

Abstract
Circulating endothelial progenitor cells (EPC) contribute to endothelial replenishment. Telmisartan is an angiotensin-receptor blocker with PPARγ-agonistic properties. PPARγ-agonists and HMG-CoA reductase inhibitors have been shown to enhance EPC number and function. We focused on the effects of telmisartan alone or in combination with simvastatin on EPC. EPC were isolated from healthy human volunteers, cultured and stimulated with telmisartan, simvastatin, or the combination of telmisartan and simvastatin. Telmisartan significantly increased the number of acLDL/lectin double-positive early EPC, the number of colony forming units (EC-CFU) as well as EPC migratory capacity, inhibited TNFα-induced EPC apoptosis and reduced glucose-induced oxidative stress. The telmisartan effect was dose-dependent and could be inhibited by GW9662, indicating a PPARγ-dependent mechanism. The combination of telmisartan and simvastatin led to a significant additive increase in EPC count and function. In wild-type mice, systemic treatment with either telmisartan or simvastatin elevated the number of sca-1/flk-1-positive EPC in bone marrow and peripheral blood, spleen-derived acLDL/lectin double-positive EPC, EPC migration and EC-CFU. Consistent with the in vitro findings, the combination of telmisartan and simvastatin resulted in a further enhancement of EPC counts. Re-endothelialization after carotid injury was significantly enhanced by telmisartan, simvastatin and the combination. Telmisartan increases EPC number and function mediated by a PPARγ-dependent mechanism. This effect is further enhanced by combination with simvastatin, suggesting a synergistic activation of potentially diverse intracellular pathways.
Keywords: PPARγ, endothelial progenitor cells, angiotensin receptor blockers, telmisartan, statins
Introduction
Endothelial progenitor cells (EPC) have recently emerged as a pivotal cell type in endothelial homeostasis and endothelial dysfunction, which is an early and crucial stage of atherosclerosis [1, 2]. Mature endothelial cells have only limited regenerative capacities, therefore EPC have been assumed to essentially determine endothelial replenishment. EPC are defined according to their surface markers and functional properties. One recent classification distinguishes between early and late outgrowth EPC by differentiating between cellular functional capacities and time of appearance. After culture of mononuclear cells for 4 to 7 days, early EPC are detectable, which are closely related to the EPC defined by Asahara and colleagues, and feature paracrine improvement of neovasculogenesis, endothelial repair and endothelial vasomotor function. Early EPC are subdivided into circulating angiogenic cells (CAC) and endothelial cell colony forming units (EC-CFU) or CFU-Hill, by specific cell culture protocols and their functional properties [3]. Late outgrowth EPC are found in cobblestone shape after 14 to 21 days. They have a high proliferative potential and contribute to tubulogenesis and neovasculogenesis [4–9]. The precise role of these EPC subsets in atherosclerosis is not yet entirely understood.
In clinical investigations, number and function of circulating and early EPC correlate negatively with cardiovascular risk factors, and have been found to be decreased and functionally impaired in association with severity of coronary heart disease and myocardial infarction. EPC are positively influenced by various physiological stimuli, for example physical activity and weight reduction [2, 10–14]. Moreover, several drugs have been identified to beneficially affect EPC number and function in vivo and in vitro, for example angiotensin converting enzyme inhibitors, 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (statins), and thiazolidinediones (TZD) [15–18].
The multiple extra- and intracellular signalling pathways involved in EPC mobilization, recruitment, activation and function are only partly identified. In addition to the phosphatidylinositol-3-kinase (PI3K) pathway, which mediates the effects of statins on EPC, TZD exert their effects via a peroxisome proliferator-activated receptor γ (PPARγ)-dependent mechanism [18, 19].
The impact of nuclear transcription factor PPARγ on biological processes remains controversial. PPARγ has mainly been studied in adipocytes, but is expressed in all relevant cell types of the cardiovascular system, including endothelial cells. After binding of specific ligands to the ligand-binding domain, the DNA-binding site of PPARγ initiates the expression of its target genes. Physiological ligands include the prostaglandin D2 derivative, 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2) and oxidized linoleic acid (9[S]- and 13[S]-HODE) as components of oxidized LDL [20].
The previously mentioned TZD are one group of pharmacological PPARγ stimulators and have been shown to have beneficial effects on EPC and to improve endothelial function in type 2 diabetes mellitus [18, 21, 22]. Several angiotensin-receptor blockers (ARB) also stimulate PPARγ activity, namely irbesartan, losartan and telmisartan [23]. Among them, the highly lipophilic telmisartan appears to have the strongest activating effect on the PPARγ pathway [24].
In this study, we investigated in vitro and in a murine in vivo model whether (i) the ARB telmisartan enhances early EPC number and function, (ii) this effect is mediated by PPARγ, and (iii) the combination with simvastatin partly leads to an additional enhancement of EPC number and function by potentially activating alternative pathways.
Methods
Materials
Angiotensin II, simvastatin, GW9662, PD123319, TNFα, salts and other chemicals were purchased from Sigma. Simvastatin was chemically activated by alkaline hydrolysis. Telmisartan was kindly provided by Boehringer Ingelheim.
Peripheral blood samples
Human peripheral blood samples were obtained from adult healthy volunteers by venopuncture and collected into sodium citrate containing tubes. All samples were obtained with informed consent and in accordance with the Declaration of Helsinki.
Isolation of EPC
EPC were isolated from peripheral blood or murine spleen and cultured in accordance with previously published methods [25]. Briefly, mononuclear cells (MNC) were isolated using Ficoll density gradient (Biocoll, Biochrom, Berlin, Germany) and seeded into 24-well tissue culture plates pre-coated with human fibronectin (Sigma, Steinheim am Albuch, Germany) at 4 × 106 (acLDL/lectin staining) or 1 × 106 (EC-CFU) cells/ml in endothelial basal medium (EBM) and supplements as recommended by the manufacturer (Promocell, Heidelberg, Germany).
Stimulation of EPC
Cells were incubated for 72 hrs with telmisartan in concentrations of 1, 10 or 100 μmol/l; angiotensin II 1 μmol/l; telmisartan 10 μmol/l and angiotensin II 1 μmol/l; simvastatin 1 μmol/l; telmisartan 10 or 100 μmol/l and simvastatin 1 μmol/l; GW9662 5 μmol/l; PD123319 1 μmol/l; TNFα 25 ng/well.
AcLDL/lectin staining
After 7 days of culture, fibronectin-adherent cells were washed and incubated with 1,1’-dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine (Dil)-labelled acetylated low-density lipoprotein (acLDL; CellSystems, St. Katharinen, Germany) and stained with FITC-labeled Ulex europaeus agglutinin (lectin; Sigma). Cells double-positive for acLDL and lectin staining were judged to be EPC [17]. At least three high-power field fluoroscopy microscopic scans of each well were analyzed, and mean values were calculated.
Endothelial cell colony forming units (EC-CFU)
For EC-CFU, cells were isolated and cultured as described previously. After 2 days of initial culture, 1 × 106 cells derived from the supernatant were re-seeded per well, cultured for additional 7 days in EBM on human fibronectin pre-coated wells [11, 14] and stimulated as described previously. The number of colonies was manually counted using light microscopy at the end of the incubation period.
Migration assay
EPC migratory function was evaluated using a modified Boyden chamber (BD Biosciences, Heidelberg, Germany). A polycarbonate filter with 8-μm pore size (BD Biosciences) was placed between the upper and lower chambers. After 4 days of initial culture, cell suspensions (1 × 105 cells per well) were placed in the upper chamber, and the lower chamber was filled with EBM containing 50 ng/ml of human recombinant stromal cell-derived factor-1 (SDF-1; R&D Systems, Minneapolis, MN, USA). Cells were then incubated for additional 24 hrs at 37°C and 5% CO2. The migrated cells on the lower side of the filter were fixed and stained for acLDL/lectin. Migration activity was evaluated by the mean number of cells counted in at least three high-power fields per filter.
Apoptosis assay
To determine EPC apoptosis, the Cell Death Detection ELISA Kit (Roche, Mannheim, Germany) was used according to the instructions of the manufacturer. Briefly, EPC were cultured and stimulated as mentioned previously. Cells were pelleted, lysed and transferred to a streptavidin-coated microplate well. Anti-histone (biotin-labelled) and anti-DNA (peroxidase-conjugated) antibodies were added leading to the binding of nucleosome complexes to streptavidin. Samples were incubated with peroxidase substrate (ABTS) and the coloured product was measured spectrophotometrically. Apoptosis was calculated in enrichment factor, which equals the millimass unit of sample (dying or dead cells) distributed to millimass unit of corresponding control (viable cells); mU = absorbance (10−3). The results are shown in relative proportion to the control group.
Measurement of reactive oxygen species
Intracellular reactive oxygen species (ROS) production in cultured EPC was measured by 2’,7’-dichlorofluorescein (DCF; 10 μmol/l) fluorescence using fluorescence microscopy techniques. EPC were stimulated with telmisartan 100 μmol/l; angiotensin II 1 μmol/l; glucose 30 mmol/l; telmisartan 100 μmol/l and angiotensin II 1 μmol/l; simvastatin 1 μmol/l; telmisartan 100 μmol/l and simvastatin 1 μmol/l.
Stimulation was followed by incubation with DCF for 30 min. in the dark at 37°C. Images were collected by single rapid scans and identical parameters, such as contrast and brightness, for all samples. Five groups of 40 cells for each sample were randomly selected from the image and fluorescent intensity was taken.
Animal studies
C57BL/6 (wild-type) mice (Charles River, Wilmington, NC, USA) were used for this study. The animals were maintained in a 22°C room with a 12-hr light/dark cycle and received drinking water ad libitum. The animals were divided into four groups. Group 1 served as control group and received standard diet (Ssniff, Soest, Germany) and saline injections (equal volume as simvastatin injections). Group 2 was fed standard diet supplemented with telmisartan (40 mg/kg body weight/d). Group 3 was fed standard diet and received simvastatin injections subcutaneously (20 mg/kg body weight/d). Group 4 was treated with a combination of telmisartan diet and simvastatin injections. Pharmacological treatment was performed for 10 days. The mice were killed after the indicated treatment. Spleen, blood and bone marrow samples (femur and tibia) were collected and processed immediately after sacrifice. All animal experiments were performed in accordance with institutional guidelines and the German animal protection law.
Flow cytometry
Murine blood samples were analyzed as previously described [17]. Following red cell lysis, the viable lymphocyte population was analyzed for sca-1-FITC (Becton Dickinson, Heidelberg, Germany) and flk-1-PE (Becton Dickinson). Isotype-identical antibodies and unstained samples served as controls in every experiment (Becton Dickinson). Cell fluorescence was measured immediately after staining using a FACSCalibur instrument (Becton Dickinson). Data were analyzed using CellQuest software (Becton Dickinson). Units of all measured components are specific events obtained after measuring 50,000 (bone marrow) or 20,000 (peripheral blood) events in a pre-specified lymphocyte gate during FACS analysis.
Carotid artery denudation
Carotid artery injury was induced as described previously [26]. Briefly, a subset of the mice was anaesthetized with 150 mg/kg body weight ketamine hydrochloride (ketanest®, Pharmacia, Karlsruhe, Germany) and 0.1 mg/kg body weight xylazine hydrochloride (rompun®2%, Bayer, Leverkusen, Germany). The common carotid artery was exposed and submitted to an electric injury starting at the bifurcation and continuing to the proximal part of the artery (in total 4 mm denudation). The denuded area was determined at day 3 after surgery following intravenous injection of 50 μl Evans blue in an enface preparation of the vessel. Evans blue stained denuded areas and the complete vessel area was measured using AxioVision (Zeiss, Munich, Germany) version 4.5.0 software. The percentage of re-endothelialization 3 days after injury is provided.
Statistical analysis
Data are expressed as mean ± SEM. Groups were compared by a one-way analysis of variance (ANOVA) with post hoc analyses for pair wise comparisons (Newman–Keuls multiple comparison). Statistical significance was assumed when a null hypothesis could be rejected at P < 0.05. Statistical analysis was performed with the use of SPSS software (SPSS Inc., Chicago, USA), version 11.5, for Microsoft Windows.
Results
EPC numbers in vitro
The uptake of acLDL and presence of lectin is a specific characteristic of EPC. AcLDL/lectin double-positive cells were counted to assess the number of EPC in vitro. Compared to the vehicle-treated control group (100%), the number of EPC was decreased after stimulation with angiotensin II (64 ± 14% of control, P < 0.05 versus control). This effect was antagonized by co-incubation with telmisartan (134 ± 9% of control, P < 0.05 versus angiotensin II), and EPC numbers appeared to be higher than in the control group (P < 0.05 versus control). Telmisartan dose-dependently increased EPC numbers in the absence of angiotensin II (1 μmol/l: 121 ± 11% of control; 10 μmol/l: 184 ± 12% of control, P < 0.05 versus control; 100 μmol/l: 275 ± 9% of control, P < 0.05 versus control) (Fig. 1A).
Fig 1.
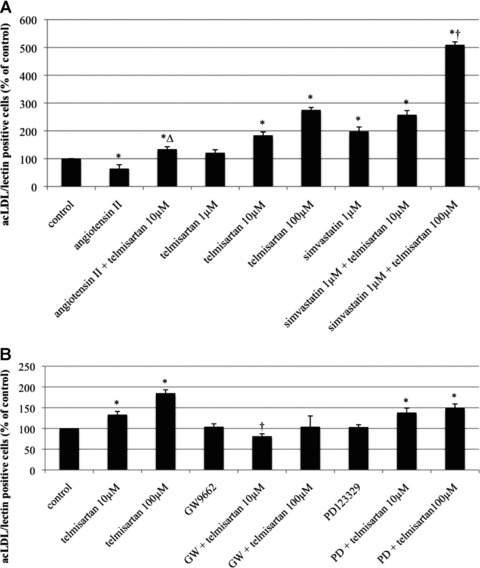
(A) Number of acLDL/lectin double-positive EPC after stimulation with angiotensin II, telmisartan, and/or simvastatin in vitro. AcLDL/lectin double-positive EPC per high-power field in per cent of control, mean ± SEM, n= 4, *P < 0.05 versus control, ΔP < 0.05 versus angiotensin II, †P versus telmisartan 10 and 100 μmol, simvastatin 1 μmol, simvastatin 1 μmol and telmisartan 10 μmol. (B) Number of acLDL/lectin double-positive EPC after stimulation with telmisartan, the PPARγ antagonist GW 9662, and/or the AT2-receptor antagonist PD 123329 in vitro. AcLDL/lectin double-positive EPC per high-power field in per cent of control, mean ± SEM, n= 4–8, *P < 0.05 versus control, †P < 0.05 versus telmisartan 10 and 100 μmol, respectively.
Stimulation with 1 μmol/l simvastatin increased the absolute EPC number to 198 ± 16% of control (P < 0.05 versus control). Combined stimulation with telmisartan and simvastatin further enhanced EPC count, with a dose-dependent effect for telmisartan (simvastatin 1 μmol/l and telmisartan 10 μmol/l: 258 ± 15% of control, P < 0.05 versus control; simvastatin 1 μmol/l and telmisartan 100 μmol/l: 509 ± 11% of control, P < 0.05 versus control, P < 0.05 versus telmisartan 10 and 100 μmol, simvastatin 1 μmol, simvastatin 1 μmol and telmisartan 10 μmol) (Fig. 1A).
Co-treatment of EPC with telmisartan and the specific PPARγ antagonist GW9662 abolished the telmisartan-induced effect on EPC counts (telmisartan 10 μmol/l: 133 ± 7% of control, P < 0.05 versus control; telmisartan 100 μmol/l: 185 ± 8% of control, P < 0.05 versus control; GW: 104 ± 7% of control; telmisartan 10 μmol/l and GW: 81 ± 6% of control, P < 0.05 versus telmisartan 10 μmol/l; telmisartan 100 μmol/l and GW: 104 ± 26% of control), whereas co-stimulation with the specific AT2-receptor antagonist PD123329 did not affect the telmisartan effect on EPC numbers (PD: 103 ± 6% of control; telmisartan 10 μmol/l and PD: 138 ± 11% of control, P < 0.05 versus control; telmisartan 100 μmol/l and PD: 149 ± 10% of control, P < 0.05 versus control) (Fig. 1B).
Endothelial cell colony forming units in vitro
Compared to the vehicle-treated control group (100%), the number of EC–CFU was significantly diminished by angiotensin II (48 ± 23% of control, P < 0.05 versus control). This was neutralized by co-treatment with telmisartan 10 μmol/l (120 ± 30% of control). Stimulation with telmisartan alone dose-dependently enhanced CFU count (1 μmol/l: 122 ± 19% of control; 10 μmol/l: 168 ± 19% of control; 100 μmol/l: 225 ± 14% of control, P < 0.05 versus control). The increase of CFU counts by stimulation with simvastatin (148 ± 20% of control) was significantly enhanced by co-incubation of simvastatin with telmisartan 10 μmol/l (194 ± 16% of control, P < 0.05 versus control) and telmisartan 100 μmol/l (367 ± 10% of control, P < 0.05 versus control) (Fig. 2).
Fig 2.
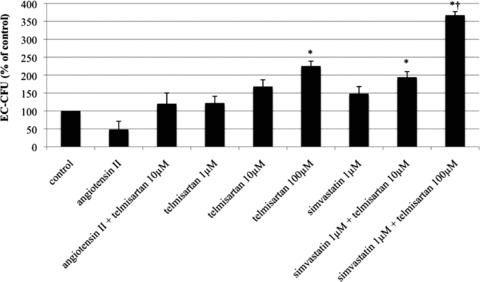
Number of endothelial cell colony forming units (EC-CFU) after stimulation with angiotensin II, telmisartan and/or simvastatin in vitro. EC-CFU per well in per cent of control, mean ± SEM, n= 4, *P < 0.05 versus control, †P <0.05 versus telmisartan 10 or 100 μmol, simvastatin 1 μmol or telmisartan 10 μmol and simvastatin 1 μmol.
Migration in vitro
The migratory capacity was assessed to determine a functional parameter of EPC. Compared to the vehicle-treated control group (100%), angiotensin II deteriorated migratory capacity (74 ± 8% of control, P < 0.05 versus control). This effect was antagonized by co-stimulation with telmisartan 10 μmol/l (125 ± 9% of control, P < 0.05 versus angiotensin II). Treatment with telmisartan alone dose-dependently improved the migratory capacity of EPC (1 μmol/l: 115 ± 8% of control; 10 μmol/l: 158 ± 13% of control, P < 0.05 versus control; 100 μmol/l: 178 ± 12% of control, P < 0.05 versus control). The significant effect of simvastatin treatment (154 ± 7% of control, P < 0.05 versus control) was further enhanced by the co-application of telmisartan (10 μmol/l: 168 ± 12% of control, P < 0.05 versus control; 100 μmol/l: 191 ± 10% of control, P < 0.05 versus control) (Fig. 3).
Fig 3.
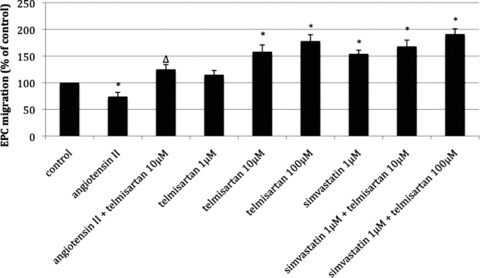
Migratory capacity of EPC after stimulation with angiotensin II, telmisartan and/or simvastatin in vitro. Migrated cells in per cent of control, mean ± SEM, n= 4, *P < 0.05 versus control, ΔP < 0.05 versus angiotensin II.
Apoptosis in vitro
TNFα-induced apoptosis resulted in the highest rate of cell death and was used as reference (100%). Vehicle-treated cells served as negative control (19 ± 14% of TNFα, P < 0.05 versus TNFα). Angiotensin II significantly enhanced EPC apoptosis, but to a lesser degree than TNFα (69 ± 6% of TNFα, P < 0.05 versus TNFα, P < 0.05 versus negative control). The angiotensin II effect was abolished by co-stimulation with telmisartan 10 μmol/l (24 ± 13% of TNFα, P < 0.05 versus angiotensin II, P < 0.05 versus TNFα). Telmisartan alone dose-dependently inhibited TNFα-induced apoptosis (1 μmol/l: 75 ± 7% of TNFα; 10 μmol/l: 58 ± 5% of TNFα, P < 0.05 versus TNFα; 100 μmol/l: 45 ± 9% of TNFα, P < 0.05 versus TNFα). Expectedly, simvastatin inhibited TNFα-induced apoptosis of EPC (50 ± 19% of TNFα, P < 0.05 versus TNFα). The combined stimulation of EPC with simvastatin and telmisartan led to an additive anti-apoptotic effect (simvastatin and telmisartan 10 μmol/l: 43 ± 16% of TNFα, P < 0.05 versus TNFα; simvastatin and telmisartan 100 μmol/l: 35 ± 14% of TNFα, P < 0.05 versus TNFα) (Fig. 4).
Fig 4.
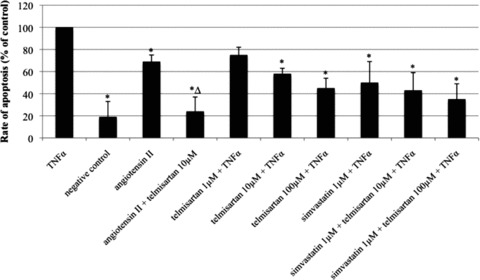
Rate of EPC apoptosis after stimulation with TNFα, angiotensin II, telmisartan and/or simvastatin in vitro. Negative control was not treated with any substance. TNFα was used as an inducer of apoptosis and used as reference. Rate of apoptosis in per cent of TNFα, mean ± SEM, n= 4, *P < 0.05 versus TNFα, ΔP < 0.05 versus angiotensin II.
ROS production in vitro
DCF fluorescence was used to measure intracellular ROS production. Compared to vehicle-treated control (100%), ROS formation was significantly enhanced by angiotensin II (145 ± 14% of control, P < 0.05 versus control), which was diminished by the addition of telmisartan (86 ± 4% of control). Because we sought to evaluate the PPARγ-associated telmisartan effect on oxidative stress beyond angiotensin-receptor blocking capacity, we additionally used glucose to generate oxidative stress. Glucose-mediated ROS production (170 ± 14% of control, P < 0.05 versus control) was comparable to that of angiotensin II. Addition of telmisartan normalised oxidative stress to control levels (98 ± 6% of control, P < 0.05 versus glucose), as did simvastatin (114 ± 4% of control, P < 0.05 versus glucose). Co-incubation of telmisartan and simvastatin decreased oxidative stress even further below control levels (79 ± 5% of control, P < 0.05 versus control, P < 0.05 versus glucose, P < 0.05 versus glucose + telmisartan) (Fig. 5).
Fig 5.
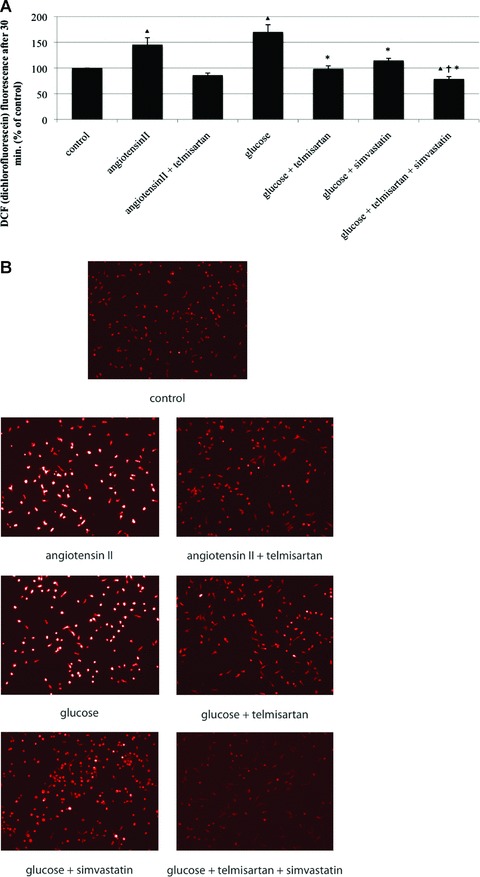
ROS production of EPC in vitro. Intracellular ROS production after angiotensin II and glucose-induced oxidative stress and after co-stimulation with telmisartan, simvastatin and a combination of telmisartan and simvastatin was visualized by DCF fluorescence. (A) DCF fluorescence in per cent of control, mean ± SEM, n= 4, *P < 0.05 versus glucose, P < 0.05 versus control, P < 0.05 versus glucose and telmisartan, or glucose and simvastatin. (B) Representative images of DCF fluorescence.
EPC counts in vivo
Wild-type mice were treated with the study drugs for 10 days, and EPC counts in bone marrow and peripheral blood were analyzed by flow cytometry. Compared to the vehicle-treated control group (100%), both telmisartan and simvastatin treatment significantly increased the number of EPC in bone marrow and peripheral blood (telmisartan: 185 ± 13% of control in bone marrow and 132 ± 8% of control in peripheral blood; simvastatin: 210 ± 9% of control in bone marrow and 159 ± 12% of control in peripheral blood; all P < 0.05 versus control). Consistent with the in vitro findings, the combination of telmisartan and simvastatin led to a synergistic enhancement of EPC counts in bone marrow (439 ± 28% of control, P < 0.05 versus control, P < 0.05 versus telmisartan, P < 0.05 versus simvastatin) and peripheral blood (195 ± 16% of control, P < 0.05 versus control) (Fig. 6).
Fig 6.
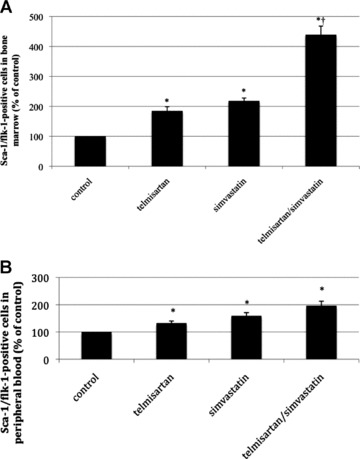
EPC counts in vivo. Wild-type mice were treated for 10 days with either vehicle, telmisartan, simvastatin or a combination of telmisartan and simvastatin. (A) Sca-1/flk-1-positive cells per 50,000 gated events in bone marrow. Numbers are given in per cent of control, mean ± SEM, n= 5 per group, *P < 0.05 versus control. (B) Sca-1/flk-1-positive cells per 20,000 gated events in peripheral blood. Numbers are given in per cent of control, mean ± SEM, n= 5 per group, *P < 0.05 versus control, †P < 0.05 versus telmisartan or simvastatin.
EPC numbers ex vivo
After cultivation of spleen-derived mononuclear cells for 7 days, the number of acLDL/lectin double-positive EPC was evaluated. Compared to the vehicle-treated control group (100%), telmisartan and simvastatin led to a significant increase of double-positive cells (138 ± 6% and 163 ± 27% of control, P < 0.05 versus control). The combination of telmisartan and simvastatin further enhanced EPC numbers (235 ± 18% of control, P < 0.05 versus control, P < 0.05 versus telmisartan) (Fig. 7).
Fig 7.
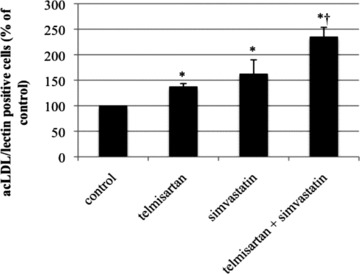
Number of spleen-derived acLDL/lectin double-positive EPC ex vivo. Wild-type mice were treated for 10 days with either vehicle, telmisartan, simvastatin or a combination of telmisartan and simvastatin. AcLDL/lectin double-positive cells in per cent of control, mean ± SEM, *P < 0.05 versus control, †P < 0.05 versus telmisartan.
Endothelial cell colony forming units ex vivo
Compared to control group (100%), the number of murine EC–CFU was significantly increased in telmisartan (162 ± 9% of control, P < 0.05 versus control) or simvastatin (219 ± 15% of control, P < 0.05 versus control) treated mice. Combined administration of telmisartan and simvastatin increased EC–CFU not only compared to control but also beyond single treatment with telmisartan or simvastatin (291 ± 24% of control, P < 0.05 versus control, P < 0.05 versus telmisartan, P < 0.05 versus simvastatin) (Fig. 8).
Fig 8.
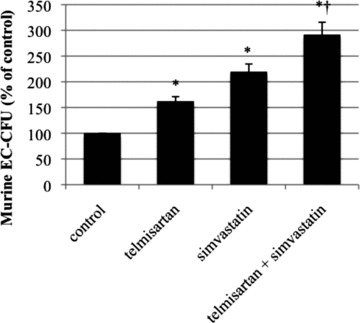
EC-CFU counts ex vivo. Wild-type mice were treated for 10 days with either vehicle, telmisartan, simvastatin or a combination of telmisartan and simvastatin. EC-CFU in per cent of control, mean ± SEM, n= 5, *P < 0.05 versus control, †P < 0.05 versus telmisartan and versus simvastatin.
Migration ex vivo
EPC migration was significantly improved in mice treated with telmisartan (130 ± 9% of control, P < 0.05 versus control) or simvastatin (135 ± 9% of control, P < 0.05 versus control), as compared to the control group (100%). The combination of telmisartan and simvastatin also significantly enhanced migratory capacity (151 ± 8% of control, P < 0.05 versus control), which was, however, not significantly increased compared to monotherapy with either telmisartan or simvastatin (Fig. 9).
Fig 9.
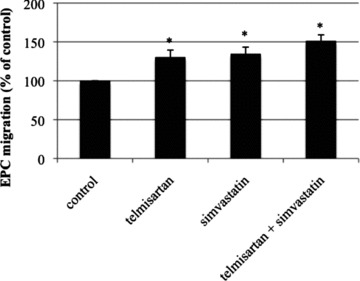
Migratory capacity of EPC ex vivo. Wild-type mice were treated for 10 days with either vehicle, telmisartan, simvastatin or a combination of telmisartan and simvastatin. Migrated cells in per cent of control, mean ± SEM, n= 5, *P < 0.05 versus control.
Re-endothelialization in vivo
Carotid arteries were electrically denuded, and the remaining lesion and re-endothelialization were assessed 3 days later. In the control group, 23 ± 3% of the total lesion area were re-endothelialized. In mice treated with telmisartan or simvastatin, this area was increased to 38 ± 3% for telmisartan (P < 0.05 versus control) and 40 ± 4% for simvastatin (P < 0.05 versus control). The combination of telmisartan and simvastatin resulted in a further but not significant improvement of re-endothelialization compared to monotherapy (44 ± 3%, P < 0.05 versus control) (Fig. 10).
Fig 10.
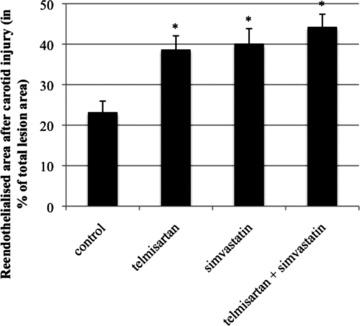
Re-endothelialization in vivo. Re-endothelialized area of carotid arteries 3 days after electrical denudation. Re-endothelialized area in per cent of entirely denuded area, mean ± SEM, n= 8–10, *P < 0.05 versus control.
Discussion
Focus of the present study was the cellular effect of the ARB telmisartan and combination of telmisartan with the HMG-CoA reductase inhibitor simvastatin on early EPC number and function in vitro and in vivo. Telmisartan blocked the deleterious effects of angiotensin II on early EPC and dose-dependently enhanced EPC number and function in the absence of angiotensin II. Furthermore, combined treatment of telmisartan with simvastatin led to, or at least suggested a trend towards, an additive, synergistic effect on EPC number and function.
It has been shown, that angiotensin II accelerates EPC senescence via AT1-receptor activation through induction of oxidative stress [27, 28]. In turn, angiotensin II blockade has been associated with improvement of EPC number and function in hypertension via antioxidant mechanisms [29–31]. As expected, the AT1-receptor antagonist telmisartan blocked angiotensin II effects on early EPC number and function, and reduced angiotensin II-induced oxidative stress in our study. Our in vitro data indicate that telmisartan exerts a direct, dose-dependent effect on EPC independent of angiotensin II blockade, namely enhancement of EPC number, colony formation and migratory capacity, and inhibition of TNFα-induced apoptosis. The reduction of glucose-induced oxidative stress likewise points to a mechanism independent of AT1-receptor blockade. Importantly, telmisartan increased EPC numbers in bone marrow, peripheral blood and spleen of wild-type mice, confirming our in vitro data in an in vivo setting.
The direct effect of telmisartan is mediated by a PPARγ-dependent mechanism, because co-incubation of telmisartan with the PPARγ antagonist GW9662 inhibited the effect of telmisartan on EPC numbers in our cell culture experiments. Our finding is in line with studies that demonstrated PPARγ-activating properties of telmisartan and showed an increase of EPC numbers and functional capacities after treatment with PPARγ-activating TZD in vitro and in vivo[18, 21, 23, 24].
Zhao and colleagues reported that angiotensin II induces PPARγ expression and ligand-mediated PPARγ activity via AT2-receptor activation, which is involved in neuronal cell differentiation [32]. Another study demonstrated a PPARγ-dependent down-regulation of the AT1 receptor by telmisartan independent of angiotensin II [33]. Telmisartan is a highly selective AT1-receptor blocker with strikingly less affinity to the AT2 receptor. However, we sought to exclude an involvement of AT2-receptor stimulation in the observed direct effects of telmisartan on EPC. In our analysis, telmisartan increased the number of EPC in the absence of angiotensin II, which could not be blocked by the specific AT2-receptor antagonist PD123319. In contrast to a possible angiotensin II effect on AT2 receptors in the presence of an AT1-receptor blocker, an involvement of AT2-receptor activation in the telmisartan-induced effect on EPC numbers can be excluded in this experimental setting.
Expectedly, simvastatin treatment improved EPC numbers and function in vitro and in vivo in our study. This finding is consistent with previous reports that demonstrated increased EPC mobilization, proliferation and migration as well as decreased EPC apoptosis and senescence after statin treatment [16, 17, 34–38]. The most important intracellular mechanism for these effects seems to be the PI3K/Akt axis. Simvastatin-induced phosphorylation and activation of Akt lead to enhanced EPC numbers, differentiation and inhibition of apoptosis, which can be effectively inhibited by PI3K blockers or over-expression of dominant-negative Akt [16, 19, 36, 38].
Interestingly, in the present study, the combined application of telmisartan and simvastatin led to an additive enhancement of EPC numbers and partly function. As described previously, telmisartan activates PPARγ, whereas simvastatin leads to activation of the PI3K/Akt pathway, indicating stimulation of diverse signalling pathways. In addition, experimental studies have shown that some TZD effects on EPC can be blocked by inhibitors of PI3K, demonstrating that PPARγ activation may lead to additional activation of the PI3K pathway [21]. Moreover, it has been demonstrated that statins inhibit premature EPC senescence by restoration of telomere repeat-binding factor-2 (TRF-2) and preventing telomere uncapping, a process not related to PI3K activation [37]. Both simvastatin and telmisartan attenuate TNFα-mediated apoptosis of EPC, which is in line with recent studies investigating the effect of statins and PPARγ-activating TZD on EPC apoptosis [21, 35, 38]. Again, the involvement of PI3K has been demonstrated for statins. TNFα-mediated apoptosis involves activation of NF-kB, and it has been shown that PPARγ-activating TZD may inhibit the phosphorylation of the p65 subunit of this signalling protein [39]. The anti-apoptotic effect of telmisartan may involve this PPARγ-mediated signalling cascade, too. Finally, it is possible that telmisartan activates additional signalling pathways that have not yet been investigated. Taken together, a synergistic stimulation of multiple intracellular pathways by telmisartan and simvastatin is most likely responsible for the additive effect on EPC.
As anticipated, monotherapy with telmisartan or simvastatin significantly enhanced re-endothelialization after a defined carotid injury in mice. The combination of both substances improved re-endothelialization with a modest trend towards further enhancement. However, this latter effect did not reach statistical significance compared to the single drugs. One may speculate that the regenerative capacity for endothelial damage was already saturated by the sole application of telmisartan or simvastatin in otherwise healthy wild-type mice, and significant differences may be obtained in other models of vascular disease. On the other hand, the combination of telmisartan and simvastatin might primarily affect numbers of early EPC but not further augment functionality such as migratory capacity.
Although telmisartan plasma concentrations of 10 μmol/l, as used in our in vitro experiments, may be achieved with therapeutic doses of this ARB in clinical settings in humans [40], higher doses may not reflect therapeutic in vivo levels in humans. However, the observed effect of telmisartan was dose-dependent and, importantly, the effect of telmisartan and the synergistic effect of combined treatment with simvastatin was confirmed in vivo in mice. Future clinical trials will have to clarify the relevance of our findings in humans.
EPC are important for endothelial replenishment, endothelial repair after vascular damage, neovasculogenesis, and endothelial function [2, 4–9]. Given the fact that endothelial dysfunction is a marker of early atherosclerosis and predicts future cardiovascular events, and EPC counts correlate not only with endothelial vasomotor function but also inversely with cardiovascular morbidity and mortality, these cells may not only reflect a cellular marker of the severity of vascular disease but may play an important role in the prevention of atherosclerosis [1, 2, 9, 11, 13, 14]. It may be speculated that EPC-enhancing treatment strategies resemble an interesting therapeutic option for the prevention and treatment of vascular disease. Statins, PPARγ agonists, and ARB have been shown to improve endothelial function, inhibit development and progression of atherosclerosis and reduce cardiovascular events, as demonstrated in numerous animal studies and human clinical trials [22, 41–50]. The mobilization, increase of cell numbers and functional improvement of EPC mediated by the aforementioned agents, as shown in our study, may possibly contribute to their therapeutic benefits. Our study demonstrates that the combination of telmisartan and simvastatin exerts synergistic effects on mobilization and specific functional characteristics beyond the respective monotherapies. These results encourage further studies to characterize the pharmacological modulation of EPC and their effect on endothelial health and regeneration.
Acknowledgments
The excellent technical assistance of Isabel Paez-Maletz, Annika Bohner and Kathrin Paul is greatly appreciated. This study was supported by the Deutsche Forschungsgemeinschaft (DFG).
References
- 1.Giannotti G, Landmesser U. Endothelial dysfunction as an early sign of atherosclerosis. Herz. 2007;32:568–72. doi: 10.1007/s00059-007-3073-1. [DOI] [PubMed] [Google Scholar]
- 2.Werner N, Nickenig G. Endothelial progenitor cells in health and atherosclerotic disease. Ann Med. 2007;39:82–90. doi: 10.1080/07853890601073429. [DOI] [PubMed] [Google Scholar]
- 3.Hirschi KK, Ingram DA, Yoder MC. Assessing identity, phenotype, and fate of endothelial progenitor cells. Arterioscler Thromb Vasc Biol. 2008;28:1584–95. doi: 10.1161/ATVBAHA.107.155960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sieveking DP, Buckle A, Celermajer DS, et al. Strikingly different angiogenic properties of endothelial progenitor cell subpopulations: insights from a novel human angiogenesis assay. J Am Coll Cardiol. 2008;51:660–8. doi: 10.1016/j.jacc.2007.09.059. [DOI] [PubMed] [Google Scholar]
- 5.Asahara T, Murohara T, Sullivan A, et al. Isolation of putative progenitor endothelial cells for angiogenesis. Science. 1997;275:964–7. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 6.Lin Y, Weisdorf DJ, Solovey A, et al. Origins of circulating endothelial cells and endothelial outgrowth from blood. J Clin Invest. 2000;105:71–7. doi: 10.1172/JCI8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wassmann S, Werner N, Czech T, et al. Improvement of endothelial function by systemic transfusion of vascular progenitor cells. Circ Res. 2006;99:e74–83. doi: 10.1161/01.RES.0000246095.90247.d4. [DOI] [PubMed] [Google Scholar]
- 8.Werner N, Junk S, Laufs U, et al. Intravenous transfusion of endothelial progenitor cells reduces neointima formation after vascular injury. Circ Res. 2003;93:e17–24. doi: 10.1161/01.RES.0000083812.30141.74. [DOI] [PubMed] [Google Scholar]
- 9.Werner N, Wassmann S, Ahlers P, et al. Endothelial progenitor cells correlate with endothelial function in patients with coronary artery disease. Basic Res Cardiol. 2007;102:565–71. doi: 10.1007/s00395-007-0680-1. [DOI] [PubMed] [Google Scholar]
- 10.Heeschen C, Lehmann R, Honold J, et al. Profoundly reduced neovascularization capacity of bone marrow mononuclear cells derived from patients with chronic ischemic heart disease. Circulation. 2004;109:1615–22. doi: 10.1161/01.CIR.0000124476.32871.E3. [DOI] [PubMed] [Google Scholar]
- 11.Hill JM, Zalos G, Halcox JP, et al. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348:593–600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- 12.Laufs U, Werner N, Link A, et al. Physical training increases endothelial progenitor cells, inhibits neointima formation, and enhances angiogenesis. Circulation. 2004;109:220–6. doi: 10.1161/01.CIR.0000109141.48980.37. [DOI] [PubMed] [Google Scholar]
- 13.Vasa M, Fichtlscherer S, Aicher A, et al. Number and migratory activity of circulating endothelial progenitor cells inversely correlate with risk factors for coronary artery disease. Circ Res. 2001;89:E1–7. doi: 10.1161/hh1301.093953. [DOI] [PubMed] [Google Scholar]
- 14.Werner N, Kosiol S, Schiegl T, et al. Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med. 2005;353:999–1007. doi: 10.1056/NEJMoa043814. [DOI] [PubMed] [Google Scholar]
- 15.Min TQ, Zhu CJ, Xiang WX, et al. Improvement in endothelial progenitor cells from peripheral blood by ramipril therapy in patients with stable coronary artery disease. Cardiovasc Drugs Ther. 2004;18:203–9. doi: 10.1023/B:CARD.0000033641.33503.bd. [DOI] [PubMed] [Google Scholar]
- 16.Dimmeler S, Aicher A, Vasa M, et al. HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. J Clin Invest. 2001;108:391–7. doi: 10.1172/JCI13152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Werner N, Priller J, Laufs U, et al. Bone marrow-derived progenitor cells modulate vascular reendothelialization and neointimal formation: effect of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibition. Arterioscler Thromb Vasc Biol. 2002;22:1567–72. doi: 10.1161/01.atv.0000036417.43987.d8. [DOI] [PubMed] [Google Scholar]
- 18.Werner C, Kamani CH, Gensch C, et al. The peroxisome proliferator-activated receptor-gamma agonist pioglitazone increases number and function of endothelial progenitor cells in patients with coronary artery disease and normal glucose tolerance. Diabetes. 2007;56:2609–15. doi: 10.2337/db07-0069. [DOI] [PubMed] [Google Scholar]
- 19.Li X, Xu B. HMG-CoA reductase inhibitor regulates endothelial progenitor function through the phosphatidylinositol 3’-kinase/akt signal transduction pathway. Appl Biochem Biotechnol. 2008;157:545–53. doi: 10.1007/s12010-008-8263-7. [DOI] [PubMed] [Google Scholar]
- 20.Walcher D, Marx N. Insulin resistance and cardiovascular disease: the role of PPARgamma activators beyond their anti-diabetic action. Diab Vasc Dis Res. 2004;1:76–81. doi: 10.3132/dvdr.2004.011. [DOI] [PubMed] [Google Scholar]
- 21.Gensch C, Clever YP, Werner C, et al. The PPAR-gamma agonist pioglitazone increases neoangiogenesis and prevents apoptosis of endothelial progenitor cells. Atherosclerosis. 2007;192:67–74. doi: 10.1016/j.atherosclerosis.2006.06.026. [DOI] [PubMed] [Google Scholar]
- 22.Caballero AE, Saouaf R, Lim SC, et al. The effects of troglitazone, an insulin-sensitizing agent, on the endothelial function in early and late type 2 diabetes: a placebo-controlled randomized clinical trial. Metabolism. 2003;52:173–80. doi: 10.1053/meta.2003.50023. [DOI] [PubMed] [Google Scholar]
- 23.Schupp M, Janke J, Clasen R, et al. Angiotensin type 1 receptor blockers induce peroxisome proliferator-activated receptor-gamma activity. Circulation. 2004;109:2054–7. doi: 10.1161/01.CIR.0000127955.36250.65. [DOI] [PubMed] [Google Scholar]
- 24.Benson SC, Pershadsingh HA, Ho CI, et al. Identification of telmisartan as a unique angiotensin II receptor antagonist with selective PPARgamma-modulating activity. Hypertension. 2004;43:993–1002. doi: 10.1161/01.HYP.0000123072.34629.57. [DOI] [PubMed] [Google Scholar]
- 25.Walenta K, Friedrich EB, Sehnert F, et al. In vitro differentiation characteristics of cultured human mononuclear cells-implications for endothelial progenitor cell biology. Biochem Biophys Res Commun. 2005;333:476–82. doi: 10.1016/j.bbrc.2005.05.153. [DOI] [PubMed] [Google Scholar]
- 26.Petoumenos V, Nickenig G, Werner N. High density lipoprotein exerts vasculoprotection via endothelial progenitor cells. J Cell Mol Med. 2009;13:4623–35. doi: 10.1111/j.1582-4934.2008.00472.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kobayashi K, Imanishi T, Akasaka T. Endothelial progenitor cell differentiation and senescence in an angiotensin II-infusion rat model. Hypertens Res. 2006;29:449–55. doi: 10.1291/hypres.29.449. [DOI] [PubMed] [Google Scholar]
- 28.Imanishi T, Hano T, Nishio I. Angiotensin II accelerates endothelial progenitor cell senescence through induction of oxidative stress. J Hypertens. 2005;23:97–104. doi: 10.1097/00004872-200501000-00018. [DOI] [PubMed] [Google Scholar]
- 29.Benndorf RA, Gehling UM, Appel D, et al. Mobilization of putative high-proliferative-potential endothelial colony-forming cells during antihypertensive treatment in patients with essential hypertension. Stem Cells Dev. 2007;16:329–38. doi: 10.1089/scd.2006.0074. [DOI] [PubMed] [Google Scholar]
- 30.Yao EH, Fukuda N, Matsumoto T, et al. Losartan improves the impaired function of endothelial progenitor cells in hypertension via an antioxidant effect. Hypertens Res. 2007;30:1119–28. doi: 10.1291/hypres.30.1119. [DOI] [PubMed] [Google Scholar]
- 31.Yu Y, Fukuda N, Yao EH, et al. Effects of an ARB on endothelial progenitor cell function and cardiovascular oxidation in hypertension. Am J Hypertens. 2008;21:72–7. doi: 10.1038/ajh.2007.5. [DOI] [PubMed] [Google Scholar]
- 32.Zhao Y, Foryst-Ludwig A, Bruemmer D, et al. Angiotensin II induces peroxisome proliferator-activated receptor gamma in PC12W cells via angiotensin type 2 receptor activation. J Neurochem. 2005;94:1395–401. doi: 10.1111/j.1471-4159.2005.03275.x. [DOI] [PubMed] [Google Scholar]
- 33.Imayama I, Ichiki T, Inanaga K, et al. Telmisartan downregulates angiotensin II type 1 receptor through activation of peroxisome proliferator-activated receptor gamma. Cardiovasc Res. 2006;72:184–90. doi: 10.1016/j.cardiores.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 34.Assmus B, Urbich C, Aicher A, et al. HMG-CoA reductase inhibitors reduce senescence and increase proliferation of endothelial progenitor cells via regulation of cell cycle regulatory genes. Circ Res. 2003;92:1049–55. doi: 10.1161/01.RES.0000070067.64040.7C. [DOI] [PubMed] [Google Scholar]
- 35.Henrich D, Seebach C, Wilhelm K, et al. High dosage of simvastatin reduces TNF-alpha-induced apoptosis of endothelial progenitor cells but fails to prevent apoptosis induced by IL-1beta in vitro. J Surg Res. 2007;142:13–9. doi: 10.1016/j.jss.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 36.Llevadot J, Murasawa S, Kureishi Y, et al. HMG-CoA reductase inhibitor mobilizes bone marrow-derived endothelial progenitor cells. J Clin Invest. 2001;108:399–405. doi: 10.1172/JCI13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spyridopoulos I, Haendeler J, Urbich C, et al. Statins enhance migratory capacity by upregulation of the telomere repeat-binding factor TRF2 in endothelial progenitor cells. Circulation. 2004;110:3136–42. doi: 10.1161/01.CIR.0000142866.50300.EB. [DOI] [PubMed] [Google Scholar]
- 38.Urbich C, Knau A, Fichtlscherer S, et al. FOXO-dependent expression of the proapoptotic protein Bim: pivotal role for apoptosis signaling in endothelial progenitor cells. FASEB J. 2005;19:974–6. doi: 10.1096/fj.04-2727fje. [DOI] [PubMed] [Google Scholar]
- 39.Lee S, Kim W, Moon SO, et al. Rosiglitazone ameliorates cisplatin-induced renal injury in mice. Nephrol Dial Transplant. 2006;21:2096–105. doi: 10.1093/ndt/gfl194. [DOI] [PubMed] [Google Scholar]
- 40.Miura M, Satoh S, Inoue K, et al. Telmisartan pharmacokinetics in Japanese renal transplant recipients. Clin Chim Acta. 2008;399:83–7. doi: 10.1016/j.cca.2008.09.020. [DOI] [PubMed] [Google Scholar]
- 41.Dormandy JA, Charbonnel B, Eckland DJ, et al. Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): a randomised controlled trial. Lancet. 2005;366:1279–89. doi: 10.1016/S0140-6736(05)67528-9. [DOI] [PubMed] [Google Scholar]
- 42.Ikejima H, Imanishi T, Tsujioka H, et al. Effects of telmisartan, a unique angiotensin receptor blocker with selective peroxisome proliferator-activated receptor-gamma-modulating activity, on nitric oxide bioavailability and atherosclerotic change. J Hypertens. 2008;26:964–72. doi: 10.1097/HJH.0b013e3282f52c36. [DOI] [PubMed] [Google Scholar]
- 43.Li AC, Brown KK, Silvestre MJ, et al. Peroxisome proliferator-activated receptor gamma ligands inhibit development of atherosclerosis in LDL receptor-deficient mice. J Clin Invest. 2000;106:523–31. doi: 10.1172/JCI10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nissen SE, Nicholls SJ, Sipahi I, et al. Effect of very high-intensity statin therapy on regression of coronary atherosclerosis: the ASTEROID trial. JAMA. 2006;295:1556–65. doi: 10.1001/jama.295.13.jpc60002. [DOI] [PubMed] [Google Scholar]
- 45.Nissen SE, Nicholls SJ, Wolski K, et al. Comparison of pioglitazone vs glimepiride on progression of coronary atherosclerosis in patients with type 2 diabetes: the PERISCOPE randomized controlled trial. JAMA. 2008;299:1561–73. doi: 10.1001/jama.299.13.1561. [DOI] [PubMed] [Google Scholar]
- 46.Wassmann S, Czech T, van Eickels M, et al. Inhibition of diet-induced atherosclerosis and endothelial dysfunction in apolipoprotein E/angiotensin II type 1A receptor double-knockout mice. Circulation. 2004;110:3062–7. doi: 10.1161/01.CIR.0000137970.47771.AF. [DOI] [PubMed] [Google Scholar]
- 47.Wassmann S, Hilgers S, Laufs U, et al. Angiotensin II type 1 receptor antagonism improves hypercholesterolemia-associated endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2002;22:1208–12. doi: 10.1161/01.atv.0000022847.38083.b6. [DOI] [PubMed] [Google Scholar]
- 48.Wassmann S, Nickenig G. Interrelationship of free oxygen radicals and endothelial dysfunction-modulation by statins. Endothelium. 2003;10:23–33. doi: 10.1080/10623320303357. [DOI] [PubMed] [Google Scholar]
- 49.Yusuf S, Teo K, Anderson C, et al. Effects of the angiotensin-receptor blocker telmisartan on cardiovascular events in high-risk patients intolerant to angiotensin-converting enzyme inhibitors: a randomised controlled trial. Lancet. 2008;372:1174–83. doi: 10.1016/S0140-6736(08)61242-8. [DOI] [PubMed] [Google Scholar]
- 50.Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. 2008;359:2195–207. doi: 10.1056/NEJMoa0807646. [DOI] [PubMed] [Google Scholar]