

Abstract
Deficiency in the signal adaptor protein sequestosome 1 (SQSTM1/A170/p62) in mice is associated with mature-onset obesity, accompanied by insulin and leptin resistance. We previously established that redox sensitive transcription factor Nrf2 up-regulates SQSTM1 expression in response to atherogenic stimuli or laminar shear stress in vascular cells, and here examine the role of SQSTM1 in neointimal hyperplasia and vascular remodelling in vivo following carotid artery ligation. Neointimal hyperplasia was markedly enhanced at ligation sites after 3 weeks in SQSTM1–/– compared with wild-type (WT) mice. The intimal area and stenotic ratio were, respectively, 2.1- and 1.7-fold higher in SQSTM1−/– mice, indicating enhanced proliferation of vascular smooth muscle cells (SMCs). When aortic SMCs were isolated from WT and SQSTM1−/– mice and cultured in vitro, we found that SQSTM1–/– SMCs proliferated more rapidly in response to foetal calf serum (FCS) and attained 2–3-fold higher cell densities compared to WT SMCs. Moreover, migration of SQSTM1–/– SMCs was enhanced compared to WT SMCs. Early and late phases of p38MAPK activation in response to FCS stimulation were also more enhanced in SQSTM1–/– SMCs, and inhibitors of p38 and ERK1/2 signalling pathways significantly attenuated SMC proliferation. In summary, SQSTM1–/– mice exhibit enhanced neointimal hyperplasia and vascular remodelling following arterial ligation in vivo. The enhanced proliferation of SQSTM1–/– aortic SMCs in vitro highlights a novel role for SQSTM1 in suppressing smooth muscle proliferation following vascular injury.
Keywords: sequestosome 1, carotid artery, neointimal hyperplasia, vascular injury, vascular remodelling, smooth muscle proliferation, p38MAPK, ERK1/2
Introduction
The cytoplasmic signal adaptor protein designated sequestosome1 (SQSTM1) [1] (A170 in mouse [2], ZIP in rat [3] or p62 in human beings [4]) plays a key role in modulating signal transduction via membrane receptors. It interacts with different protein kinases, such as tyrosine kinase p56lck[4] and PKC-ζ[3], and with other types of adaptors such as RIP [5] and TRAF6 [6]. SQSTM1 has a UBA ubiquitin-binding domain in the C-terminus [7] and participates in the assembly of ubiquitinated protein aggregates termed sequestosome [8] and modulation of ubiquitination pathways involved in NF-κB activity and receptor trafficking [6]. SQSTM1 gene-targeted knockout (KO) mice exhibited mature-onset obesity and insulin resistance [9], providing a unique model to study the molecular mechanisms underlying endothelial and smooth muscle dysfunction in obesity and insulin resistance.
We previously reported that SQSTM1/A170 is an oxidative stress- or electrophile-inducible protein [2], regulated by the redox sensitive transcription factor NF-E2-related factor 2 (Nrf2) [2, 10, 11]. Nrf2 activates ARE/EpRE (antioxidant/electrophile responsive element) mediated expression of phase II detoxifying and antioxidant genes [10]. In murine aortic smooth muscle cells (SMCs), we demonstrated that Nrf2 mediates transcriptional activation of SQSTM1 in cells challenged with 4-hydroxynonenal [11], an n-6 polyunsaturated fatty acid oxidation product detected in atherosclerotic lesions [12]. In human umbilical vein endothelial cells, expression of sqstm1 gene is up-regulated by shear stress [13]. Thus SQSTM1 may act as a novel regulator of both antioxidant and anti-inflammatory signalling pathways in the vasculature.
The aim of the present study was to determine whether SQSTM1 confers protection against vascular injury in vivo and, if so, whether SQSTM1-deficient mice exhibit alterations in vascular SMC proliferation. We examined the effects of vascular injury in wild-type (WT) and SQSTM1–/– mice by ligating the common carotid artery [14]. Complete ligation of the vessel near the carotid bifurcation induced rapid proliferation of medial SMCs, leading to extensive neointimal formation [14]. It has previously been reported that selectins, inflammatory cytokines [15] and p38MAPK[16–20] play roles in the development of neointimal hyperplasia. Because the magnitude of neointimal formation correlates with enhanced proliferation of SMCs in vitro[21], we compared proliferation of aortic SMCs from WT and SQSTM1–/– mice in vitro. Based on our in vivo and in vitro findings, we conclude that SQSTM1 is a novel key adaptor protein involved in coordinating vascular remodelling in response to vascular injury by suppressing the neointimal SMC proliferation.
Materials and methods
Reagents
Antibodies against pan-ERK1/2, p∼ERK1/2, pan-ERK5, p∼ERK5, pan-p38, p∼p38 were purchased from Cell Signaling Technology, Inc. (Beverly, MA, USA); protein kinase inhibitors, PD098059, SB203580, genistein, PD153035, from Calbiochem Corp. (Tokyo, Japan); SP600125 from Tocris Cookson Ltd. (Northpoint, UK); DMEM and anti-α-actin from Sigma-Aldrich Corp (Tokyo, Japan); foetal calf serum (FCS) from HyClone (Logan, UT, USA); immunoblotting reagents from Amersham (Tokyo, Japan), RNeasy Micro Kit from QIAGEN (Tokyo, Japan) and other chemicals from Nacalai Tesque (Kyoto, Japan) or Wako Chemical Co. (Osaka, Japan).
Wild-type and SQSTM1 knockout mice
Homologues of the signal modulator or adaptor protein SQSTM1 in human beings and mice are termed p62 and A170, respectively. We generated SQSTM1-deficient mice (sqstm1-gene KO) using gene-targeting procedures [22] approved by the University of Tsukuba Animal Research Committee and Safety Committee for Gene Recombination Research. KO mice, crossed with C57BL/6J mice (Charles River Co., Yokohama, Japan) for more than 10 generations, were fertile and grew normally. Mice were housed under specific pathogen-free conditions at 24°C with a daily cycle of 12 hrs light and darkness. Mice had free access to drinking water and standard chow containing 0.9% calcium and 0.7% phosphorus (MF: 360 kcal/100 g; Oriental Yeast, Tokyo, Japan). Male mice were used for all experiments.
Common carotid artery ligation and analyses of remodelling
Animals (13–17 weeks of age) were anaesthetized with sodium pentobarbital (50 mg/kg IP) and the right common carotid artery was surgically isolated and ligated for 3 weeks, as described previously [14, 21]. Animals were killed with pentobarbital and diethyl ether before removing the right and left (control) common carotid arteries for morphometric and histological analysis. Bilateral carotid arteries were fixed in 4% paraformaldehyde and embedded in paraffin. The ligated right carotid artery was cut five transverse sections of 4-μm-thick slices from the site of ligation. The non-ligated left carotid artery (control) was cut five transverse sections of 4-μm-thick slices from the similar position. The tissue slices were stained with haematoxylin and eosin and Masson’s trichrome. We assessed the right carotid artery morphometrically using one of the five sections near the site of ligation displaying the smallest lumen, and used the contralateral position in the left carotid artery as an internal control. The borders of the internal lumen, internal elastic lamina (IEL), and external elastic lamina (EEL) were traced on a digitizing board and analysed using Macintosh image software (NIH Image version 1.61). The intimal area was calculated by subtracting the luminal area from the IEL area, and the medial area was calculated by subtracting the IEL area from the EEL area. The vessel size was defined by EEL area in the ligated specimen. In the case of the flattened non-ligated artery, vessel size, IEL area and luminal area were, respectively, calculated from the length of each perimeter of area assuming circular dimensions. The ratio of intimal/medial area (I/M ratio) was calculated by dividing the intimal area by medial area and the stenotic ratio by dividing the intimal area by IEL area. Average values were obtained from morphometric analysis of individual sections from 11 to 16 animals per group [23].
Isolation and culture of murine aortic smooth muscle cells
Aortae were isolated from mice (8–10 weeks) and adipocyte and connective tissues removed. The aortae were cut open, endothelial cells scraped off and then cut into 2–3 mm pieces, which were placed into culture dishes [10]. The aortic fragments were pressed down with a round glass cover slip and a plastic ring until SMCs migrated from the fragments. Explant cultures of aortic SMCs from WT and SQSTM1–/– mice were maintained under 95% air/5% CO2 in Dulbecco’s modified minimum essential medium containing 25 mM glucose and 10% (v/v) FCS [11]. Several weeks were required to obtain sufficient SMCs for subculture in culture flasks. We reproducibly observed enhanced migration and proliferation of aortic SMCs derived from SQSTM1-deficient mice. SMCs from WT and SQSTM1-deficient mice were sub-cultured twice a week and passage 5–15 cells expressing SMC phenotype [11] were used for experiments in vitro.
Smooth muscle cell proliferation assay
SMCs were suspended at 2.0 × 104 cells/ml and seeded either in 12-well plates (1 ml/well) for determining cell number using a haemocytometer or in 96-well plates (100 μl/well) to determine bromodeoxyuridine (BrdU) incorporation into DNA (Roche Diagnostics, Basel, Switzerland). SMCs were serum deprived (0.5%) for 48 hrs and then challenged with 10% FCS in the absence or presence of inhibitors.
Carotid artery and aortic smooth muscle cells protein lysates and immunoblotting
Arteries were carefully isolated from mice under a dissection microscope and proteins extracted using a buffer containing 20 mM Tris-HCl (pH7.5), 5 mM EDTA, 10 mM deoxycolate, 100 mM NaF, 2 mM NaVO4, 1% nonidet P-40, 1 mM p-APMSF, protease inhibitor cocktail (Sigma, Tokyo, Japan). Approximately 2 mm sections of the ligated site in right common carotid arteries and entire left non-ligated arteries were used for protein analysis. After homogenization, the mixtures were centrifuged at 14,000 rpm for 10 min. at 4°C and the supernatants used for protein assays. In case of cultured SMCs, SDS sample buffer was used to solubilize whole cell proteins [10]. Whole cell protein lysates were fractionated by SDS-PAGE and electrotransferred onto an Immobilon membrane (Millipore, Tokyo, Japan) [10]. Immunoreactive proteins were detected with specific antibodies against A170 (SQSTM1), pan-ERK1/2 and p∼ERk1/2, pan-p38MAPK and p∼p38MAPK or pan-JNK and p∼JNK using ECL-plus immunoblotting (GE Healthcare, Little Chalfont, UK) [10, 11].
Quantitative real-time PCR
Total RNA was isolated from SMCs and steady-state mRNA levels determined by qPCR using 25 ng RNA in 25 μl reaction mixture per assay. PCR thermocycling parameters were 48°C for 30 min., 95°C for 10 min. and 40 cycles (95°C for 15 sec., 60°C for 1 min.). RT-PCR was carried out using an Applied Biosystems 7000 sequence detector. Primers and probes for MAPK phosphatase-1 (MKP-1) were designed with Primer Express (Applied Biosystems, Tokyo, Japan):
forward 5′-TGCCTGACAGTGCAGAATCC-3′ and reverse 5′-TCCTTCCGAGAAGCGTGATAG -3′.
GAPDH, primers and the MKP-1 probe in the pre-developed TaqMan assay reagents (Applied Biosystems) were used, reverse-transcribed with qPCR Master Mix (Eurogentec SA, Seraing, Belgium), assayed in duplicate and normalized to GAPDH.
Statistics
Data for the morphometric analyses of arteries are expressed as means ± S.E. of measurements in tissue sections from WT and SQSTM1-deficient mice (n= 11 to 16). For assays of SMC proliferation and BrdU incorporation data are expressed as means ± S.D. of triplicate measurements and are representative of similar results in SMC cultures derived from at three WT and three SQSTM1–/– mice. RT-PCR data are means ± S.D. of three different SMC cultures. Data were analysed using an unpaired Student’s t-test and two-way ANOVA for comparison of multiple groups. Statistical significance between data sets was established at P < 0.05 to P < 0.005.
Results
SQSTM1 deficiency enhanced vascular remodelling in a blood flow cessation model
To examine the role of SQSTM1 in carotid artery remodelling, the right common carotid artery was ligated in mice aged 15 ± 2 weeks. The body weights of WT and SQSTM1–/– mice after 3 weeks ligation were 28.5 ± 2.4 and 31.8 ± 2.6 g, respectively. Thickening of the arterial wall was not observed in the non-ligated common carotid arteries of WT and KO mice (Fig. 1A, left panels). In contrast, the right ligated common carotid artery exhibited marked intimal hyperplasia (Fig. 1A, right panels). The maximal stenosis was observed proximal to the ligation site and we observed much less stenosis at 200-μm distances from the ligation site in both WT and KO mice. We selected one of five transverse sections of 4-μm-thick serial tissue sections to illustrate maximal stenosis in each specimen. As shown in three representative tissue sections, the extent of neointimal hyperplasia or vascular stenosis in the ligated artery was significantly enhanced in SQSTM1–/– mice (Fig. 1A, right panels).
Fig 1.
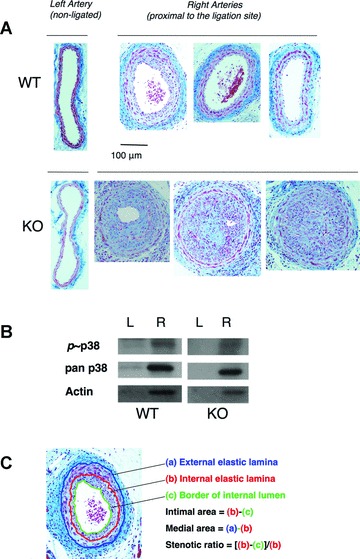
Neointimal hyperplasia and vascular remodelling in WT versus SQSTM1-deficient mice in vivo. (A) Representative photomicrographs of non-ligated left carotid artery from WT and KO (left panels); ligated right arteries from three WT (right upper images) and KO (right lower images) mice. Haematoxylin and eosin staining, bar denotes 100 μm. Neointimal hyperplasia and vascular stenosis in ligated carotid arteries was greater in KO compared to WT mice. Arteries were fixed after 3 weeks of ligation. (B) Representative immunoblotting of phosphorylated p38MAPK (p38), total p38MAPK (pan p38) and actin in carotid artery homogenates from WT and KO mice after 1 week of ligation; L (left non-ligated) and R (right ligated) artery. These proteins were detected only in the right ligated arteries. Proteins (10μg/well) were separated by SDS-PAGE and blotted as described in ‘Materials and methods’. (C) Illustration of the morphometric analyses of tissue sections.
We detected enhanced immunostaining for actin and phosphorylated and non-phosphorylated p38MAPK in ligated common carotid artery homogenates in both WT and SQSTM1–/– (Fig. 1B), most likely reflecting an increased the number of SMCs in these stenotic regions.
Morphometric analyses (Fig. 1C) were conducted by measuring the EEL (a), IEL (b) and borders of the internal lumen (c). The vessel size estimated from the EEL or vessel perimeter (flat non-ligated artery) was significantly decreased after ligation (Fig. 2A) although medial areas (see Fig.1C) increased significantly (Fig. 2B). There were no significant differences in vessel size or medial area in non-ligated arteries from WT and KO mice. Notably the ligated artery of KO mice showed 2.1- and 1.7-fold higher intimal area and stenotic ratio, respectively (see Fig. 2C and D).
Fig 2.
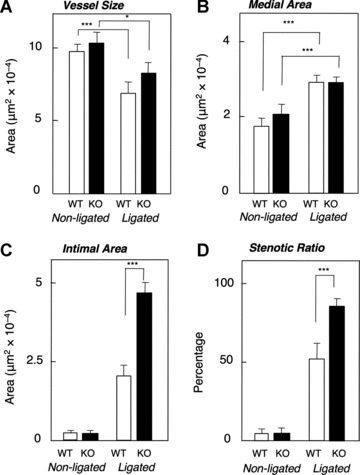
Morphometric analysis of carotid artery neointimal thickening and remodelling in WT and SQSTM1-deficient mice after 3 weeks of ligation. (A) Vessel size (inside of EEL (a) area or calculated from perimeter of (a) in case of non-ligated flat artery); (B) medial area ((a) – (b)); (C) intimal area ((b) – (c)); (D), stenotic ratio ([(b) – (c)]/(b)). Values are means ± S.E. of measurements in tissue sections from WT (non-ligated left artery, n= 16; ligated right artery, n= 13) and SQSTM1-deficient (non-ligated left artery, n= 11; ligated right artery, n= 11) mice. *P < 0.05, ***P < 0.005, between non-ligated and ligated (A and B) or WT and SQSTM1–/– mice (C and D).
Accelerated proliferation of cultured aortic smooth muscle cells from SQSTM1–/– mice
To investigate whether SQSTM1 affects SMC proliferation, aortic SMCs from WT and SQSTM1–/– mice were cultured in medium containing 10% (v/v) FCS. SQSTM1–/– SMCs proliferated faster and attained more than 2-fold higher cell densities compared to WT SMCs over 3–7 days (Fig. 3A). Enhanced proliferation of SQSTM1–/– SMCs was more marked in cells challenged with 5% and 10 % FCS compared to a lower FCS concentration (Fig. 3B). The dependence of cell proliferation on FCS was determined by assessing incorporation of BrdU into DNA (Fig. 3C). When SMCs were serum deprived (0.5%) for 24 hrs and then challenged with 10% FCS, BrdU incorporation for 48 hrs was ∼2-fold higher in SQSTM1–/– compared to WT SMCs (Fig. 3C, left panel). When the serum-deprived cells were challenged with growth factors platelet-derived growth factor (PDGF) and bFGF, respectively instead of 10% FCS, BrdU incorporation was enhanced to a much lesser extent compared to 10% FCS and there was no significant difference between WT and SQSTM1–/– SMCs (Fig. 3C, middle and right panels). These results suggest that SQSTM1 may not be directly linked with PDGF and bFGF signalling pathways. Supplementing each epidermal growth factor (EGF; 5–100 ng/ml), nerve growth factor (NGF; 5–100 ng/ml), angiotensin II (10–150 ng/ml), insulin-like growth factor (IGF; 5–100 ng/ml) and insulin (5–100 ng/ml) to the serum-deprived cultures hardly enhanced BrdU incorporation in both WT and SQSTM1–/– SMCs (data not shown).
Fig 3.
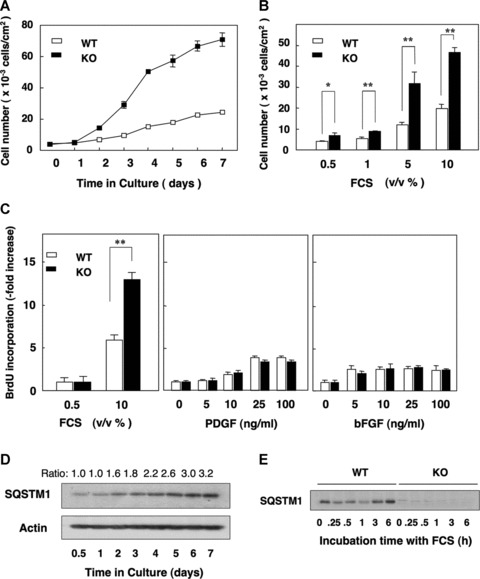
Proliferation properties of aortic SMCs from WT and SQSTM1-deficient mice in culture. (A) Comparison of the time course of proliferation in 10%(v/v) FCS containing fresh medium of WT (open columns) and KO (SQSTM1–/–, filled columns) SMCs after seeding at 1.5 × 104 cells/ml. (B) Concentration dependent effects of FCS (0.5–10%) on proliferation of WT (open columns) and SQSTM1–/– (filled columns) SMCs after 72 hrs of seeding cells at 3.0 × 104 cells/ml. (C) Effects of FCS (0.5%versus 10%, left panel), PDGF (0 to 100 ng/ml in the presence of 0.5% FCS, middle panel) and bFGF (0 to 100 ng/ml in the presence of 0.5%FCS, right panel) on BrdU incorporation into DNA in WT (open columns) and KO (filled columns) SMCs. Data expressed as fold increase compared to WT SMCs. Both SMCs types were seeded at 2.0 × 104 cells/ml and cultured in 0.5% FCS for 24 hrs. Then, FCS or other agents were added to culture media at concentrations indicated and BrdU incorporation was measured for 48 hrs. (A–C), Data denote means ± S.D. of triplicate measurements in a representative experiment, *P < 0.05, **P < 0.01. Similar results were obtained using different cultures from WT (n= 3) and KO (n= 3) mice. (D) Representative immunoblot showing a time-dependent increase in SQSTM1 protein expression during culture under the same conditions as shown in (A) in WT SMCs. Whole cell protein lysates (20 μg/well) were analysed using actin as an internal loading control. (E) Representative immunoblot showing a rapid transient down-regulation of SQSTM1 expression in WT SMCs (left) after exposure to 10% FCS. SMCs were previously cultured in 0.5% FCS containing medium for 48 hrs to down-regulate growth stimulation before exposure to 10% FCS. KO (SQSTM1–/–) SMCs (right) show no SQSTM1 immunostained band. The internal control pan p38 MAPK band of this blot is shown in Fig. 4A (same blot).
Interestingly, expression levels of SQSTM1 were quite low in WT SMCs seeded in fresh medium at low cell density and then gradually increased with time in culture and increased with cell density (Fig. 3D, same culture conditions as in Fig. 3A). Exposure of serum starved WT SMCs to 10% FCS induced rapid (0.25–1 hrs) decrease of SQSTM1 expression, and a gradual recovery after 3–6 hrs (Fig. 3E, same conditions as in Fig. 5A).
Fig 5.
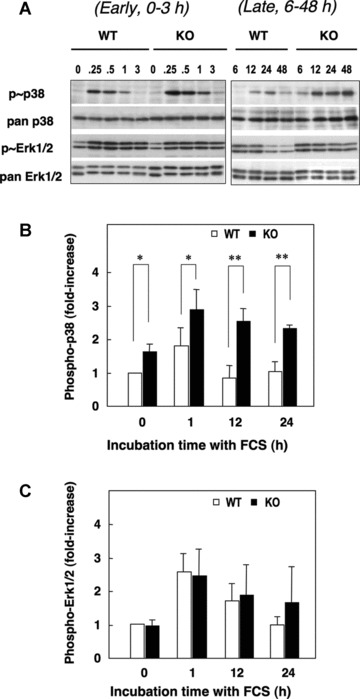
Serum-induced early and late phases of phosphorylation of p38MAPK and ERK1/2 in aortic SMCs from WT and SQSTM1-deficient mice. SMCs seeded at 2.0 × 104 cells/ml were serum deprived (0.5% FCS, 48 hrs) and then challenged with 10% FCS. (A) Early (0–3 hrs, left) and late (6–48 hrs, right) phosphorylation of p38MAPK and ERK1/2. (B–C) Densitometric analysis of p38MAPK and ERK1/2 phosphorylation in response to 10% FCS expressed relative to total pan-p38 or total pan-ERK1/2. Changes in p38MAPK or ERK1/2 phosphorylation were then expressed relative to basal values in WT SMCs cultured in 0.5% FCS. Data denote means ± S.D. of measurements in SMCs from three different cultures, *P < 0.05; **P < 0.01.
Enhanced migration of SMCs from SQSTM1–/– mice
SMCs migrated much faster from aortic tissue pieces from SQSTM1–/– compared to WT mice. Moreover, using a scratch wound assay, we observed that migration of SMCs in response to 10% FCS was enhanced in cultures from SQSTM1–/– compared to WT mice (Fig. 4). Increased migration of SQSTM1–/– SMCs was also observed under conditions of serum deprivation (data not shown). Thus, SQSTM1–/– SMCs exhibit different characteristics from WT SMCs in relation to both proliferation and migration.
Fig 4.

Scratch wound assay to monitor SMC migration. WT and SQSTM1–/– SMCs were plated into 10-cm-diameter dishes and cultured in DMEM containing 10% FCS until they reached confluence. Cells were then cultured in serum deprived (0.5% FCS) DMEM for 24 hrs, and wounds were introduced using a plastic pipette tip (0 hr). Cells were then cultured in DMEM in the presence of 10% FCS. The photographs (×100) of wound area were taken after 8 hrs.
Enhanced activation of p38MAPK by FCS in smooth muscle cells from SQSTM1 KO mice
As activation of p38MAPK and ERK1/2 are involved in the regulation of SMCs proliferation following vascular injury [24–26], we compared FCS-induced activation of MAPKs in WT and SQSTM1–/– SMCs. Cells were adapted to 0.5% FCS for 48 hrs and then challenged with 10% FCS. Basal levels and early (0–3 hrs) activation of p38MAPK in response to 10% FCS were significantly elevated in SQSTM1–/– SMCs (Figs 5A and B). Upon longer stimulation with 10% FCS (6–48 hrs), phosphorylation of p38MAPK was more prominent in SQSTM1–/– compared to WT cells (Fig. 5A and B). Serum-stimulated activation of ERK1/2 was rapid but then declined towards basal levels over 24 hrs in both WT and SQSTM1–/– cells (Fig. 5A and C), and ERK1/2 phosphorylation was similar in both WT and SQSTM1–/– cells.
We subsequently examined induction of MKP-1 gene expression by qPCR to examine feedback inhibition of p38MAPK activation [27]. MKP-1 gene expression increased over 0.25–1 hrs in both WT (7.5 ± 3.5 fold, n= 3) and SQSTM1–/– (8.8 ± 2.4 fold, n= 3) SMCs in response to 10% FCS. Expression of mRNA returned to basal levels within 3 hrs and remained low over the next 45 hrs. The similar pattern of MKP-1 mRNA expression in WT and SQSTM1–/– SMCs suggests that increased p38MAPK phosphorylation in SQSTM1–/– cells was not due to the defect of MKP-1 gene expression.
Inhibition of aortic smooth muscle cell proliferation by MAPK and tyrosine kinase inhibitors
We next examined the effects of MAPK inhibitors on the proliferation of SMCs from WT and SQSTM1–/– mice. Growth-arrested SMCs (0.5% FCS, 48 hrs) were challenged with 10% FCS in the absence or presence of specific MAPK inhibitors, and incorporation of BrdU into DNA determined. Inhibition of p38MAPK (SB: SB203580) or ERK1/2 (PD: PD98059) activation alone had negligible effects on BrdU incorporation, whereas co-treatment of cells with SB203580 (1 μM) and PD98059 (10 μM) significantly decreased the elevated BrdU incorporation in SQSTM1–/– SMCs (Fig. 6A). A JNK inhibitor (SP: SP600125, 1 and 2 μM) had no effect on BrdU incorporation in either WT or SQSTM1–/– SMCs (Fig. 6A).
Fig 6.
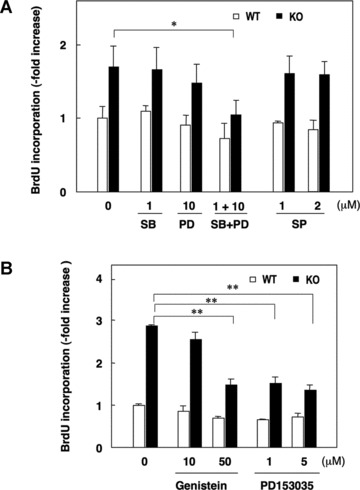
Effects of kinase inhibitors on serum induced DNA synthesis in SMCs from WT and SQSTM1-deficient mice. (A) Serum-deprived (0.5% FCS, 48 hrs) SMCs were stimulated with 10% FCS in the absence or presence (30 min. pre-treatment) of inhibitors of p38MAPK (SB; SB203580), ERK1/2 (PD; PD98059) and/or JNK (SP; SP600125). BrdU incorporation was measured over 24 hrs in WT (open columns) and KO (SQSTM1–/–, filled columns) SMCs. (B) Effects of genistein and EGF receptor kinase inhibitor (PD153035) on BrdU incorporation in WT (open columns) and KO (filled columns) SMCs. Data denote means ± S.D. of triplicate measurements in an experiment representative of similar findings in three different SMC cultures, *P < 0.05; **P < 0.01.
Because tyrosine kinases play an integral role in serum stimulated MAPK activation [28], we examined the effect of the general tyrosine kinase inhibitor genistein and the EGF receptor targeted tyrosine kinase inhibitor PD153035 [29] on BrdU incorporation. We found that 1 and 5 μM PD153035 and 50 μM genistein significantly reduced elevated BrdU incorporation in SQSTM1–/– SMCs (Fig. 6B).
Discussion
This study provides the first evidence in vivo that the signal adaptor protein SQSTM1 plays a fundamental role in attenuating neointimal hyperplasia and carotid artery remodelling after vascular injury. Notably, aortic SMCs from SQSTM1 KO mice proliferated faster and attained a higher cell density compared to WT SMCs upon stimulation with FCS in vitro (Fig. 3), implicating SQSTM1 as a suppressor of SMC proliferation. Additionally, SQSTM1-deficient SMCs migrated faster compared to WT SMCs, implicating SQSTM1 as a suppressor of migration (Fig. 4). The ability of SQSTM1 to inhibit vascular SMC proliferation and migration may explain the protection afforded against pro-atherogenic stimuli. Although obesity and insulin resistance affect vascular function [30–32], we used young KO mice aged 8–17 weeks in order to obviate potential confounding effects of metabolic syndrome associated with KO mice aged >25 weeks. In younger mice, there were no significant changes in blood glucose, cholesterol ester or total cholesterol levels and no sign of inflammation in the liver [33]. Therefore, we conclude that enhanced proliferation and migration of SQSTM1-deficient vascular SMCs in vivo or ex vivo is not a consequence of obesity.
Vascular responses to mechanical injury involve inflammatory and fibro-proliferative processes that result in neointima formation and vascular remodelling. Ligation of the carotid artery of mice is widely used to characterize factors involved in vascular remodelling [14–21]. The complex cellular interactions initiated by vascular injury are coordinated and modulated by the elaboration of cytokines and growth factors, requiring sustained activation of p38MAPK[19, 26]. Following balloon injury of the rabbit ilio-femoral artery, phosphorylation of p38MAPK is increased in SMCs in lamina of the vessel wall [20]. In the present study, we detected phosphorylated p38MAPK immunostaining in homogenates of the ligated arteries (Fig. 1B), although quantitative comparison between WT and SQSTM1–/– mouse was difficult. Our experiments in vitro established that activation of p38MAPK after serum stimulation was significantly higher and prolonged in SQSTM1–/– compared to WT SMCs. Recent studies in vivo have reported that transgenic mice overexpressing dominant negative p38α mRNA and protein exhibit reduced neointima formation after carotid injury and decreased arterial p38MAPK activation [26]. Although serum stimulation increased activation of ERK1/2, we noted no significant differences in the profile of ERK1/2 phosphorylation between WT and SQSTM1-deficient SMCs (Fig. 5C). These results suggest that the role of SQSTM1 in growth suppression is at least partially mediated via suppression of p38MAPK activation.
Previous studies in HeLa cells have shown that SQSTM1 can interact directly with p38MAPK to enhance p38MAPK activation/phosphorylation in vitro[34]. This report contrasts with our findings in murine aortic SMCs, showing that absence of SQSTM1 enhances serum-induced activation of p38MAPK (Fig. 5). As mRNA expression profiles of MKP-1 were similar in SQSTM1–/– and WT SMCs, it seems unlikely that feedback regulation of p38MAPK activation by the phosphatase MKP-1 is modulated by SQSTM1. Notably, we found that the expression level of SQSTM1 and phosphorylated p38MAPK in WT SMCs upon FCS stimulation is reciprocally regulated; the mitogenic stimulation by FCS induces rapid down-regulation of SQSTM1 levels and activation of p38MAPK in WT SMCs (Fig. 3E and 5A). This result supports an idea that SQSTM1 suppresses activation of p38MAPK by FCS. As SQSTM1 is involved in the internalization of receptors [35] and in degradation of ubiquitinated proteins by sorting them to the endosome/lysosome [22, 36], this may in turn modulate signal transduction. Based on these reports, we suggest that the enhanced mitogenic activation in SQSTM1-deficient SMCs triggered by FCS is due to defects of receptor mediated signal transduction, resulting in enhanced p38MAPK activation and SMC proliferation.
PDGF-BB is one of the most potent mitogens and chemoattractants for vascular SMCs, activates multiple signalling pathways including MAPKs and plays the central role in the onset and development of various vascular disorders [37, 38]. We observed that PDGF and bFGF in the presence of 0.5%FCS stimulated SMC proliferation to a similar extent in WT and SQSTM1-deficient cells (Fig. 3C) and that the growth promoting effects of these factors were less compared to 10% FCS. These results suggest that SQSTM1 may not be directly linked with the PDGF and FGF signalling pathways, and the serum contains unidentified components that enhance SMC proliferation, which SQSTM1 can inhibit. Supplementing medium containing 0.2% FCS, with EGF, NGF, angiotensin II, IGF and insulin only marginally enhanced proliferation of WT and KO SMCs. Identification of the growth stimulating components in serum will be important to unravel the molecular mechanisms by which SQSTM1 regulates SMC proliferation and neointima formation.
We have demonstrated for the first time that the adaptor protein SQSTM1 plays an important role in the regulation of vascular remodelling after injury in vivo. Because proliferation and migration of SMCs is intimately involved in vascular remodelling, enhanced neointimal hyperplasia in injured carotid arteries from SQSTM1 KO mice may be the consequence of ‘removing an endogenous inhibitory action’ of SQSTM1 on SMC proliferation and migration. This conclusion is consistent with the previous report that the magnitude of neointimal formation after arterial ligation correlates with enhanced proliferation of SMC (from different mouse strains) induced by FCS in vitro[21]. As we have shown that SQSTM1/A170 gene expression in mouse aortic SMC is up-regulated by atherogenic stimuli together with HO-1 and peroxiredoxin I via the redox sensitive transcription factor Nrf2 [11], this signalling pathway may provide a therapeutic target for treatment and prevention of vascular diseases such as arteriosclerosis.
Acknowledgments
We gratefully acknowledge support from the Japan Society for Promotion of Science (H.Y. and E.W.), Great Britain Sasakawa Foundation Butterfield Award for UK–Japan Collaboration (T.I. and G.E.M.) and our association with the European Union COST ACTION B35 (G.E.M.).
References
- 1.Moscat J, Diaz-Meco MT, Wooten MW. Signal integration and diversification through the p62 scaffold protein. Trends Biochem Sci. 2007;32:95–100. doi: 10.1016/j.tibs.2006.12.002. [DOI] [PubMed] [Google Scholar]
- 2.Ishii T, Yanagawa T, Kawane T, et al. Murine peritoneal macrophages induce a novel 60-kDa protein with structural similarity to a tyrosine kinase p56lck-associated protein in response to oxidative stress. Biochem Biophys Res Commun. 1996;226:456–60. doi: 10.1006/bbrc.1996.1377. [DOI] [PubMed] [Google Scholar]
- 3.Puls A, Schmidt S, Grawe F, et al. Interaction of protein kinase C zeta with ZIP, a novel protein kinase C-binding protein. Proc Natl Acad Sci USA. 1997;94:6191–6. doi: 10.1073/pnas.94.12.6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Joung I, Strominger JL, Shin J. Molecular cloning of a phosphotyrosine-independent ligand of the p56lck SH2 domain. Proc Natl Acad Sci USA. 1996;93:5991–5. doi: 10.1073/pnas.93.12.5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanz L, Sanchez P, Lallena MJ, et al. The interaction of p62 with RIP links the atypical PKCs to NF-kappaB activation. EMBO J. 1999;18:3044–53. doi: 10.1093/emboj/18.11.3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sanz L, Diaz-Meco MT, Nakano H, et al. The atypical PKC-interacting protein p62 channels NF-kappaB activation by the IL-1-TRAF6 pathway. EMBO J. 2000;19:1576–86. doi: 10.1093/emboj/19.7.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vadlamudi RK, Joung I, Strominger JL, et al. p62, a phosphotyrosine-independent ligand of the SH2 domain of p56lck, belongs to a new class of ubiquitin-binding proteins. J Biol Chem. 1996;271:20235–7. doi: 10.1074/jbc.271.34.20235. [DOI] [PubMed] [Google Scholar]
- 8.Shin J. P62 and the sequestosome, a novel mechanism for protein metabolism. Arch Pharm Res. 1998;21:629–33. doi: 10.1007/BF02976748. [DOI] [PubMed] [Google Scholar]
- 9.Rodriguez A, Duran A, Selloum M, et al. Mature-onset obesity and insulin resistance in mice deficient in the signaling adapter p62. Cell Metab. 2006;3:211–22. doi: 10.1016/j.cmet.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 10.Ishii T, Itoh K, Takahashi S, et al. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem. 2000;275:16023–9. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- 11.Ishii T, Itoh K, Ruiz E, et al. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: activation by oxidatively modified LDL and 4-hydroxynonenal. Circ Res. 2004;94:609–16. doi: 10.1161/01.RES.0000119171.44657.45. [DOI] [PubMed] [Google Scholar]
- 12.Leonarduzzi G, Chiarpotto E, Biasi F, et al. 4-Hydroxynonenal and cholesterol oxidation products in atherosclerosis. Mol Nutr Food Res. 2005;49:1044–9. doi: 10.1002/mnfr.200500090. [DOI] [PubMed] [Google Scholar]
- 13.Warabi E, Wada Y, Kajiwara H, et al. Effect on endothelial cell gene expression of shear stress, oxygen concentration, and low-density lipoprotein as studies by a novel flow cell culture system. Free Radic Biol Med. 2004;37:682–94. doi: 10.1016/j.freeradbiomed.2004.05.020. [DOI] [PubMed] [Google Scholar]
- 14.Kumar A, Lindner V. Remodeling with neointima formation in the mouse carotid artery after cessation of blood flow. Arterioscler Thromb Vasc Biol. 1997;17:2238–44. doi: 10.1161/01.atv.17.10.2238. [DOI] [PubMed] [Google Scholar]
- 15.Rectenwald JE, Moldawer LL, Huber TS, et al. Direct evidence for cytokine involvement in neointimal hyperplasia. Circulation. 2000;102:1697–702. doi: 10.1161/01.cir.102.14.1697. [DOI] [PubMed] [Google Scholar]
- 16.Ohashi N, Matsumori A, Furukawa Y, et al. Role of p38 mitogen-activated protein kinase in neointimal hyperplasia after vascular injury. Arterioscler Thromb Vasc Biol. 2000;20:2521–6. doi: 10.1161/01.atv.20.12.2521. [DOI] [PubMed] [Google Scholar]
- 17.Izumi Y, Kim S, Namba M, et al. Gene transfer of dominant-negative mutants of extracellular signal-regulated kinase and c-Jun NH2-terminal kinase prevents neointimal formation in balloon-injured rat artery. Circ Res. 2001;88:1120–6. doi: 10.1161/hh1101.091267. [DOI] [PubMed] [Google Scholar]
- 18.Zhan Y, Kim S, Izumi Y, et al. Role of JNK, p38, and ERK in platelet-derived growth factor-induced vascular proliferation, migration, and gene expression. Arterioscler Thromb Vasc Biol. 2003;23:795–801. doi: 10.1161/01.ATV.0000066132.32063.F2. [DOI] [PubMed] [Google Scholar]
- 19.Ju H, Nerurkar S, Sauermelch CF, et al. Sustained activation of p38 mitogen-activated protein kinase contributes to the vascular response to injury. J Pharmacol Exp Ther. 2002;301:15–20. doi: 10.1124/jpet.301.1.15. [DOI] [PubMed] [Google Scholar]
- 20.Jacob T, Ascher E, Alapat D, et al. Activation of p38MAPK signaling cascade in a VSMC injury model: role of p38MAPK inhibitors in limiting VSMC proliferation. Eur J Vasc Endovasc Surg. 2005;29:470–8. doi: 10.1016/j.ejvs.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 21.Harmon KJ, Couper LL, Lindner V. Strain-dependent vascular remodeling phenotypes in inbred mice. Am J Pathol. 2000;156:1741–8. doi: 10.1016/S0002-9440(10)65045-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Komatsu M, Waguri S, Koike M, et al. Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell. 2007;131:1149–63. doi: 10.1016/j.cell.2007.10.035. [DOI] [PubMed] [Google Scholar]
- 23.Murakoshi N, Miyauchi T, Kakinuma Y, et al. Vascular endothelin-B receptor system in vivo plays a favorable inhibitory role in vascular remodeling after injury revealed by endothelin-B receptor-knockout mice. Circulation. 2002;106:1991–8. doi: 10.1161/01.cir.0000032004.56585.2a. [DOI] [PubMed] [Google Scholar]
- 24.Pyles JM, March KL, Franklin M, et al. Activation of MAP kinase in vivo follows balloon overstretch injury of porcine coronary and carotid arteries. Circ Res. 1997;81:904–10. doi: 10.1161/01.res.81.6.904. [DOI] [PubMed] [Google Scholar]
- 25.Gennaro G, Menard C, Michaud SE, et al. Inhibition of vascular smooth muscle cell proliferation and neointimal formation in injured arteries by a novel, oral mitogen-activated protein kinase/extracellular signal-regulated kinase inhibitor. Circulation. 2004;110:3367–71. doi: 10.1161/01.CIR.0000147773.86866.CD. [DOI] [PubMed] [Google Scholar]
- 26.Proctor BM, Jin X, Lupu TS, et al. Requirement for p38 mitogen-activated protein kinase activity in neointima formation after vascular injury. Circulation. 2008;118:658–66. doi: 10.1161/CIRCULATIONAHA.107.734848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jacob A, Smolenski A, Lohmann SM, et al. MKP-1 expression and stabilization and cGK Ialpha prevent diabetes- associated abnormalities in VSMC migration. Am J Physiol Cell Physiol. 2004;287:C1077–86. doi: 10.1152/ajpcell.00477.2003. [DOI] [PubMed] [Google Scholar]
- 28.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y, Poon RT, Shao W, et al. Blockage of epidermal growth factor receptor by quinazoline tyrosine kinase inhibitors suppresses growth of human hepatocellular carcinoma. Cancer Lett. 2007;248:32–40. doi: 10.1016/j.canlet.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 30.Fox CS, Massaro JM, Hoffmann U, et al. Abdominal visceral and subcutaneous adipose tissue compartments: association with metabolic risk factors in the Framingham Heart Study. Circulation. 2007;116:39–48. doi: 10.1161/CIRCULATIONAHA.106.675355. [DOI] [PubMed] [Google Scholar]
- 31.Rodford JL, Torrens C, Siow RC, et al. Endothelial dysfunction and reduced antioxidant protection in an animal model of the developmental origins of cardiovascular disease. J Physiol. 2008;586:4709–20. doi: 10.1113/jphysiol.2008.156976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Despres JP, Lemieux I, Bergeron J, et al. Abdominal obesity and the metabolic syndrome: contribution to global cardiometabolic risk. Arterioscler Thromb Vasc Biol. 2008;28:1039–49. doi: 10.1161/ATVBAHA.107.159228. [DOI] [PubMed] [Google Scholar]
- 33.Okada K, Yanagawa T, Warabi E, et al. The α-glucosidase inhibitor acarbose prevents obesity and simple steatosis in sequestosome 1/A170/p62 deficient mice. Hepatol Res. 2009;39:490–500. doi: 10.1111/j.1872-034X.2008.00478.x. [DOI] [PubMed] [Google Scholar]
- 34.Kawai K, Saito A, Sudo T, et al. Specific regulation of cytokine-dependent p38 MAP kinase activation by p62/SQSTM1. J Biochem. 2008;143:765–72. doi: 10.1093/jb/mvn027. [DOI] [PubMed] [Google Scholar]
- 35.Geetha T, Wooten MW. Association of the atypical protein kinase C-interacting protein p62/ZIP with nerve growth factor receptor TrkA regulates receptor trafficking and Erk5 signaling. J Biol Chem. 2003;7:4730–9. doi: 10.1074/jbc.M208468200. [DOI] [PubMed] [Google Scholar]
- 36.Pankiv S, Clausen TH, Lamark T, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–45. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 37.Sundaresan M, Yu ZX, Ferrans VJ, et al. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–9. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 38.Choi MH, Lee IK, Kim GW, et al. Regulation of PDGF signalling and vascular remodelling by peroxiredoxin II. Nature. 2005;435:347–53. doi: 10.1038/nature03587. [DOI] [PubMed] [Google Scholar]