

Abstract
Among the stress proteins that are up-regulated in the heart due to imposed biomechanical stress, αB-crystallin (CryAB) is the most abundant and pivotal in rendering protection against stress-induced cell damage. Cardiomyocyte-specific expression of the CryAB gene was shown to be dependent upon an intact αBE4 cis-element located in the CryAB enhancer. To date, there is no evidence on the identity of regulatory proteins and associated signalling molecules that control CryAB expression in cardiomyocytes. In this study, we define a mechanism by which the calcineurin/NFAT and Jak/STAT pathways regulate CryAB gene expression in response to a hypertrophic agonist endothelin-1 (En-1), in hypertrophic hearts of mice with pressure overload (TAC) and in heart-targeted calcineurin over-expressing mice (MHC-CnA). We observed that in response to various hypertrophic stimuli the transcription factors NFAT, Nished and STAT3 form a dynamic ternary complex and interact with the αBE4 promoter element of the CryAB gene. Both dominant negative NFAT and AG490, an inhibitor of the Jak2 phosphorylation, inhibited CryAB gene transcription in transient transfection assays. AG490 was also effective in blocking the nuclear translocation of NFAT and STAT3 in cardiomyocytes treated with En-1. We observed a marked increase in CryAB gene expression in MHC-CnA mouse hearts accompanied with increased phosphorylation of STAT3. We conclude that hypertrophy-dependent CryAB gene expression can be attributed to a functional linkage between the Jak/STAT and calcineurin/NFAT signalling pathways, each of which are otherwise known to be involved independently in the deleterious outcome in cardiac hypertrophy.
Keywords: cardiac hypertrophy, αB-crystallin, calcineurin/NFAT, Jak/STAT
Introduction
Cardiomyocytes respond to stress by stimulating adaptive intracellular events that foster preservation of cardiomyocyte viability and function. However, if the stress persists, the protection is subdued leading to a maladaptive response, that is apoptosis, necrosis and heart failure. Among the stress proteins, also known as heat shock proteins (Hsp’s), that are up-regulated in response to stress stimuli, the lens protein αB-crystallin (CryAB) is the most abundant in cardiomyocytes [1]. CryAB functions as a negative regulator of apoptosis [2, 3] and protects against disruption of myofibrillar structure by its localization to the Z-bands of sarcomeres [4, 5]. Gain- and loss-of-function studies have documented a protective role of CryAB in myocardial ischemia and reperfusion injury [6–8].
Srivastava et al.[8] have identified an enhancer element, αBE4, with a conserved sequence (5′-AGGAATCTTC-3′) in the CryAB gene that is required for its cardiac-specific expression. We subsequently noticed a similarity of this sequence with the intronic regulatory element (IRE) in the ventricular myosin light chain-2 (MLC2v) gene, which mediates its cardiac-specific up-regulation in response to hypertrophic stress [9]. Stress-induced expression of the MLC2v gene was dependent upon binding of transcription factors Nished and nuclear factor of activated T cells (NFAT) to the IRE [9]. This correlation led us to speculate that these two transcription factors might be involved in αBE4-mediated up-regulation of the CryAB gene.
Calcineurin-dependent gene expression is mediated by the activation of NFAT through its dephosphorylation and nuclear translocation [10]. Activation of Jak2 on the other hand triggers phosphorylation of STAT proteins and translocation into the nucleus as homo- or heterodimers [11]. Both STAT and NFAT induce expression of target genes via binding to their respective target sequences. Emerging evidence suggests that besides mediating maladaptive hypertrophy in cardiomyocytes, calcineurin/NFAT and Jak/STAT signalling cascades, exert a pro-survival regulatory effect as well [12, 13]. However, little is known on the downstream events underlying this cytoprotection mechanism. Here, we show that transcription factors Nished, NFAT and STAT3 activate CryAB gene expression in cultured cardiomyocytes exposed to the hypertrophic agonist endothelin (En-1) and in hypertrophic hearts of calcineurin over-expressing transgenic mice (MHC-CnA) [14]. Cain, an inhibitor of calcineurin, and dominant negative NFAT (dnNFAT) reduce CryAB gene transcription in transient transfection assays. Likewise, AG490, an inhibitor of Jak2 phosphorylation, effectively blocks the up-regulation of CryAB gene expression caused by constitutively active Jak2 kinase in cultured cardiomyocytes and in hypertrophic hearts induced by transverse aortic constriction (TAC). AG490 also blocks En-1-induced translocation of NFAT into the nucleus. An increase in CryAB gene expression in MHC-CnA mice was accompanied by phosphorylation of STAT3. These results support the notion that a coactive linkage between calcineurin/NFAT and Jak/STAT signalling is required for up-regulation of the CryAB gene as part of a program that acts to protect cardiomyocytes against deleterious hypertrophic stimuli.
Materials and methods
Animals
Transverse aortic constriction of mice was done in our laboratory as described previously [15]. Male mice C57/BL6 of 7 to 8 weeks old matched for weight, sex and age were subjected to TAC for 15 days, and injected daily intraperitoneally with vehicle solution (0.05% DMSO in PBS) only as a control, or AG490 (Alexis) resuspended in 0.05% DMSO 48 μg/kg/day. Sham mice were subjected to median sternotomy without constriction. MHC-CnA mice, kindly provided by Dr. Molkentin, were bred and the colony was maintained according to the Guidelines for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85–23, revised 1996).
Antibodies and chemicals
Polyclonal rabbit antibodies to Nished protein were described previously [9]. Monoclonal anti-phospho-STAT3 (Tyr705), polyclonal anti-STAT3 and anti-acetyl-histone H3 antibodies were from Upstate Biotechnology. Polyclonal anti-NFATc3, anti-NFATc4 and Lamin B were from Santa Cruz Biotechnology. Endothelin-1 was from Sigma and AG490 was from Alexis Biochemicals.
Construction of reporter genes
Mouse CryAB cDNA and pRD15 plasmid, containing the 6.5-kbp PstI fragment of the mouse CryAB gene, were a kind gift from Dr. Piatigorsky [8]. The plasmid was constructed by restricting with SmaI/NaeI and religation into the promoter-less vector, pGL2Basic (Promega). The resulting plasmid CryAB-Luc retained a 647-bp fragment of the CryAB gene from –661 to –14 in the correct orientation with respect to the vector. The αBE4 sequence in CryAB-Luc was mutated (GAAGCTTC→GtAcCccg) according to the Quick Change II Site-Directed Mutagenesis kit (Stratagene).
Gel mobility shift assays
Gel mobility shift assays (GMSA) was done as described previously [9]. The following commercial oligonucleotides were utilized: upper αBE4 (5′-GCCTAGGAAGATTCCAGTCCCT GC-3′), lower αBE4 (5′-GCAGGGACTGGAATCTTCCTAGGC-3′), upper mutαBE4 (5′-GCCTAGtgAGATTCCAGTCCCTGC-3′), lower mutαBE4 (5′-GCA GGGACTGGAATCTcaCTAGGC-3′).
Quantitative RT-PCR
Total RNA was isolated with the QIAGEN Real-time polymerase chain reaction (Valencia, CA, USA) RNeasy kit, treated with RNase-free DNase (Ambion, Applied Biosystems, Foster City, CA, USA) and reverse transcribed performed with the SuperScript® III First-Strand Synthesis RT system (Invitrogen, Carlsbad, CA, USA). Quantitative RT-PCR was performed on a GeneAmp 5700 Sequence Detection System (Applied Biosystems, Foster City, CA, USA) performed with the Real-Time SYBR Green PCR Master Mix (SA Biosciences, Frederic, MD, USA) and specific primers for CryAB and β-actin (SA Biosciences). All measurements were performed in triplicate.
Chromatin immunoprecipitation
Chromatin immunoprecipitation (ChIP) assays were performed according to the protocol of ChIP assay kit (Upstate Biotechnology, Lake Placid, NY, USA), with ChIP grade specific antibodies and primers flanking αBE-4 or the GAPDH promoter.
Immunoprecipitation and western blotting
Mouse heart nuclear protein extracts were prepared as described earlier [16]. Total cell lysates from hearts were prepared in RIPA as described previously [17] and immunoprecipitates were fractionated on SDS-polyacrylamide gels. After transfer, the nitrocellulose membranes (Amersham, GE Healthcare, Piscataway, NJ, USA) were incubated with primary antibodies followed by horseradish peroxidase-conjugated secondary antibodies. The immune complexes were visualized by the ECL technique (Amersham).
Immunocytochemistry
Rat primary cardiac cells were isolated from 2-day-old WKY rats performed with the neonatal cardiomyocyte isolation system (Worthington Biochemical Corp., Lakewood, NJ, USA). AG490 (5 μM) was added when needed 5 min. before treatment with 50 nM Endothelin-1 for 10 min. DMSO was used for treatment of control cells. Following fixing with 4% paraformaldehyde for 20 min., treatment with 0.2% triton X-100 for 10 min. and blocking in 1% bovine serum albumin/0.1% triton-X-100, cells were incubated with primary antibody (1:100) overnight at 4°C. Then cells were washed and incubated with fluorescence labelled donkey–anti-rabbit Alexa fluor 594 and goat anti-mouse FITC (Invitrogen) secondary antibodies (1:200) in blocking buffer for 30 min. Cells were washed and cover slips were mounted on slides performed with Prolong Gold anti-fade mounting medium with Dapi (Invitrogen) to visualize nuclei by Fluorescence microscopy.
Transient transfections and luciferase assay
The murine cardiomyocyte-derived HL-1 cell line was obtained from Dr. Claycomb (Louisiana State University). HL-1 cardiomyocytes were transiently transfected using FuGENE 6 reagent (Roche Diagnostics, Indianapolis, IN, USA) 24 hrs after plating in 12-well plates with 500 ng of reporter gene constructs and 5 ng of thymidine kinase promoter-driven Renilla luciferase-thymidine kinase vector, which was used to normalize the transfection efficiency. Luciferase activity was assayed by the dual luciferase assay kit (Promega, Madison, WI, USA) according to the manufacturer’s protocol.
RNA extraction
Total RNA was extracted from cardiomyocytes using Trizol reagent Invitrogen).
Northern hybridization
Northern hybridization and size-fractionation was done as described previously [9].
Semi-quantitative RT-PCR
The SuperScript One-Step RT-PCR with Platinum Taq cycle kit from Invitrogen was used for cDNA synthesis.
Statistical analysis
Data were expressed as means ± SE. To compare two means/groups for significance, a paired t-test was used, and P < 0.05 or P < 0.01 were considered significant. Statistical analyses were performed with the Prism software for Windows version 4 (GraphPad Inc., La Jolla, CA, USA).
Results
αBE4 is required for constitutive and En-1-induced CryAB expression in HL-1 cardiomyocytes
We have previously shown that the cis-acting IRE sequence in the MLC2v promoter is required for cardiac-specific, hypertrophy-induced up-regulation of MLC2v gene expression [9]. IRE and αBE4 share a distinct sequence similarity (Fig. 1A). In order to confirm whether αBE4 is functionally important in stress-induced CryAB gene expression, we generated plasmids containing 647 bp of upstream wild type (WT) or mutated promoter of the CryAB gene fused to a luciferase expression vector. First, the WT CryAB-Luc and mutated mutBE4-Luc plasmids were tested for promoter activity by transient transfection assays in HL-1 cardiomyocytes (Fig. 1B). We observed that the mutation in αBE4 reduced transcription by about 35%. Based on the similarity between the sequences of IRE and αBE4, we speculated that Nished might interact with αBE4 to enhance transcription of the CryAB gene. Co-transfection of CryAB-Luc or mutBE4-Luc plasmids with a Nished expression vector [9] in HL-1 cells increased luciferase reporter activity of the WT reporter by 40%, and was dependent upon an intact αBE4 enhancer (Fig. 1C). GMSA performed with heart nuclear extracts and (−392 to −367 bp) αBE4 DNA as a probe yielded the formation of a single protein/DNA complex that was super-shifted by pre-incubation of extracts with antibody to Nished (Fig. 1D).
Fig 1.
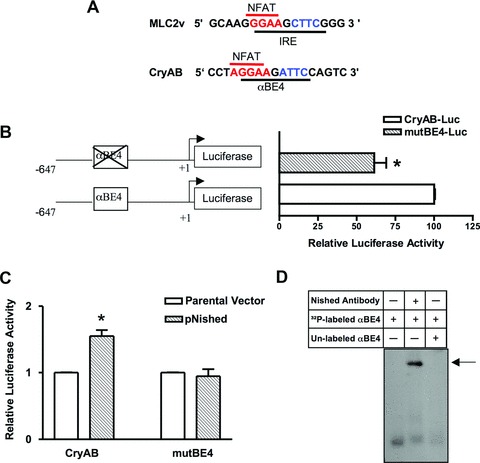
αBE4 enhancer activity. (A) Comparison of sequences of cardiac specific elements, IRE in the MLC2v and the αBE4 in the CryAB gene. The IRE and the αBE4 sequences contain a NFAT binding site, shown in red, 5′ to the Nished binding element, shown in blue. (B) Schematic representation of WT (CryAB-Luc) and mutant (mutBE4-Luc) gene constructs. Numbers denote the position of 5′ and 3′ ends of the gene relative to the transcription initiation site. HL-1 cardiomyocytes were transfected with CryAB-Luc or mutBE4-Luc plasmids. (C) HL-1 cells were co-transfected with CryAB-Luc or mutBE4-Luc plasmids along with parental vector (pcDNAV5) or Nished expression plasmid (pcDNAV5-Nished). Luciferase activities are expressed relative to the mean value derived from cells transfected with CryAB-Luc plasmids (B). In cells co-transfected with pcDNAV5-Nished, luciferase activities are expressed relative to the mean value derived from cells cotransfected with parental vector (pcDNAV5) and CryAB-Luc or mutBE4-luc plasmids. Renilla luciferase activity was used for normalization of transfection efficiency. Experiments have been repeated at least three times. Data are mean ± SE. *P < 0.05 and **P < 0.01. (D) Nuclear extracts from heart tissue of WT mice were incubated with a 32P-labeled oligonucleotide encompassing the αBE4 sequence and analyzed by gel mobility shift assay. Fifty-fold of unlabeled αBE4 DNA was used as a specific competitor. Preincubation of cardiac nuclear extracts with anti-Nished antibody shows a supershift indicated by arrow (n= 3).
Next, we examined if CryAB gene expression is induced in cardiomyocytes by the hypertrophic agonist endothelin (En-1) and whether this response is dependent on αBE4 and Nished. HL-1 cardiomyocytes were collected at 12, 24 and 48 hrs after pre-treatment with different concentrations of En-1 (25, 50, 100 nM) and RNA was examined by Northern blot performed with radiolabeled CryAB cDNA as a probe. There was a concentration and time-dependent increase in endogenous CryAB mRNA that reached a peak level with 50 nM En-1 at 48 hrs of En-1 treatment (Fig. 2A). HL-1 cells were transfected with WT or mutated αBE4 luciferase reporters and treated with 50nM En-1 (Fig. 2B). Treatment caused a twofold increase in WT promoter activity, which was further increased by co-transfection with Nished expression vector. The increase in reporter activity with mutated αBE4 was negligible. Thus, En-1 and Nished mediated an increase in promoter activity of the CryAB gene that was dependent upon an intact αBE4 element.
Fig 2.
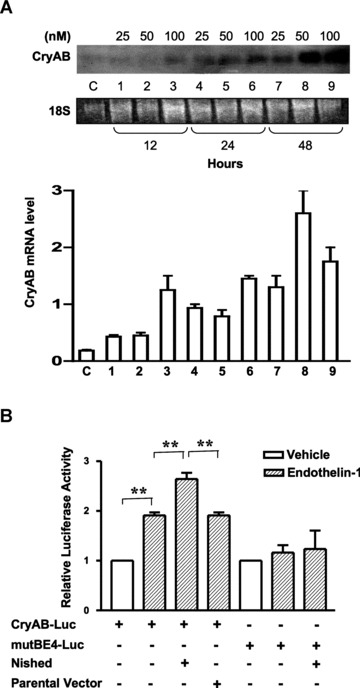
αBE4 enhancer activity in response to En-1 treatment. (A) Serum-starved HL-1 cells were treated with either different concentrations of En-1 (25, 50, 100 nM) for 12, 24 and 48 hrs before collection or vehicle (water). Total RNA levels were examined by Northern blotting with a CryAB cDNA probe. 18S ribosomal RNA was used as a loading control, (lower panel). Quantification of time and concentration dependent effect of En-1 on CryAB mRNA levels in HL-1 cells (n= 2). (B) HL-1 cells were cotransfected with an expression plasmid encoding Nished or parental vector where indicated along with CryAB-Luc, or mutBE4-Luc plasmids. After preincubation in serum-free medium cells were treated with 50 nM En-1. Luciferase activities in cells treated with En-1 are expressed to the mean value obtained from cells exposed to vehicle (water). In cells co-transfected with pcDNAV5-Nished (Nished) treated with En-1, luciferase activities are expressed relative to the mean value derived from cells cotransfected with pcDNAV5 (parental vector) and CryAB-Luc or mutBE4-luc plasmids and treated with En-1. Renilla luciferase expression was used for normalization. Experiments have been repeated at least three times. Data are mean ± SE. **P < 0.01.
αBE4/protein interaction is induced in pressure-overload hypertrophy
We have previously reported that pressure-overload cardiac hypertrophy in mice subjected to transverse aortic constriction (TAC) is blocked by AG490, implicating Jak/STAT signalling in the hypertrophic response [15]. Here, we examined hypertrophic ventricular tissue from TAC hearts for expression of the CryAB gene and found that there was a marked increase in CryAB mRNA, which was inhibited by AG490 treatment (Fig. 3A). Cardiac hypertrophy in mice subjected to TAC was evaluated by increase in heart-to-body weight ratios (HBWR) compared to that of weight- and age-matched sham animals (data not shown) consistent with our previous observations [15]. We also observed an induction of the mRNA for the known hypertrophic gene marker, atrial natriuretic factor (ANF) (Fig. 3A). We then examined the response of αBE4-binding protein(s) to TAC-induced pressure overload myocardial hypertrophy. GMSA performed with nuclear extracts and the αBE4 probe showed a robust increase in a DNA/protein complex in TAC heart extracts relative to sham heart extracts. The complex was competed out with 50-fold excess of unlabelled αBE4 oligonucleotide (S), but not with non-specific randomized oligomer (NS). Incubation of nuclear extract with antibody against Nished resulted in a shift of the complex (Fig. 3B). These data indicate that the hypertrophy-associated increase in CryAB expression is mediated by Jak/STAT signalling and that Nished is a part of the protein/DNA complex.
Fig 3.
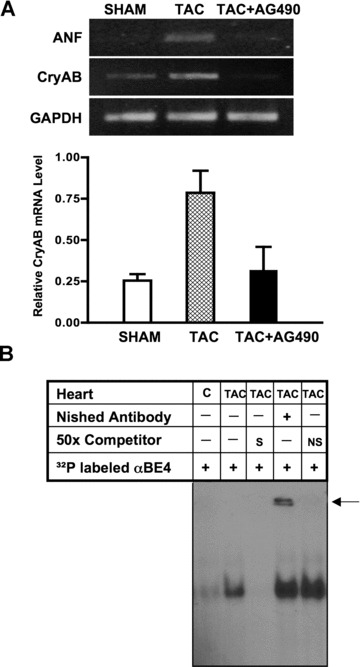
αBE4 enhancer activity in response to pressure-overload hypertrophy. Mice were subjected to transverse aortic constriction (TAC). (A) Semi-quantitative RT-PCR was performed with primers from the coding region of CryAB, ANF and GAPDH genes. Quantification of the CryAB mRNA level relative to the GAPDH mRNA level is shown in the lower panel. Data shown are representative of two separate experiments, with three individual mice per experiment. (B) Nuclear extracts were prepared from hearts of sham (C) and TAC mice. GMSA was performed with radiolabeled αBE4 oligonucleotide. Competition with 50× molar excess of unlabeled αBE4 (S) and nonspecific (NS) oligonucleotides is shown. Preincubation of cardiac nuclear extracts with anti-Nished antibody shows a supershift, indicated by arrow. Data shown are one representative of two separate experiments, with two mice per experiment.
Calcineurin/NFAT signalling in CryAB gene expression
The presence of a NFAT binding site [18] in the αBE4 region (Fig. 1A) and the demonstrated cooperativity between Nished and NFAT observed previously [9] suggest a possible involvement of calcineurin/NFAT signalling in regulation of CryAB gene expression under hypertrophic conditions. To test this, a plasmid overexpressing calcineurin inhibitory peptide, cain [19], was co-transfected along with the CryAB-Luc reporter plasmid into HL-1 cells treated with En-1. We observed that cain effectively inhibited the En-1-stimulated expression of the reporter (Fig. 4A). A primary substrate for activated calcineurin is the NFAT family of transcription factors. As seen in Figure 4B, co-transfection of a dominant negative form of NFAT (dnNFAT) [20] inhibited En-1-stimulated expression of the CryAB-Luc reporter by about 65% and the mutBE4-Luc promoter by 28%, while the inhibition in cells treated with vehicle alone was negligible.
Fig 4.
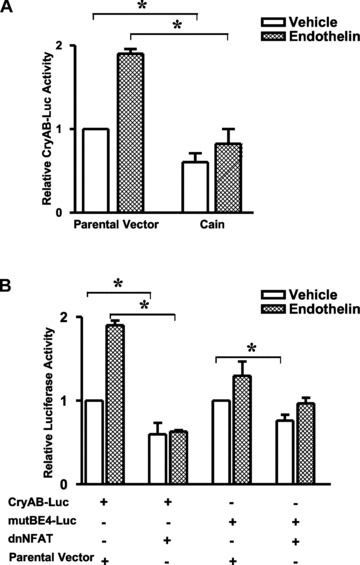
Calcineurin and NFAT are necessary for constitutive and En-1 inducible CryAB gene transcription. HL-1 cells were transfected either with CryAB-Luc (A), or with mutBE4-Luc (B). Reporter constructs were cotransfected with an expression plasmid encoding either cain or pECE-flag (parental vector) (A), or either dominant negative NFAT (dnNFAT) plasmid or pcDNA3. (parental vector) (B). Cells were treated with 50 nM En-1 or vehicle (water). Renilla luciferase expression was used for normalization. Luciferase activities in cells cotransfected with cain or dnNFAT are expressed relative to the mean value obtained from cells cotransfected with parental vector and treated with the vehicle only. Cells cotransfected with cain or dnNFAT and exposed to En-1 versus cotransfected with parental vector exposed to En-1. Experiments have been repeated at least three times. Data are mean ± SE. *P < 0.05.
To examine whether CryAB gene expression is up-regulated in the hearts of transgenic mice with heart-targeted over-expression of calcineurin (MHC-CnA) [14], total RNA was isolated from WT and MHC-CnA ventricular tissue and CryAB mRNA levels were measured by quantitative PCR (Fig. 5A). There was a 2.5-fold increase in CryAB mRNA in MHC-CnA ventricle compared to WT mice. GMSA showed robust induction of αBE4/protein interaction in MHC-CnA hypertrophic hearts compared to WT hearts (Fig. 5B). Prior incubation with antibody against Nished caused a shift, as before. Incubation with antibodies against NFATc3 and c4 caused a distinct reduction of αBE4 complex formation indicating that both NFATs are part of the αBE4/protein complex (Fig. 5C). These results were confirmed by ChIP assays (Fig. 5D). Chromatin was prepared from WT and MHC-CnA hearts, cross-linked and immunoprecipitated with antibodies specific for NFATc3, NFATc4 and acetylated histone H3. The precipitated DNA was analyzed by PCR performed with primers flanking the αBE4 sequence. There was an interaction of both NFATc3 and NFATc4 with αBE4 DNA (Fig. 5D). ChIP grade antibody for Nished is not available; hence the association of Nished with αBE4, that was evident in GMSA, could not be confirmed by ChIP assay.
Fig 5.
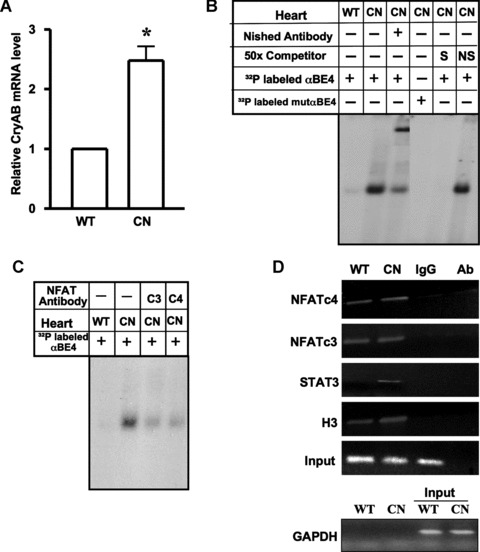
MHC-CnA mice have enhanced levels of CryAB mRNA and induced αBE-4/protein complex formation. (A) Real time PCR was done for quantification of CryAB mRNA in the hearts of WT C57BL/6 (WT) (n= 7) and transgenic MHC-CnA (CN) (n= 7) mice. β-actin was used for normalization. Data are mean ± SE. *P < 0.05. (B) Nuclear extracts were prepared from hearts of WT and MHC-CnA (CN) littermates. Gel mobility assays were performed with radiolabeled αBE4 or mutated αBE4 oligonucleotides. Binding reactions were performed in the presence of anti-Nished antibody, 50x molar excess unlabeled αBE4 (S) and nonspecific (NS) oligonucleotides. (C) Preincubation of cardiac nuclear extracts isolated from MHC-CnA mice (CN) with anti-NFATc3 (c3) and c4 (c4) antibodies were shown. Data shown are representative of three separate experiments, with two individual mice per experiment. (D) Chromatin isolated from WT and MHC-CnA mice (CN) hearts was subjected to ChIP. The chromatin DNA was immunoprecipitated with antibody against NFATc4, c3, STAT3 and acetylated histone H3 and analyzed by semiquantitative PCR with primers flanking the αBE4 sequence. A non-specific IgG was used as a negative control for immunoprecipitation and antibody alone (Ab), without lysate, served as a control for cross-contamination. Input DNA represents 10% of total chromatin used in each reaction. Primers specific to GAPDH were used before (input) and after immunoprecipitation as a control to monitor immunoprecipitation specificity. Data shown are representative of three separate experiments, with two individual mice per experiment.
Signal transduction by STAT3 has been implicated in both hypertrophy and cardioprotection [12, 13]. We therefore examined the participation of STAT3 in the αBE4 interacting protein complex by ChIP analysis. Binding to αBE4 was observed in extracts from MHC-CnA hearts but not from WT hearts (Fig. 5D) indicating that STAT3 is also a part of the αBE4/protein complex in calcineurin-induced cardiac hypertrophy. This is supported by increased phosphorylation of STAT3 on Tyr705 in the nuclear extracts of MHC-CnA hearts compared to WT (Fig. 6A). We also looked at the levels of the other proteins that interact with αBE4. Both NFATc3 and c4 proteins were higher in MHC-CnA hearts compared to WT hearts. There was no discernable difference in the level of Nished protein (Fig. 6A). We confirmed the participation of STAT3 in the protein complex interacting with the αBE4 element in MHC-CnA hearts by co-immunoprecipitation assay. Lysates from WT and MHC-CnA hearts were immunoprecipitated with anti-STAT3 antibody and western blotted with antibodies for NFATc3 and NFATc4 (Fig. 6B). Both NFATc3 and NFATc4 interact with STAT3 in MHC-CnA hearts, but not in WT hearts, suggesting that there is a direct interaction between the components of the calcineurin/NFAT and Jak/STAT signalling pathways.
Fig 6.
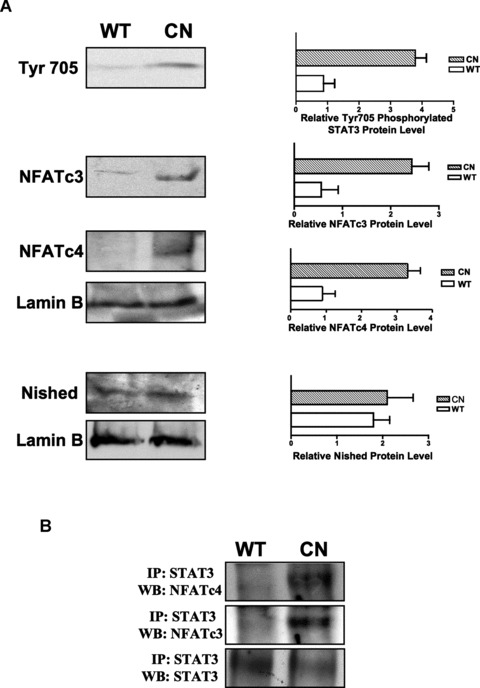
Interaction between the αBE4 binding proteins is induced in MHC-CnA mice. (A) Cardiac nuclear protein extracts from WT and MHC-CnA (CN) mice were subjected to SDS-PAGE fractionation followed by western blot with anti-phospho-Stat3 (Tyr705) antibody (that detects only Stat3 phosphorylated at tyrosine 705), anti-NFATc3, anti-NFATc4 and anti-Nished antibodies (n= 2). Lamin B was used as a loading control. Quantification of protein levels in the (WT) and MHC-CnA (CN) mice. (B) Cardiac cell lysates from WT and MHC-CnA (CN) mice were immunoprecipitated (IP) with anti-STAT3 antibody and western blotted (WB) with anti-NFATc4, anti-NFATc3 and anti-STAT3 antibodies. Data shown represent one of two separate experiments, with two individual mice per experiment.
Jak2 signalling regulates CryAB gene expression
To further confirm the role of Jak2 signalling in regulation of CryAB gene expression, the CryAB-Luc reporter and Tel-Jak2 (a kind gift from Dr. Barber), a fusion gene that encodes a Tel-Jak2, chimeric protein with constitutive tyrosine kinase activity [21], were co-transfected into HL-1 cells. There was a 14-fold increase in CryAB-Luc reporter activity. Tel-Jak2-induced luciferase activity was abrogated by treatment with 50 μM AG490 (Fig. 7A). Together, these results suggest that Jak/STAT signalling has a role in regulation of CryAB gene expression.
Fig 7.
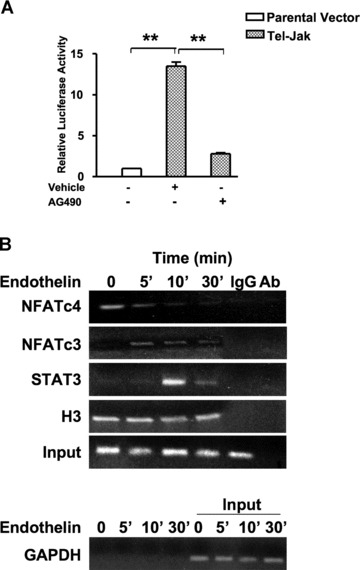
Jak2 regulates the transcriptional activity of the CryAB promoter in cardiomyocytes. (A) HL-1 cells were co-transfected with CryAB-Luc plasmid along with parental vector (pcDNAV5) or pTel-Jak2. Cells were serum-starved and treated with vehicle (DMSO) or 50 μM AG490 for 24 hrs. Luciferase activities in cells treated with AG490 and co-transfected with Tel-Jak are expressed relative to the mean value of the co-transfected cells exposed to the vehicle. Renilla luciferase expression was used for normalization. Experiments have been repeated at least three times. Data are mean ± SE. *P < 0.05 and **P < 0.01. (B) Serum-starved HL-1 cells were treated for 5, 10 and 30 min. with either En-1 (50 nM) or vehicle (water) and subjected to ChIP assays. Soluble chromatin from cross-linked cells was immunoprecipitated with indicated antibodies. A non-specific IgG was used as a negative control for immunoprecipitation and the antibody alone (Ab), without lysate, served as a control for cross-contamination. Input DNA represents 10% of total chromatin used in each reaction. DNA was amplified performed with primers flanking the αBE4 sequence. Primers specific to GAPDH were used before (input) and after immunoprecipitation as a control to monitor immunoprecipitation specificity. Data shown are one representative of three separate experiments.
Next, the assembly of STAT3, NFATc4 and NFATc3 proteins on αBE4 was examined by ChIP assay performed with HL-1 cells treated with En-1 (Fig. 7B). We observed that NFATc4 is present on αBE4 in non-stimulated HL-1 cells. The recruitment of NFATc3 and STAT3 to αBE4 occurs at 5 and 10 min. of En-1 administration, respectively. To examine En-1-induced nuclear translocation of NFATc3 and STAT3, rat primary cardiomyocytes were immunostained with antibodies against NFATc3 and STAT3 (Fig. 8). There was a significant enhancement of NFATc3 and STAT3 localization to the nucleus within 10 min. of En-1 administration. Importantly, however, AG490 (5 μM) treatment blocked En-1 induced translocalization, suggesting that calcineurin/NFAT and Jak/STAT pathways interact outside of the nucleus.
Fig 8.
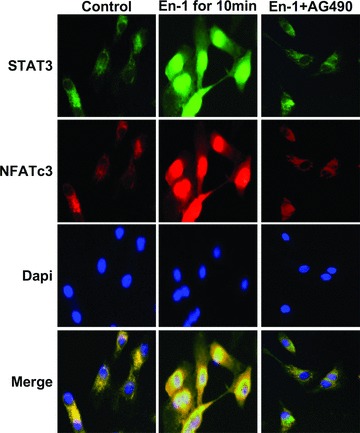
Immunocytochemical localization of STAT3 and NFATc3 in En-1 induced and AG490 treated rat primary cardiomyocytes. Cultured neonatal primary cardiomyocytes were treated with vehicle (DMSO) (left panel), En-1 (50 nM) (middle panel), or En-1 (50 nM) with AG490 (5 μM) (right panel), then immunostained for sub-cellular localization of STAT3 and NFATc3. Nuclei were stained with 4′, 6-diamidino-2-phenylindole (Dapi) and merged patterns are shown in the bottom panel. All images were obtained at 40× magnification.
Discussion
Recent studies implicate calcineurin/NFAT and Jak/STAT signalling in both beneficial and detrimental responses in the heart [12, 13]. In this report, we demonstrated a link between these signalling pathways and expression of the CryAB gene in response to hypertrophic stress. Transcription factors NFAT, STAT3 and Nished, participate in the formation of a dynamic ternary complex influenced by an interplay between the calcineurin/NFAT and Jak/STAT signalling pathways. In this context, induction of CryAB gene expression by these signalling molecules may mediate the protective outcomes. Srivastava et al.[8] have shown that chromatin at the αBE4 site is in an “open” configuration suggesting that αBE4 is accessible for transcription factors. Additional evidence supporting this comes from our observation of histone H3 acetylation in non-stimulated cardiomyocytes. Our GMSA and ChIP data confirmed the association of NFAT, STAT3 and Nished with the αBE4 enhancer element. Of the two NFAT isoforms, NFAT3 is involved in CryAB expression in response to hypertrophic stimuli, while NFAT4 appears to be part of the constitutive expression of the CryAB gene in unstimulated cardiomyocytes. A role for NFAT isoforms in non-stressed adult myocardium has been suggested previously [22–24]. It is known that calcineurin responds to the intracellular calcium levels that cycle in contracting cardiomyocytes [23]. These calcium levels could sustain a level of calcineurin activity that maintains a level of NFAT4 sufficient to activate the target CryAB gene, even in the absence of the hypertrophic stimulus. A deletion mutant of NFAT3, which renders it nuclear-localized, was sufficient to induce hypertrophy in transgenic mice supporting the importance of NFAT3 in hypertrophy [14]. Both isoforms of NFAT are, nonetheless, required for development of the heart and its function, as loss of both in a genetically manipulated animal model resulted in embryonic lethality while loss of either did not [25].
Induction of CryAB gene expression in MHC-CnA mice is accompanied by a concomitant increase in STAT3 phosphorylation. The inhibition of Jak2 signalling by AG490 effectively abrogates CryAB mRNA expression. These observations, combined with our data on the association of NFAT with STAT3 in MHC-CnA hearts suggest that these two signalling pathways are functionally linked. Treatment of cardiomyocytes with AG490 resulted in effective blockage of En-1-induced nuclear import of not only STAT3 but that of NFATc3 as well, supporting the notion that the cooperative activity of NFAT and STAT pathways is physiologically relevant. Previous studies on AG490-mediated blockage of the antiapoptotic effects of En-1 [26] and the demonstration that En-1 activates calcineurin in apoptotic cardiomyocytes [27] suggest that En-1 activation of the En-A receptor in cardiomyocytes activates Jak2 kinase, which in turn, regulates the activation of the calcineurin/NFAT pathway [28]. One mechanism through which Jak2 kinase can cross-activate calcineurin/NFAT signalling is by raising intracellular Ca++ level through phosphorylation of the En-1 receptor, or by regulation of modulatory calcineurin-interacting proteins (MCIPs) proposed earlier [29].
The cooperation of calcineurin with Jak2 signalling in response to hypertrophic stimuli may promote the association of NFAT/Nished with αBE4. The role of STAT3, in this case, may possibly be via interaction with NFAT rather than direct protein/DNA binding. Alternatively, STAT3 could facilitate the anchorage of the Nished/NFAT interacting complex on αBE4. This scenario may work at the initial compensatory stages of cardiac hypertrophy. However, if a strong and sustained hypertrophic stimulus persists, one may envision that the protective properties of Jak/STAT and calcineurin/NFAT can be overridden by interference with other transcription factors that are known to interact with NFAT or STAT3.
CryAB is the most studied chaperone protein that plays a critical role in protein quality control (PQC) in the heart. Human genetic studies and experiments performed with mouse transgenic models have demonstrated that a R120G mutation in CryAB (CryABR120G) is sufficient to cause aberrant protein aggregation and heart failure [30, 31]. Inadequacy of PQC in cardiomyocytes can lead to accumulation of misfolded and damaged proteins that, in turn, impairs PQC and deteriorates cardiomyocyte function [32]. The study here on the regulatory control of CryAB gene expression was based on the premise that CryAB is a component of the PQC system and an important determinant of tolerance against stress. Unravelling the molecular basis governing the mechanism of CryAB expression may provide insights into cell survival programs that could inform ways of manipulating CryAB gene expression to improve heart performance.
Acknowledgments
We thank Dr. J. Piatigorsky for his gift of mouse αB-crystallin cDNA. We are grateful to Drs J. Molkentin and R. Davies for providing cain and dnNFAT expression plasmids, respectively. We thank Dr. M. Wagner for his help in reviewing the manuscript and Inna Rozenberg for technical assistance.
Source of funding
This work was supported by NIH grant HL07339 to M. A. Q. Siddiqui.
References
- 1.Xiao X, Benjamin IJ. Stress-response proteins in cardiovascular disease. Am J Hum Genet. 1999;64:685–90. doi: 10.1086/302305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kamradt MC, Chen F, Cryns VL. The small heat shock protein alpha B-crystallin negatively regulates cytochrome c- and caspase-8-dependent activation of caspase-3 by inhibiting its autoproteolytic maturation. J Biol Chem. 2001;276:16059–63. doi: 10.1074/jbc.C100107200. [DOI] [PubMed] [Google Scholar]
- 3.Kamradt MC, Lu M, Werner ME, Kwan T, et al. The small heat shock protein alpha B-crystallin is a novel inhibitor of TRAIL-induced apoptosis that suppresses the activation of caspase-3. J Biol Chem. 2005;280:11059–66. doi: 10.1074/jbc.M413382200. [DOI] [PubMed] [Google Scholar]
- 4.Golenhofen N, Ness W, Koob R, et al. Ischemia-induced phosphorylation and translocation of stress protein alpha B-crystallin to Z lines of myocardium. Am J Physiol. 1998;274:H1457–64. doi: 10.1152/ajpheart.1998.274.5.H1457. [DOI] [PubMed] [Google Scholar]
- 5.Bullard B, Ferguson C, Minajeva A, et al. Association of the chaperone alphaB-crystallin with titin in heart muscle. J Biol Chem. 2004;279:7917–24. doi: 10.1074/jbc.M307473200. [DOI] [PubMed] [Google Scholar]
- 6.Ray PS, Martin JL, Swanson EA, et al. Transgene overexpression of alphaB crystallin confers simultaneous protection against cardiomyocyte apoptosis and necrosis during myocardial ischemia and reperfusion. FASEB J. 2001;15:393–402. doi: 10.1096/fj.00-0199com. [DOI] [PubMed] [Google Scholar]
- 7.Morrison LE, Whittaker RJ, Klepper RE, et al. Roles for alphaB-crystallin and HSPB2 in protecting the myocardium from ischemia-reperfusion-induced damage in a KO mouse model. Am J Physiol Heart Circ Physiol. 2004;286:H847–55. doi: 10.1152/ajpheart.00715.2003. [DOI] [PubMed] [Google Scholar]
- 8.Gopal-Srivastava R, Haynes JI, 2nd, Piatigorsky J. Regulation of the murine alpha B-crystallin/small heat shock protein gene in cardiac muscle. Mol Cell Biol. 1995;15:7081–90. doi: 10.1128/mcb.15.12.7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mathew S, Mascareno E, Siddiqui MAQ. A ternary complex of transcription factors, Nished and NFATc4, and co-activator p300 bound to an intronic sequence, intronic regulatory element, is pivotal for the up-regulation of myosin light chain-2v gene in cardiac hypertrophy. J Biol Chem. 2004;279:41018–27. doi: 10.1074/jbc.M403578200. [DOI] [PubMed] [Google Scholar]
- 10.Crabtree GR. Generic signals and specific outcomes: signaling through Ca2+, calcineurin, and NF-AT. Cell. 1999;96:611–4. doi: 10.1016/s0092-8674(00)80571-1. [DOI] [PubMed] [Google Scholar]
- 11.Kaptein A, Paillard V, Saunders M. Dominant negative stat3 mutant inhibits interleukin-6-induced Jak-STAT signal transduction. J Biol Chem. 1996;271:5961–4. doi: 10.1074/jbc.271.11.5961. [DOI] [PubMed] [Google Scholar]
- 12.van Empel VP, De Windt LJ. Myocyte hypertrophy and apoptosis: a balancing act. Cardiovasc Res. 2004;63:487–99. doi: 10.1016/j.cardiores.2004.02.013. [DOI] [PubMed] [Google Scholar]
- 13.Baines CP, Molkentin JD. STRESS signaling pathways that modulate cardiac myocyte apoptosis. J Mol Cell Cardiol. 2005;38:47–62. doi: 10.1016/j.yjmcc.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 14.Molkentin JD, Lu JR, Antos CL, et al. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93:215–28. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beckles DL, Mascareno E, Siddiqui MAQ. Inhibition of Jak2 phosphorylation attenuates pressure overload cardiac hypertrophy. Vascul Pharmacol. 2006;45:350–7. doi: 10.1016/j.vph.2006.05.006. [DOI] [PubMed] [Google Scholar]
- 16.Liew CC, Jackowski G, Ma T, et al. Nonenzymatic separation of myocardial cell nuclei from whole heart tissue. Am J Physiol. 1983;244:C3–10. doi: 10.1152/ajpcell.1983.244.1.C3. [DOI] [PubMed] [Google Scholar]
- 17.Mascareno E, Manukyan I, Das DK, et al. Downregulation of cardiac lineage protein (CLP-1) expression in CLP-1 +/− mice affords cardioprotection against ischemic stress. J Cell Mol Med. 2008 doi: 10.1111/j.1582-4934.2008.00404.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Macian F, Lopez-Rodriguez C, Rao A. Partners in transcription: NFAT and AP-1. Oncogene. 2001;20:2476–89. doi: 10.1038/sj.onc.1204386. [DOI] [PubMed] [Google Scholar]
- 19.Taigen T, De Windt LJ, Lim HW, et al. Targeted inhibition of calcineurin prevents agonist-induced cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A. 2000;97:1196–201. doi: 10.1073/pnas.97.3.1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chow CW, Rincon M, Davis RJ. Requirement for transcription factor NFAT in interleukin-2 expression. Mol Cell Biol. 1999;19:2300–7. doi: 10.1128/mcb.19.3.2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ho JM, Beattie BK, Squire JA, et al. Fusion of the ets transcription factor TEL to Jak2 results in constitutive Jak-Stat signaling. Blood. 1999;93:4354–64. [PubMed] [Google Scholar]
- 22.van Rooij E, Doevendans PA, de Theije CC, et al. Requirement of nuclear factor of activated T-cells in calcineurin-mediated cardiomyocyte hypertrophy. J Biol Chem. 2002;277:48617–26. doi: 10.1074/jbc.M206532200. [DOI] [PubMed] [Google Scholar]
- 23.Wilkins BJ, De Windt LJ, Bueno OF, et al. Targeted disruption of NFATc3, but not NFATc4, reveals an intrinsic defect in calcineurin-mediated cardiac hypertrophic growth. Mol Cell Biol. 2002;22:7603–13. doi: 10.1128/MCB.22.21.7603-7613.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilkins BJ, Molkentin JD. Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem Biophys Res Commun. 2004;322:1178–91. doi: 10.1016/j.bbrc.2004.07.121. [DOI] [PubMed] [Google Scholar]
- 25.Bushdid PB, Osinska H, Waclaw RR, et al. NFATc3 and NFATc4 are required for cardiac development and mitochondrial function. Circ Res. 2003;92:1305–13. doi: 10.1161/01.RES.0000077045.84609.9F. [DOI] [PubMed] [Google Scholar]
- 26.Ogata Y, Takahashi M, Ueno S, et al. Antiapoptotic effect of endothelin-1 in rat cardiomyocytes in vitro. Hypertension. 2003;41:1156–63. doi: 10.1161/01.HYP.0000064342.30653.24. [DOI] [PubMed] [Google Scholar]
- 27.Kakita T, Hasegawa K, Iwai-Kanai E, et al. Calcineurin pathway is required for endothelin-1-mediated protection against oxidant stress-induced apoptosis in cardiac myocytes. Circ Res. 2001;88:1239–46. doi: 10.1161/hh1201.091794. [DOI] [PubMed] [Google Scholar]
- 28.De Windt LJ, Lim HW, Taigen T, et al. Calcineurin-mediated hypertrophy protects cardiomyocytes from apoptosis in vitro and in vivo: An apoptosis-independent model of dilated heart failure. Circ Res. 2000;86:255–63. doi: 10.1161/01.res.86.3.255. [DOI] [PubMed] [Google Scholar]
- 29.Bush E, Fielitz J, Melvin L, et al. A small molecular activator of cardiac hypertrophy uncovered in a chemical screen for modifiers of the calcineurin signaling pathway. Proc Natl Acad Sci U S A. 2004;101:2870–5. doi: 10.1073/pnas.0308723101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rajasekaran NS, Connell P, Christians ES, et al. Human alpha B-crystallin mutation causes oxido-reductive stress and protein aggregation cardiomyopathy in mice. Cell. 2007;130:427–39. doi: 10.1016/j.cell.2007.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Osinska H, Klevitsky R, et al. Expression of R120G-alphaB-crystallin causes aberrant desmin and alphaB-crystallin aggregation and cardiomyopathy in mice. Circ Res. 2001;89:84–91. doi: 10.1161/hh1301.092688. [DOI] [PubMed] [Google Scholar]
- 32.Wang X, Su H, Ranek MJ. Protein quality control and degradation in cardiomyocytes. J Mol Cell Cardiol. 2008;45:11–27. doi: 10.1016/j.yjmcc.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]