

Abstract
Histone acetylation is a fundamental mechanism in the regulation of local chromatin conformation and gene expression. Research has focused on the impact of altered epigenetic environments on the expression of specific genes and their pathways. However, changes in histone acetylation also have a global impact on the cell. In this study we used digital texture analysis to assess global chromatin patterns following treatment with trichostatin A (TSA) and have observed significant alterations in the condensation and distribution of higher-order chromatin, which were associated with altered gene expression profiles in both immortalised normal PNT1A prostate cell line and androgen-dependent prostate cancer cell line LNCaP. Furthermore, the extent of TSA-induced disruption was both cell cycle and cell line dependent. This was illustrated by the identification of sub-populations of prostate cancer cells expressing high levels of H3K9 acetylation in the G2/M phase of the cell cycle that were absent in normal cell populations. In addition, the analysis of enriched populations of G1 cells showed a global decondensation of chromatin exclusively in normal cells.
Keywords: histone acetylation, prostate cancer, chromatin, epigenetics, image analysis, trichostatin A
Introduction
Microscopic changes in chromatin pattern within the nucleus are associated with changes in the underlying physiology, functional activity and molecular biology of the cell. Chromatin is known to be visibly rearranged in association with the onset of malignancy and is routinely used by diagnostic pathologists as a visual microscopic clue in the histological diagnosis, grading and prognosis of cancer. The study of chromatin phenotype in human tissue and cells in this way has been significantly enhanced through the use of digital image analysis and the measurement of chromatin distribution. This has been extensively studied in prostate neoplasia and shown to be a strong potential biomarker of diagnosis and prognosis of prostate cancer [1–4].
Chromatin-based mechanisms are now recognized as having a major role in tumorigenesis. Alterations in the higher-order structure of chromatin may be caused by the dysregulation of epigenetic enzyme dynamics within the cell nucleus, which in turn leads to globally altered epigenetic signatures which impact gene expression [5]. Histone acetylation at key lysine residues disrupts the structure of chromatin due to the addition of an acetyl group to the positively charged amino acid tail of the histone protein, which neutralizes the electrostatic interactions that bind the chromatin together, and in turn causes chromatin to adopt an ‘open’ conformation. DNA in these regions is more readily accessible to transcription factors and for this reason acetylation is thought to encourage gene expression [6–9]. The dysregulation of these epigenetic controls may contribute to the aberrant expression or silencing of specific genes that have been linked to the initiation and progression of prostate cancer [10]. Understanding the relationship between morphological phenotype and the underlying molecular alterations effecting chromatin structure is important but infrequently studied.
In normal cells an equilibrium of histone acetyltransferases (HATs) and histone deacetylases (HDAC) regulate the acetylation and deacetylation of specific internal lysine residues located on the exposed amino terminals of histone proteins [11–12]. One strategy for analysing the precise effects of histone acetylation is to block the activities of these acetylating or deacetylating enzymes by specific inhibitors. Trichostatin A (TSA) is a potent, non-competitive inhibitor that is highly specific for HDACs. TSA has been found to exhibit anti-proliferative properties at nanomolar concentrations in transformed cells by inducing cell cycle arrest, differentiation and apoptosis [13–15]. TSA treatment has been associated with global hyperacetylation that may lead to the activation of transcription of a selection of genes that inhibit tumour growth [16–19]. Recent studies have shown specific gene expression changes in response to TSA treatment that subsequently impact cell cycle and apoptotic pathways [20–22]. However, in this study we have focused on measuring the impact of TSA treatment on the global acetylation status of the cell and the morphological phenotype of the nucleus in terms of chromatin condensation and distribution using quantitative methods. This study has focused exclusively on acetylation of histone 3 at the lysine 9 residue due to the identification of H3K9 acetylation as a primary step in the sequence of acetylation at histone 3, and its potential role as an epigenetic switch in the re-expression of genes, in an environment of repressive heterochromatin [23–24].
Materials and methods
Cell culture and TSA treatment
PNT1A prostate cell line was purchased from European Collection of Cell Culture. LNCaP and DU145 cells were purchased from DSMZ (Braunschweig, Germany). All cell lines were grown and maintained in RPMI 1640 nutrient medium (Sigma, Dorset, UK) supplemented with foetal calf serum (Labtech International, East Sussex, UK), 4 mM/l L-glutamine (Sigma) 0.06 mg/ml benzylpenicillin and 0.1 mg/ml streptomycin (Sigma). TSA (Sigma) was administered at 12 ng/ml and 100 ng/ml concentrations for 24 hrs.
Flow cytometric cell cycle analysis
Adherent cells were harvested by trypsinization and fixed in methanol. Cells were stained with 500 μl of 30 μg/ml propidium iodide (PI) (Sigma) containing 100 μg/ml RNase (Sigma) and 5 μg/ml triton (Sigma). All flow cytometry was performed on a Coulter Elite flow cytometer (Beckman Coulter, London, UK) equipped with a 488 nm argon laser. Calibration was conducted using FLOW-CHECK™ Fluorospheres (Beckman Coulter). The orientation of the laser, stream and optics were calibrated such that the Half CV for fluorescence within each photomultiplier tube was <2. A DNA content analysis protocol was used and samples were run at a data rate between 150 and 200 cells per second. PI fluorescence was detected using a band pass filter of 675 nm. Gates were placed on histograms between channel 50 and 300 in order to measure sub-G1 events. Cell cycle characteristics were analysed using WinCycle version 3.0 software (Phoenix Flow Systems, San Diego, CA, USA).
Flow cytometric quantitation of H3K9 acetylation by single and dual parameter methods
Cells were fixed in methanol and stained in suspension with 5 μg/ml monoclonal mouse anti-AcH3K9 antibody (Abcam, Cambridge, UK) and 5 μg/ml IgG1 isotype control (Dako), followed by 1:50 dilution of FITC conjugated rabbit antimouse secondary antibody (Dako). For dual staining, cells were subsequently stained with PI. FITC fluorescence was detected using a band pass filter of 525 nm. Voltage parameters were set based on isotype controls and subsequently maintained for the assessment of replicate samples. Red/green compensation was applied for the assessment of dual stained cells and PI (675 nm band pass filter) and FITC (525 nm band pass filter) were measured simultaneously. Mean channel fluorescence (MCF) values were calculated using Immuno-4 analysis software (Beckman Coulter).
Digital microscopy and calibration procedures
A calibrated digital video photometer, consisting of a Sony DXC-930P 3 chip CCD Camera (Surrey, UK), a Kingshill CL6123 regulated power supply and a Leitz orthoplan microscope (LeitzMicroscopes.com), was used to record high-resolution colour images. In-house software developed in KS400 macro programming language (Carl Zeiss Vision, Carl Zeiss, Hertfordshire, UK) was used and chromatin texture analysis of haematoxylin stained nuclei was performed in the red colour channel. Fifty nuclei from each sample were analysed for nuclear texture using this software and a total of 65 texture features (Table 1) were computed for each nucleus (as previously described by Mohamed et al.[25]). For the fluorescence texture analysis of H3K9 distribution, a series of fluorescent images (1392 × 1040 pixels) were captured using a Leica (Milton Keynes, UK) CTR6000 Fluorescent Microscope (×63 objective lens) and FW4000 Leica software for each treatment group incorporating a minimum of 50 cell nuclei. Texture analysis had to be adjusted for the analysis of fluorescent images by measuring transmission pixel values rather than optical density. Subsequently, for easy interpretation, the discrete feature values were inverted so that high-density objects correspond to the lightest and therefore most densely stained regions. The remainder of the texture analysis parameters and statistical analysis of data was carried out as previously described [25].
Table 1.
This table lists all of the nuclear texture features measured for both chromatin organization and H3K9 spatial expression [26, 27]. Many ofthese are second- and third-order statistical calculations (e.g. Haralick features [26]) from the spatial domain. These allow the measurement of subvisual features not visible to the naked eye in addition to spatial alterations readily visible as changes in chromatin organization
| Abbreviation | Texture feature description |
|---|---|
| Geometric features | |
| Area | Area occupied by nucleus |
| Ellipsea | Major axis length |
| Ellipseb | Minor axis length |
| Perim | Perimeter of the nucleus |
| Feretmin | Minimum ferret length |
| Feretmax | Maximum ferret length |
| Feretratio | Ratio of ferret length |
| Fcircle | Form circle |
| Dcircle | Equivalent circle |
| Densitimetric features | |
| Meand | Mean optical density within the nucleus |
| Stdd | Standard deviation of mean optical densities |
| Skewd | Skewness of optical densities (distribution around the mean) |
| Kurtd | Kurtosis of optical densities (extremes from the mean) |
| Energyd | Uniformity of chromatin |
| Entropyd | Disorganization of chromatin |
| Mind | Minimum optical density within the nucleus |
| Maxd | Maximum optical density within the nucleus |
| Sumd | Sum of the optical densities within the nucleus |
| Sumqd | Sum of squared optical densities within the nucleus |
| Haralick features | |
| Haram 1–11 | Mean values in four principle directions (Haralick) |
| Harar 1–11 | Range values in four principle directions (Haralick) |
| DISCRETE FEATURES | |
| LDarea | Area covered by low-density regions within the nucleus (Doudkine) |
| MDarea | Area covered by medium-density regions within the nucleus (Doudkine) |
| HDarea | Area covered by high-density regions within the nucleus (Doudkine) |
| Lamount | Proportion of sum optical density due to low-density regions (Doudkine) |
| Mamount | Proportion of sum optical density due to medium-density regions (Doudkine) |
| Hamount | Proportion of sum optical density due to high-density regions (Doudkine) |
| LVM | Comparison of mean optical densities of low-density regions with those of medium-density regions (Doudkine) |
| LVH | Comparison of mean optical densities of low-density regions with those of high-density regions (Doudkine) |
| LVMH | Comparisons of mean optical densities of low-density regions with those of medium and high-density regions (Doudkine) |
| LDO | Number of low-density objects (Doudkine) |
| MDO | Number of medium-density objects (Doudkine) |
| HDO | Number of high-density objects (Doudkine) |
| LDC | Compactness (shape) of low-density regions (Doudkine) |
| MDC | Compactness (shape) of medium-density regions (Doudkine) |
| HDC | Compactness (shape) of high-density regions (Doudkine) |
| MHDC | Compactness (shape) of medium and high-density regions (Doudkine) |
| LAD | Average distance of low-density regions from the geometric centre of the nucleus (Doudkine) |
| MAD | Average distance of medium-density regions from the geometric centre of the nucleus (Doudkine) |
| HAD | Average distance of high-density regions from the geometric centre of the nucleus (Doudkine) |
| MHAD | Average distance of medium and high-density regions from the geometric centre of the nucleus (Doudkine) |
| LCOM | Distance between the centre of mass of low-density regions and the geometric centre of the nucleus (Doudkine) |
| MCOM | Distance between the centre of mass of medium-density regions and the geometric centre of the nucleus (Doudkine) |
| HCOM | Distance between the centre of mass of high-density regions and the geometric centre of the nucleus (Doudkine) |
| MHCOM | Distance between the centre of mass of medium and high regions and the geometric centre of the nucleus (Doudkine) |
Flow cytometric G1 cell sorting
Calibration was carried out using FLOW-CHECK™ Fluorospheres (Beckman Coulter). PI fluorescence was measured using a 575 nm band pass filter and cell sorting was based on cells detected within the two inner quartiles of the G0/G1 peak. In order to maximize the purity of the sorted sample the ‘coincidence abort’ function was switched on during sorting to exclude distorted droplets. The G0/G1 sorted cells were collected and rerun to check for sort purity. G0/G1 sorted cells were cytospun onto glass slides using a cytospin centrifuge at 1000 rpm for 1 min. (Cytospin 3, Shandon, Inc., Thermo Scientific, Waltham, MA, USA). Cells were then incubated with 5 μg/ml monoclonal mouse anti-AcH3K9 antibody followed by FITC conjugated rabbit antimouse secondary antibody (1:50 dilution). Slides were then mounted using fluorescent mounting medium (Vectashield Mounting Medium, Vector Laboratories, Inc., Peterborough, UK).
Statistical analysis
All statistical analysis was performed with the SPSS statistical package (Version 12 for Windows). Analysis of feature distribution for each treatment group showed some non-normal distributions and so the Mann-Whitney U-test was used for the comparison between groups. Information regarding specific texture features was illustrated by plotting the mean and 95% confidence intervals for each treatment group.
DNA microarray experiments
The DNA microarray was performed with an MWG Human 30K array A which incorporates oligonucleotides representing 9850 genes, including 30 replicas and 104 Arabidopsis controls. Total RNA was isolated from samples prepared from PNT1A and LNCaP cells, comparing untreated cultures with those treated with TSA at low (12 ng/ml) and high (100 ng/ml) doses. Therefore, four different microarray experiments were conducted, each in triplicate. PolyA+ RNA from control and treated cells were purified and retro-transcribed in the presence of either Cy3-dCTP or Cy5-dCTP to produce pairs of separately-labelled cDNAs. Replicates included 1 dye swap control per experiment. Labelled probes were mixed and competitively hybridized overnight to a microarray slide. Microarray scanning was performed with a GenePix Axon 4000A microarray scanner (Axon Instruments, Inc.). Using mining software (Acuity 2.0, Axon Instruments, Inc.) distorted or low-quality spots were excluded from further analysis and a range of quantitative measures were automatically calculated and recorded for each array spot. Of these, we have consistently referred to the ‘ratio of means’ values. To allow direct comparison of results between replicate arrays the data were normalized to correct for differences in signal intensity.
Results
The impact of trichostatin A treatment on cell growth
Our results have shown a differential response to TSA treatment in ‘immortalized normal’ prostatic cell line PNT1A and androgen-dependent prostate cancer cell line LNCaP. TSA-induced cell cycle disruption was observed in both PNT1A and LNCaP cell lines in a dose-dependent manner with the most pronounced effects occurring at exposure to high doses (100 ng/ml) of TSA treatment. However, this disruption occurred following differential treatment doses and arrest occurred at different phases of the cell cycle. In PNT1A cells evidence of a cell cycle arrest at both S and G2/M phases of the cell cycle was observed in response to TSA doses exceeding 25 ng/ml. These effects were characterized by a reduction in the G0/G1 peak and significant increases in the proportion of the cells in the S phase and G2/M phases. An increase in the sub-G1 apoptotic cell fraction was also observed following doses of 100 ng/ml (Fig. 1A). Alternatively, LNCaP cells remained largely unaltered at doses of TSA treatment lower than 50 ng/ml. Following 100 ng/ml TSA treatment a significant G2/M arrest was identified and this was signified by an emptying of the cell cycle S phase fraction and a subsequent increase in the proportion of cells in G2/M (Fig. 1A). An apoptotic response was also evident in LNCaP cells in response to high doses of TSA although this effect was not as pronounced as in PNT1A cells (Fig. 1B).
Fig 1.

High doses of TSA treatment (>12 ng/ml) induce differential cell cycle responses in PNT1A and LNCaP cells, as assessed by flow cytometric cell cycle analysis. (A) Flow cytometry histograms representing cell cycle profiles of PNT1A and LNCaP cells following treatment with low (12 ng/ml) and high (100 ng/ml) doses of TSA for 24 hrs. Both cell lines showed minimal cell cycle disruption following 12 ng/ml TSA while exhibiting different patterns of cell cycle arrest and apoptotic induction following 100 ng/ml TSA treatment. (B) Shows a graph displaying the proportion of apoptotic cells and cells present in each stage of the cell cycle (Multicycle software). (C) This shows an EC50 doseresponse curve based on levels of apoptosis in PNT1A and LNCaP cell lines and EC50 values were calculated as 25.73 and 57.82, respectively.
The EC50 values based on apoptotic response for the TSA treatment of PNT1A and LNCaP cells were calculated as 25.73 and 57.82, respectively (Fig. 1C). Hereafter in this study, in order to assess the cellular response to TSA treatment at both low and high doses, assessment was carried out on cells exposed to 12 ng/ml (low) doses of TSA, where minimal cell cycle disruption is evident, and 100 ng/ml (high) TSA doses where cell cycle arrest and apoptotic pathways have been induced.
TSA induced H3K9 hyperacetylation using H3K9 loci as a key marker of acetylation status
In order to establish if the inhibition of HDACs by TSA induces a global hyperacetylation at a key residue histone 3 lysine 9 (H3K9), PNT1A and LNCaP cells were stained with an anti-AcH3K9 antibody and assessed by flow cytometric protein quantitation, and confocal microscopy following exposure to high (100 ng/ml) and low (12 ng/ml) doses of TSA (Fig. 2).
Fig 2.
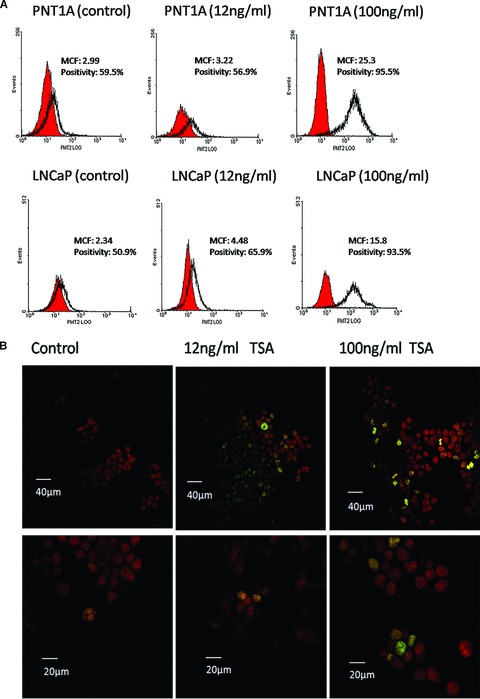
TSA treatment induces a global H3K9 hyperacetylation in PNT1A and LNCaP cells. (A) Flow cytometry histograms of AcH3K9 fluorescence in PNT1A and LNCaP cell samples following TSA treatment doses of 12 ng/ml and 100 ng/ml. Histograms were generated to represent levels of AcH3K9 (red peaks) in relation to appropriate IgG1 isotype controls (clear peaks). MCF and percentage positivity values were calculated for each treatment group using Immuno-4 analysis software (Beckman Coulter). Increased MCF values for AcH3K9 were observed in both PNT1A and LNCaP cell lines in a dose-dependent manner. (B) Confocal microscopy images showing the distribution of TSA-induced hyperacetylation across the cell population. Treated cells were stained with anti-AcH3K9 (green) and PI (red) and images captured at ×100 magnification. In the low power images scale bars represent 40 μm and 20 μm in the high power images H3K9 acetylation was found to be highly present in a subpopulation of mitotic figures and this phenomenon became more pronounced following TSA treatment showing an increase in the fluorescence intensity of AcH3K9 and in the number of hyperacetylated cells.
Flow cytometric analysis enabled the detection of changes in H3K9 acetylation relative to an IgG1 isotype control. Using this technique PNT1A and LNCaP cells showed a progressive increase in H3K9 acetylation that was TSA dose dependent. At low TSA doses H3K9 acetylation showed an increase, and this is shown by a rise in MCF values from 2.99 to 3.22 in PNT1A cells and from 2.34 to 4.48 in LNCaP cells. Furthermore, this effect became more severe with higher TSA treatments in both cell lines when MCF values increased further to 25.3 in PNT1A cells and 15.8 in LNCaP cells (Fig. 2A). In general, the positive AcH3K9 peaks were found to cover a large range of fluorescence values, suggesting the presence of differential H3K9 acetylation states within individual cells throughout the cell populations. Confocal microscopy demonstrated a small subpopulation of cells highly expressing AcH3K9 in LNCaP cell populations, a phenomenon which became more distinct following TSA treatment (Fig. 2B).
TSA induces chromatin disruption in PNT1A and LNCaP cells
The aim of this study was to study the impact of induced histone hyperacetylation on chromatin phenotype using high-resolution cellular imaging. Visually, TSA treatment led to dose-dependent changes in the general morphology of both cell lines, where prominent heterochromatic regions in PNT1A cells were reduced to lightly stained regions defined only by a remnant densely stained chromatin boundary (as indicated by red arrows in Fig. 3A). Chromatin outside of these heterochromatic boundaries became substantially more granular following TSA treatment compared with the more homogenous nature of chromatin distribution in untreated nuclei. Furthermore, TSA-induced cell cycle disruption was also evident due to an observed increase in the number of PNT1A apoptotic bodies and a marked increase in mitotic figures within treated LNCaP cell populations (Fig. 3A). This provides morphological confirmation for the cell cycle disruptions seen using flow cytometry.
Fig 3.
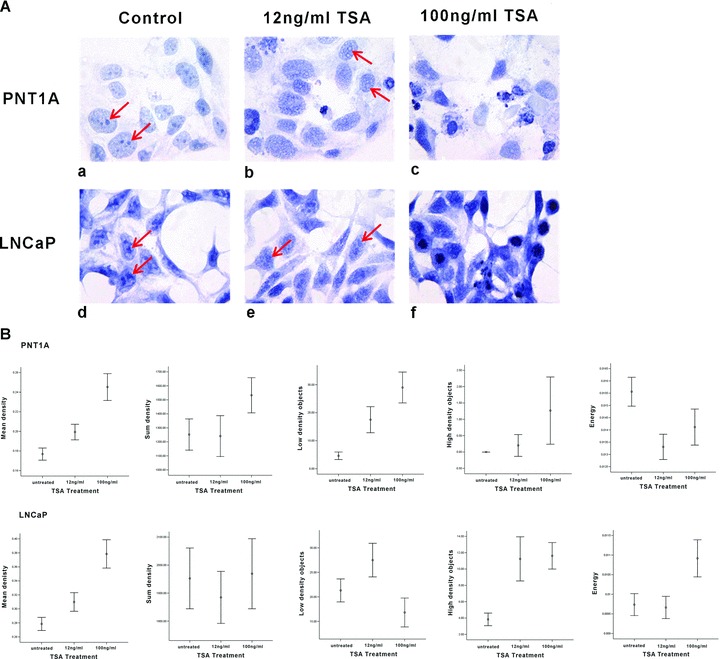
TSA-induced hyperacetylation results in the disruption of chromatin condensation and distribution. (A) Haematoxylin stained PNT1A and LNCaP cells were visually assessed for chromatin changes and red arrows highlight areas of heterochromatin where chromatin appears to disperse following TSA treatment. In general chromatin was more granular in both cell lines following low doses of TSA treatment whereas after high doses of TSA cell cycle perturbations were evident with the increased presence of apoptotic bodies in PNT1A cells and a large number of mitotic bodies in LNCaP populations. (B) High-resolution digital texture analysis of haematoxylin stained cells allowed the quantitative analysis of chromatin changes through the assessment of 65 chromatin texture features for each cell nucleus. Graphs were plotted representing 95% confidence intervals for a subset of chromatin texture features significantly altered by TSA treatment including; mean density, sum density, low-density objects, highdensity objectsand energy.
These visual observations of altered global chromatin folding were supported quantitatively by the application of digital texture analysis, where significant changes in a range of nuclear texture features were detected and statistically assessed following TSA treatment (Fig. 3B). Some of these features (e.g. mean density) can be directly related to visually evident changes in nuclear chromatin [26]. Others (e.g. energy) cannot always be easily described in visual terms. Features such as the Haralick feature set (see Table 1) are higher-order statistical measures of pixel distribution which allow us to measure ‘sub-visual’ changes in pixel (chromatin) organization and distribution [27]. Quantitative digital texture analysis also allowed the measurement of subtle distributional changes in chromatin architecture induced by TSA. Using a Mann Whitney U test of significance between untreated PNT1A cells and those treated with 100 ng/ml TSA, 62/65 chromatin texture features analysed were found to be significantly altered in response to high doses of TSA, and in the analysis of LNCaP nuclei 64/65 texture features were found to be significantly altered between untreated and those cells treated with 100 ng/ml TSA.
H3K9 acetylation variation across the phases of the cell cycle
This study has so far shown that although TSA induces hyperacetylation, this is also accompanied by cell cycle disruptions. For this reason it was important to assess if TSA-induced changes in acetylation were consistent across the cell cycle or if certain phases of the cell cycle were more sensitive to induced hyperacetylation. DU145 metastatic prostate cancer cells were also assessed in the following experiments in order to further understand the differential response of normal and androgen dependent and independent prostate cancer cells to altered global acetylation patterns induced by TSA treatment.
PNT1A, LNCaP and DU145 cells were fluorescently stained for AcH3K9 and labelled for DNA by PI. Dual parameter flow cytometry was adopted in order to disentangle H3K9 acetylation from cell cycle phase and allowed the determination of MCF values and percentage positivity of cells corresponding to H3K9 acetylation within specific phases of the cell cycle. In all cell lines, an increase in H3K9 acetylation was detected as the cell progresses through the cell cycle from G1 to mitosis (Fig. 4A). This may be indicative of the increase in available acetylation sites that occurs as the cell progresses through the cell cycle. Interestingly, a subpopulation of cells highly expressing AcH3K9 was evident exclusively in the G2/M phase of the cell cycle in the prostate cancer cell lines LNCaP and DU145 and was absent in normal prostatic PNT1A cells. Therefore, high levels of H3K9 acetylation in LNCaP and DU145 cells may be predominantly due to hyperacetylated G2/M cell populations and cells in G2M phase of the cell cycle were much more susceptible to TSA-induced H3K9 hyperacetylation (Fig. 4A).
Fig 4.

Global H3K9 acetylation patterns are altered across the cell cycle. Following TSA treatment with 12 ng/ml and 100 ng/ml doses PNT1A, LNCaP and DU145 cells were assessed using dual parameter flow cytometry for the analysis of AcH3K9 and concurrent cell cycle changes. (A) Dual parameter scatterplots representing cellular levels of AcH3K9 ( y-axis) against corresponding DNA content/cell cycle phase ( x-axis). These plots show a differential distribution of AcH3K9 across the cell cycle which is intensified following TSA treatment and demonstrated the presence of a subpopulation of cells within the G2/M cell population in DU145, and to a lesser extent in LNCaP cells, that highly express AcH3K9. (B) Graphs showing MCF values generated from the dot plots in (A) for each TSA treatment group. These show variations in the levels of AcH3K9 within each cell cycle phase (G0/G1, S phase and G2/M) in addition to differential patterns of H3K9 acetylation between cell lines.
Furthermore, in androgen-independent DU145 cells, a significant increase in the MCF values for each cell cycle phase was also observed in response to treatment with 12 ng/ml doses of TSA and this response was not observed in any of the other cell lines at this low dose. The impact of high doses of TSA was also noticeably different in DU145 cells. Global acetylation levels were significantly reduced while simultaneously the subpopulation of highly expressing cells in G2/M became more pronounced, indicating that TSA-induced hyperacetylation in these cells exclusively occurs in a subpopulation of G2/M cells that correspond to the mitotic fraction of the DU145 cell population (Fig. 4B).
Selective analysis of chromatin organization in flow sorted G1 cells following TSA induced hyperacetylation
It was clear from the above studies that the analysis of induced chromatin rearrangement must be carried out on cell cycle specific populations, otherwise results may be confounded by the variable cell cycle response to TSA. In a new approach to measuring chromatin textural changes, quantitative fluorescence texture analysis was performed on cells treated with TSA and subsequently flow cytometrically sorted G1 cell populations stained with PI for the assessment of chromatin and simultaneously with anti-AcH3K9/FITC in order to detect the spatial characteristics of TSA-induced global hyperacetylation in G1 cells (Fig. 5A). This analysis was performed in order to study the direct impact of low and high doses of TSA treatment on the global density and distribution of nuclear chromatin in PNT1A, LNCaP and DU145 cells, in the absence of confounding chromatin remodelling events that occur between cell cycle phases. As before, 65 texture features were measured in order to quantitatively evaluate visual and sub-visual changes in chromatin organization and H3K9 distribution with the nucleus.
Fig 5.
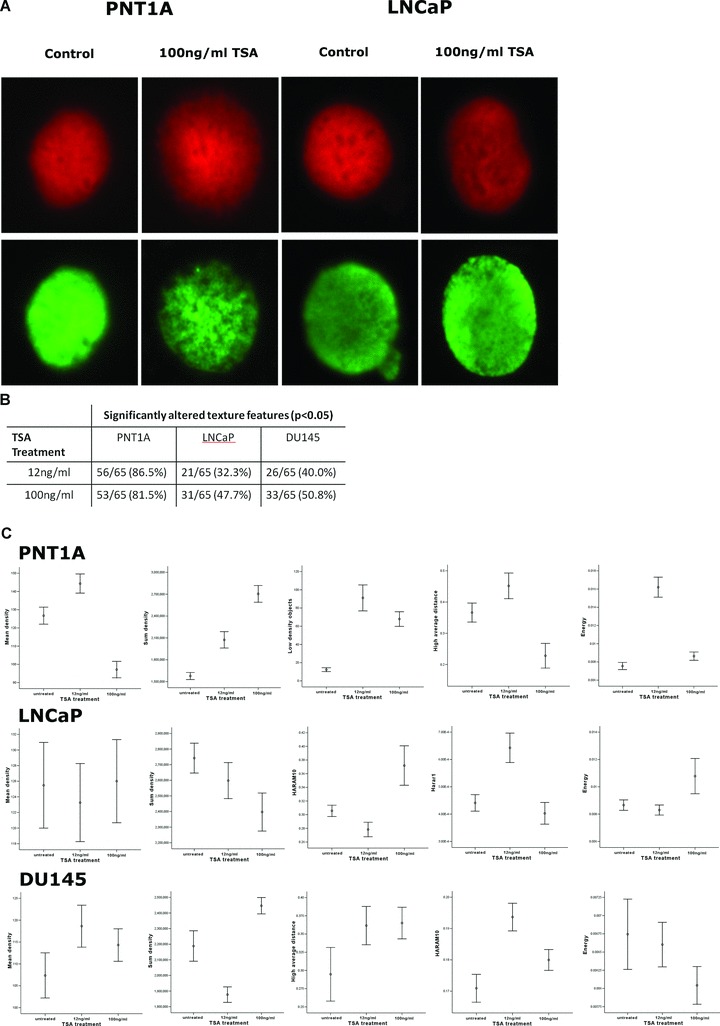
Fig. 5 TSA-induced chromatin changes in G1 enriched cell populations, as measured by quantitative digital texture analysis (A) Images show DNA distribution in red (PI) and AcH3K9 in green (FITC conjugated antibody) in G0/G1 sorted populations of PNT1A, LNCaP and DU145 cells treated with TSA (Leica CTR6000, ×63 objective magnification). (B) Quantitative Fluorescence texture analysis was performed on these images and texture feature measurements were based on transmission pixel values. A table representing the total number of chromatin texture features altered in each cell line following low and high doses of TSA showed the most significant impact of TSA on chromatin phenotype in PNT1A cells. (C) To summarize the chromatin results, graphs were plotted (summarized using the mean and 95% confidence intervals) for a subset of significantly altered chromatin texture features within each cell line in response to TSA treatment in G0/G1 sorted cells. The texture features plotted here represent changes in both density and distribution of chromatin. Although distributional features such as energyare significantly disrupted in all cell lines, the graphs representing mean densityindicate a global chromatin decondensation in PNT1A cells but not in LNCaP and DU145 cells.
As expected, an increase in H3K9 acetylation was observed in G1 cells from all three cell lines in response to TSA treatment. In addition, TSA at both low and high doses was also found to induce significant differences in the distribution of H3K9 acetylation throughout the nuclei of PNT1A, LNCaP and DU145 cells, with low doses of TSA inducing significant changes in >35% of texture features in all three cell lines and >55% in response to high doses of TSA treatment (data not shown). In tandem with TSA-induced alterations in global acetylation a significant disruption in chromatin architecture was identified in response to treatment. Mann-Whitney U-tests were applied to assess overall changes in chromatin phenotype and demonstrated a range of chromatin texture features within each dataset that were significantly altered by TSA treatment. Significant but differential changes in the nuclear chromatin phenotype were observed between PNT1A, LNCaP and DU145 cells.
PNT1A normal cells exhibited highly significant changes in chromatin phenotype in response to TSA treatment at both low and high doses compared to prostate cancer cells LNCaP and DU145 cells. By applying a Mann-Whitney U statistical test to summarize global chromatin changes in response to low doses of TSA treatment relative to untreated cells 56/65 (86.2%) of the texture features were significantly altered (P > 0.05) and 53/65 (81.5%) were altered in response to high doses of TSA (Fig. 5B). Measurements for a selection of those texture features, which were significantly altered in response to TSA treatment in PNT1A cells, were plotted using mean and 95% confidence limits in order to identify specific changes in chromatin organization (Fig. 5C). In response to TSA treatment PNT1A chromatin was found to be dramatically decondensed, as illustrated by the low values of mean density texture feature following 100 ng/ml TSA treatment and the significant increase in the number of low-density objects following both low and high doses of TSA. Significant distributional changes were also induced by TSA and these alterations were also identified in response to low doses. Combined quantitative information from several distributional features showed that following low doses of TSA a more uniform chromatin phenotype was evident in PNT1A G0/G1 nuclei (energy) and areas of high-density chromatin were predominantly localized to the nuclear periphery (high average distance), whereas at high doses chromatin was less uniform (energy) and regions of high density (high average distance) appeared most often within the nuclear interior (Fig. 5C).
When considering the chromatin architecture of LNCaP and DU145 prostate cancer cells, a comparison of untreated cells and those treated with low doses of TSA treatment showed that, of the 65 texture features analysed, 21/65 (32.3%) and 26/65 (40%), respectively, were significantly altered. Furthermore, in an independent analysis of cells treated with 100 ng/ml TSA this number of features increased to 31/65 (47.7%) and 33/65 (50.8%) relative to untreated nuclei (Fig. 5B). Following the assessment of specific changes in LNCaP chromatin organization, TSA-treated nuclei were found to exhibit little change in chromatin density (as illustrated by mean density). However, in DU145 cells both low and high doses of TSA were associated with a disruption in chromatin density. Moreover, it has become clear based on textural analysis results for the distributional and sub-visual texture features measured that TSA alters the organization of chromatin in both LNCaP and DU145 cells even at low doses (Fig. 5C). In LNCaP cells these changes are illustrated by alterations in chromatin uniformity, and distributional features Haram10 and Harar1. In DU145 a significant change in the high average distance texture feature may suggest the relocation of highly condensed chromatin to peripheral nuclear regions, distributional chromatin changes are best illustrated by significant changes in the Haram 10 and energy texture features (Fig. 5C).
Altered gene expression profiles in response to TSA induced hyperacetylation
DNA microarray analysis performed on TSA-treated cells using an MWG Human 30K array incorporating 9850 genes showed that TSA induces global gene expression changes in PNT1A and LNCaP cells at both low (12 ng/ml) and high (100 ng/ml) doses. A comprehensive list of all the genes altered in response to TSA treatment within the 1.75-fold change filter are presented in Tables S1–4. These results are summarized in Fig. 6 by gene ontology studies which show that the greatest proportion of genes altered by TSA treatment both at low and high doses are those involved in metabolic processes within the cell. Furthermore the proportion of metabolic genes altered was found to be enhanced following high (100 ng/ml) doses of TSA when compared to low doses in both PNT1A and LNCaP cells. Also in response to high doses of TSA a greater impact was evident in the proportion of genes involved in the cellular response to stress, cell cycle, death and chromosome segregation. This effect may be due to the known cell cycle arrest and apoptotic response associated with high doses of TSA treatment.
Fig 6.
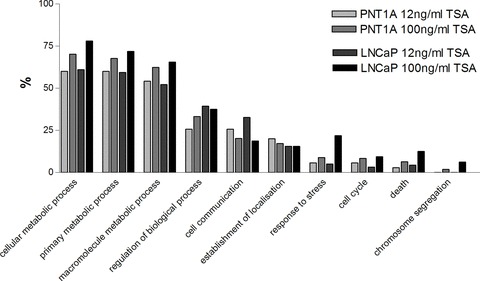
Fig. 6 A gene ontology summary of global changes in gene expression induced in PNT1A cells and LNCaP cells following treatment with 12 ng/ml and 100 ng/ml TSA. All genes were assigned gene functions by the Babelomics functional enrichment tool for gene ontology analysis (http://babelomics.bioinfo.cipf.es/).
Discussion
Studies on the molecular basis of prostate cancer increasingly suggest a significant role for global histone acetylation in the development of this disease [28]. Furthermore, it has been suggested that signatures of global histone modifications, chromatin phenotype and alterations in the expression of chromatin remodelling enzymes may represent novel diagnostic and prognostic markers of prostate cancer [29–31].
By initially using a cell line model of normal (PNT1A) and prostate cancer (LNCaP) cells we have shown that TSA induces significant changes in H3K9 acetylation together with concurrent perturbations in cell cycle characteristics and in chromatin organization. In this study, hyperacetylation of the key residue H3K9, was used as a marker for TSA-induced hyperacetylation and was shown to be clearly hyperacetylated following TSA treatment using a range of techniques including immunocytochemistry, flow cytometry, digital image analysis and confocal microscopy. Although the impact of TSA-induced hyperacetylation on the cell cycle is well recognized in a wide range of cell types [32–36], this is the first extensive study involving the analysis of a normal prostate cell line in comparison to the commonly used metastatic prostate cell lines. Here we have shown that the prostatic cell line PNT1A and prostate cancer cell line LNCaP exhibit differential responses to TSA treatment with PNT1A cells showing increased cell cycle perturbations. This was supported by the overall EC-50 results. The different responses of the cell lines may be explained by the subsequent efficiency of major cell cycle checkpoints, which defines the tolerance of cells to changes in acetylation status and the point at which detection of hyperacetylation initiates an arrest at functional cell cycle checkpoints.
The exact mechanism of cell cycle arrest induced by HDAC inhibition is still debatable. Although it is thought that the overexpression of certain key cell cycle regulators such as cyclin B1 and p21 Waf1/Cip1 have a role in TSA-induced arrest [37], it is impossible to relate such changes to the action of a single gene or protein due to the genome-wide influence of TSA on gene expression profiles. In this study, PNT1A cells were found to arrest in the G2 phase of the cell cycle. This is supported by Qiu et al., who suggested the presence of a checkpoint in G2 that is specifically sensitive to levels of HDAC activity, and is found exclusively in normal cells [38]. Alternatively, some evidence suggests that TSA-induced G2/M cell cycle arrest in cancer cells is attributed to the induction of the mitotic checkpoint in early anaphase due to improper kinetochore localization [39, 40]. This may provide an explanation for the TSA-induced arrest in LNCaP cells.
In addition to cell cycle arrest we have also shown that TSA-induced H3K9 hyperacetylation is strongly correlated with cell cycle phase, showing increasing levels of H3K9 acetylation as the cells move through the cell cycle. This was only possible using dual parameter flow cytometry and double labelling the cells for DNA content and H3K9 acetylation. This strong association may be indicative of the increase in available acetylation sites that occurs as the cell progresses through the cell cycle. However, we also observed a distinct subpopulation of G2/M cells highly expressing AcH3K9 that was observed in prostate cancer cell lines and appeared to be absent in normal PNT1A cells. This effect was more pronounced in the more aggressive DU145 cell line and at higher doses of TSA. Subsequent visual analysis of confocal images confirmed the presence of this AcH3K9 rich population and confirmed these to be mitotic figures. Therefore, mitotic cells appear to demonstrate higher levels of AcH3K9 than the rest of the cell cycle, and TSA induces selective hyperacetylation of mitotic cells. This was an unusual finding because histone acetylation is most commonly associated with transcription, and it is generally accepted that transcription is terminated during mitosis [41, 42]. Moreover, in mitosis, chromatin is present in its most condensed form where histone acetylation levels would be expected to be low. Several studies have demonstrated maintained histone acetylation during mitosis, and a specific role for H3K9 acetylation during this process [38, 43]. We suspect that the same phenomenon is at play in this study, although further work is necessary to elucidate the exact mechanisms involved.
One of the key goals of this study was to determine the relationship between hyperacetylation and chromatin phenotype. Because we have shown that hyperacetylation induces cell cycle anomalies, and cell cycle and chromatin organization are very strongly associated, it would be easy to confound the results and explain changes in chromatin in terms of hyperacetylation rather than underlying cell cycle alterations. This has been largely ignored in previous studies of this type. Two different experimental strategies were adopted in an attempt to disentangle these phenomenon: (i) a sufficiently low dose of TSA was identified that had no major impact on cell cycle distribution and (ii) flow cytometric cell sorting was used to selectively sample specific phases of the cell cycle. Therefore, we demonstrated that the disruption of global histone acetylation directly alters the nuclear chromatin configuration even when there are no major changes to the cell cycle. Moreover, we have established that subtle alterations in chromatin phenotype can be accurately quantified using high-resolution digital texture analysis, and by utilizing this method we were able to detect differential chromatin responses to TSA treatment in normal and prostate cancer cells. To our knowledge this is the first study to have used computerized texture analysis to concurrently measure fluorescent signals representing AcH3K9 and chromatin organization in G0/G1 cell populations. Our results have demonstrated that histone acetylation has a major role in the regulation of higher-order chromatin structure, as even in a well defined cell cycle population of G0/G1 cells, at low TSA doses, specific changes in chromatin phenotype were identified together with changes in the spatial distribution of AcH3K9 foci. Interestingly, PNT1A cells exhibited a significant global chromatin decondensation which was not observed in LNCaP and DU145 cells. This would suggest that nuclear architecture and its response to TSA-induced histone hyperacetylation reflects underlying genomic differences in the cell types and may reflect the subtle nuclear phenotypic changes seen in human prostate tissue undergoing neoplastic progression. Indeed subsequent studies on human tissue support these observations [25, 29].
TSA-induced changes in the chromatin architecture impacts the functioning of the cell through the dysregulation of key cellular pathways through the altered expression of many genes and their targets. In this regard, the impact of TSA treatment on gene expression profiles was explored in this study and was shown to be different between normal and prostate cancer cells. DNA microarray analysis of TSA-treated normal (PNT1A) and prostate cancer (LNCaP) cells revealed alterations in the expression of many genes throughout the genome, with 4% of genes on this array responding to treatment. In the current study these genome-wide effects of TSA were also identified in response to low TSA doses where cell cycle arrest and apoptosis were not induced. In a recent study by Roy et al. the impact of TSA on LNCaP cells was also studied and a widespread disruption in gene expression was observed suggesting that TSA exhibits its effects on gene expression through the acetylation of transcription factors, such as p53, which subsequently impact an array of downstream cellular pathways [20]. In this current study the most evident response to TSA treatment was in PNT1A normal prostate cells which displayed an increased number of significant changes in gene expression than in LNCaP cells.
In summary, in this study we have elucidated the global impact of the chromatin modifying agent TSA on normal and prostate cancer cell lines by identifying cell-specific responses in chromatin phenotype, cell cycle disruptions and gene expression profiles. Such differential responses may be utilized for targeted therapeutic purposes. However, the detailed study of these factors requires careful dissection of cell cycle alterations from dependent histone hyperacetylation and associated chromatin organizational changes. Such careful cellular studies are essential as a means of informing more applied studies on HDAC inhibitors in phase I/II clinical trials and their potential for future prostate cancer therapies.
Supporting Information
Microarray data for PNT1A cells following treatment with 12 ng/ml TSA
Table S2 Microarray data for PNT1A cells following treatment with 100 ng/ml TSA
Table S3 Microarray data for LNCaP cells following treatment with 12 ng/ml TSA
Table S4 Microarray data for LNCaP cells following treatment with 100 ng/ml TSA
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Bartels PH, Montironi R, Hamilton PW, et al. Chromatin texture signatures in nuclei from prostate lesions. Anal Quant Cytol Histol. 1998;20:407–16. [PubMed] [Google Scholar]
- 2.Bartels PH, Montironi R, Hamilton PW, et al. Nuclear chromatin texture in prostatic lesions. I. PIN and adenocarcinoma. Anal Quant Cytol Histol. 1998;20:389–96. [PubMed] [Google Scholar]
- 3.Bartels PH, da Silva VD, Montironi R, et al. Nuclear chromatin texture in prostatic lesions II: PIN and malignancy associated changes. Anal Quant Cytol Histol. 1998;20:397–406. [PubMed] [Google Scholar]
- 4.da Silva VD, Montironi R, Thompson D, et al. Chromatin texture in high grade prostatic intraepithelial neoplasia and early invasive carcinoma. Anal Quant Cytol Histol. 1999;21:113–20. [PubMed] [Google Scholar]
- 5.Jia L, Shen HC, Wantroba M, et al. Locus-wide chromatin remodeling and enhanced androgen receptor-mediated transcription in recurrent prostate tumor cells. Mol Cell Biol. 2006;26:7331–41. doi: 10.1128/MCB.00581-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Allfrey VG, Faulkner R, Mirsky AE. Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc Natl Acad Sci. USA. 1964;51:786–94. doi: 10.1073/pnas.51.5.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Turner BM. Histone acetylation and control of gene expression. J Cell Sci. 1991;99:13–20. doi: 10.1242/jcs.99.1.13. [DOI] [PubMed] [Google Scholar]
- 8.Lee DY, Hayes JJ, Pruss D, et al. A positive role for histone acetylation in transcription factor access to nucleosomal DNA. Cell. 1993;72:73–84. doi: 10.1016/0092-8674(93)90051-q. [DOI] [PubMed] [Google Scholar]
- 9.Ehrenhofer-Murray AE. Chromatin dynamics at DNA replication, transcription and repair. Eur J Biochem. 2004;271:2335–49. doi: 10.1111/j.1432-1033.2004.04162.x. [DOI] [PubMed] [Google Scholar]
- 10.Lui Y, Hong Y, Zhao Y, et al. Histone H3 (lys-9) deacetylation is associated with transcriptional silencing of E-cadherin in colorectal cancer cell lines. Cancer Invest. 2008;26:575–82. doi: 10.1080/07357900701837168. [DOI] [PubMed] [Google Scholar]
- 11.Wade PA. Transcriptional control at regulatory checkpoints by histone deacetylases: molecular connections between cancer and chromatin. Hum Mol Genet. 2001;10:693–8. doi: 10.1093/hmg/10.7.693. [DOI] [PubMed] [Google Scholar]
- 12.De Ruijter AJ, Van Gennip AH, Caron HN, et al. Histone deacetylases: characterization of the classical HDAC family. Biochem. 2002;J370:737–49. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoshikawa Y, Kwon HJ, Yoshida M, et al. Trichostatin A induces morphological changes and gelsolin expression by inhibiting histone deacetylase in human carcinoma cell lines. Exp Cell Res. 1994;214:189–97. doi: 10.1006/excr.1994.1248. [DOI] [PubMed] [Google Scholar]
- 14.Yoshida M, Kijima M, Akita M, et al. Potent and specific inhibition of mammalian histone deacetylase both in vivo and in vitro by trichostatin A. J Biol Chem. 1990;265:17174–9. [PubMed] [Google Scholar]
- 15.Vigushin DM, Ali S, Pace PE, et al. Trichostatin A is a histone deacetylase inhibitor with potent antitumor activity against breast cancer in vivo. Clin Cancer Res. 2001;7:971–6. [PubMed] [Google Scholar]
- 16.Archer SY, Meng S, Shei A, et al. p21(WAF1) is required for butyrate-mediated growth inhibition of human colon cancer cells. Proc Natl Acad Sci USA. 1998;95:6791–6. doi: 10.1073/pnas.95.12.6791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee BI, Park SH, Kim JW, et al. MS-275, a histone deacetylase inhibitor, selectively induces transforming growth factor beta type II receptor expression in human breast cancer cells. Cancer Res. 2001;61:931–4. [PubMed] [Google Scholar]
- 18.Richon VM, Sandhoff TW, Rifkind RA, et al. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci USA. 2000;97:10014–9. doi: 10.1073/pnas.180316197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsubaki J, Hwa V, Twigg SM, et al. Differential activation of the IGF binding protein-3 promoter by butyrate in prostate cancer cells. Endocrinology. 2002;143:1778–88. doi: 10.1210/endo.143.5.8766. [DOI] [PubMed] [Google Scholar]
- 20.Roy S, Jeffery R, Tenniswood M. Array-based analysis of the effects of trichostatin A and CG-1521 on cell cycle and cell death in LNCaP prostate cancer cells. Mol Cancer Ther. 2008;7:1931–9. doi: 10.1158/1535-7163.MCT-07-2353. [DOI] [PubMed] [Google Scholar]
- 21.Taghiyev AF, Guseva NV, Glover RA, et al. TSA-induced cell death in prostate cancer cell lines is caspase-2 dependent and involves the PIDDosome. Cancer Biol Ther. 2006;5:e1–7. doi: 10.4161/cbt.5.9.3168. [DOI] [PubMed] [Google Scholar]
- 22.Rokhlin OW, Glover RB, Guseva NV, et al. Mechanisms of cell death induced by histone deacetylase inhibitors in androgen receptor-positive prostate cancer cells. Mol Cancer Res. 2006;4:113–23. doi: 10.1158/1541-7786.MCR-05-0085. [DOI] [PubMed] [Google Scholar]
- 23.Chicoine LG, Allis CD. Regulation of histone acetylation during macronuclear differentiation in Tetrahymena: evidence for control at the level of acetylation and deacetylation. Dev Biol. 1986;116:477–85. doi: 10.1016/0012-1606(86)90148-x. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Reinberg D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 2001;15:2343–60. doi: 10.1101/gad.927301. [DOI] [PubMed] [Google Scholar]
- 25.Mohamed MA, Greif PA, Diamond J, et al. Epigenetic events, remodeling enzymes and their relationship to chromatin organization in prostatic intraepithelial neoplasia and prostatic adenocarcinoma. BJU Int. 2007;99:908–15. doi: 10.1111/j.1464-410X.2006.06704.x. [DOI] [PubMed] [Google Scholar]
- 26.Doudkine A, McCaulay C, Poulin N, et al. Nuclear texture measurements in image cytometry. Pathologica. 1995;87:286–99. [PubMed] [Google Scholar]
- 27.Haralick RM, Shanmugan K, Dinstein I. IEEE Transactions on system, man and cybernetics. St Louis: Elsevier Science; 1973. Texture features for image classification. pp. 610–21. [Google Scholar]
- 28.Li LC, Carroll R, Dahiya R. Epigenetic changes in prostate cancer: implication for diagnosis and treatment. J Nat Cancer Instit. 2005;97:103–14. doi: 10.1093/jnci/dji010. [DOI] [PubMed] [Google Scholar]
- 29.Seligson DB, Horvath S, Shi T, et al. Global histone modification patterns predict risk of prostate cancer recurrence. Nature. 2005;435:1262–6. doi: 10.1038/nature03672. [DOI] [PubMed] [Google Scholar]
- 30.Huisman A, Ploeger LS, Dullens HF, et al. Discrimination between benign and malignant prostate tissue using chromatin texture analysis in 3-D by confocal laser scanning microscopy. Prostate. 2007;67:248–54. doi: 10.1002/pros.20507. [DOI] [PubMed] [Google Scholar]
- 31.Wang Z, Zang C, Rosenfeld JA, et al. Combinatorial patterns of histone acetylations and methylations in the human genome. Nat Genet. 2008;40:897–903. doi: 10.1038/ng.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim MS, Blake M, Baek JH, et al. Inhibition of histone deacetylase increases cytotoxicity to anticancer drugs targeting DNA. Cancer Res. 2003;63:7291–300. [PubMed] [Google Scholar]
- 33.Heerdt BG, Houston MA, Mariadason JM, et al. Dissociation of staurosporine-induced apoptosis from G2-M arrest in SW620 human colonic carcinoma cells: initiation of the apoptotic cascade is associated with elevation of the mitochondrial membrane potential (deltapsim) Cancer Res. 2000;60:6704–13. [PubMed] [Google Scholar]
- 34.Sambucetti LC, Fischer DD, Zabludoff S, et al. Histone deacetylase inhibition selectively alters the activity and expression of cell cycle proteins leading to specific chromatin acetylation and antiproliferative effects. J Biol Chem. 1999;274:34940–7. doi: 10.1074/jbc.274.49.34940. [DOI] [PubMed] [Google Scholar]
- 35.Siavoshian S, Segain JP, Kornprobst M, et al. Butyrate and trichostatin A effects on the proliferation/differentiation of human intestinal epithelial cells: induction of cyclin D3 and p21 expression. Gut. 2000;46:507–14. doi: 10.1136/gut.46.4.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Strait KA, Dabbas B, Hammond EH, et al. Cell cycle blockade and differentiation of ovarian cancer cells by the histone deacetylase inhibitor trichostatin A are associated with changes in p21, Rb, and Id proteins. Mol Cancer Ther. 2002;1:1181–90. [PubMed] [Google Scholar]
- 37.Noh EJ, Lee JS. Functional interplay between modulation of histone deacetylase activity and its regulatory role in G2-M transition. Biochem Biophys Res Commun. 2003;310:267–73. doi: 10.1016/j.bbrc.2003.09.013. [DOI] [PubMed] [Google Scholar]
- 38.Qiu L, Burgess A, Fairlie DP, et al. Histone deacetylase inhibitors trigger a G2 checkpoint in normal cells that is defective in tumor cells. Mol Biol Cell. 2000;11:2069–83. doi: 10.1091/mbc.11.6.2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cimini D, Mattiuzzo M, Torosantucci L, et al. Histone hyperacetylation in mitosis prevents sister chromatid separation and produces chromosome segregation defects. Mol Biol Cell. 2003;14:3821–33. doi: 10.1091/mbc.E03-01-0860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shin HJ, Baek KH, Jeon AH, et al. Inhibition of histone deacetylase activity increases chromosomal instability by the aberrant regulation of mitotic checkpoint activation. Oncogene. 2003;22:3853–8. doi: 10.1038/sj.onc.1206502. [DOI] [PubMed] [Google Scholar]
- 41.Prescott DM. Cellular sites of RNA synthesis. Prog Nucleic Acid Res Mol Biol. 1964;3:33–57. doi: 10.1016/s0079-6603(08)60738-2. [DOI] [PubMed] [Google Scholar]
- 42.Gebrane-Younes J, Fomproix N, Hernandez-Verdun D. When rDNA transcription is arrested during mitosis, UBF is still associated with non-condensed rDNA. J Cell Sci. 1997;110:2429–40. doi: 10.1242/jcs.110.19.2429. [DOI] [PubMed] [Google Scholar]
- 43.Kruhlak MJ, Hendzel MJ, Fischle W, et al. Regulation of global acetylation in mitosis through loss of histone acetyltransferases and deacetylases from chromatin. J Biol Chem. 2001;276:38307–19. doi: 10.1074/jbc.M100290200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Microarray data for PNT1A cells following treatment with 12 ng/ml TSA
Table S2 Microarray data for PNT1A cells following treatment with 100 ng/ml TSA
Table S3 Microarray data for LNCaP cells following treatment with 12 ng/ml TSA
Table S4 Microarray data for LNCaP cells following treatment with 100 ng/ml TSA