

Abstract
Podosomes and invadopodia are actin-based structures at the ventral cell membrane, which have a role in cell adhesion, migration and invasion. Little is known about the differences and dynamics underlying these structures. We studied podosome-like structures of oral squamous carcinoma cells and invadopodia of their invasive variant that has undergone a spontaneous epithelial-mesenchymal transition (EMT). In 3D imaging, podosomes were relatively large structures that enlarged in time, whereas invadopodia of invasive cells remained small, but were more numerous, degraded more extracellular matrix (ECM) and were morphologically strikingly different from podosomes. In live-cell imaging, highly dynamic, invadopodia-embedded actin tails were frequently released and rocketed through the cytoplasm. Resembling invadopodia, we found new club-ended cell extensions in EMT-experienced cells, which contained actin, cortactin, vinculin and MT1-matrix metalloproteinase. These dynamic cell extensions degraded ECM and, in field emission scanning electron microscopy, protruded from the dorsal cell membrane. Plectin, αII-spectrin, talin and focal adhesion kinase immunoreactivities were detected in podosome rings, whereas they were absent from invadopodia. Tensin potentially replaced talin in invadopodia. Integrin α3β1 surrounded both podosomes and invadopodia, whereas integrin αvβ5 localized only to invadopodia heads. Pacsin 2, in conjunction with filamin A, was detected early in podosomes, whereas pacsin 2 was not found in invadopodia and filamin A showed delayed accumulation. Fluorescence recovery after photobleaching indicated faster reorganization of actin, cortactin and filamin A in podosomes compared to invadopodia. In conclusion, EMT affects the invasion machinery of oral squamous carcinoma cells. Non-invasive squamous carcinoma cells constitutively organize podosomes, whereas invasive cells form invadopodia. The club-ended cell extensions, or externalized invadopodia, are involved in ECM degradation and maintenance of contact to adhesion substrate and surrounding cells during invasion.
Keywords: epithelial-mesenchymal transition, filamin A, fluorescence recovery after photobleaching, invadopodia, pacsin 2, podosome, oral squamous cell carcinoma, total internal reflection fluorescence microscopy
Introduction
In order for carcinoma cells to metastasize to distant sites, they have to disrupt cell–cell adhesion junctions, break through the basement membrane and migrate across the extracellular matrix (ECM) [1]. At subcellular level, special actin-rich membrane microdomains located at the ventral surface, i.e. podosomes and invadopodia, have been connected to cancer invasion [2, 3].
Podosomes are dynamic structures which have been found in macrophages, endothelial cells, transformed fibroblasts, osteoclasts, malignant B lymphocytes and carcinoma cells [4–9]. They have an actin-based core surrounded by a ring of adhesion molecules and a cloud of unpolymerized actin. Podosomes have a role in cell adhesion and ECM degradation, but their function in cell invasion has not been verified [10]. Some reports suggest that podosomes could provide local anchorage in order to stabilize cellular protrusions and thus function in directional migration [3], and that they may be the precursors of invadopodia [11].
Invadopodia have been detected mostly in invasive carcinoma cells and are more directly linked to invasion and metastasis [2, 11]. Invadopodia appear as irregular actin-based dots without a definable ring structure. Invadopodia contain proteins related to actin dynamics, cell adhesion, membrane remodelling, cell signalling and ECM degradation [12]. Implications of their functions in vivo exist in malignancy, as essential components of invadopodia, e.g. cortactin and N-WASP are up-regulated or amplified in carcinomas, such as head and neck squamous cell carcinoma (SCC) [12–14]. The formation and maintenance of invadopodia depend on continuous actin assembly, and they are relatively labile structures with variable life-spans [2, 15]. However, the molecular and functional mechanisms between podosomes and invadopodia still remain a controversy, and it is currently not clear whether these two phenomena truly represent distinct structures. Recently, it has been proposed that the differentiation of and definition for podosomes and invadopodia should be restricted to include podosomes in non-cancerous cells and invadopodia in malignant cells [3].
Epithelial-mesenchymal transition (EMT) occurs at early stages of development and is essential for gastrulation and mesoderm formation. EMT has been proposed to operate in pathological situations, such as in the acquisition of an invasive phenotype in epithelial tumours, in which it may enable the first steps for metastasis [16]. EMT plays a role in, e.g. oral SCC, which is considered a highly invasive disease with poor patient prognosis [14, 17]. Carcinoma cells undergoing EMT attain a fibroblastoid phenotype followed by an ability to migrate and invade the surrounding tissues. EMT induces a massive rearrangement of the actin cytoskeleton, as well as changes in cell–cell and cell-ECM junctions, including tight and adherens junctions, desmosomes and hemidesmosomes [18–20]. Recently, it was suggested that EMT could provoke an assembly of podosomes or invadopodia, although no such evidence yet exists [21].
In this study, we have investigated podosome-like structures and invadopodia of oral SCC cells and their aggressive descendants, which have undergone EMT [18, 20]. We hypothesized that the characteristics gained through EMT, such as increased invasion and cell migration, would be reflected also in the adhesion and invasion machinery and thus could result in a further distinction of the differences between the podosomes and invadopodia. In the present study, we show that the actin-based structures in invasive SCC cells differ from those of non-invasive SCC cells by their protein composition, organization and rate of turnover.
Materials and methods
Cell lines, cell culture and transfections
Oral SCC cell line UT-SCC-43A (43A) is derived from a primary gingival tumour of a 75-year-old Caucasian female. UT-SCC-43B (43B) is derived from a recurrent tumour of the same patient. 43A and 43B cells have been previously characterized [18, 20]. The cells were cultured in RPMI 1640 medium (Sigma-Aldrich, St. Louis, MO, USA) with 10% foetal calf serum (FCS) and antibiotics. Transfections (Table 1) were performed with Fugene HD reagent (Roche, Mannheim, Germany), and second passage cells after transfection were used in the experiments.
Table 1.
Gene constructs, antibodies and antisera used*
| Target | Antibody | Reference |
|---|---|---|
| EGFP-actin | BD Biosciences, San Jose, CA, USA | |
| EGFP-cortactin | [22] | |
| EGFP-filamin A | [23] | |
| Arp 2/3 | Pc rabbit antiserum | Upstate/Millipore, Charlottesville, VA |
| Cortactin | 4F11 | Upstate/Millipore |
| F-actin | rhodamine phalloidin | Molecular Probes/Invitrogen, Eugene, OR |
| Filamin A | PM6/317 | Chemicon/Millipore, Temecula, CA, USA |
| FAK | 2A7 | Upstate/Millipore |
| Integrin α3 subunit | J143 | [24] |
| Integrin αv subunit | LM142.69 | [25] |
| Integrin β1 subunit | 102DF5 | [26] |
| Integrin β5 subunit | 1A9 | [27] |
| MT1-MMP | LEM-2/15 | [28] |
| Nucleus | TO-PRO-3 | Molecular Probes/Invitrogen |
| C-terminal region of human pacsin 2 | Pc rabbit antiserum | Abgent, San Diego, CA |
| Phosphotyrosine | PY20 | Molecular Probes/Invitrogen |
| Plectin | HD-121 | [29] |
| αII-spectrin | 101AA6 | [30] |
| Talin | MCA725S | AbD Serotec/MorphoSys, Oxford, UK |
| Tensin | Clone 5 | BD Biosciences |
| β-tubulin | DM3B3 | [31] |
| Vinculin | Pc rabbit antiserum | [4] |
Antibodies are monoclonal unless otherwise stated. Pc: polyclonal.
Immunofluorescence and confocal microscopy
The cells were grown on glass cover slips and fixed in methanol at –20°C or 4% paraformaldehyde in phosphate-buffered saline at room temperature (RT) for 15 min. Primary antibodies (Table 1) were applied for 1 hr followed by Alexa Fluor® 488 or 594 conjugates (Molecular Probes/Invitrogen, Eugene, OR, USA) for 30 min. The specimens were studied either with Olympus AX70 (Olympus Corporation, Hamburg, Germany) or Leica DM RXA2 (Leica Microsystems AG, Wetzlar, Germany) microscope. Confocal microscopy was carried out using a Leica TCS SP2 AOBS system with argon 488 or DPSS 561 nm excitation lines and HCX PL APO CS 63 ×/1.40 NA or 40 ×/1.25 NA oil immersion objective. Image stacks were collected through the specimen using sequential scanning and a standardized 120 nm z-sampling density. Selected image stacks were further subjected to deconvolution and restoration using theoretical point spread function and iterative maximum likelihood estimation algorithm of Huygens Professional software (Scientific Volume Imaging BV, Hilversum, the Netherlands).
Wound-healing assay
Confluent cells grown on glass cover slips were wounded with a rubber policeman, washed with phosphate-buffered saline and the wounded areas were cleared from inviable cells with suction. Cells were allowed to migrate at 37°C for 2–24 hrs, and were paraformaldehyde-fixed and labelled with rhodamine phalloidin (Molecular Probes/Invitrogen). In a subset of experiments, 100 ng/ml epidermal growth factor (EGF; Sigma-Aldrich), was used in attempts to induce migration in 43A cells.
Random cell migration
The cells were labelled with 20 μM CellTracker Orange (Molecular Probes/Invitrogen) at 37°C for 30 min. and 1 × 105 cells were seeded on coverglass bottom dishes (MatTek, Ashland, MA, USA) in pre-warmed RPMI supplemented with 10% FCS, and allowed to attach for 20 min. Epifluorescence imaging was performed with an Olympus IX71 inverted microscope with UPlanFl 10×/0.30 NA objective and TILL Photonics imaging system (TILL Photonics/Agilent Technologies, Munich, Germany) at 37°C. Images were acquired at 5-min. intervals for a total duration of 10 hrs. The trajectories were analysed with public domain ImageJ version 1.41e software (Rasband WS: ImageJ, National Institutes of Health, Bethesda, MD, http://rsb.info.nih.gov/ij/, 1997–2008) using MTrackJ plugin (by Erik Meijering). The experiments were repeated at least three times.
Cell invasion assay
Cell invasion was examined with modified Boyden chambers using Falcon FluoroBlok Individual Cell Culture Inserts (8 μm pore size; BD Biosciences) as previously described [20]. The filters were coated with 5 mg/ml Matrigel (BD Biosciences) for 1 hr. 5 × 104 cells suspended in RPMI was added to the inserts and RPMI alone was added to the lower chamber. The cells were allowed to grow and invade at 37°C for 24 hrs, after which the filters were fixed, labelled with rhodamine phalloidin, detached from the inserts with a scalpel and mounted on objective slides. The cells that had invaded through the matrix and filter pores were photographed using Olympus AX70 microscope with UPlanFl 40×/0.75 NA objective. The experiments were repeated at least three times.
Determination of podosome/invadopodia number and morphology on different ECM substrata
Cells were seeded on glass cover slips coated with 4 μg/ml type I collagen (Sigma-Aldrich), fibronectin, 2 μg/ml laminin-332, laminin-511, or left uncoated, and let to adhere at 37°C for 48 hrs. Fibronectin was purified from human plasma by gelatin-Sepharose affinity chromatography (Amersham Biosciences, Uppsala, Sweden) by standard methods. Purified human laminin-332 was obtained from Patricia Rousselle (Institut de Biologie et Chimie des Protéines, Unité Mixte de Recherche, Université Lyon, France), and purified human laminin-511 was from Kiyotoshi Sekiguchi (Institute for Protein Research, Osaka University, Japan). The cells were fixed and labelled with rhodamine phalloidin. The percentage of cells that had assembled at least two podosomes/invadopodia per cell were counted in 10 visual fields using 40×/0.75 NA objective as above.
In situ zymography for ECM degradation
Glass cover slips were coated with fluorescein-conjugated gelatin (0.2 mg/ml in 2% sucrose buffer; Molecular Probes/Invitrogen) for 2 hrs, crosslinked with 0.5% glutaraldehyde at RT for 15 min. and treated with 5 mg/ml NaBH4 for 3 min. The cover slips were quenched with two washes with RPMI at 37°C and coated with 1 μg/ml fibronectin for 1 hr. The cells were seeded on coated cover slips, incubated at 37°C for 2, 4, 6 or 15 hrs, and the samples were fixed and stained with rhodamine phalloidin or appropriate antibodies (Table 1). Foci of degraded matrix were visible as dark areas devoid of fluorescence. Images were acquired as above with Leica TCS SP2 AOBS confocal microscope (Leica Microsystems AG), deconvolved with Huygens Professional software (Scientific Volume Imaging) and 3D reconstructions and volume calculations of actin fluorescence intensity were performed with Imaris software (Bitplane, Zurich, Switzerland). The results were normalized against a standard curve generated using 0.5, 1.0 and 2.0 μm diameter carboxylate-modified microspheres (FluoroSpheres Size Kit, Molecular Probes/Invitrogen). Degradation areas were measured after maximum entropy thresholding and background subtraction with ImageJ.
Field emission scanning electron microscopy (FESEM)
The cells were cultured on glass cover slips and fixed with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.2) for 30 min. The samples were washed with cacodylate buffer, dehydrated through a graded series of ethanol, and dried with hexamethyldisilazane. The samples were sputter-coated with 20 μM chromium (Emitech K575X; Emitech, Kent, UK), and examined with a field emission scanning electron microscope (JEOL JSM-6335F; JEOL, Tokyo, Japan) at 5–15 kV and 0–45° inclination.
Immunoprecipitation and Western blot
For immunoprecipitation experiments of focal adhesion kinase (FAK), the cells were exposed to 60 mM KCl, 1 mM ethylenediaminetetraacetic acid, 2 mM EGTA, 1 mM cysteine, 40 mM imidazole, pH 7.0, supplied with 0.5% Triton X-100, 1 mM PMSF and 1 mM Na3VO4, on ice for 30 min. Elute was collected, centrifuged and used for immunoprecipitation. After pre-absorption with uncoupled GammaBind Plus Sepharose beads (Amersham Biosciences, Uppsala, Sweden), the samples were incubated with antibody-coupled Sepharose beads at 4°C overnight. For detection of integrins, the cells were surface-labelled with 0.2 mg/ml NHS-SS-Biotin (Pierce, Rockford, IL, USA), solubilized in 100 mM Tris, 150 mM NaCl, 1 mM CaCl2, 1 mM MgCl2, 1% Triton X-100, 0.1% SDS, 0.1% Nonidet P-40, pH 7.4, and applied to Sepharose beads. For detection of other proteins, the cells were lysed in Laemmli’s sample buffer. The proteins were separated with reducing or non-reducing 5–8% SDS-PAGE gels. For Western blots, the samples were transferred onto nitrocellulose filters and blocked with 5% dry milk in phosphate-buffered saline. Equal loading was verified with Ponceau S stainings or with MAb to β-tubulin. The immunoreactive bands were detected either with horseradish peroxidase-coupled anti-mouse or anti-rabbit immunoglobulins (Dako, Glostrup, Denmark), or with anti-phosphotyrosine MAb (Table 1) using Vectastain Elite® ABC Kit (Vector Laboratories, Burlingame, CA, USA) or SuperSignal® West Pico Chemiluminescent Substrate (Pierce).
Live cell imaging and total internal reflection fluorescence (TIRF) microscopy
EGFP-actin or EGFP-cortactin transfected cells were seeded on coverglass bottom dishes in pre-warmed CO2-Independent Medium (Gibco/Invitrogen) supplemented with 10% FCS. Images were acquired at 37°C using Olympus IX71 inverted microscope with PlanAPO 60×/1.20 NA water immersion objective and TILL Photonics imaging system. Cells were exposed to 480 nm monochromatic light for 20–100 ms with 10 sec. intervals for 30 min. or with 5-min. intervals for 15 hrs, and observed with longpass filter 520 nm. The image series were 2D deconvolved with Huygens Professional and analysed with ImageJ. In a subset of experiments, the cells were treated with cycloheximide, cytochalasin B, demecolcine (10 μg/ml; Sigma-Aldrich), or their combination. TIRF microscopy was performed with Olympus IX71 inverted microscope and CellR TIRF imaging system, 476 nm solid state 20 mW laser, PLAPON 60×/1.45 NA TIRF objective and Hamamatsu Orca ER CCD camera. For prolonged TIRF acquisition we used motorized Nikon Eclipse Ti-E TIRF imaging system with TI-ND6-Perfect Focus Unit and NIS-Elements AR software, Coherent Sapphire 488 nm solid state 20 mW laser, CFI APO 100×/1.49 NA TIRF objective, and Nikon DS-Qi1MC camera (Nikon Corporation, Toyko, Japan). Images were acquired at 37°C with 10 sec. intervals for 30–60 min., or two frames per minute for 6–12 hrs.
Fluorescence recovery after photobleaching (FRAP)
FRAP was performed at 37°C with Leica TCS SP2 AOBS confocal microscope (Leica Microsystems AG) with argon excitation line 488 nm and HCX PL APO LU-V-I 63×/0.9 NA water immersion objective, and a zoom factor of 4. With 512 × 512 pixel resolution and 1000 Hz scanning speed, bleaching was carried out with 5 high intensity short pulses (3.3 sec. total) and zoom-in function to increase the bleaching power. Fluorescence recovery was monitored for a total duration of 135 sec. under low intensity illumination. After raw data measurement, the background was subtracted, and the data was corrected and normalized taking into account laser intensity fluctuations and loss of fluorescence during recording [32]. Nonlinear regression analysis was performed for the relative intensity, and plateau recovery, half-time of recovery, mobile and immobile fractions were calculated with GraphPad Prism software (Prism 4, GraphPad Software, La Jolla, CA, USA). For each FRAP experiment, an area of 10 μm2 was bleached and the fluorescence recovery was measured from a 1.35 μm2 region covering the immediate vicinity of podosomes, invadopodia or cell extensions. When cell extensions were imaged, the average distance between the centre of mass of the extension and cell surface was approximately 3 μm.
Statistical analysis
Statistical analyses were performed with a two-tailed, unpaired t-test, the ANOVA with Bonferroni’s post hoc test for intergroup analyses, and nonlinear regression analysis (Prism 4.0 software). P < 0.05 was regarded as statistically significant.
Results
To study the mechanisms of cell adhesion and invasion of oral SCC cells, we used a previously characterized EMT cell model [18, 20]. Primary tumour cell line 43A presents an epithelial phenotype with E-cadherin at the cell–cell junctions and hemidesmosomes, and its endogenously EMT-experienced 43B from the recurrent tumour shows an E-cadherin to N-cadherin switch and a more fibroblastoid phenotype. EMT-driven 43B cells invade through the constituents of basement membrane (Matrigel) in significantly higher numbers compared with 43A cells. Trypsinization of 43A cells requires 45 min., thus resembling the podosome-mediated adhesion strength of, e.g. macrophages, compared to 5 min. required for 43B cells. Thus, in this model 43A cells represent a non-invasive and 43B cells an invasive phenotype of oral SCC [20].
EMT modulates cell migration and invasion capacity
Cell migration and cell invasion are key features underlying metastatic ability. In order to elucidate migration capabilities of 43A and 43B cells, we performed wound-healing experiments and evaluated the organization of actin by rhodamine phalloidin labelling. At 2 hrs after seeding, 43A cells showed tight cell–cell junctions without any indication of migration (Fig. 1A). In contrast, 43B cells showed cellular protrusions towards the wound (Fig. 1C). Dot-like actin accumulations resembling podosomes were detected in 43A cells (Fig. 1A and B, arrows). After 24 hrs, 43A cells migrated as a homogenous front towards the wound and maintained cell–cell contacts (Fig. 1B). In contrast, 43B cells migrated as single cells and presented an elongated phenotype. In addition, actin-rich accumulations and club-ended membrane extensions were detected in 43B cells (Fig. 1D and H, arrows). The actin-based structures could be detected in confluent cells as well, excluding a triggering effect for their formation by the wound. When EGF was applied to 43A cells to see whether augmented migration activity influences the actin structures, the cells migrated again as a common cell front, and the morphology of the actin structures remained the same. We also monitored random cell migration using time-lapse live-cell imaging. When the cells were allowed to migrate for 10 hrs, 43A cells showed mostly rotational motion with barely any true migration. In 43B cells, however, the migration trajectories were an average of fivefold longer compared with 43A (43A 66.0 ± 9.0 μm versus 43B 339.0 ± 23.7 μm, P < 0.0001) (Fig. 1E and F). This was also reflected in the ratio between the total length and the distance between the start and end positions of the trajectories, i.e. 43B cell trajectories showed higher directionality compared with 43A (P= 0.0050) (Fig. 1G). In invasion assays, only 43B cells invaded through the Matrigel to the lower side of the filter pores in Boyden chambers [20]. The actin-rich accumulations could also be detected in the invaded 43B cells (Fig. 1I and J). In a 48-hr study, 86 ± 1.9% of 43A cells and 98 ± 1.8% of 43B cells formed actin accumulations when seeded on glass (Fig. 1K). Seeding of cells on different ECM substrata, i.e. fibronectin, type I collagen, laminin-332, or laminin-511, did not significantly change either the number or the morphology of actin accumulations. However, 43B cells formed less actin-based structures on fibronectin versus glass (90.2 ± 2.5%versus 97.9 ± 1.8%, P < 0.05). Taken together, EMT-experienced 43B cells have higher migration and invasion capacity compared with their parent 43A SCC cells. Induction of migration in 43A cells does not change the morphology of the actin-based structures to resemble those in 43B cells.
Fig 1.

Wound-healing, random cell migration and cell invasion assays of oral SCC 43A and 43B cells. In wound-healing experiments, rhodamine phalloidin-labelled 43A cells presented tight cell–cell junctions but no migration after 2 hrs. After 24 hrs, 43A cells showed some migration as a homogenous front and maintained a close relationship with their neighbouring cells (A, B). Dot-like accumulations of actin that resembled podosomes could be seen in 43A cells (arrows). In contrast, EMT-experienced 43B cells showed membrane protrusions towards the wound area after 2 hrs (C). After 24 hrs, elongated 43B cells migrated as individual cells and presented long, filamentous cell extensions with actin accumulations (D, H, arrows). Time-lapse imaging of random cell migration of fluorophore-labelled cells (E–G) showed that the trajectory length of 43B cells was on average 339 ± 23.7 μm in 10 hrs, whereas in 43A cells it was only 66 ± 9.0 μm (E). The trajectories of 43B cells were fivefold longer than those of 43A cells. The migrated distance of both cells increased linearly with time (F). The ratio of the trajectory length and the distance between the start and end-point was significantly lower in 43B cells, indicating higher directionality compared with 43A cells. ***P < 0.0001; **P= 0.0050. In 24-hr invasion assay, 43A cells did not invade through the Matrigel (I). In contrast, 43B cells invaded through the Matrigel to the lower Boyden chambers and presented dot-like actin accumulations (J, arrows). Filter pores can be seen in photographs (I) and (J) as round rings. In 48-hr assay, the percentage of cells showing actin-based structures was high in both cells regardless of the ECM substrate. The only difference was that in 43B cells, the amount of actin-based structures was diminished when the cells were seeded on fibronectin compared with glass (K). #P < 0.05. Scale bars: 20 μm.
Morphological difference of adhesion structures at sites of ECM degradation
To study the cytoskeletal organization of oral SCC cells, we cultured the cells on fluorescein-conjugated gelatin and performed rhodamine phalloidin labelling. In non-invasive 43A cells, we detected polymerized actin along the cell cortex, but also at their ventral surfaces in phase-dense, podosome-like structures associated with foci of degraded ECM cavities after 2 hrs of plating (Fig. 2A, arrows). In contrast, in invasive 43B cells, strong stress fibres were found after 2 hrs, and notably, the actin-dense podosome-like structures at the cell-substratum interface were irregular (Fig. 2B, arrows). Based on the morphological differences and the facts that podosomes have been detected in keratinocytes and non-invasive carcinoma cells, and invadopodia in invasive carcinoma cells [7, 11, 15, 33], we call the matrix-degrading structures in 43A cells podosome-like structures and those in 43B cells invadopodia. Furthermore, 43B cells had long cell surface protrusions with actin rich club-ended tips. These extensions had accumulated actin throughout the filamentous stems to the broad substrate attached tip. The extensions in 43B cells were detected after 2 hrs of plating, but the club-like ends appeared after 5 hrs (Fig. 2C, arrowheads). The club-ended extensions localized within cavities of ECM, indicating gelatinase activity and a role in ECM degradation. The extensions and irregular invadopodia of 43B cells were also detectable in EGFP-actin transfected cells (Fig. 2D).
Fig 2.

Morphology of podosome-like structures and invadopodia in oral SCC 43A and 43B cells. The cells were seeded on fluorescein-conjugated ECM for 2–5 hrs, labelled with rhodamine phalloidin, and analysed with confocal microscope. 43A cells showed round, dot-like podosomes at 2 hrs after seeding (A, arrows), which produced ECM degradation beneath them. In 43B cells, actin accumulated as multiple, irregular, punctate structures, which also colocalized with the fluorescence-devoid ECM cavities (B, arrows). At 2 hrs, filopodia-like cell membrane extensions (arrowheads) were seen in 43B cells that matured at 5 hrs into club-ended, actin-containing cell extensions (C, arrowheads). Also these club-ended extensions localized to the of fluorescein-conjugated ECM cavities. EGFP-actin transfections and phase contrast images showed the complex invadopodia and club-ended extensions in 43B cells (D). Scale bar: 20 μm.
Club-ended extensions may originate from the dorsal cell membrane
To gain further structural knowledge on the club-ended extensions in 43B cells, we studied the cells under field emission scanning electron microscope. FESEM showed that 43A cells were flat, round epithelioid cells with broad lamellipodia (Fig. 3A). In contrast, 43B cells presented complex projections at their dorsal surface (Fig. 3B) that formed elongated extensions with club-like endings that adhered to the substratum (Fig. 3C and D).
Fig 3.

Field emission scanning electron microscopy of 43A and 43B cells. In FESEM, 43A cells presented a round, flat, epithelioid phenotype with broad lamellipodia (A, arrows). In contrast, 43B cells were covered with multiple and complex membrane projections (B–D). These cell extensions appeared to originate from granular buds at their dorsal cell membrane (C, arrows). In some occasions, the tips of the extensions were broad and flat (B–D), differing from those of, e.g. microspikes and filopodia. Scale bars: (A): 100 μm, (B): 10 μm, (C) and (D): 1 μm.
Differential spatial organization of podosome associated structural proteins in EMT-modulated invadopodia
Immunolocalization, immunoprecipitations and Western blots were used to characterize the possible differences in structural proteins between the 43A podosome-like structures and 43B invadopodia (Figs 4–6 and Table 2). Plectin, a cytolinker protein that connects the intermediate filaments to the basement membrane, localized in 43A cells at the podosome rings surrounding the actin cores (Fig. 4A) and at the cell membrane. In 43B cells, plectin did not localize to invadopodia, instead it was found associated with cytoplasmic fibrils resembling intermediate filaments [18]. αII-spectrin, another member of the spectraplakin superfamily with a role in crosslinking and anchoring of actin to the cell membrane, was detected at podosome rings of 43A cells; however, in 43B cells it was found only as a diffuse and faint immunoreactivity at the cell surface (Fig. 4B). Talin, a protein with a role in integrin-mediated cell adhesion, was detected in focal adhesions and podosome rings in 43A cells (Fig. 4C). In double-labelling experiments, Arp2/3 was detected at the cores of podosomes with talin at the podosome rings. In 43B cells, talin was associated only with focal adhesions but not with invadopodia. Vinculin, a protein that functions as a stabilizer for focal adhesions by crosslinking talin and actin, colocalized with cortactin in 43A cells in podosome cores (Fig. 4D). In 43B cells, vinculin was detected in focal adhesions and both the invadopodia and club-ended cellular extensions. As talin is a major structural adaptor between integrins and the actin cytoskeleton, we next studied which other proteins could replace it in invadopodia. Interestingly, tensin localized to invadopodia in 43B cells, whereas it was not found in podosomes of 43A cells (Fig. 4E). FAK, a catalytic adaptor of integrins, was localized at the podosome rings in 43A cells, but only in focal adhesions in 43B cells (Fig. 4F). Immunoprecipitations and Western blots showed equal expression levels of all these proteins in 43A and 43B cells (Fig. 4G). Only levels of FAK were slightly increased in 43A cells compared with 43B cells.
Fig 4.
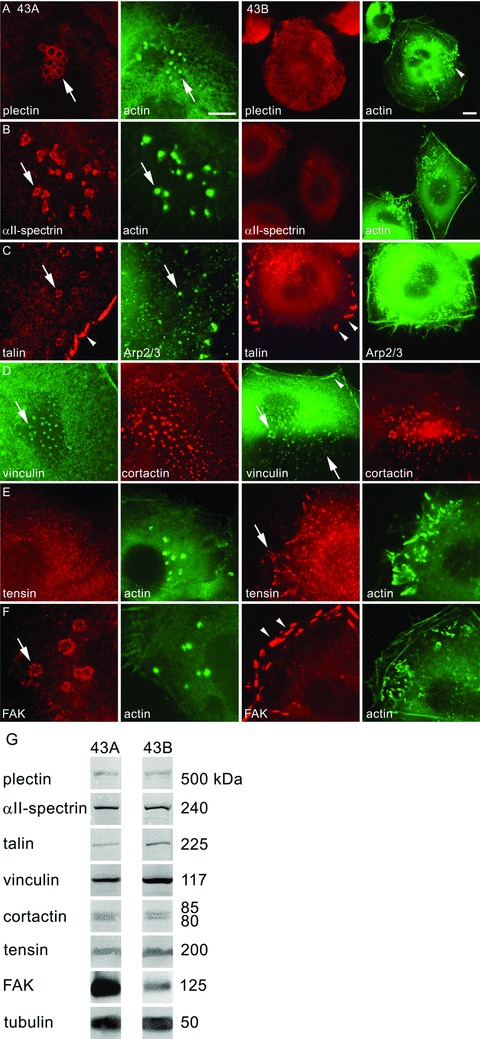
Localization of plectin, αII-spectrin, talin, vinculin, tensin and FAK, in 43A podosome-like structures and 43B invadopodia. Plectin immunoreactivity localized in peripheral rings of podosomes in EGFP-actin transfected 43A cells (A, arrow). However, in 43B cells, plectin did not localize to invadopodia that were characterized by EGFP-actin accumulation (arrowhead), but associated with cytoplasmic filaments. αII-spectrin immunoreactivity was found in the peripheral rings surrounding the podosome cores (B, arrow). In 43B cells, αII-spectrin was found as diffuse cell surface immunoreactivity. In double-labelling with Arp 2/3 (C), talin was detected both in podosome rings (arrow) and focal adhesions (arrowhead) in 43A cells, whereas in 43B cells, talin was found only in focal adhesions (arrowheads). Vinculin colocalized in double-labelling with cortactin in podosome cores, invadopodia and club-ended cell extensions (D, arrows). Vinculin immunoreactivity was found also in focal adhesions in 43B cells (arrowhead). In 43A cells, tensin reactivity was diffuse, but in 43B cells, it localized to invadopodia (E, arrow). FAK was confined to the peripheral ring of podosomes (F, arrow), but in 43B cells it localized only to focal adhesions (arrowheads). Scale bars: 10 μm. In Western blots of 43A and 43B cell lysates, the protein levels of plectin, αII-spectrin, talin, vinculin, cortactin and tensin were equal (G). In immunoprecipitation with Mab against FAK followed by Western blot with MAb against phosphotyrosine, FAK expression levels were somewhat stronger in 43A cells compared with 43B cells. Tubulin was used as a loading control.
Table 2.
Localization of different proteins to podosome-like structures and invadopodia
| Protein | 43A cell podosome-like structures | 43B cell invadopodia |
|---|---|---|
| Plectin | Ring | - |
| α-spectrin | Ring | - |
| Talin | Ring | - |
| Arp 2/3 | Core | Head and tail |
| Vinculin | Core | Head and tail |
| Cortactin | Core | Head and tail |
| Tensin | - | Head and tail |
| FAK | Ring | - |
| Integrin α3 subunit | Ring | Surrounding head and tail |
| Integrin β1 subunit | Ring | Surrounding head and tail |
| Integrin αv subunit | - | Head |
| Integrin β5 subunit | - | Head |
| Pacsin 2 | Ring | - |
| Filamin A | Core | -/late accumulation to head and tail |
| MT1-MMP | Core | Head |
Since talin and tensin bind to integrin β subunits, we also studied the expression of different integrin subunits in podosomes and invadopodia. Integrin α3 subunit was detected at the outer ring of 43A cell podosomes, and it surrounded 43B cell invadopodia at the cell membrane (Fig. 5A). A similar pattern was detected in the expression of integrin β1 subunit (Fig. 5B). Integrin αv subunit localized only to focal adhesions in 43A cells, whereas a specific localization to the heads of invadopodia was found in 43B cells (Fig. 5C). Only faint cytoplasmic immunoreactivity for integrin β5 subunit was found in 43A cells, whereas strong reactivity was found at the heads of invadopodia in 43B cells (Fig. 5D). 43B cells also contained abundant integrin αvβ5-positive focal and point adhesions (not shown). Immunoprecipitations showed prominent expression of Mr 150,000 integrin α3 subunit as well as its binding partner, Mr 110,000 integrin β1 subunit in both cells (Fig. 5E, lanes 1–2, 6–7). MAb against integrin αv subunit showed in 43A cells a single faint Mr 150,000 band corresponding to the size of integrin αv subunit (Fig. 5E, lane 3). MAb against integrin β5 subunit showed a similar single Mr ca. 100,000 β5 band in 43A cells (Fig. 5E, lane 4). In 43B cells, much stronger bands corresponding to integrin αv and β5 subunits were detected together, corroborating their high expression and colocalization as seen in immunofluorescence studies (Fig. 5E, lanes 8–9).
Fig 5.

Localization of integrin α3, β1, αv and β5 subunits. Immunoreactivity for integrin α3 subunit was detected diffusely at the cell surface, but especially in the podosome ring of EGFP-actin transfected 43A cells (A, arrow). In 43B cells, integrin α3 subunit localized at the plasma membrane enveloping the invadopodia and cell extensions (arrow). Integrin β1 subunit, a potential binding partner of integrin α3 subunit, also localized in the podosome ring in 43A cells and enveloped the invadopodia heads and tails in 43B cells (B, arrow). Integrin αv subunit immunoreactivity was not found in 43A podosomes (C), but localized to focal adhesions (arrowhead). However, in EGFP-transfected 43B cells, integrin αv subunit immunoreactivity was found at, or occasionally around (arrow) the invadopodia heads attached to the plasma membrane. Integrin β5 subunit reactivity was faint in 43A cells and absent in podosomes (D), whereas in 43B cells, it was found strictly at the actin head-plasma membrane interface (arrow). Scale bars: 10 μm. Immunoprecipitations with MAbs against integrin α3 (E, lanes 1, 6) and β1 (lanes 2, 7) subunits showed strong bands of integrin α3β1 in both cells, indicating heterodimer formation. MAb against αv detected only a single band corresponding to the size of integrin αv in 43A cells (lane 3), but detected two strong bands of integrin αvβ5 heterodimer in 43B cells (lane 8). MAb against integrin β5 subunit showed again a single band in 43A cells, but two strong bands of αvβ5 in 43B cells (lanes 4, 9). Lanes 5, 10: negative controls without primary antibody.
Pacsin 2 has a dual role as an inductor of membrane curvature and organizer of cortical actin network. Therefore, we hypothesized that pacsin 2 might function as part of the maintenance machinery of podosome-like structures and invadopodia. As in previous experiments, the cells were grown on fluorescein-conjugated ECM for 2–15 hrs and analysed under confocal microscope. In 43A, pacsin 2 was found only in podosomes where it localized at the ring structures (Fig. 6A). In 43B, only faint immunoreactivity at the cell surface was detected with no localization to invadopodia. Filamin A, which crosslinks actin filaments and has been to shown to bind chicken homolog of pacsin 2, was concentrated in 43A cells at cores of podosomes already at 2 hrs after plating filling also the ECM degradation cavities (Fig. 6C). However, filamin A did not accumulate to invadopodia until 15 hrs after plating. Therefore, there is a difference also in the organization of both pacsin 2 and filamin A between the podosomes and invadopodia. These results were confirmed with EGFP-filamin A transfections (Fig. 6E). EGFP-filamin A fusion protein showed colocalization with pacsin 2 in 43A cells, but it did not lead to invadopodial pacsin 2 accumulation in 43B cells. Finally, and in concordance with the noted ECM degradation activity of both the podosomes and invadopodia, immunoreactivity for MT1-matrix metalloproteinase (MMP), a protease known to confer proteolytic degradation activity to invadopodia, was found to associate with podosomes, invadopodia and the club-ended cell extensions, together with the ECM degradation cavities in both 43A and 43B cells (Fig. 6F). However, either MMP-2 or MMP-9 immunoreactivity were not detected in association with the degradation cavities (not shown). Western blots showed equal expression levels of pacsin 2, filamin A and MT1-MMP in 43A and 43B cells (Fig. 6G).
Fig 6.

Localization of pacsin 2, filamin A and MT1-MMP in 43A podosome-like structures and 43B invadopodia. Confocal images showed membrane-bending protein pacsin 2 immunoreactivity at the podosome rings that were associated also with ECM degradation cavities after 2 hrs of plating (A). Pacsin 2 immunoreactivity was not found in or near invadopodial structures (B). Pacsin-binding protein filamin A was found in 43A podosome cores after 2 hrs, but it accumulated to invadopodia only after 15 hrs of plating (C, D, arrows). Even then, filamin A was detected only in a small proportion of invadopodia. EGFP-filamin A transfections showed that filamin A and pacsin 2 colocalized in 43A podosomes, but the transfections did not result in pacsin 2 expression in 43B invadopodia (E). MT1-MMP was found as dot-like accumulations in the proximity of podosomes and invadopodia and in association with ECM cavities (F). MT1-MMP also localized to cell extensions of 43B cells, indicating that they possess gelatinase activity and thus ECM degradation capability. Scale bars: 20 μm. Western blots indicated the presence of pacsin 2, filamin A and MT1-MMP in both 43A and 43B cells (G). Tubulin was used as a loading control.
Podosome-like structures and invadopodia are characterized by different size and ECM degradation capacity
Confocal imaging and 3D reconstructions showed phalloidin-labelled podosomes in 43A cells as relatively large and wide columns, whereas the more numerous (43A 12.3 ± 0.7 versus 43B 32.4 ± 1.6 per cell, P < 0.0001) invadopodia in 43B cells were characteristically narrow funnel-shaped columns, agreeing with the notion that actin tails localize to invadopodia [34] (Fig. 7A and B). Volume measurements of deconvolved isosurface reconstructions demonstrated significantly smaller invadopodial volumes compared with podosomes (Fig. 7E). Podosome volumes in 43A cells increased steadily between 2–6 hrs of plating (P < 0.05). In comparison, invadopodia in 43B showed much less increase and were an average of 2.6-fold smaller after 4 and 6 hrs of plating (P < 0.01 for both). As in previous experiments both the podosomes and invadopodia were found to protrude through the fluorescein-conjugated gelatin matrix, we next performed a modified fluorescent in situ zymography assay for ECM degradation. In both cells, ECM degradation was specific to the immediate vicinity of podosomes and invadopodia (Fig. 7C, D, G and H). The number of degradation cavities per cell was significantly higher in 43B compared with 43A cells (126 ± 13.6 versus 70 ± 12.1, P < 0.01). Consequently, the total degradation area per cell was significantly larger in 43B compared with 43A cells (231 ± 36.1 μm2versus 118 ± 18.1 μm2, P < 0.01) (Fig. 7F) although the individual degradation cavity areas did not differ significantly (43A 2.1 ± 0.2 μm2versus 43B 1.7 ± 0.1 μm2). At 4 hrs after plating, the cavities under 43A cells were predominantly rounded, whereas in 43B cells, the shapes and sizes were substantially diverse (Fig. 7G and H). The cavities under 43B cells were larger than the actin cores of invadopodia, whereas in 43A cells, the actin cores filled the cavities completely. In addition, some small degradation areas without the associated actin core were found underneath 43B cells that may represent the footprints of short-lived structures that sensor the surrounding substrate [15]. Taken together, these results suggest that podosomes of primary oral SCC are relatively large structures that produce a restricted degree of ECM degradation, whereas invadopodia of the recurrent EMT-experienced SCC have smaller volumes but are more numerous and hence exhibit more gelatinase activity and ECM degradation.
Fig 7.

Podosome and invadopodia size and ECM degradation activity. 3D reconstructions of confocal images obtained from cells grown on fluorescein-conjugated gelatin showed dome-like, wide podosome columns in phalloidin-labelled 43A cells, whereas invadopodia in 43B cells were narrow funnel-shaped structures (A, B). Podosomes and invadopodia protruded through the fluorescein-labelled ECM (C, D). Nucleus, blue; gelatin, green; isosurface rendering of actin structures, grey; actin, red. Podosomes increased their relative sizes at 2–6 hrs (n= 5–9 cells/time-point) (**P < 0.01 by t-test and #P < 0.05 by ANOVA) (E). The mean relative volumes of 43A podosomes were significantly higher than those of 43B invadopodia at 4–6 hrs (**P < 0.01 for both), being at 6 hrs 1.05 ± 0.14 μm3, whereas the volumes of invadopodia were approximately 2.6-fold less (0.40 ± 0.03 μm3). In situ zymography for ECM degradation showed that 43B cells produced significantly more degradation cavities per cell, and the resorption area per cell was also larger compared with 43A cells (**P < 0.01; n= 33 cells for 43A and n= 32 cells for 43B) (F). After 4 hrs, the cavities of 43A cells were predominantly round, whereas substantial diversity in cavity shape and size was detected for 43B cells (G, H). Scale bars: (A), (B), (G), (H): 10 μm; (C), (D): 2 μm.
Podosomes are highly stable but are changed in EMT to actin comet-based invadopodia
To evaluate the dynamics underlying the function of podosome-like structures and invadopodia in more thorough, we used live-cell imaging for EGFP-actin and EGFP–cortactin transfected cells. Podosomes of 43A cells were highly stable and had a life-span well over 60 min. (Fig. 8A, Supplementary Movie 1). Cortactin colocalized into these structures (not shown). Strikingly, actin and cortactin were detected in 43B cells in tail-resembling structures originating from invadopodia (actin comets) (Fig. 8A, Supplementary Movies 2 and 3). Actin tails moved rapidly around in a circular manner and the heads were embedded in the ventral cell membrane. In some occasions, we could see that actin tails were dissociated from the membrane moving across the cytoplasm and eventually disappearing (Supplementary Movie 4). However, invadopodia were predominantly long-lived, although occasional rapid disassembly and new assembly could be detected (Supplementary Movie 5). Actin and cortactin were detected in 43B cells in the club-ended cell extensions, or externalized invadopodia, where they were found in rapid motion at the retractive side of migrating cells sometimes also reaching the boundaries of other cells (Supplementary Movies 3 and 6). EGF treatment induced migration in 43A cells, during which they assembled podosome-like structures (Supplementary Movie 7). This result suggests that the switch from podosomes to invadopodia is not due to increased migration activity. Next, we studied the podosomes and invadopodia with live-cell TIRF microscopy that allows high contrast visualization of fluorescent molecules and adhesion structures at the cell-substrate contact. The podosomes and invadopodia were located within the narrow TIRF evanescent field that extends only approximately 100–200 nm into the cell providing evidence of immediate proximity to the substratum (Fig. 8B). During pro-longed 12-hr TIRF acquisition, podosomes were practically immobile and extremely long-lived with a surrounding slowly moving fluorescent halo possibly consisting of actin monomers surrounding the podosomes. However, during cell migration the externalized podosomes were quickly disassembled at the retractive side of 43A cells (Supplementary Movie 8). In 43B cells, the invadopodial heads were also immobile but showed some degree of disassembly during the acquisition period. Growing actin filaments moved rigorously within the cell-substrate interface but no similar fluorescent halo around the invadopodia could be detected as in 43A podosomes (Supplementary Movie 9). Lateral movement of invadopodia tails and transient motion of 43B cell extensions could also be detected (Supplementary Movie 10). Consistently occurring fusion or fission events could not be detected in either the 43A podosomes or 43B invadopodia. These observations suggest that podosomes and invadopodia have a relatively long life-span, and that the actin tails truly originate from the heads of invadopodia. The substructures of podosomes and invadopodia appear to undergo constant remodelling possibly from different pools of origin.
Fig 8.

Dynamics of podosome-like structures and invadopodia. EGFP-actin transfected cells were followed with wide-field time-lapse imaging for 30 min. 43A podosomes were immobile dot-like structures, whereas 43B invadopodia were complex structures with rapid motion of propelling actin tails (A). Arrows indicate single podosomes or actin comet-embedded invadopodia. Inset: A magnified image of the actin tail marked by an arrow. Time-lapse TIRF imaging showed that podosome-like structures, invadopodia and cell extensions were within the narrow TIRF evanescent field and thus in immediate proximity to the substrate (B). In prolonged 12-hr time-lapse, trafficking of EGFP-actin fluorescence to podosomes seemed sparse and no new podosomes were formed. A halo of fluorescent EGFP-actin (arrows) was detected around podosomes. In contrast, rapid motion was detected in 6-hr time-lapse at the basal surface of 43B cells, including actin filaments (arrowheads). Actin comet-based invadopodia were long-lived (arrow). Some propelling movement of the actin tails could also be detected at this level, as well as undulating movement of the club-ended cell extensions. No similar fluorescent EGFP-actin halo as in 43A cells was detected around invadopodia. Scale bar: 20 μm.
Podosome-like structures and invadopodia are resistant to inhibitors of actin and tubulin polymerization
In order to extend these notions on the mechanisms underlying the turnover of podosomes and invadopodia, we used inhibitors of actin and tubulin polymerization and followed the events in EGFP-actin transfected cells with wide-field live-cell imaging. As expected, cytochalasin B led to a rapid distortion of the actin cytoskeleton and resulted in cytoplasmic aggregates within seconds. In contrast to the previously presented hypothesis that invadopodia would be rapidly abolished, cytochalasin B barely affected the morphology of podosomes or invadopodia (Fig. 9). In fact, their sizes appeared to increase as a result of the treatment. Only after several hours, both podosomes and invadopodia were seen to disappear. Disruption of the microtubule system with demecolcine for 30 min. did not affect either the podosomes or invadopodia (Fig. S1A). Simultaneous treatment with cytochalasin B and demecolcine led to a collapse of the cellular organization, but had a similar effect as cytochalasin B alone to podosomes and invadopodia (Fig. S1B). However, when adhered cells were treated with cycloheximide that blocks the synthesis and secretion of proteins, no podosomes or invadopodia developed within the 60-min. follow-up period, suggesting that de novo synthesis of proteins is required for the organization.
Fig 9.

Podosome-like structures, invadopodia and invadopodial cell extensions are resistant to inhibitors of actin and tubulin polymerization. The dynamics of EGFP-actin were monitored using wide-field time-lapse microscopy for 30 min. after applying the inhibitors. Treatment with cytochalasin B (10 μM), an inhibitor of actin polymerization, did not change the morphology of podosomes or invadopodia, although cell shape and cytoskeletal actin organization underwent major alterations. The club-ended cell extensions could still be seen in 43B cells after disruption of the actin cytoskeleton (arrows). Scale bar: 20 μm.
Fluorescence recovery after photobleaching indicates faster protein turnover in podosome-like structures compared with invadopodia
The finding that both podosomes and invadopodia are highly stable with relatively long life-span led us to investigate the dynamics and turnover of actin, cortactin and filamin A in transfected primary and EMT-experienced SCC cells. FRAP analyses indicated that EGFP-actin fusion protein had a rapid but differential turnover in podosomes and invadopodia. Plateau of fluorescence intensity recovery after photobleaching was reached significantly faster in 43A compared with 43B cells (64.4 ± 5.2s versus 80.6 ± 5.5s, P < 0.05) (Fig. 10A and B). In 43A cells, there was a complete EGFP-actin recovery, however, in 43B cells the mobile fraction was less (92%) (P < 0.001). Calculations of the half-time of recovery showed significantly faster corresponding characteristic times in podosomes than in invadopodia (8.0 ± 0.6s versus 10.5 ± 1.0s, P < 0.05). We also measured fluorescent recovery of EGFP-actin for the club-like extensions found in 43B cells. As expected, the recovery after photobleaching was slower compared with the internal cytoplasmic invadopodia, but was still surprising rapid. The plateau of recovery was reached in 74.4 ± 16.5s with mobile fraction of 63%, and the half-time of recovery in 11.9 ± 3.2s (Fig. 10B). These findings fit well with the morphological restriction of actin diffusion in these extracellular filamentous structures, and further suggest that they represent a dynamic structure that may contribute to localized ECM degradation and possibly cell invasion. EGFP-cortactin recovery showed even more profound differences between the podosomes and invadopodia. The plateau of fluorescence recovery of EGFP-cortactin in 43A cells was reached in 22,04,7s with half-time of recovery in 2,40,5s. In EMT-experienced 43B cells the plateau was reached only after 103.1 ± 6.2s, and the half-time of recovery was also significantly slower, 20.1 ± 2.3s (P < 0.0001 for both). The mobile fraction was 100% for podosomes and 95% for invadopodia resembling the difference for the two cell types in polymerization rate of EGFP-actin (P < 0.05). However, as the bleaching efficiency of EGFP-cortactin in 43A cells was less compared with the other FRAP analyses (Fig. 9B), we believe that the true EGFP-cortactin recovery in podosomes is even more rapid due to simultaneous bleaching and fast recovery. Finally, we also addressed the possibly different temporal organization and turnover rate of filamin A in 43A and 43B cells. As for EGFP-actin and EGFP–cortactin, podosomes showed more rapid recovery of EGFP-filamin A compared with invadopodia (35.2 ± 3.0s versus 44.2 ± 4.1s). Half-time of recovery in podosomes was significantly faster compared with invadopodia (3.8 ± 0.2s versus 5.1 ± 0.5s, P < 0.05). Mobile fractions were 100% and 98%, respectively, representing almost complete recovery in both podosomes and invadopodia (Fig. 10B). Thus, in 43A podosomes the rate of recovery of EGFP-filamin A was more rapid compared with EGFP-actin but slower than that of EGFP-cortactin. In contrast, in 43B invadopodia the rate of recovery of EGFP-filamin A was fastest followed by EGFP-actin with EGFP-cortactin being much slower compared with all the other FRAP analyses. Taken together, wide-field and TIRF imaging indicated slower structural 43A podosome turnover compared with 43B invadopodia. In contrast, FRAP analyses indicated higher actin, cortactin and filamin A recovery in 43A podosomes compared with 43B invadopodia. These findings are implicative for significantly different turnover rates between the podosomes and invadopodia, that is likely to include less rigid cortactin-mediated maintenance of actin polymerization and hence the shorter life-span of the invadopodial complex in invasive recurrent 43B SCC cells.
Fig 10.

FRAP of EGFP-actin, -cortactin and -filamin A in transfected cells. Actin fluorescence intensity in podosome-like structures, invadopodia and cell extensions before photobleaching, immediately after photobleaching, at half-time of recovery (t 1/2), and at plateau of recovery (A). Rectangle, magnified areas show the FRAP analyses of single podosomes and invadopodia; circle, photobleached area. Kinetics of EGFP-actin, -cortactin and -filamin A in podosome-like structures, invadopodia and cell extensions (B). Plateau of recovery in 43A podosomes was reached in 64,45,2s and in 43B invadopodia in 80.6 ± 5.5s (n= 25; P < 0.05). Half-time of recovery of EGFP-actin was 8,00,6s in podosomes, whereas it was significantly longer (10.5 ± 1.0s) in invadopodia (P < 0.05). The mobile fraction of EGFP-actin in invadopodia was less (P < 0.001) compared with the total recovery in podosomes. In 43B cell extensions (n= 9), only 63% recovery was gained, and plateau was reached at 74.4 ± 16.5s with t 1/2 at 11.9 ± 3.2s. Half-time of recovery for EGFP-cortactin was extremely rapid in podosomes (2.4 ± 0.5s), whereas it was 8.4-fold slower (20.1 ± 2.3s) in invadopodia (n= 25; P < 0.0001). Plateau was reached in 22.0 ± 4.7s in 43A and 103.1 ± 6.2s in 43B cells with 100% and 95% mobile fractions. As for EGFP-actin and -cortactin, EGFP-filamin A showed more rapid turnover in podosomes compared with invadopodia (35.2 ± 3.0s versus 44.2 ± 4.1s). The corresponding half-time of recovery was significantly faster in podosomes than in invadopodia (3.8 ± 0.2s versus 5.1 ± 0.5s, n= 25, P < 0.05). The S.E.M. are shown with non-linear regression fit. Scale bars: 10 μm, in small figures: 2 μm.
Discussion
The present study shows that podosome-like structures of non-invasive SCC cells differ by composition and dynamics from invadopodia of invasive, EMT-experienced cells obtained from their recurrence. Invadopodia with propelling actin comet-like tails were detected in migrating and invading cells, thus suggesting a functional role for them. Podosome volumes increased after adherence; however, invadopodia remained smaller but produced more ECM degradation cavities that involved the presence of MT1-MMP. Although both podosomes and invadopodia had a long life-span, FRAP analysis indicated a rapid turnover of actin, cortactin and filamin A in both types of SCC cells. The club-ended cell extensions that were characterized to contain similar constituents as invadopodia, maintained the influx of new proteins, indicating that they were not merely deserted organelles but active and functional structures likely to represent part of the invasion machinery of EMT-experienced SCC cells.
Podosomes are depicted as ring structures, in which adhesion molecules, such as integrins, define the ring that surrounds the vertical actin-rich core [10, 35]. In contrast, invadopodia generally appear as puncta with no strict separation between the components [2]. In line with this definition, podosome-like structures in 43A cells were characterized with actin, cortactin and Arp2/3 within the core structures, and several of previously characterized proteins within the outer rings. The morphology and protein contents of 43A cell podosome-like structures highly corresponded to those depicted in podosomes of endothelial cells, epithelial carcinoma cells and keratinocytes [7, 33, 36]. During the present study, several previously unrecognized proteins were characterized for their association with podosomes, including filamin A in core structures, and plectin, αII-spectrin and pacsin 2 within the outer rings. Characteristic to 43B cells was the lack of plectin, αII-spectrin, talin, FAK, and pacsin 2 in the invadopodia complexes. Although it is somewhat unclear whether podosomes and invadopodia exist in vivo, studies describing podosomes in leukocytes migrating through endothelial vessel walls and invadopodia in extravasating tumour cells support their existence [8, 37]. We detected podosomes and invadopodia in functional wound-healing and cell invasion assays. Podosomes and invadopodia assembled spontaneously and shortly after seeding, whereas the club-ended cell extensions emerged later. These club-ended structures could originate from the highly similar short extensions detected with field emission scanning electron microscope near the cell surface, or from left-behind invadopodia in migrating cells. Apart from the clubbed ends, the slender extensions resembled filopodia, which also contain a core of actin filaments and may function as sensors that explore the cell surroundings [38]. However, as the molecular structure resembled that of the invadopodia, we gather that these structures are ‘externalized invadopodia’.
Plectin interacts with F-actin, microtubules and intermediate filaments [39]. In stratified epithelia, plectin localizes to hemidesmosomes and mediates adhesion with cytokeratin filaments, integrin α6β4 and basement membrane. We found plectin in podosome rings, but also on the cell surface, where it localized to hemidesmosomes. In contrast, invadopodia were completely devoid of plectin. The structural role of plectin, as well as its putative role as scaffolding and signalling protein [39], go well together with the characteristics of oral SCC podosomes. In smooth muscle cells, plectin may provide molecular guidance for podosome formation and positioning through its dual interaction with intermediate and actin filaments [40]. Our results support these suggestions and further propose that plectin could have a role in stabilization of podosomes.
By crosslinking actin and linker proteins to cell membrane, spectrins organize the submembraneous cytoskeleton and maintain cell shape [39, 41]. In 43A cells, we found αII-spectrin solely in podosome rings, whereas it was not found in invadopodia. In 43B cells, αII-spectrin immunoreactivity was found diffusely on the cell surface. Recently, another member of the spectraplakin family, ACF7, was shown in keratinocytes to guide microtubule tips along actin filaments to focal adhesions [42]. Spectraplakins were thus suggested to promote a dynamic link between the intermediate and actin filaments, an essential requirement for cell migration. These findings suggest that αII-spectrin may operate in membrane scaffolding, protein accumulation, signal transduction and even in microtubule guidance in podosomes.
Recruitment of talin and vinculin in focal adhesions regulates adhesion dynamics and cell migration [43]. We found talin in podosome rings. In EMT-driven 43B cells, talin localized only to focal adhesions, whereas vinculin localized to both podosomes and invadopodia. As talin is an important structural adaptor between integrins and actin cytoskeleton, we studied other candidate proteins that might have substituted its function. In our cell model, tensin immunoreactivity was found in 43B cell invadopodia and focal adhesions but not in 43A cell podosomes. Talin and tensin competitively bind the tails of, e.g. integrin β1 and β5 subunits and mediate adhesion strength to actin filaments [44]. The talin-tensin switch may even transform a structural adhesion to an adhesion that participates in signalling [44]. FAK facilitates signal transduction pathways from adhesion sites to the cytoplasm [44]. In 43A cells, podosome rings showed strong immunoreactivity for FAK, which was absent in 43B cell invadopodia. Supporting our findings, the presence of FAK has been shown in podosomes of Src-transformed fibroblasts [45], but it is neither necessary for invadopodia assembly in colon carcinoma cells nor localizes to invadopodia in breast carcinoma cells [46, 47].
Integrins mediate adhesion and function in outside-in and inside-out signalling between the cytoskeleton and ECM. We found enriched immunoreactivity for integrin α3β1 in podosome rings and, in a unique pattern, at the cell membrane enveloping the invadopodia. Previously, integrin α3β1 immunoreactivity has been detected in podosomes of bladder carcinoma cells [33], and it participates in docking of gelatinolytic enzyme seprase to invadopodia of melanoma cells [48]. Integrin αvβ5 is associated with migration in several cell types, including keratinocytes in wound healing [49]. In our cell model, 43A cells showed low levels of both integrin αv and β5 subunits that did not localize to podosomes. In contrast, EMT-driven 43B cells had up-regulated levels of integrin αvβ5 that localized sharply to the heads of invadopodia. The novel finding of integrin αvβ5 localization to invadopodia may relate to the transformed mesenchymal phenotype of 43B cells, as αvβ5 has been implicated previously in EMT occurring in tooth development [50].
Pacsins function in vesicle formation and transport by gathering interacting proteins, such as N-WASP and dynamin, to sites of endocytosis [51]. F-BAR domain enables them to induce and regulate membrane curvature and conduct membrane tubulation [52, 53]. In the present study, pacsin 2 confined topologically to the peripheral podosome ring, but it was not found in 43B cells. These results suggest that pacsin 2 may operate as an inductor or regulator of membrane curvature for podosomes. Filamin A, a potential downstream effector of pacsin 2, crosslinks actin into orthogonal networks or bundles [54]. We detected filamin A in podosomes after 2 hrs adhesion, whereas it emerged in invadopodia only after 15 hrs. These results suggest a sequential model, in which pacsin 2 recruits filamin A to podosomes and induces stabilization via crosslinking actin fibres that may lead to decreased motility. Invadopodia lacked pacsin 2, and possibly therefore the recruitment of filamin A was delayed by several hours. Pacsin 2 or filamin A were not, however, needed for the initial organization of invadopodia, or for the ability of ECM degradation.
Actin comets or tails detected in 43B cells represented a dynamic pattern of actin remodelling. The tails could also be seen in 3D reconstructions of invadopodia. Similar structures have been described in propelling tails of, e.g. Listeria monocytogenes pathogen, which exploits the host’s actin polymerization machinery to induce actin-based motility [55]. Actin comet-based invadopodia in 43B cells were long-lived, and produced degraded ECM areas underneath them [34]. With time-lapse microscopy, we captured events of actin shuttling, where an actin comet head was released from its attachment to the ventral cell membrane and moved across the cytoplasm. As actin comets are suggested to produce the force that enables movement of intracellular organelles [55], they could de facto generate the membrane protrusions in invadopodia. An actin cloud surrounding podosomes contains a reservoir of G- and F-actin that is thought to serve as a storage site for actin filament formation [56]. With TIRF microscopy, we detected a fluorescent halo possibly consisting of actin monomers surrounding the podosomes, whereas no such halos were found in the vicinity of invadopodia in EGFP-actin transfected cells. In contrast, replenishment of invadopodia in FRAP experiments seemed to occur through the actin tails. Therefore, the organization of actin filaments differs between podosomes and invadopodia, which may explain the slower recovery rates and the turnover of actin, cortactin and filamin A in the invadopodia complex.
Microtubules influence cell adhesion and direction of migration by modulating cell adhesion stability. In macrophages, podosome clustering is microtubule-dependent, although they are not required for podosome formation in all systems [57, 58]. In our studies, podosome-like structures and invadopodia were highly resistant to inhibitors of tubulin and actin polymerization. Cytochalasin B treatment led to a rapid collapse of actin cytoskeleton but did not abolish either the podosomes or invadopodia. Podosomes and invadopodia disappeared only after several hours. Similarly, demecolcine, an inhibitor of tubulin polymerization did not affect the existing podosomes or invadopodia. However, during these experiments no new podosomes or invadopodia developed. These results may be due to the complex, multiprotein adhesion structure localized to the cell membrane, and the strong invaginating or protruding contacts with the substrate. Initial assembly of podosomes and invadopodia may still require intact microtubules, as no new structures emerged during the follow-up period.
There are some controversies between previous reports and the present study concerning the dynamics and life-span of podosomes and invadopodia. The majority of macrophage podosomes fragment from pre-existing podosome precursors or assemble from existing mature podosomes at the leading edge [57]. We did not detect such events in oral SCC podosomes, since they all formed de novo, and required new protein synthesis. Also, the life-span of podosomes in osteoclasts and macrophages are short [56, 57], whereas oral SCC podosomes were stationary for over 12 hrs. Life-span of invadopodia, on the other hand, apparently can vary from few minutes to several hours [15]. The prolonged life-span of 43A cell podosomes is the only characteristic that resembles those affiliated to invadopodia. The long-lived invadopodia of mammary carcinoma cells were considered as more mature compared with the ones that had a short life-span, as they had a higher content of actin. Furthermore, invadopodia with short life-span had the ability to move [15]. In our wide-field and TIRF time-lapse studies, invadopodia generally had a long life-span. However, we could not detect movement of individual invadopodia in the immediate contact of the substrate, although actin dynamics inside the EMT-experienced SCC cells were intense. In addition, ECM degradation studies showed several small degradation cavities without the presence of invadopodia, as well as elongated, irregular cavities that are likely to result from the turnover and even lateral movement of small short life-span invadopodia probing the environment.
Photobleaching of osteoclast podosomes have shown that the recovery to plateau for actin is approximately 1 min., whereas cortactin recovery is only through the formation of new podosomes [56]. Our results are somewhat contradictory, as we found that in 43A SCC cells actin, cortactin and filamin A all recovered extremely quickly (half-time of recovery <10 sec.) after photobleaching of podosomes. However, in EMT-experienced 43B cells the recovery of actin, cortactin and filamin A to invadopodia was constantly slower than that of podosomes. Concerning cortactin, the half-time of recovery was 8.4-fold slower in invadopodia, and was not derived from organization of new invadopodia complexes but from cytoplasmic diffusion into the existing structure [56]. Furthermore, the present results show the kinetic actin turnover in 43B cell extensions, providing evidence that they are actively modulated. Our FRAP and TIRF experiments suggest that actin polymerization in invadopodia is through the tails. The differences between previous and the present study may stem from different cell systems, cell dynamics, or experimental settings. In contrast to osteoclast podosomes, oral SCC podosomes and invadopodia are stable, easily detectable structures, which therefore gives strength to the results. Furthermore, osteoclast podosomes have been photobleached in large colonies [56], whereas we focused on the dynamics of single podosomes and invadopodia, thus aiming not to disturb the surrounding cytoplasmic milieu and allowing a free access for the EGFP fusion proteins to diffuse and polymerize. Taken together, the results suggest that podosomes represent more complex structures with highly regulated turnover and hence with longer life-span, whereas the composition of invadopodia is more labile and they are assembled in higher numbers during invasion.
It is unclear whether podosomes and invadopodia develop from a common precursor or whether podosomes can transform into invadopodia. It has been proposed that cells may have the basic ability to assemble both podosomes and invadopodia, depending on whether the cells reside on digestible matrices, or on the signalling cascades activated at certain time-points [3, 59, 60]. In our experiments, podosomes and invadopodia remained unaltered regardless of the cell substratum. However, the fundamental difference between our cell lines is EMT, which is linked to up-regulated expression of transcription factors ZEB-1 and ZEB-2 [18, 20]. Shared pathways regulating EMT as well as podosomes and invadopodia are controlled by, e.g. receptor tyrosine kinases, Src, transforming growth factor-β/BMP, PI3 kinase and Rho GTPases [16, 19, 21, 58]. Furthermore, major targets of EMT pathways include cell–cell and cell-ECM adhesion and invasion machineries, thus, it is reasonable that podosomes and invadopodia could be affected. In 43A cells, induction of cell migration in wound-healing experiments or in time-lapse imaging of individual cells did not change the appearance of podosomes to those of invadopodia, suggesting a more complex effect induced by EMT. Therefore, our results suggest that invadopodia may resemble aggressive or modified versions of podosomes, as podosomes are replaced in EMT by invadopodia. The difference between podosomes and invadopodia may not necessarily rely only on their molecular composition, but also on their functional differences. To our knowledge, this is the first study depicting a link between EMT, podosomes and invadopodia.
In conclusion, podosome-like structures of non-invasive oral SCC cells and invadopodia of their EMT-experienced, invasive recurrent are morphologically and dynamically different, although both were able to degrade ECM but to a different degree. Proteins common to invadopodia were found also in dynamic, probing extensions of 43B cells, which enabled constant contact with ECM in migration. Plectin, αII-spectrin, talin and FAK were only found in podosome rings, which were possibly related to the stable appearance of podosomes. A potential talin to tensin switch occurred in invadopodia, and integrin αvβ5 appeared only in the tips of invadopodia. Oral SCC podosomes contained both pacsin 2 and filamin A, whereas invadopodia totally lacked pacsin 2 and accumulated filamin A only after several hours. FRAP analysis showed rapid, although not similar, recovery rates of actin and filamin A in both podosomes and invadopodia, and a markedly delayed recovery of cortactin in invadopodia. These results provide new insights and further distinction into the structure, composition and function of podosomes and invadopodia, and strengthen their roles as active participants in cytoskeletal regulation, membrane transport and cell invasion in carcinomas.
Acknowledgments
We thank Alicia G. Arroyo, Stephen Blose, Martin Hemler, L.J. Old, Katsushi Owaribe, Patricia Rousselle and Kiyotoshi Sekiguchi for antibodies and reagents, Kari Lounatmaa for assistance with FESEM, and Pipsa Kaipainen, Hannu Kamppinen, Reijo Karppinen, Marja-Leena Piironen, Outi Rauanheimo, Anne Reijula and Hanna Wennäkoski for technical assistance. Minna Takkunen received support from the Finnish Medical Foundation Duodecim, the Finnish-Norwegian Medical Foundation, the K. Albin Johansson Stiftelse and the Helsinki University Medicine Fund; and Ismo Virtanen from the Helsinki University Central Hospital (EVO Grant no. TYH6269) and the Diabetes Research Foundation. The Academy of Finland, Biocenter Finland, Biocentrum Helsinki and the University of Helsinki, are acknowledged for support for the equipment and infrastructure.
Supporting Information
EGFP-actin transfected 43A cells showed that podosome-like structures are stable structures with long life-spans. Wide-field images were acquired at 10-sec. intervals for a total duration of 30 min. (180 frames).
EGFP-actin transfected 43B cells showed intense motion of actin filaments and actin comet-embedded invadopodia. Inset: Magnified actin tail marked by arrow. Wide-field images were acquired at 10-sec. intervals for a total duration of 30 min. (180 frames).
EGFP-cortactin transfected 43B cells showed tails of invadopodia that maintained substratum contact after retraction of the cell. Wide-field images were acquired at 10-sec. intervals for a total duration of 30 min. (180 frames).
Propelling actin tail is released from the ventral cell membrane in 7.8-min. time-lapse movie of EGFP-actin transfected 43B cells. Inset: magnification of actin tail with strongly reduced background. Cropped images of 47 frames from original wide-field images acquired at 10-sec. intervals for a total duration of 30 min.
Large numbers of invadopodia are rapidly disassembled, whereas one invadopodium is assembled in EGFP-actin transfected 43B cells. Wide-field images were acquired at 10-sec. intervals for a total duration of 30 min. (180 frames).
Externalized invadopodia can be detected at the reaching boundaries between the two cells in EGFP-actin transfected 43B cells. Wide-field images were acquired at 10-sec. intervals for a total duration of 30 min. (180 frames).
Induction of migration with EGF (100 ng/ml) leads EGFP-actin transfected 43A cells to organize multiple new podosome-like structures. When podosomes are left outside the cell membrane, they quickly disappear. Wide-field images were acquired at 5-min. intervals for a total duration of 15 hrs (180 frames).
In 12-hr time-lapse total internal reflection fluorescence (TIRF) microscopy, EGFP-actin transfected 43A cell slowly migrates towards the right. Podosome-like structures are immobile, and a halo of fluorescent EGFP-actin is detected around the podosomes. When podosomes are externalized, they are rapidly disassembled. Some podosomes are also disassembled from the middle section of the cell during the migration. TIRF images were acquired at 2-min. intervals for a total duration of 12 hrs (360 frames).
In 6-hr time-lapse TIRFmicroscopy, invadopodia in EGFP-actin transfected 43B cells arerelatively stable, and rigorous movement of actin filaments isdetected. The actin tails are not visible due to the narrowevanescent field of TIRF microscopy, i.e. they are located above the focal plane. TIRF images were acquired at 2-min. intervals for a total duration of 6 hrs (180 frames).
In 60-min. time-lapse TIRF microscopy, the cell extensions show continuous movement in EGFP-actin transfected 43B cells. TIRF images were acquired at 12-sec. intervals for a total duration of 60 min (180 frames).
Fig. S1 Treatment with microtubule inhibitor demecolcine(10 μM) for 30 min. had only a mild effect on podosome-likestructures and invadopodia (A). The combination ofcytochalasin B and demecolcine disrupted the cytoarchitecturerapidly, but the podosomes and invadopodia persisted (B).Scale bars: 20 μm.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting information supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Cavallaro U, Christofori G. Cell adhesion and signalling by cadherins and Ig-CAMs in cancer. Nat Rev Cancer. 2004;4:118–32. doi: 10.1038/nrc1276. [DOI] [PubMed] [Google Scholar]
- 2.Weaver AM. Invadopodia: specialized cell structures for cancer invasion. Clin Exp Metastasis. 2006;23:97–105. doi: 10.1007/s10585-006-9014-1. [DOI] [PubMed] [Google Scholar]
- 3.Linder S. The matrix corroded: podosomes and invadopodia in extracellular matrix degradation. Trends Cell Biol. 2007;17:107–17. doi: 10.1016/j.tcb.2007.01.002. [DOI] [PubMed] [Google Scholar]
- 4.Lehto VP, Hovi T, Vartio T, et al. Reorganization of cytoskeletal and contractile elements during transition of human monocytes into adherent macrophages. Lab Invest. 1982;47:391–9. [PubMed] [Google Scholar]
- 5.Tarone G, Cirillo D, Giancotti FG, et al. Rous sarcoma virus-transformed fibroblasts adhere primarily at discrete protrusions of the ventral membrane called podosomes. Exp Cell Res. 1985;159:141–57. doi: 10.1016/s0014-4827(85)80044-6. [DOI] [PubMed] [Google Scholar]
- 6.Caligaris-Cappio F, Bergui L, Tesio L, et al. Cytoskeleton organization is aberrantly rearranged in the cells of B chronic lymphocytic leukemia and hairy cell leukemia. Blood. 1986;67:233–9. [PubMed] [Google Scholar]
- 7.Spinardi L, Marchisio PC. Podosomes as smart regulators of cellular adhesion. Eur J Cell Biol. 2006;85:191–4. doi: 10.1016/j.ejcb.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 8.Carman CV, Sage PT, Sciuto TE, et al. Transcellular diapedesis is initiated by invasive podosomes. Immunity. 2007;26:784–97. doi: 10.1016/j.immuni.2007.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kocher HM, Sandle J, Mirza TA, et al. Ezrin interacts with cortactin to form podosomal rosettes in pancreatic cancer cells. Gut. 2009;58:271–84. doi: 10.1136/gut.2008.159871. [DOI] [PubMed] [Google Scholar]
- 10.Stylli SS, Kaye AH, Lock P. Invadopodia: at the cutting edge of tumour invasion. J Clin Neurosci. 2008;15:725–37. doi: 10.1016/j.jocn.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 11.Buccione R, Orth JD, McNiven MA. Foot and mouth: podosomes, invadopodia and circular dorsal ruffles. Nat Rev Mol Cell Biol. 2004;5:647–57. doi: 10.1038/nrm1436. [DOI] [PubMed] [Google Scholar]
- 12.Yamaguchi H, Condeelis J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim Biophys Acta. 2007;1773:642–52. doi: 10.1016/j.bbamcr.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodrigo JP, Garcia LA, Ramos S, et al. EMS1 gene amplification correlates with poor prognosis in squamous cell carcinomas of the head and neck. Clin Cancer Res. 2000;6:3177–82. [PubMed] [Google Scholar]
- 14.Kelley LC, Shahab S, Weed SA. Actin cytoskeletal mediators of motility and invasion amplified and overexpressed in head and neck cancer. Clin Exp Metastasis. 2008;25:289–304. doi: 10.1007/s10585-008-9154-6. [DOI] [PubMed] [Google Scholar]
- 15.Yamaguchi H, Lorenz M, Kempiak S, et al. Molecular mechanisms of invadopodium formation: the role of the N-WASP-Arp2/3 complex pathway and cofilin. J Cell Biol. 2005;168:441–52. doi: 10.1083/jcb.200407076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Thiery JP. Epithelial-mesenchymal transitions in development and pathologies. Curr Opin Cell Biol. 2003;15:740–6. doi: 10.1016/j.ceb.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 17.Yanjia H, Xinchun J. The role of epithelial-mesenchymal transition in oral squamous cell carcinoma and oral submucous fibrosis. Clin Chim Acta. 2007;383:51–6. doi: 10.1016/j.cca.2007.04.014. [DOI] [PubMed] [Google Scholar]
- 18.Takkunen M, Grenman R, Hukkanen M, et al. Snail-dependent and -independent epithelial-mesenchymal transition in oral squamous carcinoma cells. J Histochem Cytochem. 2006;54:1263–75. doi: 10.1369/jhc.6A6958.2006. [DOI] [PubMed] [Google Scholar]
- 19.Peinado H, Olmeda D, Cano A. Snail, Zeb and bHLH factors in tumour progression: an alliance against the epithelial phenotype? Nat Rev Cancer. 2007;7:415–28. doi: 10.1038/nrc2131. [DOI] [PubMed] [Google Scholar]
- 20.Takkunen M, Ainola M, Vainionpaa N, et al. Epithelial-mesenchymal transition downregulates laminin alpha5 chain and upregulates laminin alpha4 chain in oral squamous carcinoma cells. Histochem Cell Biol. 2008;130:509–25. doi: 10.1007/s00418-008-0443-6. [DOI] [PubMed] [Google Scholar]
- 21.Yilmaz M, Christofori G. EMT, the cytoskeleton, and cancer cell invasion. Cancer Metastasis Rev. 2009;28:15–33. doi: 10.1007/s10555-008-9169-0. [DOI] [PubMed] [Google Scholar]
- 22.Zhu J, Yu D, Zeng XC, et al. Receptor-mediated endocytosis involves tyrosine phosphorylation of cortactin. J Biol Chem. 2007;282:16086–94. doi: 10.1074/jbc.M701997200. [DOI] [PubMed] [Google Scholar]
- 23.Nakamura F, Pudas R, Heikkinen O, et al. The structure of the GPIb-filamin A complex. Blood. 2006;107:1925–32. doi: 10.1182/blood-2005-10-3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fradet Y, Cordon-Cardo C, Thomson T, et al. Cell surface antigens of human bladder cancer defined by mouse monoclonal antibodies. Proc Natl Acad Sci USA. 1984;81:224–8. doi: 10.1073/pnas.81.1.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheresh DA, Spiro RC. Biosynthetic and functional properties of an Arg-Gly-Asp-directed receptor involved in human melanoma cell attachment to vitronectin, fibrinogen, and von Willebrand factor. J Biol Chem. 1987;262:17703–11. [PubMed] [Google Scholar]
- 26.Ylanne J, Virtanen I. The Mr 140,000 fibronectin receptor complex in normal and virus-transformed human fibroblasts and in fibrosarcoma cells: identical localization and function. Int J Cancer. 1989;43:1126–36. doi: 10.1002/ijc.2910430628. [DOI] [PubMed] [Google Scholar]
- 27.Pasqualini R, Bodorova J, Ye S, et al. A study of the structure, function and distribution of beta 5 integrins using novel anti-beta 5 monoclonal antibodies. J Cell Sci. 1993;105:101–11. doi: 10.1242/jcs.105.1.101. [DOI] [PubMed] [Google Scholar]
- 28.Galvez BG, Matias-Roman S, Albar JP, et al. Membrane type 1-matrix metalloproteinase is activated during migration of human endothelial cells and modulates endothelial motility and matrix remodeling. J Biol Chem. 2001;276:37491–500. doi: 10.1074/jbc.M104094200. [DOI] [PubMed] [Google Scholar]
- 29.Hieda Y, Nishizawa Y, Uematsu J, et al. Identification of a new hemidesmosomal protein, HD1: a major, high molecular mass component of isolated hemidesmosomes. J Cell Biol. 1992;116:1497–506. doi: 10.1083/jcb.116.6.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ylikoski J, Pirvola U, Narvanen O, et al. Nonerythroid spectrin (fodrin) is a prominent component of the cochlear hair cells. Hear Res. 1990;43:199–203. doi: 10.1016/0378-5955(90)90228-h. [DOI] [PubMed] [Google Scholar]
- 31.Blose SH, Meltzer DI, Feramisco JR. 10-Nm filaments are induced to collapse in living cells microinjected with monoclonal and polyclonal antibodies against tubulin. J Cell Biol. 1984;98:847–58. doi: 10.1083/jcb.98.3.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rabut G, Ellenberg J. Photobleaching techniques to study mobility and molecular dynamics of proteins in live cells: FRAP, iFRAP, and FLIP. In: Goldman RD, Spector DL, editors. Live cell imaging: a laboratory manual. New York: Cold Spring Harbor Laboratory Press; 2005. pp. 101–126. [Google Scholar]
- 33.Spinardi L, Rietdorf J, Nitsch L, et al. A dynamic podosome-like structure of epithelial cells. Exp Cell Res. 2004;295:360–74. doi: 10.1016/j.yexcr.2004.01.007. [DOI] [PubMed] [Google Scholar]
- 34.Baldassarre M, Ayala I, Beznoussenko G, et al. Actin dynamics at sites of extracellular matrix degradation. Eur J Cell Biol. 2006;85:1217–31. doi: 10.1016/j.ejcb.2006.08.003. [DOI] [PubMed] [Google Scholar]
- 35.Gimona M, Buccione R. Adhesions that mediate invasion. Int J Biochem Cell Biol. 2006;38:1875–92. doi: 10.1016/j.biocel.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 36.Varon C, Tatin F, Moreau V, et al. Transforming growth factor beta induces rosettes of podosomes in primary aortic endothelial cells. Mol Cell Biol. 2006;26:3582–94. doi: 10.1128/MCB.26.9.3582-3594.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Condeelis J, Segall JE. Intravital imaging of cell movement in tumours. Nat Rev Cancer. 2003;3:921–30. doi: 10.1038/nrc1231. [DOI] [PubMed] [Google Scholar]
- 38.Faix J, Rottner K. The making of filopodia. Curr Opin Cell Biol. 2006;18:18–25. doi: 10.1016/j.ceb.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 39.Sonnenberg A, Liem RK. Plakins in development and disease. Exp Cell Res. 2007;313:2189–203. doi: 10.1016/j.yexcr.2007.03.039. [DOI] [PubMed] [Google Scholar]
- 40.Gad A, Lach S, Crimaldi L, et al. Plectin deposition at podosome rings requires myosin contractility. Cell Motil Cytoskeleton. 2008;65:614–25. doi: 10.1002/cm.20287. [DOI] [PubMed] [Google Scholar]
- 41.Roper K, Gregory SL, Brown NH. The ‘spectraplakins’: cytoskeletal giants with characteristics of both spectrin and plakin families. J Cell Sci. 2002;115:4215–25. doi: 10.1242/jcs.00157. [DOI] [PubMed] [Google Scholar]
- 42.Wu X, Kodama A, Fuchs E. ACF7 regulates cytoskeletal-focal adhesion dynamics and migration and has ATPase activity. Cell. 2008;135:137–48. doi: 10.1016/j.cell.2008.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Critchley DR. Cytoskeletal proteins talin and vinculin in integrin-mediated adhesion. Biochem Soc Trans. 2004;32:831–6. doi: 10.1042/BST0320831. [DOI] [PubMed] [Google Scholar]
- 44.Legate KR, Fassler R. Mechanisms that regulate adaptor binding to beta-integrin cytoplasmic tails. J Cell Sci. 2009;122:187–98. doi: 10.1242/jcs.041624. [DOI] [PubMed] [Google Scholar]
- 45.Hauck CR, Hsia DA, Ilic D, et al. v-Src SH3-enhanced interaction with focal adhesion kinase at beta 1 integrin-containing invadopodia promotes cell invasion. J Biol Chem. 2002;277:12487–90. doi: 10.1074/jbc.C100760200. [DOI] [PubMed] [Google Scholar]
- 46.Bowden ET, Onikoyi E, Slack R, et al. Co-localization of cortactin and phosphotyrosine identifies active invadopodia in human breast cancer cells. Exp Cell Res. 2006;312:1240–53. doi: 10.1016/j.yexcr.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 47.Vitale S, Avizienyte E, Brunton VG, et al. Focal adhesion kinase is not required for Src-induced formation of invadopodia in KM12C colon cancer cells and can interfere with their assembly. Eur J Cell Biol. 2008;87:569–79. doi: 10.1016/j.ejcb.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 48.Mueller SC, Ghersi G, Akiyama SK, et al. A novel protease-docking function of integrin at invadopodia. J Biol Chem. 1999;274:24947–52. doi: 10.1074/jbc.274.35.24947. [DOI] [PubMed] [Google Scholar]
- 49.Gailit J, Welch MP, Clark RA. TGF-beta 1 stimulates expression of keratinocyte integrins during re-epithelialization of cutaneous wounds. J Invest Dermatol. 1994;103:221–7. doi: 10.1111/1523-1747.ep12393176. [DOI] [PubMed] [Google Scholar]
- 50.Salmivirta K, Gullberg D, Hirsch E, et al. Integrin subunit expression associated with epithelial-mesenchymal interactions during murine tooth development. Dev Dyn. 1996;205:104–13. doi: 10.1002/(SICI)1097-0177(199602)205:2<104::AID-AJA2>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 51.Modregger J, Ritter B, Witter B, et al. All three PACSIN isoforms bind to endocytic proteins and inhibit endocytosis. J Cell Sci. 2000;113:4511–21. doi: 10.1242/jcs.113.24.4511. [DOI] [PubMed] [Google Scholar]
- 52.Halbach A, Morgelin M, Baumgarten M, et al. PACSIN 1 forms tetramers via its N-terminal F-BAR domain. FEBS J. 2007;274:773–82. doi: 10.1111/j.1742-4658.2006.05622.x. [DOI] [PubMed] [Google Scholar]
- 53.Heath RJ, Insall RH. F-BAR domains: multifunctional regulators of membrane curvature. J Cell Sci. 2008;121:1951–4. doi: 10.1242/jcs.023895. [DOI] [PubMed] [Google Scholar]
- 54.van der Flier A, Sonnenberg A. Structural and functional aspects of filamins. Biochim Biophys Acta. 2001;1538:99–117. doi: 10.1016/s0167-4889(01)00072-6. [DOI] [PubMed] [Google Scholar]
- 55.Gouin E, Welch MD, Cossart P. Actin-based motility of intracellular pathogens. Curr Opin Microbiol. 2005;8:35–45. doi: 10.1016/j.mib.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 56.Destaing O, Saltel F, Geminard JC, et al. Podosomes display actin turnover and dynamic self-organization in osteoclasts expressing actin-green fluorescent protein. Mol Biol Cell. 2003;14:407–16. doi: 10.1091/mbc.E02-07-0389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Evans JG, Correia I, Krasavina O, et al. Macrophage podosomes assemble at the leading lamella by growth and fragmentation. J Cell Biol. 2003;161:697–705. doi: 10.1083/jcb.200212037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Linder S, Aepfelbacher M. Podosomes: adhesion hot-spots of invasive cells. Trends Cell Biol. 2003;13:376–85. doi: 10.1016/s0962-8924(03)00128-4. [DOI] [PubMed] [Google Scholar]
- 59.Jurdic P, Saltel F, Chabadel A, et al. Podosome and sealing zone: specificity of the osteoclast model. Eur J Cell Biol. 2006;85:195–202. doi: 10.1016/j.ejcb.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 60.Furmaniak-Kazmierczak E, Crawley SW, Carter RL, et al. Formation of extracellular matrix-digesting invadopodia by primary aortic smooth muscle cells. Circ Res. 2007;100:1328–36. doi: 10.1161/CIRCRESAHA.106.147744. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
EGFP-actin transfected 43A cells showed that podosome-like structures are stable structures with long life-spans. Wide-field images were acquired at 10-sec. intervals for a total duration of 30 min. (180 frames).
EGFP-actin transfected 43B cells showed intense motion of actin filaments and actin comet-embedded invadopodia. Inset: Magnified actin tail marked by arrow. Wide-field images were acquired at 10-sec. intervals for a total duration of 30 min. (180 frames).
EGFP-cortactin transfected 43B cells showed tails of invadopodia that maintained substratum contact after retraction of the cell. Wide-field images were acquired at 10-sec. intervals for a total duration of 30 min. (180 frames).
Propelling actin tail is released from the ventral cell membrane in 7.8-min. time-lapse movie of EGFP-actin transfected 43B cells. Inset: magnification of actin tail with strongly reduced background. Cropped images of 47 frames from original wide-field images acquired at 10-sec. intervals for a total duration of 30 min.
Large numbers of invadopodia are rapidly disassembled, whereas one invadopodium is assembled in EGFP-actin transfected 43B cells. Wide-field images were acquired at 10-sec. intervals for a total duration of 30 min. (180 frames).
Externalized invadopodia can be detected at the reaching boundaries between the two cells in EGFP-actin transfected 43B cells. Wide-field images were acquired at 10-sec. intervals for a total duration of 30 min. (180 frames).
Induction of migration with EGF (100 ng/ml) leads EGFP-actin transfected 43A cells to organize multiple new podosome-like structures. When podosomes are left outside the cell membrane, they quickly disappear. Wide-field images were acquired at 5-min. intervals for a total duration of 15 hrs (180 frames).
In 12-hr time-lapse total internal reflection fluorescence (TIRF) microscopy, EGFP-actin transfected 43A cell slowly migrates towards the right. Podosome-like structures are immobile, and a halo of fluorescent EGFP-actin is detected around the podosomes. When podosomes are externalized, they are rapidly disassembled. Some podosomes are also disassembled from the middle section of the cell during the migration. TIRF images were acquired at 2-min. intervals for a total duration of 12 hrs (360 frames).
In 6-hr time-lapse TIRFmicroscopy, invadopodia in EGFP-actin transfected 43B cells arerelatively stable, and rigorous movement of actin filaments isdetected. The actin tails are not visible due to the narrowevanescent field of TIRF microscopy, i.e. they are located above the focal plane. TIRF images were acquired at 2-min. intervals for a total duration of 6 hrs (180 frames).
In 60-min. time-lapse TIRF microscopy, the cell extensions show continuous movement in EGFP-actin transfected 43B cells. TIRF images were acquired at 12-sec. intervals for a total duration of 60 min (180 frames).
Fig. S1 Treatment with microtubule inhibitor demecolcine(10 μM) for 30 min. had only a mild effect on podosome-likestructures and invadopodia (A). The combination ofcytochalasin B and demecolcine disrupted the cytoarchitecturerapidly, but the podosomes and invadopodia persisted (B).Scale bars: 20 μm.