

Abstract
Gene targeting by single-stranded oligodeoxyribonucleotides (ssODNs) is emerging as a powerful tool for the introduction of subtle gene modifications in mouse embryonic stem (ES) cells and the generation of mutant mice. Here, we have studied the role of ssODN composition, transcription and replication of the target locus, and DNA repair pathways to gain more insight into the parameters governing ssODN-mediated gene targeting in mouse ES cells. We demonstrated that unmodified ssODNs of 35–40 nt were most efficient in correcting a chromosomally integrated mutant neomycin reporter gene. Addition of chemical modifications did not further enhance the efficacy of these ssODNs. The observed strand bias was not affected by transcriptional activity and may rather be caused by the different accessibility of the DNA strands during DNA replication. Consistently, targeting frequencies were enhanced when cells were treated with hydroxyurea to reduce the rate of replication fork progression. Transient down-regulation of various DNA repair genes by RNAi had no effect on the targeting frequency. Taken together, our data suggest that ssODN-mediated gene targeting occurs within the context of a replication fork. This implies that any given genomic sequence, irrespective of transcriptional status, should be amenable to ssODN-mediated gene targeting. The ability of ES cells to differentiate into various cell types after ssODN-mediated gene targeting may offer opportunities for future therapeutic applications.
Keywords: single-stranded oligonucleotides, gene targeting, targeted gene alteration, embryonic stem cells, DNA replication
Introduction
Gene modification by synthetic single-stranded oligodeoxyribonucleotides (ssODNs) is a promising alternative to existing strategies for the generation of subtle mutations in mouse ES cells [1, 2]. These strategies are based on targeting vectors that upon homologous recombination (HR) integrate a selectable marker gene together with the desired genetic alteration into the gene of interest. Construction of these targeting vectors can be laborious and time-consuming. Moreover, although the marker gene is subsequently removed by Cre/lox-mediated recombination, traces of exogenous DNA are inevitably left behind in the target locus [3]. Alternatively, short ssODNs of 40 nt can be used to modify a single or a few bases in a specific location of the genome without altering the genomic organization.
In mouse ES cells, ssODN-mediated gene targeting frequencies range from 10−7 to 10−4 and are relatively low compared to those found in other mammalian cell lines [4–7]. Previously, we have demonstrated that the DNA mismatch repair (MMR) system strongly impaired the introduction of genetic alterations by ssODNs in mouse ES cells [2, 4]. Transfer of the genetic information requires base pairing between the ssODN and the chromosomal target locus, resulting in the formation of DNA mismatches. The MMR system recognizes and removes these mismatches, thereby preventing stable inheritance of the desired genetic alteration. Knock-down of the central MMR gene Msh2 by RNA interference dramatically improved the targeting frequency in mouse ES cells [1]. This effect has been confirmed in human hepatocytes [8] and in an episomal reporter system in mouse embryonic fibroblasts (MEFs) [9]. Thus, by transiently disabling the MMR system we could develop a simple and rapid procedure for the generation of mutant mouse ES cells. Importantly, the resulting mutant ES cells retained their pluripotency and mutant alleles could successfully be transmitted through the mouse germline [1, 2]. Further improvement of the targeting frequency in mouse ES cells may greatly enhance the applicability of ssODN-mediated gene targeting, not only as a tool for biomolecular research but also for therapeutic purposes.
Thus far, parameters governing ssODN-mediated gene targeting have been studied in bacteria, yeast and a variety of mammalian cell lines. This has yielded conflicting data that may be ascribed to differences in ssODN composition, type of reporter system and model organism (reviewed in [10]). Many reports have shown that antisense ssODNs (i.e. complementary to the non-transcribed strand) are more effective than sense ssODNs [5, 11–13]. Protection of ssODN against nucleolytic degradation by 2′-O-methyl RNA residues [5, 11], phosphorothioate (PTO) linkages [12, 13] or locked nucleic acid (LNA) bases [14, 15] seemed to enhance targeting frequencies. Transcriptional activity promoted ssODN-mediated gene targeting in inducible episomal reporter systems in Escherichia coli[16, 17], Saccharomyces cerevisiae[12] and mammalian cells [18]. However, in chromosomal reporter systems in E. coli it has been shown that the direction of replication rather than the direction of transcription influenced the targeting frequency [16, 17, 19, 20]. By changing the orientation of the reporter gene towards the replication fork, higher targeting frequencies were observed when ssODNs with the same polarity as the nascent lagging strand were used. DNA replication also seems to play an important role in ssODN-mediated gene targeting in mammalian cells. Synchronization of cells in the S phase of the cell cycle or reducing the rate of replication fork progression improved the frequency of ssODN-mediated gene correction [21–24]. On the other hand, targeting frequencies decreased when the replication machinery was blocked, indicating that DNA synthesis is required for effective ssODN-mediated gene correction. These findings suggest that the ssODN anneals to its single-stranded target region within the context of a replication fork. After annealing, the ssODN is extended by the replication machinery, resulting in incorporation of the ssODN into the nascent DNA strand [20, 25]. Nonetheless, DNA repair proteins may be involved in promoting various steps of the targeting process, because gene targeting frequencies were elevated upon overexpression of HR proteins [26, 27] or endonucleases [28] and upon treatment with DNA-damaging agents [29, 30].
In this report, we have systematically investigated the role of various parameters in the targeting process in mouse ES cells, such as ssODN composition, transcription and replication of the target locus, and DNA repair pathways.
Materials and methods
Cell lines and culture conditions
129/Ola-derived E14-IB10 ES cells [31] were cultured on MEF feeders in Glasgow minimal essential medium supplemented with 10% foetal calf serum, 1 mM sodium pyruvate, 1× non-essential amino acids, 1 mM 2-mercaptoethanol and 1000 U/ml of leukaemia inhibitory factor. For transfections and antibiotic selections, ES cells were cultured onto gelatin-coated plates in BRL (Buffalo rat liver cells)-conditioned medium [31].
Neo reporter cell lines
We developed a selectable mutant neomycin (neo) reporter cell line in which the start codon of the neo resistance gene was mutated from ATG to AAG (Fig. 1). A single copy of the mutant neo reporter gene was stably integrated into the Rosa26 locus of Msh2-deficient and wild-type ES cells as described previously [4]. To create a Rev neo cell line that carries the neo gene in the opposite direction relative to the Rosa26 promoter, we cloned the neo gene in the opposite orientation into the Rosa26-His targeting vector. Single-copy integration was confirmed by Southern blot analysis.
Fig 1.
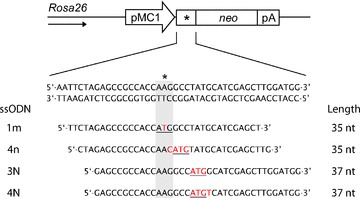
Mutant neomycin reporter gene and ssODN sequences. Sequence of the mutant neomycin (neo) reporter gene in which the start codon (ATG) is replaced for AAG (*). Activity of the neo gene can be restored by ssODNs that substitute one to four nucleotides (indicated in red) to create a new ATG start codon (underlined). Sequences of sense ssODNs complementary to the transcribed strand are shown. The arrow indicates the direction of the Rosa26 promoter.
ROSA-TET cell lines
To establish ES cell lines that express the neo gene under the control of the Tc-controllable promoter, we used the ROSA-TET system described by Masui et al.[32]. E14tg2a-derived EBRTcH3 ES cells (carrying the tetracycline-controllable transactivator tTA and IRES-Venus ORFs in the Rosa26 locus) and plasmids pPthC-Oct3/4 and pCAGGS-Cre were a kind gift from Dr Masui. To construct an exchange vector with the mutant neo cDNA, the neo gene was amplified by PCR using forward primer 5′-CCGCTCGAGCGACCCAATTCTA-3′ and reverse primer 5′-ATATGCGGCCGCTCAGAAGAACTCGTCAAG-3′ and the Rosa26-His-neo targeting vector as template to introduce XhoI and NotI sites (underlined). The XhoI/NotI digested neo fragment was inserted into exchange vector pPthC-Oct-3/4 to obtain pPthC-(AAG)neo. Similarly, a pPthC-(ATG)neo exchange vector with the wild-type neo start codon was constructed for control purposes (PCR amplification with forward primer 5′-CCGCTCGAGATGGCCTATGCATCG-3′ to introduce a correct ATG start codon). We established ROSA-TET ES cells by co-transfection of 5 μg exchange vector pPthC-neo and 2 μg pCAGGS-Cre with 10 μl of Lipofectamine 2000 (Invitrogen, Breda, The Netherlands) following previously described procedures [32]. Single copy-integration was confirmed by Southern blot analysis. The recombinant cell lines were maintained in 1.5 mg/ml puromycin (Sigma-Aldrich, Zwijndrecht, The Netherlands).
For targeting experiments, 7 × 105 ROSA-TET ES cells were seeded onto a gelatin-coated 6-well in BRL-conditioned medium supplemented with 1 μg/ml tetracycline (+Tc; Sigma-Aldrich) or without tetracycline (−Tc) the day before ssODN transfection. Cells were exposed to ssODN in +Tc or −Tc medium for 24 hrs as described elsewhere. Following transfection, cells were washed twice with PBS, counted and replated onto three gelatin-coated 100 mm dishes in medium without Tc.
Transfection
ssODNs and pSUPER plasmids were transfected following the TransFast-mediated transfection method described earlier [1]. Generally, 7 × 105 ES cells were seeded onto a gelatin-coated 6-well in BRL-conditioned medium the day before transfection. For one well, 3 μg of ssODN and 27 μl of TransFast transfection agent (Promega, Leiden, The Netherlands) were diluted in 1.4 ml serum-free medium and incubated for 15 min at room temperature. After 75 min of exposure to the transfection mixture at 37°C, 4 ml of BRL-conditioned medium with serum was added to the cells. The day after ssODN transfection, cells were counted and replated onto three gelatin-coated 100 mm dishes. G418 selection (800 μg/ml for Fwd neo and Rev neo cell lines, 2.5 mg/ml for ROSA-TET cell line; GIBCO-Invitrogen) was started 48 hrs after transfection. After 10 days, G418-resistant colonies were stained with Leishman’s eosin methylene blue solution (Merck, Darmstadt, Germany) and counted. Transfection efficiencies were calculated by dividing the number of G418-resistant colonies by the number of cells that were plated 24 hrs after ssODN transfection.
Oligonucleotides
ssODNs were designed to correct the mutant start region of the neo reporter gene by introducing an in-frame ATG start codon (sequences of sense ssODN are listed in Fig. 1). Antisense ssODNs introduce identical base changes but have opposite polarity (i.e. are complementary to the non-transcribed strand). When indicated, ssODNs were modified with either three PTO linkages or a single LNA base on each end to protect them against nucleolytic degradation. Unmodified and PTO-modified ssODNs, deprotected and desalted, were obtained from Sigma-Aldrich, Inc. LNA-modified ssODNs were obtained from Sigma-Proligo and Eurogentec.
Fluorescence microscopy
ROSA-TET ES cells carrying the mutant or wild-type neo reporter gene were plated at a density of 1.5 × 106 cells per well onto 35 mm glass-bottom Willco dishes in both Tc+ and Tc− medium. After 24 hrs, dishes were transferred to a Zeiss Axiovert 200M microscope, equipped with a 0.55 numeric aperture condenser and a 40X Plan-Neo DIC objective (N.A. 1.3) using a Photometrics Coolsnap HQ CCD camera (Roper Scientific, Vianen, The Netherlands) with a CFP/YFP dual-band pass filter set to select specific fluorescence. Images were processed using MetaMorph software (Universal Imaging, Downington, PA, USA).
Drug treatment and colony survival
The sensitivity of ES cells to increasing doses of replication inhibitors was determined by measuring their colony-forming ability. ES cells were plated at 7 × 105 cells per well onto gelatin-coated 6-well plates 24 hrs prior to drug treatment. Cells were exposed to increasing doses of aphidicolin (100 μM stock in DMSO; Sigma-Aldrich) or hydroxyurea (100 mM stock in PBS; Sigma-Aldrich) for 6 hrs, transfected with ssODN as described above and exposed to drugs for another 22 hrs. Then, cells were counted and plated at 500 cells/100 mm dish to determine colony survival, whereas the remainder of the cells were plated for G418 selection. After 8 days, surviving colonies were stained and counted.
RNA interference
We used the pSUPER vector containing a puromycin resistance gene for expression of shRNAs [33]. For each target gene, three different pSUPER vectors were constructed (sequences available upon request). An empty vector without gene-specific 19-nucleotide sequence served as a non-silencing control (pS), whereas luciferase (pP-Luc) and pS-MSH2 [1] vectors served as additional controls. For transient down-regulation of the various DNA repair proteins, ES cells were first transfected with 3 μg of pSUPER vector followed by selection with 20 μg/ml of puromycin (Sigma-Aldrich) for 2 days. Cells were washed twice with PBS, trypsinized and plated at a density of 7 × 105 cells per well for ssODN transfection as described above.
RNA isolation and quantitative real-time PCR
ES cells were transfected with the different pSUPER constructs as described above. After puromycin selection for 2 days, total RNA was extracted using RNA-Bee Total RNA Isolation Reagent (Campro Scientific, Veenendaal, The Netherlands). For each reaction, 1 μg of RNA was used for first-strand cDNA synthesis using SuperScript II Reverse Transcriptase (Invitrogen) and random primers.
Quantitative real-time PCR was performed in an ABI Prism 7000 Sequence Detection System (Applied Biosystems, Nieuwerkerk a/d IJssel, The Netherlands) using the standard amplification program. Amplification was performed in a final volume of 25 μl, containing 15–30 ng of cDNA from the reversed transcribed reaction, 0.4 μM each of forward and reverse primers, and 12.5 μl of 2x SYBR Green PCR Master Mix (Applied Biosystems). Where possible, primers were designed to overlap adjacent exon boundaries to exclude detection of genomic DNA (Primer Express 1.0 software, Applied Biosystems). The final mRNA levels of the studied genes were normalized to mouse β-actin mRNA levels.
Results
Reporter system
As a read-out for the frequency of ssODN-mediated gene targeting, we used our previously described neomycin (neo) reporter system in which the neo gene was mutated by a single-base substitution in the start codon (ATG to AAG; Fig. 1). A single copy of this mutant neo gene was stably integrated into the Rosa26 locus of Msh2-deficient and wild-type mouse embryonic stem (ES) cells [4]. ssODNs of 35–37 nt containing one, three or four centrally located nucleotide substitutions were used to correct the mutant neo sequence (i.e. generate a novel ATG start codon) resulting in G418 resistance. Targeting efficiencies were calculated by dividing the number of G418-resistant colonies by the number of cells that were plated after ssODN exposure. Using unmodified ssODNs, Msh2-deficient ES cells showed targeting efficiencies of approximately 3 × 10−5, whereas wild-type ES cells typically showed targeting efficiencies of less than approximately 5 × 10−7[2, 4].
Chemical ssODN modifications
The length of the ssODN and its chemical composition may influence the targeting efficiency. To investigate this, we designed three series of ssODNs varying in length and chemical composition: ssODN 4n (Fig. 2A), ssODNs 1m (Fig. 2B) and ssODN 4N (Fig. 2C). For all these ssODNs, the optimal length of unmodified ssODNs was 35–40 nt and sense ssODNs were more effective than antisense ssODNs (Fig. 2, black bars). We previously found a similar optimal length for an ssODN, correcting a frameshift neo reporter [4].
Fig 2.
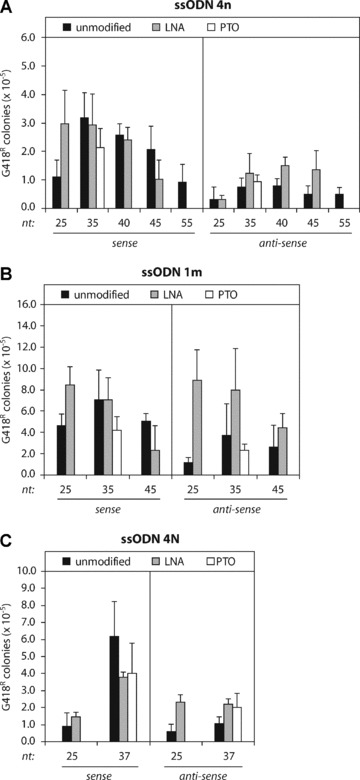
Gene targeting by different types of ssODNs in Msh2-deficient ES cells. (A) Unmodified (black bars), LNA-modified (grey bars) and PTO-modified (white bars) ssODN 4n of various length (nt, nucleotides) and polarity (sense or antisense) were used to correct the mutant neo gene in Msh2-deficient ES cells. (B) Correction of the mutant neo gene by unmodified (black bars), LNA-modified (grey bars) and PTO-modified (white bars) ssODN 1m. (C) Correction of the mutant neo gene by unmodified (black bars), LNA-modified (grey bars) and PTO-modified (white bars) ssODN 4N. Targeting efficiency is the number of G418-resistant colonies per 105 cells that were plated after ssODN exposure. Error bars represent the standard deviation (S.D.) of at least three independent experiments.
Many reports have shown that gene targeting was only efficient when ssODNs were protected against nucleolytic degradation by flanking PTO linkages or LNA bases [6, 15, 24]. We have tested ssODNs that were modified with a single LNA base at the 5′ and 3′ end position (Fig. 2, grey bars). Addition of LNA modifications had no significant effect on the efficiency of 35–40 nt sense ssODNs. However, the targeting frequencies of 35–40 nt antisense ssODNs were increased by LNA modifications, but they did not exceed the frequencies obtained with sense ssODNs. Short 25 nt ssODNs benefited most strongly from the addition of LNA modifications, reaching frequencies similar to those obtained with 35–40 nt unmodified sense ssODNs (Fig. 2A and B), except for ssODNs 4n antisense and 4N sense.
The effect of PTO modifications was tested by adding three PTO linkages to the 5′ and 3′ ends of ssODNs of 35 and 37 nt (Fig. 2, white bars). PTO modifications decreased the targeting efficiency of sense ssODNs, as previously shown using the frameshift neo reporter [4], and hardly affected the efficiency of antisense ssODNs. Similar results were obtained when LNA- or PTO-modified ssODNs were tested in wild-type ES cells, excluding MMR-dependent effects (data not shown).
Taken together, we have shown that unmodified sense ssODNs of 35–40 nt were most effective in correcting the mutant neo sequence in Msh2-deficient ES cells. Chemical ssODN modifications did not further improve the targeting efficiency and were not required for optimal ssODN-mediated gene targeting.
Transcription
We showed that ssODNs in the sense orientation were more effective than ssODNs in the antisense orientation (Fig. 2). In contrast, others have shown that antisense ssODNs, that is, complementary to the non-transcribed strand, were more effective than sense ssODNs [5, 11–13]. This strand bias has been attributed to the increased accessibility of the non-transcribed strand for ssODN annealing during transcription, suggesting that transcription has a beneficial effect on ssODN-mediated gene targeting [18]. To gain insight into the role of transcription in ssODN-mediated gene targeting in ES cells, we used the ROSA-TET system described by Masui et al.[32]. In this Tet-off system, the neo gene is under the control of the hCMV*-1 promoter containing a tetracycline response element and the transcriptional transactivator tTA, stably integrated into the Rosa26 locus of wild-type ES cells (Fig. 3A). In culture medium without tetracycline (Tc), tTA binds to the hCMV*-1 promoter and induces the expression of the neo reporter gene as well as Venus, whose fluorescence is used as an internal control for Tc-responsiveness. Msh2-proficient ROSA-TET ES cell lines were constructed containing either a wild-type (ATG) or a mutant (AAG) neo reporter gene. To test the Tc-responsiveness, ROSA-TET ES cells were cultured in −Tc (transcription ‘on’) or +Tc (transcription ‘off’) culture medium for 24 hrs. Addition of Tc to the culture medium resulted in a 20-fold reduced expression of neo in both the wild-type and the mutant neo ROSA-TET cell lines, as determined by Venus fluorescence (Fig. 3B) and quantitative RT-PCR analysis of neo mRNA levels (Fig. 3C).
Fig 3.
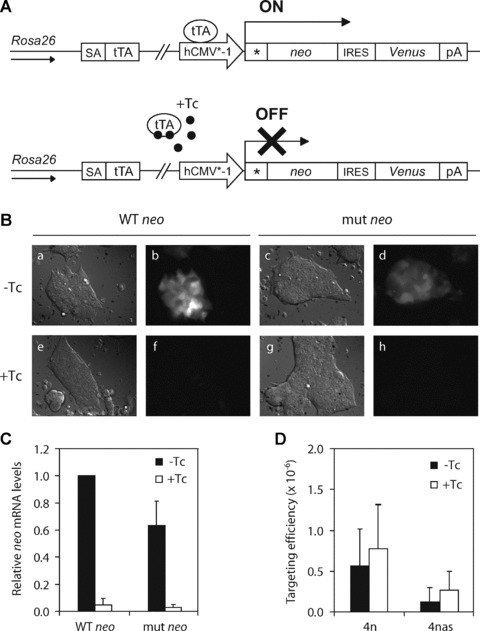
Effect of transcription on ssODN-mediated gene targeting in wild-type ES cells. (A) A single copy of a tetracycline (Tc)-controllable mutant neo reporter gene was stably integrated into the Rosa26 locus of wild-type ES cells. In the resulting ROSA-TET cell line, binding of tTA to the hCMV*-1 promoter induces expression of a neo-IRES-Venus reporter gene in culture medium without Tc. (B) Tc-induced inhibition of Venus expression in ROSA-TET ES cells carrying a wild-type (WT) or mutant (mut) neo reporter gene. Cells were cultured for 24 hrs in medium without tetracycline (−Tc; panels a, b, c and d) or medium plus tetracycline (+Tc; panels e, f, g and h) before visualization of Venus fluorescence through a YFP filter (panels b, d, f and h). Corresponding bright field images are shown in panels a, c, e and g, respectively. (C) Quantitative RT-PCR showing Tc-induced inhibition of neo expression in ROSA-TET ES cells carrying a wild-type (WT) or mutant (mut) neo reporter gene. Mouse β-actin mRNA levels were used for normalization. (D) Correction of the mutant neo gene in ROSA-TET ES cells by ssODN 4n (sense orientation) or ssODN 4nas (antisense orientation) in −Tc medium (transcription ‘on’) or +Tc medium (transcription ‘off’). Targeting efficiency is the number of G418-resistant colonies per 106 cells that were plated after ssODN exposure. Error bars represent the S.D. of nine independent experiments.
To investigate whether the correction frequency of the mutant neo reporter gene was influenced by the level of transcription, ROSA-TET ES cells were cultured in −Tc or +Tc culture medium from 24 hrs before to 24 hrs after ssODN transfection. Then, all cells were cultured in −Tc medium to induce expression of the neo gene before G418 selection was started (48 hrs after transfection).
Although Tc addition resulted in a 20-fold decrease in neo expression levels, the targeting efficiencies of sense ssODN 4n and antisense ssODN 4nas were not significantly altered (Fig. 3D). Independent of the level of transcriptional activity, sense ssODNs were still more effective than antisense ssODNs. Our findings demonstrate that transcription does not play a role in ssODN-mediated gene targeting in ES cells, suggesting that poorly or even non-transcribed genes are accessible to ssODN-mediated gene targeting.
Replication
As the observed strand bias was not affected by differences in transcriptional activity, we investigated whether the different accessibility of the leading and lagging strand during DNA replication plays a role in dictating the strand bias. In E. coli, consistently higher targeting frequencies were obtained with ssODNs corresponding in sequence to the nascent lagging strand [16, 17, 19, 20]. During discontinuous lagging strand synthesis, there are more single-stranded regions available than during the more continuous leading strand synthesis, thereby possibly facilitating annealing of the ssODN to its target site. To examine whether the strand bias correlated with the direction of replication, we constructed Msh2-deficient ES cell lines that contain a single copy of the neo gene in the opposite orientation at the same chromosomal position in the Rosa26 locus (Fig. 4A). In the resulting Rev neo and original Fwd neo reporter cell lines, the transcribed and non-transcribed strands are the same, whereas leading and lagging strands have (supposedly) interchanged. If the strand bias is directed by preferred incorporation of sense ssODNs into the nascent lagging strand in the Fwd neo cell line, then antisense ssODNs should be most effective in the Rev neo cell line.
Fig 4.
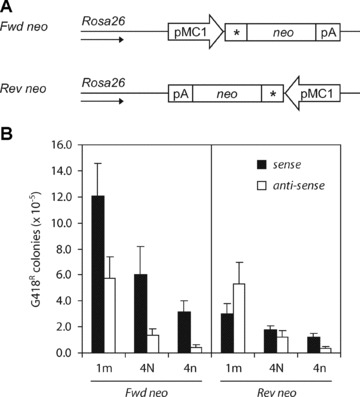
Effect of the orientation of the neo reporter gene on ssODN-mediated gene targeting. (A) Fwd and Rev neo cell lines contain a single copy of the mutant neo gene integrated in opposite directions at the same chromosomal position in the Rosa26 locus of Msh2-deficient ES cells. The direction of the Rosa26 promoter is indicated by an arrow. (B) Correction of the neo gene by sense ssODNs (black bars) or antisense ssODNs (white bars) in the Fwd and Rev neo cell lines. Targeting efficiency is the number of G418-resistant colonies per 105 cells that were plated after ssODN exposure. Error bars represent the S.D. of at least three independent experiments.
As shown in Fig. 4B, the efficiency of all sense ssODNs was three to fourfold decreased in the Rev neo cell line compared to the Fwd neo cell line. Although this may indicate that the strand bias was indeed dictated by the direction of replication, the decreased efficiency of the sense ssODN was not accompanied by an increase in efficiency of the antisense ssODN in the Rev neo cell line. In fact, the targeting efficiencies of the antisense ssODNs were not affected by reversal of the neo reporter. Consequently, the strand bias observed in the Fwd neo cell line was no longer present in the Rev neo cell line: sense and antisense ssODNs were equally effective. These findings were confirmed using cell lines carrying a frameshift neo reporter in opposite orientations into the Rosa26 locus. Our results obtained with the sense ssODNs indicate that the direction of replication is an important factor in dictating the strand bias in ssODN-mediated gene targeting; however, the results obtained with the antisense ssODNs remain enigmatic.
Slowing replication fork progression
More evidence that replication plays an important role in ssODN-mediated gene targeting comes from the observation that S phase appeared to be the preferred cell-cycle phase for gene correction [23, 24]. Synchronization of cells at the G1/S phase or slowing down replication fork progression has led to increased targeting frequencies in a variety of mammalian cell lines [21–24]. Because approximately 50% of asynchronously growing ES cells are already in S phase (unpublished observation), we attempted to reduce the rate of replication fork progression, rather than increasing the number of cells progressing through S phase. We used low doses of two different replication inhibitors: aphidicolin, which directly inhibits the replicative polymerases α, γ and by competing with dCTP binding [34], and hydroxyurea, which inactivates ribonucleotide reductase, thereby depleting cellular dNTP pools [35].
Msh2-deficient ES cells were exposed to increasing concentrations of aphidicolin or hydroxyurea between 6 hrs before and 22 hrs after transfection of ssODN 3N. Then, part of the cells were plated to assess the colony-forming ability, whereas the remainder of the cells were plated in G418-containing medium to assess the neo correction frequency. In each independent experiment, the results were normalized to the targeting efficiency of untreated control cells. As shown in Fig. 5A, aphidicolin treatment resulted in a dose-dependent decrease in targeting efficiency compared to untreated control cells (Fig. 5A, black bars). However, aphidicolin treatment also reduced the colony-forming ability of ES cells (Fig. 5B). When targeting efficiencies were corrected for this decreased survival, aphidicolin treatment had no significant effect on the targeting efficiency (Fig. 5A, white bars).
Fig 5.
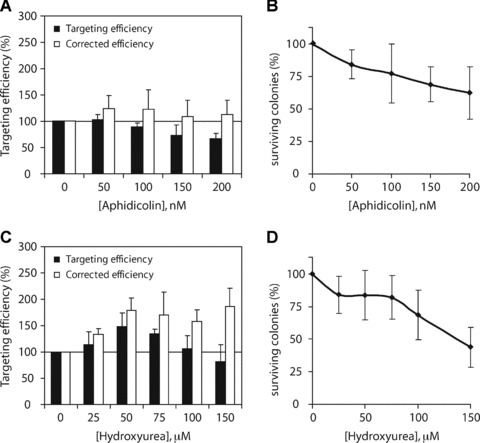
Effect of replication inhibition on ssODN-mediated gene targeting. (A) Msh2-deficient ES cells carrying the mutant neo gene were incubated with increasing concentrations of aphidicolin or hydroxyurea (C) between 6 hrs before and 22 hrs after exposure to ssODN 3N. Relative targeting efficiencies are the number of G418-resistant colonies per 105 cells that were plated after ssODN exposure, normalized to the efficiency found in untreated control cells (black bars). Corrected targeting efficiencies are adjusted for the decreased colony survival after treatment with aphidicolin or hydroxyurea (white bars). Error bars represent the S.D. of three independent experiments. (B) Colony-forming ability of Msh2-deficient ES cells after exposure to increasing doses of aphidicolin or hydroxyurea (D) between 6 hrs before and 22 hrs after ssODN exposure. Error bars represent the S.D. of three independent experiments.
In contrast, when cells were treated with hydroxyurea, targeting frequencies increased to 148% at 50 μM hydroxyurea (Fig. 5C, black bars). Higher concentrations of hydroxyurea resulted in a gradual decrease in targeting efficiency, reflecting the reduced colony survival at these doses (Fig. 5D). After correction for this decreased survival, targeting efficiencies reached levels of 179% at 50 μM hydroxyurea to 186% at 150 μM hydroxyurea (Fig. 5C, white bars). These findings suggest that the ssODN anneals to its homologous target region within the context of a replication fork.
DNA repair pathways
Treatment with DNA-damaging agents has been shown to stimulate ssODN-mediated gene targeting, which has been attributed to the induction of DNA double-strand breaks [29, 30, 36]. In addition, (over)expression of the HR proteins Rad51 and Rad54 may promote annealing of the ssODN with its target region [26, 27], whereas nucleotide excision repair (NER) endonucleases ERCC1/XPF and XPG may be involved in resection of the target region to facilitate ssODN incorporation [28].
We examined the involvement of different DNA repair pathways in the ssODN-mediated gene targeting process in ES cells, using transient down-regulation by RNA interference. The role of proteins involved in MMR (Msh3, ExoI), HR (Mre11, Rad50, Rad51, XRCC2, Rad52, Rad54), non-homologous end joining (Ku70, Ku80), NER (XPA, XPF, ERCC1), translesion synthesis (Rev1, Rev3), post-replicative repair (Rad6, Rad18, MMS2, Rad9) and DNA damage signalling (ATM, ATR, p53) was investigated as well as that of various DNA endonucleases (Mus81, Eme1, Fen1, MBD4) and DNA helicases (Blm, Wrn, Brip1). Msh2-deficient and wild-type ES cells containing the mutant neo reporter gene were transfected with a pSUPER vector expressing a short RNA hairpin, followed by puromycin selection for 2 days to eliminate untransfected cells. Then, cells were transfected with ssODN 3N to correct the neo reporter gene. In each experiment, empty pSUPER (pS) transfected ES cells were used for normalization of the targeting efficiencies. Luciferase (pP-Luc) and Msh2 knock-down vectors served as additional controls. As shown in Fig. 6, none of the tested knock-down vectors resulted in a significant change in targeting efficiency in Msh2-deficient ES cells (Fig. 6, black bars) or in wild-type ES cells (data not shown), although the median knock-down level was 79% (ranging from 96% to 43%) as determined by quantitative RT-PCR (Fig. 6, grey bars). Down-regulation of Rad51 and Blm appeared to reduce the targeting efficiency in Msh2-deficient ES cells to 30 and 60%, respectively; however, this could be fully attributed to a reduction in the percentage of surviving colonies (29 and 53% compared to pS-transfected control cells, respectively).
Fig 6.
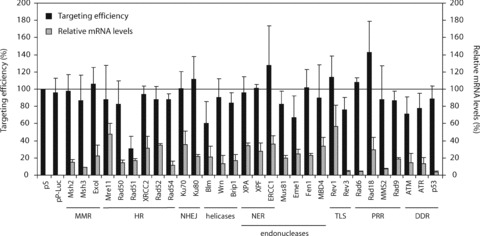
Effect of RNAi-mediated down-regulation of DNA repair genes on ssODN-mediated gene targeting. Correction of the mutant neo gene by ssODN 3N in Msh2-deficient ES cells after transient down-regulation of various DNA repair genes by vector-mediated RNAi. Relative targeting efficiencies are the number of G418-resistant colonies per 105 cells that were plated after ssODN exposure, normalized to the efficiency found in empty vector (pS)-transfected control cells (black bars). Error bars represent the S.D. of at least three independent experiments. Expression levels of the various DNA repair genes were quantified by RT-PCR and normalized using mouse β-actin mRNA levels (grey bars). MMR, mismatch repair; HR, homologous recombination; NHEJ, non-homologous end joining; NER, nucleotide excision repair; TLS, translesion synthesis; PRR, post-replicative repair; DDR, DNA damage response.
In conclusion, knock-down of the investigated DNA repair proteins had no significant effect on the targeting efficiency in either Msh2-deficient or wild-type ES cells, indicating that these proteins are neither essential for nor detrimental to ssODN-mediated gene targeting.
Discussion
Numerous reports have demonstrated that ssODNs can be used for site-specific modification of a genomic target sequence. This technique would be particularly useful in ES cells for a number of reasons. First, ES cells are diploid, requiring only two gene copies to be modified for phenotypic analyses in vitro; second, mouse ES cells can be used to generate mice, allowing the modification to be studied in the context of a living animal; third, ES cells or ES-cell-like cells may become the preferred target cell for gene correction for therapeutic purposes. These applications will strongly benefit from in-depth insight into the parameters governing ssODN-directed gene modification in ES cells. We have previously shown that DNA mismatch repair raises a strong barrier to ssODN-mediated gene targeting in ES cells and that this problem can be overcome by transient down-regulation of the central MMR gene Msh2. In the present study, we have systematically investigated the role of various parameters in the targeting process, such as transcription, replication and DNA repair pathways.
Using our neo reporter system in mouse ES cells, we have shown that ssODNs in the sense orientation (with respect to the direction of transcription of the neo gene) are more effective than ssODNs in the antisense orientation, independent of ssODN length (Fig. 2). This preference for sense ssODNs has been confirmed in some reports [7, 17, 36, 37], whereas others have shown a preference for targeting by antisense ssODNs [5, 11–13]. A possible explanation for this difference in reported strand biases could be the type of read-out used to determine the targeting frequency. The chromosomally integrated mutant EGFP[13, 22] or LacZ[5, 11] reporter genes that were used in these studies allow detection of targeted cells by flow cytometry or (fluorescence) microscopy already 24 hrs after ssODN transfection. Because antisense ssODNs are incorporated into the transcribed strand, gene correction by these ssODNs will lead to instant EGFP/LacZ expression, without the need for an extra round of DNA replication. In contrast, correction by sense ssODNs does require an extra round of replication to copy the base alterations to the transcribed strand before EGFP/LacZ expression can be detected. Consequently, reporter genes with an early read-out may skew the results towards antisense ssODNs being the most effective. In our assay, correction of the neo reporter gene is monitored by the appearance of G418-resistant colonies after 8–10 days, which eliminates this ‘early’ transcriptional strand bias.
Besides polarity, we have also investigated the effect of ssODN length and chemical composition on the targeting frequency (Fig. 2). Extending our previous results, we found that the optimal length of unmodified ssODNs was approximately 35–40 nt. LNA modifications did not affect the targeting efficiency of sense ssODNs of this size, but improved the efficiency of most antisense ssODNs. Addition of LNA modifications particularly improved the targeting efficiencies of short ssODNs of 25 nt. Shorter ssODNs may be more susceptible to nucleolytic degradation, which is prevented by the addition of chemical modifications. Additionally, LNA modifications may also increase the stability of the heteroduplex that is formed between the ssODN and its target sequence. Since 25 nt ssODNs can form fewer base pairs with the target sequence than their longer counterparts, LNA may be more beneficial for short ssODNs.
To investigate the role of transcription on the targeting frequency and the observed strand bias, we have developed a ROSA-TET cell line in which expression of the neo gene could be regulated in a reversible manner in the same clonal cell line. In this system, 20-fold increased transcription did clearly not promote ssODN-mediated gene targeting (Fig. 3D), thereby challenging previous findings. When targeting frequencies of two different CHO-K1 clones carrying a randomly integrated mutant LacZ reporter gene were compared, higher targeting frequencies were observed in the clone that showed 20-fold higher expression levels [18]. Yet, this difference in targeting frequency may reflect locus-specific differences in chromatin state or clonal variations rather than an actual effect of transcription. Although transcriptional activity also promoted ssODN-mediated gene targeting in inducible episomal reporter systems [12, 16–18], these findings may not apply to ssODN-mediated targeting of chromosomally located genes [5].
In E. coli, a clear correlation was observed between the direction of replication and preferential targeting by ssODNs with the same sequence as the nascent lagging strand [16, 17, 19, 20]. In contrast, the strand bias did not change in two CHO cell lines carrying an mEGFP reporter gene in opposite directions at the same chromosomal location [13]. In the mEGFP-rev CHO cell line, the correction frequency of antisense ssODNs was twofold lower than in the mEGFP-fwd CHO cell line, whereas correction by sense ssODNs was undetectable in both cell lines. Similarly, in our Rev neo ES cell line, sense ssODNs were less effective than in the Fwd neo ES cell line, although the frequency of antisense ssODNs remained unchanged (Fig. 4B). Moreover, the strand bias that was evident in the Fwd neo cell line was absent in the Rev neo cell line. In mammalian cells, DNA replication is initiated at numerous replication origins. However, in a single cell cycle only a fraction of all potential replication origins in the genome are activated (reviewed in [38]). Epigenetic modifications play an important role in origin choice and timing of DNA replication. Possibly, reversal of the neo gene has led to changes in chromatin structure that may interfere with replication fork movement or choice of origin activation [39], thereby obscuring our results. Although the impact on the strand bias remained somewhat unclear, changing the orientation of the neo gene clearly had an impact on the targeting frequency. This suggests that the direction of replication does play a role in ssODN-mediated gene targeting.
More evidence for the involvement of replication in the targeting process comes from the observation that hydroxyurea treatment improved the targeting efficiency in Msh2-deficient ES cells in a dose-dependent manner (Fig. 5C). Yet, slowing down replication fork movement by aphidicolin had no effect on the targeting efficiency (Fig. 5A). Replication fork stalling can induce uncoupling of the replicative polymerase and helicase activities, resulting in the formation of ssDNA [40, 41]. Perhaps, higher doses of aphidicolin were required to effectively inhibit the replicative polymerases resulting in this uncoupling and the formation of ssDNA. Unfortunately, ES cells are highly sensitive to inhibition of replication fork progression and readily undergo apoptosis, because higher doses of replication inhibitors clearly reduced the viability of the cells (Fig. 5B and D). On the other hand, even without addition of replication inhibitors or DNA-damaging agents, already a relatively large proportion of ES cells progress through S phase during ssODN exposure.
ES cells have robust mechanisms to repair DNA damage in order to prevent accumulation of mutations in their progeny. Although DNA repair proteins may facilitate ssODN incorporation by stabilizing the heteroduplex or by increasing the accessibility of the target locus [26–28], our results suggest that the DNA repair proteins tested are no major determinants in the targeting process in mouse ES cells. RNAi-mediated down-regulation of various DNA repair proteins did not significantly improve or decrease the targeting efficiency in either Msh2-deficient or wild-type ES cells (Fig. 6).
In conclusion, we have demonstrated that unmodified ssODNs of 35–40 nt are most efficient in correcting a chromosomal neo reporter gene in mouse ES cells. Addition of chemical modifications does not further enhance the efficacy of these ssODNs. The observed strand bias is likely caused by the different accessibility of the DNA strands during DNA replication, rather than during transcription of the target locus. This implies that any given genomic sequence, irrespective of transcriptional status, should be amenable to ssODN-mediated gene targeting. The optimal orientation of the ssODN seems to be dictated by the direction of the replication fork through the target locus. Because this will be different for each locus, we recommend testing ssODNs in both orientations. Although treatment with hydroxyurea improved the targeting efficiency, we argue against standard application in mouse ES cells because these cells are highly sensitive to hydroxyurea. Taken together, our data support a model in which the ssODN is physically incorporated into the genomic target locus during replication.
Acknowledgments
We thank Dr Shinji Masui (Research Institute, International Research Center of Japan) for providing the EBRTcH3 ES cells and the pPthC-Oct3/4 and pCAGGS-Cre plasmids and also for helpful suggestions. We are grateful to Edith van de Vijver for her technical assistance in construction of the ROSA-TET cell lines. The authors thank Sietske Bakker, Rob Dekker and Eva Wielders for critical reading of the manuscript. This work was supported by grants from the Netherlands Genomics Initiative (Horizon Breakthrough project 050-71-007 and Horizon project 050-71-051).
References
- 1.Aarts M, Dekker M, de Vries S, et al. Generation of a mouse mutant by oligonucleotide-mediated gene modification in ES cells. Nucleic Acids Res. 2006;34:e147. doi: 10.1093/nar/gkl896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dekker M, Brouwers C, Aarts M, et al. Effective oligonucleotide-mediated gene disruption in ES cells lacking the mismatch repair protein MSH3. Gene Ther. 2006;13:686–94. doi: 10.1038/sj.gt.3302689. [DOI] [PubMed] [Google Scholar]
- 3.Hasty P, Ramirez-Solis R, Krumlauf R, et al. Introduction of a subtle mutation into the Hox-2.6 locus in embryonic stem cells. Nature. 1991;350:243–6. doi: 10.1038/350243a0. [DOI] [PubMed] [Google Scholar]
- 4.Dekker M, Brouwers C, te Riele H. Targeted gene modification in mismatch-repair-deficient embryonic stem cells by single-stranded DNA oligonucleotides. Nucleic Acids Res. 2003;31:e27. doi: 10.1093/nar/gng027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nickerson HD, Colledge WH. A comparison of gene repair strategies in cell culture using a lacZ reporter system. Gene Ther. 2003;10:1584–91. doi: 10.1038/sj.gt.3302049. [DOI] [PubMed] [Google Scholar]
- 6.Pierce EA, Liu Q, Igoucheva O, et al. Oligonucleotide-directed single-base DNA alterations in mouse embryonic stem cells. Gene Ther. 2003;10:24–33. doi: 10.1038/sj.gt.3301857. [DOI] [PubMed] [Google Scholar]
- 7.Murphy BR, Moayedpardazi HS, Gewirtz AM, et al. Delivery and mechanistic considerations for the production of knock-in mice by single-stranded oligonucleotide gene targeting. Gene Ther. 2007;14:304–15. doi: 10.1038/sj.gt.3302866. [DOI] [PubMed] [Google Scholar]
- 8.Igoucheva O, Alexeev V, Anni H, et al. Oligonucleotide-mediated gene targeting in human hepatocytes: implications of mismatch repair. Oligonucleotides. 2008;18:111–22. doi: 10.1089/oli.2008.0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maguire KK, Kmiec EB. Multiple roles for MSH2 in the repair of a deletion mutation directed by modified single-stranded oligonucleotides. Gene. 2007;386:107–14. doi: 10.1016/j.gene.2006.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Igoucheva O, Alexeev V, Yoon K. Oligonucleotide-directed mutagenesis and targeted gene correction: a mechanistic point of view. Curr Mol Med. 2004;4:445–63. doi: 10.2174/1566524043360465. [DOI] [PubMed] [Google Scholar]
- 11.Igoucheva O, Alexeev V, Yoon K. Targeted gene correction by small single-stranded oligonucleotides in mammalian cells. Gene Ther. 2001;8:391–9. doi: 10.1038/sj.gt.3301414. [DOI] [PubMed] [Google Scholar]
- 12.Liu L, Rice MC, Drury M, et al. Strand bias in targeted gene repair is influenced by transcriptional activity. Mol Cell Biol. 2002;22:3852–63. doi: 10.1128/MCB.22.11.3852-3863.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olsen PA, Randol M, Luna L, et al. Genomic sequence correction by single-stranded DNA oligonucleotides: role of DNA synthesis and chemical modifications of the oligonucleotide ends. J Gene Med. 2005;7:1534–44. doi: 10.1002/jgm.804. [DOI] [PubMed] [Google Scholar]
- 14.Parekh-Olmedo H, Drury M, Kmiec EB. Targeted nucleotide exchange in Saccharomyces cerevisiae directed by short oligonucleotides containing locked nucleic acids. Chem Biol. 2002;9:1073–84. doi: 10.1016/s1074-5521(02)00236-3. [DOI] [PubMed] [Google Scholar]
- 15.Andrieu-Soler C, Casas M, Faussat AM, et al. Stable transmission of targeted gene modification using single-stranded oligonucleotides with flanking LNAs. Nucleic Acids Res. 2005;33:3733–42. doi: 10.1093/nar/gki686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huen MS, Lu LY, Liu DP, et al. Active transcription promotes single-stranded oligonucleotide mediated gene repair. Biochem Biophys Res Commun. 2007;353:33–9. doi: 10.1016/j.bbrc.2006.11.146. [DOI] [PubMed] [Google Scholar]
- 17.Li XT, Costantino N, Lu LY, et al. Identification of factors influencing strand bias in oligonucleotide-mediated recombination in Escherichia coli. Nucleic Acids Res. 2003;31:6674–87. doi: 10.1093/nar/gkg844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Igoucheva O, Alexeev V, Pryce M, et al. Transcription affects formation and processing of intermediates in oligonucleotide-mediated gene alteration. Nucleic Acids Res. 2003;31:2659–70. doi: 10.1093/nar/gkg360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ellis HM, Yu D, DiTizio T, et al. High efficiency mutagenesis, repair, and engineering of chromosomal DNA using single-stranded oligonucleotides. Proc Natl Acad Sci U S A. 2001;98:6742–6. doi: 10.1073/pnas.121164898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huen MS, Li XT, Lu LY, et al. The involvement of replication in single stranded oligonucleotide-mediated gene repair. Nucleic Acids Res. 2006;34:6183–94. doi: 10.1093/nar/gkl852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brachman EE, Kmiec EB. Gene repair in mammalian cells is stimulated by the elongation of S phase and transient stalling of replication forks. DNA Repair (Amst) 2005;4:445–57. doi: 10.1016/j.dnarep.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 22.Engstrom JU, Kmiec EB. DNA replication, cell cycle progression and the targeted gene repair reaction. Cell Cycle. 2008;7:1402–14. doi: 10.4161/cc.7.10.5826. [DOI] [PubMed] [Google Scholar]
- 23.Wu XS, Xin L, Yin WX, et al. Increased efficiency of oligonucleotide-mediated gene repair through slowing replication fork progression. Proc Natl Acad Sci U S A. 2005;102:2508–13. doi: 10.1073/pnas.0406991102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olsen PA, Randol M, Krauss S. Implications of cell cycle progression on functional sequence correction by short single-stranded DNA oligonucleotides. Gene Ther. 2005;12:546–51. doi: 10.1038/sj.gt.3302454. [DOI] [PubMed] [Google Scholar]
- 25.Radecke S, Radecke F, Peter I, et al. Physical incorporation of a single-stranded oligodeoxynucleotide during targeted repair of a human chromosomal locus. J Gene Med. 2006;8:217–28. doi: 10.1002/jgm.828. [DOI] [PubMed] [Google Scholar]
- 26.Liu L, Cheng S, van Brabant AJ, et al. Rad51p and Rad54p, but not Rad52p, elevate gene repair in Saccharomyces cerevisiae directed by modified single-stranded oligonucleotide vectors. Nucleic Acids Res. 2002;30:2742–50. doi: 10.1093/nar/gkf397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu L, Maguire KK, Kmiec EB. Genetic re-engineering of Saccharomyces cerevisiae RAD51 leads to a significant increase in the frequency of gene repair in vivo. Nucleic Acids Res. 2004;32:2093–101. doi: 10.1093/nar/gkh506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Igoucheva O, Alexeev V, Scharer O, et al. Involvement of ERCC1/XPF and XPG in oligodeoxynucleotide-directed gene modification. Oligonucleotides. 2006;16:94–104. doi: 10.1089/oli.2006.16.94. [DOI] [PubMed] [Google Scholar]
- 29.Ferrara L, Parekh-Olmedo H, Kmiec EB. Enhanced oligonucleotide-directed gene targeting in mammalian cells following treatment with DNA damaging agents. Exp Cell Res. 2004;300:170–9. doi: 10.1016/j.yexcr.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 30.Ferrara L, Kmiec EB. Camptothecin enhances the frequency of oligonucleotide-directed gene repair in mammalian cells by inducing DNA damage and activating homologous recombination. Nucleic Acids Res. 2004;32:5239–48. doi: 10.1093/nar/gkh822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hooper M, Hardy K, Handyside A, et al. HPRT-deficient (Lesch-Nyhan) mouse embryos derived from germline colonization by cultured cells. Nature. 1987;326:292–5. doi: 10.1038/326292a0. [DOI] [PubMed] [Google Scholar]
- 32.Masui S, Shimosato D, Toyooka Y, et al. An efficient system to establish multiple embryonic stem cell lines carrying an inducible expression unit. Nucleic Acids Res. 2005;33:e43. doi: 10.1093/nar/gni043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–3. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 34.Oguro M, Suzuki-Hori C, Nagano H, et al. The mode of inhibitory action by aphidicolin on eukaryotic DNA polymerase alpha. Eur J Biochem. 1979;97:603–7. doi: 10.1111/j.1432-1033.1979.tb13149.x. [DOI] [PubMed] [Google Scholar]
- 35.Rosenkranz HS, Levy JA. Hydroxyurea: a specific inhibitor of deoxyribonucleic acid synthesis. Biochim Biophys Acta. 1965;95:181–3. doi: 10.1016/0005-2787(65)90225-x. [DOI] [PubMed] [Google Scholar]
- 36.Radecke F, Peter I, Radecke S, et al. Targeted chromosomal gene modification in human cells by single-stranded oligodeoxynucleotides in the presence of a DNA double-strand break. Mol Ther. 2006;14:798–808. doi: 10.1016/j.ymthe.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 37.Brachman EE, Kmiec EB. Targeted nucleotide repair of cyc1 mutations in Saccharomyces cerevisiae directed by modified single-stranded DNA oligonucleotides. Genetics. 2003;163:527–38. doi: 10.1093/genetics/163.2.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aladjem MI. Replication in context: dynamic regulation of DNA replication patterns in metazoans. Nat Rev Genet. 2007;8:588–600. doi: 10.1038/nrg2143. [DOI] [PubMed] [Google Scholar]
- 39.Courbet S, Gay S, Arnoult N, et al. Replication fork movement sets chromatin loop size and origin choice in mammalian cells. Nature. 2008;455:557–60. doi: 10.1038/nature07233. [DOI] [PubMed] [Google Scholar]
- 40.Walter J, Newport J. Initiation of eukaryotic DNA replication: origin unwinding and sequential chromatin association of Cdc45, RPA, and DNA polymerase alpha. Mol Cell. 2000;5:617–27. doi: 10.1016/s1097-2765(00)80241-5. [DOI] [PubMed] [Google Scholar]
- 41.Byun TS, Pacek M, Yee MC, et al. Functional uncoupling of MCM helicase and DNA polymerase activities activates the ATR-dependent checkpoint. Genes Dev. 2005;19:1040–52. doi: 10.1101/gad.1301205. [DOI] [PMC free article] [PubMed] [Google Scholar]