migration of cells. Cells that are attracted by chemokines follow a signal of increasing chemokine concentration towards the source of the chemokine.
Several studies have reported that human MSCs constitutively express various chemokine receptors, but the results have not been consistent [37–39]. In this study, we have characterized that mBM-MSCs expressed the transcripts for Ccr1, Ccr2, Ccr7, Ccr9, Cxcr1, Cxcr3, Cxcr4 and Cxcr6, as well as a number of other receptors that have been shown to contribute to cellular migration and homing, including Fgfr1IIIc, Fgfr2IIIc and c-Met. Among these receptors, CCR9, CXCR4 and c-MET were significantly up-regulated by treatment with liver-injured mouse serum, while cellular migration was notably inhibited by blockage of these cytokine receptors. Correspondingly, the related ligands, CCL25, SDF-1 and HGF, were up-regulated under the liver-injured condition. Cytokine/cytokine receptor interactions may therefore contribute to the recruitment of mBM-MSCs to injured livers. SDF-1, CCL25 and HGF would serve, at least, as important chemotactic factors for the migration of mBM-MSCs.
To determine the liver specificity of recruitment, experimental models with acute liver injury or acute kidney injury and acute myocardial injury were used in this study. In liver injury mice, the donor-derived signals were only detected in livers but not in other tissues such as hearts or kidneys. By contrast, in kidney or myocardial injured mice, the donor-derived signals were detected in the injured kidneys or hearts as previously reported [12, 14], but not in livers (data not shown). These results suggested that MSCs may be specifically recruited to different tissues in emergencies, but that the chemokines/cytokines responsible for this process may be distinct in different tissues.
Accumulating evidence has shown that in vitro expanded BM-MSCs can achieve engraftment in various normal and damaged tissues. As well, they home to the bone marrow after systemic infusion [31]. However, the fate of injected MSCs requires elucidation. Based on our studies and other accumulating data, we speculate that the ‘PB-MSCs’ mentioned in previous studies represent systemic migration of MSCs derived from bone marrow or other MSC niches rather than the existence of common stem/progenitor cells. However, the PB-MSCs have a lower growth capacity and senesce easily. We suppose that ‘PB-MSCs’ may have begun to directionally differentiate during their migration from the bone marrow cavity towards injured liver, but the hepatic markers such as AFP and ALB were not detected either in these expanded PB-MSCs or in PBMCs from the liver-injured mice (Supplement 2). Additional studies are needed to further characterize ‘PB-MSCs’ and their relationship to BM-MSCs. Moreover, based on the existing evidence and our results, we constructed a model in which BM-MSCs in vivo would spontaneously leave the marrow cavity and enter the bloodstream, or migrate in response to systemic signals towards tumours or other tissues in need of repair (Fig. 11) [10, 13, 40, 41].
Fig 11.
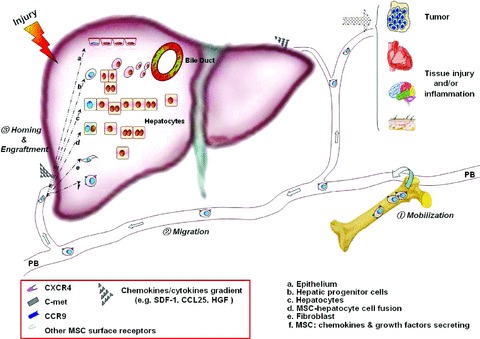
Model showing recruitment of endogenous MSCs to damaged tissues. Stress in the liver results in the release of various chemokines/cytokines including SDF-1, CCL25 and HGF. These increase MSC mobility through the cell surface receptors (e.g. CXCR4, CCR9 and c-MET), thereby, facilitating MSC mobilization into the peripheral blood and homing to sites of wound healing. Engrafted MSCs at wound sites are then able to trans-differentiate into multiple component cell types, including hepatocytes, epithelium and fibroblasts. MSCs may also secrete cytokines to stimulate the recovery of the hepatic cells, regulate the activity of HSCs, and induce the proliferation of the endogenous liver progenitor/stem cells, hence contributing to liver regeneration and wound healing.
Researchers have reported beneficial effects of MSC-based therapy on liver structural and functional repair [14, 17, 42]. However, the percentage of hepatocytes that trans-differentiated from donor-derived MSCs is at best very low [43]. We assume that engrafted MSCs at wound sites are not only able to trans-differentiate into multiple component cell types but also secrete cytokines to stimulate the recovery of the hepatic cells, regulate the activity of HSCs, and induce the proliferation of the endogenous liver progenitor/stem cells, hence contributing to liver regeneration and wound healing.
In conclusion, our study demonstrated that there is mobility of the multi-potent MSCs between bloodstream and organs. A number of receptors (including CCR9, CXCR4 and c-MET) are important for mediating the specific migration of these cells to the injured liver. Further study about the mode by which the BM-MSCs enter blood circulation and how they engraft in livers and participate in wound healing is still needed. Attracting endogenous MSCs to a defect site for wound healing and/or tissue regeneration would be an easier, safer and more efficient therapy than traditional cell therapy. Strategies to mobilize an internalized receptor and to increase its functional expression may be useful for improving engraftment of MSCs to injured tissues. We hope the findings presented here will provide new insight into the in vivo biology of MSCs and add new information to be considered when developing clinical protocols involving the MSC compartment.
Acknowledgments
This work was supported by the grant of medicine and health key project of Zhejiang Province, Science and Technology Foundation of Ministry Of Health of the People’s Republic of China (WKJ2007–2-037), and Shaoxing key project for Science and Technology (2007A23008, 2005141).
Supporting Information
Fig. S1 Expression of FGFRs and c-Met, which have been shown to contribute to cellular migration and homing, was detected by RT-PCR. Total mRNA from mice brain and lung were used as positive controls.
{kind=link}
Fig. S2 PCR analysis of AFP, ALB gene expression incultured MSCs and freshly isolated mononuclear cells. No AFP andALB expression was detected in the examined cells, total mRNA frommouse liver was used as positive control. Line 1: In vitroexpanded BM-MSCs; Line 2: In vitro expanded PB-MSCs; Line 3: bone marrow derived mononuclear cells of liver-injured mice; Line 4: peripheral blood derived mononuclear cells of liver-injured mice.
{kind=link}
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Bianco P, Robey PG, Simmons PJ. Mesenchymal stem cells: revisiting history, concepts, and assays. Cell Stem Cell. 2008;2:313–9. doi: 10.1016/j.stem.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jiang Y, Jahagirdar BN, Reinhardt RL, et al. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature. 2002;418:41–9. doi: 10.1038/nature00870. [DOI] [PubMed] [Google Scholar]
- 3.Bianco P, Riminucci M, Gronthos S, et al. Bone marrow stromal stem cells: nature, biology, and potential applications. Stem Cells. 2001;19:180–92. doi: 10.1634/stemcells.19-3-180. [DOI] [PubMed] [Google Scholar]
- 4.Chen Y, Dong XJ, Zhang GR, et al. In vitro differentiation of mouse bone marrow stromal stem cells into hepatocytes induced by conditioned culture medium of hepatocytes. J Cell Biochem. 2007;102:52–63. doi: 10.1002/jcb.21275. [DOI] [PubMed] [Google Scholar]
- 5.Chen Y, Shao JZ, Xiang LX, et al. Mesenchymal stem cells: a promising candidate in regenerative medicine. Int J Biochem Cell Biol. 2008;40:815–20. doi: 10.1016/j.biocel.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 6.Phinney DG, Prockop DJ. Concise review: mesenchymal stem/multipotent stromal cells: the state of transdifferentiation and modes of tissue repair–current views. Stem Cells. 2007;25:2896–902. doi: 10.1634/stemcells.2007-0637. [DOI] [PubMed] [Google Scholar]
- 7.Black IB, Woodbury D. Adult rat and human bone marrow stromal stem cells differentiate into neurons. Blood Cells Mol Dis. 2001;27:632–6. doi: 10.1006/bcmd.2001.0423. [DOI] [PubMed] [Google Scholar]
- 8.Hermann A, Gastl R, Liebau S, et al. Efficient generation of neural stem cell-like cells from adult human bone marrow stromal cells. J Cell Sci. 2004;117:4411–22. doi: 10.1242/jcs.01307. [DOI] [PubMed] [Google Scholar]
- 9.Cheng Z, Ou L, Zhou X, et al. Targeted migration of mesenchymal stem cells modified with CXCR4 gene to infarcted myocardium improves cardiac performance. Mol Ther. 2008;16:571–9. doi: 10.1038/sj.mt.6300374. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Deng Y, Zhou GQ. SDF-1alpha/ CXCR4-mediated migration of systemically transplanted bone marrow stromal cells towards ischemic brain lesion in a rat model. Brain Res. 2008;1195:104–12. doi: 10.1016/j.brainres.2007.11.068. [DOI] [PubMed] [Google Scholar]
- 11.Zhao J, Zhang N, Prestwich GD, et al. Recruitment of Endogenous Stem Cells for Tissue Repair. Macromol Biosci. 2008;8:836–42. doi: 10.1002/mabi.200700334. [DOI] [PubMed] [Google Scholar]
- 12.Zhang D, Fan GC, Zhou X, et al. Over-expression of CXCR4 on mesenchymal stem cells augments myoangiogenesis in the infarcted myocardium. J Mol Cell Cardiol. 2008;44:281–92. doi: 10.1016/j.yjmcc.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sasaki M, Abe R, Fujita Y, et al. Mesenchymal stem cells are recruited into wounded skin and contribute to wound repair by transdifferentiation into multiple skin cell type. J Immunol. 2008;180:2581–7. doi: 10.4049/jimmunol.180.4.2581. [DOI] [PubMed] [Google Scholar]
- 14.Morigi M, Introna M, Imberti B, et al. Human bone marrow mesenchymal stem cells accelerate recovery of acute renal injury and prolong survival in mice. Stem Cells. 2008;26:2075–82. doi: 10.1634/stemcells.2007-0795. [DOI] [PubMed] [Google Scholar]
- 15.Wu Y, Chen L, Scott PG, et al. Mesenchymal stem cells enhance wound healing through differentiation and angiogenesis. Stem Cells. 2007;25:2648–59. doi: 10.1634/stemcells.2007-0226. [DOI] [PubMed] [Google Scholar]
- 16.Sackstein R, Merzaban JS, Cain DW, et al. Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat Med. 2008;14:181–7. doi: 10.1038/nm1703. [DOI] [PubMed] [Google Scholar]
- 17.Oyagi S, Hirose M, Kojima M, et al. Therapeutic effect of transplanting HGF-treated bone marrow mesenchymal cells into CCl4-injured rats. J Hepatol. 2006;44:742–8. doi: 10.1016/j.jhep.2005.10.026. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Johnsen HE, Mortensen S, et al. Changes in circulating mesenchymal stem cells, stem cell homing factor, and vascular growth factors in patients with acute ST elevation myocardial infarction treated with primary percutaneous coronary intervention. Heart. 2006;92:768–74. doi: 10.1136/hrt.2005.069799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jin HK, Carter JE, Huntley GW, et al. Intracerebral transplantation of mesenchymal stem cells into acid sphingomyelinase-deficient mice delays the onset of neurological abnormalities and extends their life span. J Clin Invest. 2002;109:1183–91. doi: 10.1172/JCI14862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Anjos-Afonso F, Siapati EK, Bonnet D. In vivo contribution of murine mesenchymal stem cells into multiple cell-types under minimal damage conditions. J Cell Sci. 2004;117:5655–64. doi: 10.1242/jcs.01488. [DOI] [PubMed] [Google Scholar]
- 21.Wojakowski W, Kucia M, Kazmierski M, et al. Circulating progenitor cells in stable coronary heart disease and acute coronary syndromes: relevant reparatory mechanism? Heart. 2008;94:27–33. doi: 10.1136/hrt.2006.103358. [DOI] [PubMed] [Google Scholar]
- 22.Swenson S, Guest I, Ilic Z, et al. Hepatocyte nuclear factor-1 as marker of epithelial phenotype reveals marrow-derived hepatocytes, but not duct cells, after liver injury in mice. Stem Cells. 2008;26:1768–77. doi: 10.1634/stemcells.2008-0148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen Y, Dong XJ, Zhang GR, et al. Transdifferentiation of mouse BM cells into hepatocyte-like cells. Cytotherapy. 2006;8:381–9. doi: 10.1080/14653240600735800. [DOI] [PubMed] [Google Scholar]
- 24.Constandinou C, Henderson N, Iredale JP. Fibrosis research: methods and protocols. In: Varga J, Brenner DA, Phan SH, editors. Methods in molecular medicine. California: Baker & Taylor Books; 2005. pp. 237–50. [Google Scholar]
- 25.Zwezdaryk KJ, Coffelt SB, Figueroa YG, et al. Erythropoietin, a hypoxia-regulated factor, elicits a pro-angiogenic program in human mesenchymal stem cells. Exp Hematol. 2007;35:640–52. doi: 10.1016/j.exphem.2007.01.044. [DOI] [PubMed] [Google Scholar]
- 26.Kushida T, Inaba M, Hisha H, et al. Intra-bone marrow injection of allogeneic bone marrow cells: a powerful new strategy for treatment of intractable autoimmune diseases in MRL/lpr mice. Blood. 2001;97:3292–9. doi: 10.1182/blood.v97.10.3292. [DOI] [PubMed] [Google Scholar]
- 27.Yang L, Jung Y, Omenetti A, et al. Fate-mapping evidence that hepatic stellate cells are epithelial progenitors in adult mouse livers. Stem Cells. 2008;26:2104–13. doi: 10.1634/stemcells.2008-0115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schnabl B, Kweon YO, Frederick JP, et al. The role of Smad3 in mediating mouse hepatic stellate cell activation. Hepatology. 2001;34:89–100. doi: 10.1053/jhep.2001.25349. [DOI] [PubMed] [Google Scholar]
- 29.Forte G, Minieri M, Cossa P, et al. Hepatocyte growth factor effects on mesenchymal stem cells: proliferation, migration, and differentiation. Stem Cells. 2006;24:23–33. doi: 10.1634/stemcells.2004-0176. [DOI] [PubMed] [Google Scholar]
- 30.Neuss S, Becher E, Woltje M, et al. Functional expression of HGF and HGF receptor/c-met in adult human mesenchymal stem cells suggests a role in cell mobilization, tissue repair, and wound healing. Stem Cells. 2004;22:405–14. doi: 10.1634/stemcells.22-3-405. [DOI] [PubMed] [Google Scholar]
- 31.da Silva Meirelles L, Chagastelles PC, Nardi NB. Mesenchymal stem cells reside in virtually all post-natal organs and tissues. J Cell Sci. 2006;119:2204–13. doi: 10.1242/jcs.02932. [DOI] [PubMed] [Google Scholar]
- 32.He Q, Wan C, Li G. Concise review: multipotent mesenchymal stromal cells in blood. Stem Cells. 2007;25:69–77. doi: 10.1634/stemcells.2006-0335. [DOI] [PubMed] [Google Scholar]
- 33.Roufosse CA, Direkze NC, Otto WR, et al. Circulating mesenchymal stem cells. Int J Biochem Cell Biol. 2004;36:585–97. doi: 10.1016/j.biocel.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 34.Kassis I, Zangi L, Rivkin R, et al. Isolation of mesenchymal stem cells from G-CSF-mobilized human peripheral blood using fibrin microbeads. Bone Marrow Transplant. 2006;37:967–76. doi: 10.1038/sj.bmt.1705358. [DOI] [PubMed] [Google Scholar]
- 35.Piersma AH, Ploemacher RE, Brockbank KG, et al. Migration of fibroblastoid stromal cells in murine blood. Cell Tissue Kinet. 1985;18:589–95. doi: 10.1111/j.1365-2184.1985.tb00702.x. [DOI] [PubMed] [Google Scholar]
- 36.Fernandez M, Simon V, Herrera G, et al. Detection of stromal cells in peripheral blood progenitor cell collections from breast cancer patients. Bone Marrow Transplant. 1997;20:265–71. doi: 10.1038/sj.bmt.1700890. [DOI] [PubMed] [Google Scholar]
- 37.Croitoru-Lamoury J, Lamoury FM, Zaunders JJ, et al. Human mesenchymal stem cells constitutively express chemokines and chemokine receptors that can be upregulated by cytokines, IFN-beta, and Copaxone. J Interferon Cytokine Res. 2007;27:53–64. doi: 10.1089/jir.2006.0037. [DOI] [PubMed] [Google Scholar]
- 38.Honczarenko M, Le Y, Swierkowski M, et al. Human bone marrow stromal cells express a distinct set of biologically functional chemokine receptors. Stem Cells. 2006;24:1030–41. doi: 10.1634/stemcells.2005-0319. [DOI] [PubMed] [Google Scholar]
- 39.Sordi V, Malosio ML, Marchesi F, et al. Bone marrow mesenchymal stem cells express a restricted set of functionally active chemokine receptors capable of promoting migration to pancreatic islets. Blood. 2005;106:419–27. doi: 10.1182/blood-2004-09-3507. [DOI] [PubMed] [Google Scholar]
- 40.Spaeth E, Klopp A, Dembinski J, et al. Inflammation and tumor microenvironments: defining the migratory itinerary of mesenchymal stem cells. Gene Ther. 2008;15:730–8. doi: 10.1038/gt.2008.39. [DOI] [PubMed] [Google Scholar]
- 41.Belema-Bedada F, Uchida S, Martire A, et al. Efficient homing of multipotent adult mesenchymal stem cells depends on FROUNT-mediated clustering of CCR2. Cell Stem Cell. 2008;2:566–75. doi: 10.1016/j.stem.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 42.Higashiyama R, Inagaki Y, Hong YY, et al. Bone marrow-derived cells express matrix metalloproteinases and contribute to regression of liver fibrosis in mice. Hepatology. 2007;45:213–22. doi: 10.1002/hep.21477. [DOI] [PubMed] [Google Scholar]
- 43.Kallis YN, Alison MR, Forbes SJ. Bone marrow stem cells and liver disease. Gut. 2007;56:716–24. doi: 10.1136/gut.2006.098442. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Expression of FGFRs and c-Met, which have been shown to contribute to cellular migration and homing, was detected by RT-PCR. Total mRNA from mice brain and lung were used as positive controls.
Fig. S2 PCR analysis of AFP, ALB gene expression incultured MSCs and freshly isolated mononuclear cells. No AFP andALB expression was detected in the examined cells, total mRNA frommouse liver was used as positive control. Line 1: In vitroexpanded BM-MSCs; Line 2: In vitro expanded PB-MSCs; Line 3: bone marrow derived mononuclear cells of liver-injured mice; Line 4: peripheral blood derived mononuclear cells of liver-injured mice.