

Abstract
Although the peroxisome proliferator-activated receptor (PPAR) δ has been implicated in the wound healing process, its exact role and mechanism of action have not been fully elucidated. Our previous findings showed that PPARδ induces the expression of the transforming growth factor (TGF)-β1, which has been implicated in the deposit of extracellular matrix proteins. Here, we demonstrate that administration of GW501516, a specific PPARδ ligand, significantly promoted wound closure in the experimental mouse and had a profound effect on the expression of collagen types I and III, alpha-smooth muscle actin, pSmad3 and TGF-β1, which play a pivotal role in wound healing processes. Activation of PPARδ increased migration of human epidermal keratinocytes and dermal fibroblasts in in vitro scrape-wounding assays. Addition of a specific ALK5 receptor inhibitor SB431542 significantly suppressed GW501516-induced migration of human keratinocytes and fibroblasts. In these cells, activated PPARδ also induced the expression of collagen types I and III and fibronectin in a TGF-β1-dependent or -independent manner. The effect of PPARδ on the expression of type III collagen was dually regulated by the direct binding of PPARδ and Smad3 to a direct repeat-1 site and a Smad-binding element, respectively, of the type III gene promoter. Taken together, these results demonstrated that PPARδ plays an important role in skin wound healing in vivo and that it functions by accelerating extracellular matrix-mediated cellular interactions in a process mediated by the TGF-β1/Smad3 signaling-dependent or - independent pathway.
Keywords: extracellular matrix, keratinocytes, peroxisome proliferator-activated receptor γ, transforming growth factor-β1, wound healing
Introduction
Wound healing is a highly dynamic biological process that restores tissue integrity and relies on a balance of collagen deposition and remodelling to repair tissue at the site of an injury [1]. Of the cytokines excreted at the wound site, transforming growth factor (TGF)-β1 represents diverse biological activities in the regulation of extracellular matrix (ECM) deposition and tissue remodelling, cellular adhesion and proliferation and the modulation of inflammatory responses [2]. TGF-β1 signals through a complex of two membrane-associated receptors that recruit and phosphorylate Smad2 and Smad3. Phosphorylated Smad2 and Smad3 then form a heteromeric complex with Smad4 and translocate into nucleus, where they activate transcription of TGF-β1-responsive genes in a Smad-binding element (SBE)-dependent and/or SBE-independent manner [3]. Exogenous TGF-β1 exerts a predominant effect in the enhanced wound healing process through induction and deposition of ECM molecules, including collagen types I and III in excisional wounds [4]. During the early stages of wound healing, collagen synthesis is accelerated to promote scar formation, followed by a gradual decay in this high rate of collagen synthesis within a wound to normal levels in the final process of normal wound healing [5]. Numerous studies corroborate the hypothesis that TGF-β1 plays a pivotal role in this process by enhancing ECM production and reducing inflammation [6, 7]. However, much remains to be determined regarding its mechanism of action on targets like keratinocytes and skin fibroblasts, which are the critical cell types in wound healing processes and act via production and deposition of matrix proteins, predominantly collagen types I and III [5].
Peroxisome proliferator-activated receptors (PPARs) are ligand-inducible transcription factors belonging to the nuclear hormone receptor superfamily, which control many cellular and metabolic processes, including cellular differentiation and proliferation, lipid homeostasis and energy metabolism [8, 9]. Three distinct PPAR subfamilies have been identified: PPARα (NR1C1), PPARδ (NR1C2, also known as PPARβ, FAAR and NUC1) and PPARγ (NR1C3). These receptors regulate gene expression by forming dimers with the retinoid X receptor (RXR) and binding to specific recognition sequences termed PPAR response elements (PPRE) located in the regulatory regions of target genes [10]. The promoter regions of PPAR target genes contain a direct repeat (DR1 or DR2) of the hexameric nucleotide (nt) sequence AGGTCA, separated by one or two nt [11]. Among the three PPAR isoforms, PPARδ is abundantly and ubiquitously expressed in a variety of cell lineages, including keratinocytes, and has been implicated in wound healing, inflammatory responses, embryo implantation and lipid metabolism [9, 12]. Recent reports demonstrated that PPARδ plays a pivotal role in the keratinocyte response to inflammation produced immediately after a skin injury, and its inflammation-induced activation at the wound edge maintains a number of viable keratinocytes sufficient for reepithelization [13, 14]. Therefore, it has been postulated that PPARδ might represent a promising new therapeutic target for tissue injury [15]. A better understanding of the cellular mechanisms elicited by PPARδ may thus reveal its full therapeutic potential.
A recent report showed that TGF-β1 is a molecular target for PPARδ in vascular smooth muscle cells [16]. Because TGF-β1 is a well-known regulator of ECM homeostasis [2], its up-regulation by PPARδ seems to affect expression of genes related to regulation of ECM. We thus hypothesized that PPARδ plays a potential role as a key molecule in the processes of wound healing via modulation of ECM homeostasis. Here, we report that an activator of PPARδ promotes wound healing and also regulates expression of ECM-related genes, directly and/or indirectly, in a TGF-β1/Smad3-dependent or -independent manner.
Materials and methods
Materials
GW501516, WY14643, troglitazone, 15-deoxy-delta 12,14-prostaglandin J2 and TGF-β type I receptor (TβR-I) inhibitor were from Calbiochem (La Jolla, CA, USA). SB431542 (4-[4-(1,3-benzodioxol-5-yl)-5-(2-pyridinyl)-1H-imidazol-2-yl]benzamide) was obtained from Tocris Bioscience (Bristol, UK). Actinomycin D, mitomycin C and cycloheximide were obtained from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture
Human primary keratinocytes (Donor age 11; Passage 3; Welskin, Seoul, Korea) was cultured in Keratinocyte Growth Medium containing keratinocyte growth supplement, based on the manufacturer’s recommendations. Human skin fibroblasts (Donor age 9; Passage 3; Welskin, Seoul, Korea), Human keratinocyte-derived (HaCaT; Korean Cell Line Bank, Seoul, Korea) and fibroblast-derived (Detroit551; Passage 12; Korean Cell Line Bank, Seoul, Korea) cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) containing 100 U/ml penicillin and 100 μg/ml streptomycin supplemented with 10% heat-inactivated foetal bovine serum at 37 °C in an atmosphere of 95% air and 5% CO2.
Northern blot analysis
Aliquots of 5 μg of total RNA were heat-denatured at 65 °C for 15 min. in running buffer (40 mM MOPS, 10 mM sodium acetate and 1 mM EDTA, pH 7) containing 50% formamide and then electrophoresed on a 1% agarose gel containing 2.2 M formaldehyde. The size-fractionated RNA was transferred onto a Hybond-N+ nylon membrane (Amersham Biosciences UK Ltd., UK) overnight by capillary action and hybridized with a 32P-labelled specific probe at 68 °C in QuikHyb solution (Stratagene, La Jolla, CA, USA). The membranes were washed and radioactive signals were detected by a Fuji BAS-2500 Bioimaging Analyzer (Tokyo, Japan). The blots were stripped and rehybridized with a 32P-labeled GAPDH cDNA probe. cDNA probes were generated by PCR using primers that amplified nt 105–447, 555–896, 436–760 and 1039–1297 of COL3A1, COL1A1, Fibronectin and TGF-β1, respectively.
Western blot analysis
Cells treated with the reagents indicated were washed in ice-cold phosphate-buffered saline (PBS) and lysed in PRO-PREP protein extraction solution (iNtRON Biotechnology, Seoul, Korea). An aliquot of the cell lysate was subjected to SDS–polyacrylamide gel electrophoresis and transferred onto a Hybond-P+ polyvinylidene difluoride membrane (Amersham Biosciences). The membranes were probed with specific antibodies as described previously [17]. Polyclonal antibodies specific for PPARδ (dilution 1:1000), types III collagen (dilution 1:1000), as well as horseradish peroxidase (HRP)-conjugated IgG (dilution 1:1000) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Polyclonal antibodies specific for Smad3 (dilution 1:1000), phospho-Smad3 (dilution 1:1000), anti-Flag (dilution 1:1000) and anti-haemagglutinin (dilution 1:1000) were obtained from Cell Signaling (Beverly, MA, USA). Monoclonal mouse anti-TGF-β1 antibody (dilution 1:1000) was purchased from R&D Systems (Minneapolis, MN, USA). Polyclonal rabbit anti-β-actin antibody (dilution 1:2000) was obtained from Sigma-Aldrich.
Cell migration assay
Cell monolayers grown to confluence in 6-mm tissue culture plates were treated with 8-μg/ml mitomycin C in culture media for 2 hrs to eliminate proliferation. Cells were washed with PBS, and then scraped with a sterile single-edged razor blade and incubated in fresh medium containing 50 nM GW501516 or 14 nM DMSO. After incubation for the periods indicated, cells were fixed and stained with trypan blue. The fixed cells were observed under a microscope and the number of cells that migrated across the regions of the wound’s edge were counted.
Wound healing study
All animal studies were approved by the Institutional Animal Care Committee of Gyeongsang National University. Six-week-old male ICR mice (22–25 g) were purchased from Hyochang Science (Daegu, Korea) and maintained under controlled environmental conditions. Mice injected intraperitoneally with 10 mg/kg body weight of GW501516 (n= 12) or DMSO (n= 12) prior to 18–24 hrs was anaesthetized by intraperitoneal injection with ketamine. Four full-thickness 5-mm punch wounds were inflicted on the dorsal surface of the mouse, which was shaved prior to the procedure. To measure wound areas, photographs were taken from day 0 to day 14. For immunohistochemical analysis, wound tissues (2–3 mm border) surrounding the injuries were collected and placed in 4% paraformaldehyde.
Immunohistochemical analysis
All samples were verified by histological examination. After fixation with 4% paraformaldehyde and cryoprotection in 20% sucrose, tissues were embedded in OCT (Sakura Finetech Co., Tokyo, Japan) and snap-frozen in isopentene prechilled in liquid nitrogen. Serial cryosections (10 μm) were mounted on gelatin-coated slides and boiled in 10 mM sodium citrate for 3 min. Sections were blocked for 2 hrs at room temperature in 0.1 M Tris-buffered saline (TBS) containing 1% BSA, followed by incubation with the indicated antibodies at 4 °C overnight. After washing three times, staining was performed using ABC kit (Vector Labs, Burlingame, CA, USA) for diaminobenzidine (DAB) according to the manufacturer’s instructions. Colour development was achieved using 0.05 M Tris-HCl (pH 7.4), 0.2 mg/ml diaminobenzidine (DAB) and 0.003% H2O2 for 2 min. Polyclonal goat anti-Col-I (dilution 1:50), goat anti-rabbit IgG-HRP (dilution 1:100), donkey anti-goat IgG-HRP (dilution 1:100) and goat anti-mouse IgG-HRP (dilution 1:100) antibodies were obtained from Santa Cruz Biotechnology. Polyclonal rabbit anti-alpha smooth muscle actin (dilution 1:50) antibody was obtained from Abcam (Cambridge, UK). Polyclonal rabbit anti-Col-III antibody, goat anti-phospho-Smad3 antibody and monoclonal mouse anti-TGF-β1 antibody were diluted 1:50.
Gene silencing with small interfering RNA
Cells were seeded into 100 mm culture dishes 18–24 hrs prior to transfection and then transfected with 80 nM of control small interfering RNA (siRNA) or human PPARδ siRNA (Ambion, Austin, TX, USA) in serum-free medium using Welfect-Q (WelGENE, Daegu, Korea). After incubation for 6 hrs, cells were provided with fresh medium and grown for a further 3 days, which was followed by a 38-hr incubation with the reagents indicated. The effects of gene silencing were determined by Western and Northern blot analyses, and also by reporter gene assay.
Plasmid construction
The luciferase construct pGL3-COL3A1 containing the sequence of the human COL3A1 gene from nt –1685 to +68 was generously provided by Dr. H. Yoshioka (Department of Anatomy, Oita University, Oita, Japan). Constructs containing truncations of the 5′ end of the promoter but sharing the same 3′ end at position +68 were generated by self-ligation following digestion with PstI (Δ–1494/–1030-luc) and by PCR amplification using pGL3-COL3A1 as the template and the following SacI/XhoI-tagged primers: 5′–GAGCTCTTTACTGCTGAGGGGATGGGT–3′ and 5′–CTCGAGTTGAGGTGCTACTTTGAAC–3′; 5′–GAGCTCAGTTTTATGACGGGCCCGGTG–3′ and 5′– CTCGAGTTGAGGTGCTACTTTGAAC–3′. The amplified fragments were digested with SacI/XhoI and then ligated into the corresponding sites of pGL3-basic to generate the –225-luc and –115-luc constructs, respectively.
Site-directed mutagenesis
Nucleotide substitutions were introduced into the SBE and DR1 sites of the COL3A1 promoter using the QuikChange site-directed mutagenesis kit (Stratagene). PCR amplification of the wild-type human pGL3-COL3A1 luciferase reporter plasmid was performed using site-directed mutation primer sets (COL3A1-SBEmt: 5′–TGA AAT CTC GAG CGT AGA TT–3′ and 5′–AAT CTA CGG TCG AGA TTT CA–3′ or COL3A1-DR1mt: 5′–ATT TAA GAT CTA AGC AAA GGA AT–3′ and 5′–ATT CCT TTG CTT AGA TCT TAA AT–3′). The mutated bases are indicated in bold. PCR amplification was performed using 5 ng of template DNA and 12 cycles of 95 °C for 1 min., 55 °C for 1 min. and 68 °C for 7 min. PCR products were digested with DpnI for 1 hr at 37 °C prior to transformation into Escherichia coli XL-1 Blue competent cells. Colonies were screened by XhoI or BglII digestion and the integrity of the constructs was verified by sequencing.
Reporter gene assay
Luciferase reporter vectors for p6xSmadwt and p6xSmadmut and expression vectors for c-JUN, c-FOS and Sp-1 were gifts from Dr. Li Li (Department of Internal Medicine, Wayne State University, Detroit, MI, USA) and Dr. J.-H. Kim (Pochon CHA University, Seoul, Korea), respectively. The expression plasmids for Smad2, 3, 4 and 7 were obtained from Addgene (Cambridge, MA, USA). Cells were seeded into 6 well tissue culture plates 18–24 hrs prior to transfection, and then cotransfected with the luciferase reporter plasmids, the expression vectors indicated, and pSV β-Gal (SV40 β-galactosidase expression vector, Promega, Madison, WI, USA) using the SuperFect reagent (Qiagen, Valencia, CA, USA). After incubation in the presence or absence of the indicated reagents, cells were lysed in luciferase reporter lysis buffer (Promega) and luciferase activity was determined as described previously [18].
Chromatin immunoprecipitation (ChIP) assay
Chromatin immunoprecipitation was performed as described previously using anti-Smad3 or anti-PPARδ antibodies [16]. Rat genomic sequences containing the putative Smad3 binding site (SBE), PPARδ binding site (DR1) and an internal region from the COL3A1 promoter (oligo #1; –1302/–963, oligo #2; –222/–12 and oligo #3; –771/–534) were amplified using the following primer pairs: oligo #1 set 5′–GAAGTTGTGAGCTGGCTCAT–3′ and 5′–CAGATGAGGTCTTGGAACAG–3′, oligo #2 set 5′–TTACTGCTGAGGGGATGGGT–3′ and 5′–GCACAAAGAGTCTCATGTCT–3′ and oligo #3 set 5′–ACAAGGAAGCAGTCAGGGTT–3′ and 5′–TCAGGATGGACTCTGGCAAAA–3′.
Statistical analyses
The significance of differences between groups was evaluated using Student’s t-test or repeated measurement analysis of variance (ANOVA). All data represent means ± standard error (SE).
Results
Activation of PPARδ promotes wound healing processes
To explore the role of PPARδ in wound healing processes, we examined the effects of GW501516, a specific ligand of PPARδ[19], on wound closure and the expressional regulation of ECM-related proteins, including collagen types I and III, TGF-β1, alpha smooth muscle actin (α-SMA) and pSmad3, in tissues after incisional injury in the mouse. The activation of PPARδ by GW501516 significantly accelerated wound repair from an early time-point after injury, when compared with the vehicle-treated wounds (Fig. 1A and B). At day 8, closure of GW501516-treated wounds was almost completed, but not in the wounds exposed to vehicle (Fig. 1A).
Fig 1.
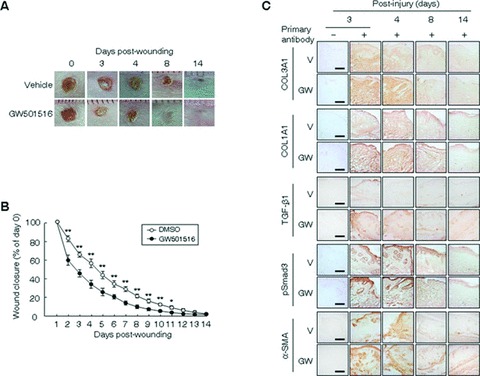
The PPARδ ligand promotes wound healing via up-regulation of the expression of TGF-β1, collagen types I and III collagen (Col-I, Col-III), α-SMA and pSmad3. (A) Four full-thickness 5-mm punch wounds were inflicted on the dorsal surface of a mouse pretreated with GW501516 or vehicle (DMSO). Photographs were acquired daily up to day 14 to measure wound areas; representative photographs are shown for days 0, 3, 4, 8 and 14. (B) Wound surface areas (n= 12) were plotted as a percentage of surface area measured at day 1. **P < 0.01 or *P < 0.05, compared with vehicle (DMSO) group. (C) Serial cryosections were prepared and stained with antibodies against the proteins indicated. Representative images (×200) of DAB staining at days indicated are shown. Immunopositive signals are denoted by a dark-brown colour. Bars, 50 μm. Immunohistochemistry was performed at least thrice using different sections and similar results were obtained each time. GW, GW501516; V, vehicle.
As the deposition of large amounts of matrix proteins (predominantly collagen types I and III) by peripheral cells has been implicated in the enhanced tensile strength of healing wounds, and because TGF-β1 is the potential stimulator of ECM production [5, 20], we examined whether the ligand-activated PPARδ affects the expression of ECM-related proteins in wound biopsies. The administration of GW501516 markedly increased the expression of collagen types I and III, TGF-β1, α-SMA and pSmad3 at days 3–8, when compared with the vehicle-treated wounds. At day 14, the enhanced expression of these molecules had gradually decayed to levels similar to those in vehicle-treated wounds (Fig. 1C). The expression levels of these proteins were concordant with the decreased wound size in post-injury days 3–8 (Fig. 1). These results indicate that activation of PPARδ accelerates wound repair through the enhancement of expression of matrix proteins and α-SMA in wounded mouse tissues.
Activation of PPARδ enhances migration of human epidermal keratinocytes and dermal fibroblasts through TGF-β1/Smad3 signalling pathway
The migration of keratinocytes and fibroblasts from surrounding tissues into wound sites is a well-known critical step in the process of epithelialization of the wound and ECM components, including collagen, seem to facilitate keratinocyte migration [21, 22]. To elucidate the effect of GW501516 on the migration of human epidermal keratinocytes and dermal fibroblast cells, we adopted an in vitro scrape-wounding model. The cultured cells grew actively after plating and typically reached confluence after 5 days, and the percentage of cells undergoing active proliferation conversely declined to minimum at days 5 when determined by [3H]-thymidine incorporation (data not shown). Using the proliferation index described previously as a reference basis, GW501516 significantly enhanced the migration of these four types of cells in a time-dependent manner, when compared with the DMSO-treated groups or untreated cells (Fig. 2 A and B). These results indicate that PPARδ promotes wound healing processes by accelerating the migration of keratinocytes and fibroblasts from surrounding tissues into the wound site.
Fig 2.
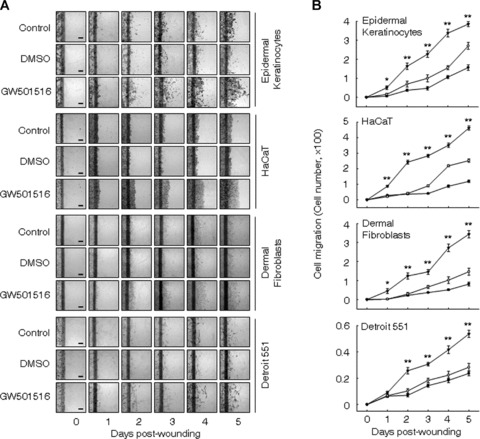
Ligand-activated PPARδ enhances the migration of human epidermal keratinocytes and dermal fibroblasts in the monolayer wound healing assay. (A and B) Confluent cells treated with mitomycin C (8 μg/ml) were wounded by scraping and were subsequently incubated in fresh medium containing 50 nM GW501516 (▾) or 14 nM DMSO (•). Representative photographs from three independent experiments are shown (A) and were used for quantifications (B). Bars, 50 μm. **P≤ 0.01 or *P≤ 0.05, compared with both the DMSO-treated and untreated cells. Open circles designate untreated cells (control).
To confirm the role of TGF-β1/Smad3 in the PPARδ-mediated cell migration, we examined the effect of SB431542, a specific ALK5 receptor inhibitor, on the GW501516-induced cell migration. As shown in Fig. 3, activation of PPARδ significantly increased cell migration, while it was significantly attenuated in SB431542-treated cells. These results clearly indicated that TGF-β1/Smad3 signalling is involved in the up-regulation of PPARδ-mediated cell migration.
Fig 3.
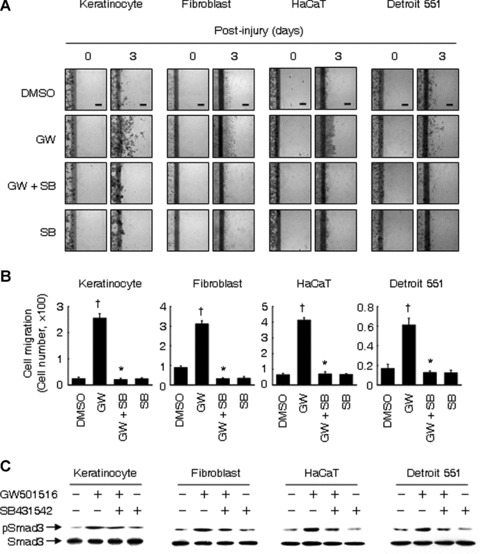
GW501516 enhances cell migration via the TGF-β1/Smad3 signaling pathway. (A and B) Confluent cells were treated with mitomycin C (8 μg/ml) for 2 hrs. After washing with PBS, cells were wounded by scraping and subsequently incubated in fresh medium containing 50 nM GW501516 (GW) or 14 nM DMSO in the presence or absence of 10 μM SB431542 (SB) for indicated time periods. Representative photographs from three independent experiments are shown (A) and were used for quantifications (B). Bars, 50 μm. †P < 0.01, compared with the DMSO-treated group; *P < 0.01, compared with the GW501516-treated group. (C) Cells were treated with mitomycin C (8 μg/ml) for 2 hrs. After washing with PBS, cells were stimulated with 50 nM GW501516 or 14 nM DMSO for 24 hrs in the presence or absence of 10 μM SB431542. Western blot analysis was performed using anti-pSmad3 antibody.
A PPARδ ligand induces the mRNA expression of ECM-related genes in a TGF-β1-dependent or -independent manner
Treatment of human keratinocyte-derived HaCaT and human dermal fibroblast-derived Detroit 551 cells with GW501516 resulted in a marked increase in the mRNA levels of collagen types I and III collagen and fibronectin in a concentration-dependent manner (Fig. 4A). Maximum levels of expression were observed after 38 hrs of exposure with 50–100 nM GW501516. Surprisingly, the levels of TGF-β1 transcript were also enhanced in a dose-dependent fashion and in parallel with the enhanced expression of the ECM-related genes, which raised the possibility that TGF-β1 is involved in the regulation of the expression of ECM-related genes.
Fig 4.
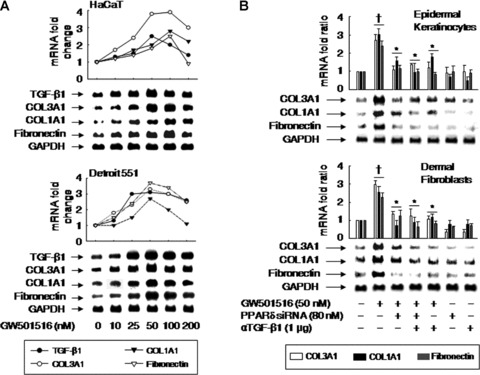
Activation of PPARδ specifically up-regulates the mRNA levels of TGF-β1, collagen types I and III (COL1A1, COL3A1), and fibronectin in human keratinocytes and fibroblasts. (A) Human keratinocyte-derived (HaCaT) and fibroblast-derived (Detroit551) cells were incubated for 38 hrs with indicated concentrations of GW501516. (B) Human primary epidermal keratinocytes and dermal fibroblasts were transfected with or without PPARδ siRNA and grown for 72 hrs, after which they were treated with GW501516 for 38 hrs in the presence or absence of anti-TGF-β1 antibody (αTGF-β1). Northern blot analyses were performed using the indicated specific cDNA probes. Membranes were stripped and reprobed with a GAPDH cDNA probe as internal controls. The radioactivity of the signals was quantified by an image analyzer and plotted as fold changes relative to untreated cells. The data were representative of two or three independent experiments. †P < 0.01, compared with the untreated group; *P < 0.05, compared with the GW501516-treated group.
To verify the role of PPARδ in the up-regulation of ECM-related genes, we examined the effect of GW501516 on cells that were treated with an siRNA against PPARδ. Although PPARδ was induced in the cells treated with either PPARδ siRNA or control siRNA, the levels of PPARδ in the four types of cells were markedly reduced after transfection with PPARδ siRNA, in the presence or absence of GW501516. However, control siRNA, consisting of a pool of nonspecific sequences, had no effect on PPARδ levels (Fig. S1). The siRNA-mediated down-regulation of PPARδ also suppressed the GW501516-induced expression of collagen types I and III collagen and fibronectin (Fig. 4B). To determine whether the increased mRNA expression of ECM-related genes was mediated by TGF-β1, we examined the effect of a neutralizing antibody against TGF-β1. Treatment of cells with anti-TGF-β1 antibody elicited a marked reduction in the GW501516-induced up-regulation of ECM-related genes (Fig. 4B).
A ligand of PPARδ, but not of PPARα or PPARγ, induces the mRNA expression of TGF-β1 and type III collagen in HaCaT and Detroit 551 cells
As type III is the major collagen species with type I collagen and TGF-β1 is the potential stimulator of these ECM components at the edge of wounds [5, 20], we investigated the expressional regulation of collagen type III and TGF-β1 by the various PPARs. Incubation of cells with specific ligands for PPARα, PPARγ or PPARδ led to an increase in the mRNA levels of type III collagen and TGF-β1 exclusively for the PPARδ-specific ligand GW501516 (Fig. 5A). The effect of GW501516 on the mRNA expression levels of type III collagen and TGF-β1 was time-dependent (Fig. 5B). These findings suggest that the PPARδ isoform is specifically involved in the up-regulation of these genes.
Fig 5.
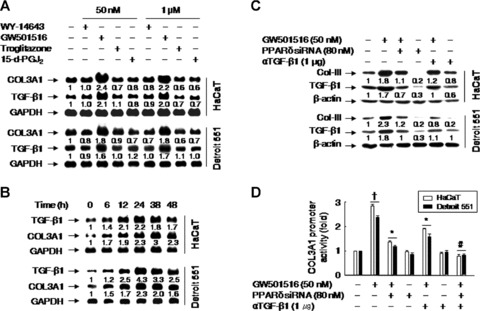
A ligand of PPARδ, but not of PPARα or γ, up-regulates type III collagen (COL3A1) in a TGF-β1-dependent or -independent manner. (A) Cells were incubated for 38 hrs in low (50 nM) or high (1 μM) concentrations of WY-14643 (a specific agonist of PPARα), GW501516 (a specific agonist of PPARγ), troglitazone (a specific agonist of PPARγ) or 15-deoxy-Delta 12,14-prostaglandin J2 (15-d-PGJ2; a specific agonist of PPARγ). (B) Cells were incubated with 50 nM GW501516 for the indicated times and levels of mRNAs were analyzed by Northern blot. (C) Cells were transfected with or without PPARδ siRNA, grown for 72 hrs, and then treated with GW501516 for 38 hrs in the presence or absence of the anti-TGF-β1 antibody. The protein levels of type III collagen (Col-III) and TGF-β1 were assessed by Western blot analysis using anti-type III collagen and anti-TGF-β1 antibodies. The band intensities were quantified using an image analyzer and fold changes relative to untreated cells are indicated below each band. The data represent the means of three independent experiments. (D) Cells pretreated for 72 hrs with or without PPARδ siRNA were transfected with a luciferase reporter plasmid (1 μg) driven by the COL3A1 promoter (containing 1685 nt upstream of the COL3A1) and with the pSV β-Gal (0.5 μg) plasmid. Transfectants were grown for 24 hrs and then treated with GW501516 for 38 hrs in the presence or absence of anti-TGF-β1 antibody. Luciferase activity was normalized to β-galactosidase activity; data represent the means ± SE of 4 to 6 independent transfections. †P < 0.01, compared with the untreated group; *P < 0.01, compared with the GW501516-treated group; #P < 0.01, compared with the GW501516 + PPARδ siRNA- or αTGF-β1-treated groups.
TGF-β1 mediates the up-regulation of type III collagen expression by PPARδ
As the transcriptional regulation of type III collagen is relatively poorly characterized when compared with type I collagen [23], we examined the PPARδ-mediated regulation of type III collagen expression in more detail. To explore whether de novo protein or RNA synthesis is required for GW501516-induced expression of type III collagen, we examined the effects of actinomycin D and cycloheximide on the GW501516-induced increase in type III collagen mRNA levels. The induction of type III collagen mRNA by the PPARδ activator was markedly reduced in the presence of actinomycin D or cycloheximide (Fig. S2), indicating that de novo synthesis of mRNA and protein(s) that act on the type III collagen gene promoter is required for the GW501516-induced expression of type III collagen.
To further characterize the role of PPARδ in the GW50156-induced up-regulation of type III collagen, cells were transfected with an siRNA against PPARδ. The down-regulation of PPARδ expression by siRNA almost completely reversed the expression pattern of type III collagen and TGF-β1 proteins induced by the PPARδ ligand. Similarly, the addition of the anti-TGF-β1 antibody suppressed the induction of GW501516-mediated type III collagen (Fig. 5C). These results suggest that TGF-β1 is involved in the up-regulation of type III collagen by GW501516.
To determine whether PPARδ activation induces the expression of type III collagen via TGF-β1 at the transcriptional level, transfection assays were performed using luciferase reporter constructs driven by the COL3A1 promoter. The activation of PPARδ by GW501516 significantly increased COL3A1 promoter activity, which was consistent with the observed increase in type III collagen protein levels. However, while the increased promoter activity induced by GW501516 was almost completely suppressed by cotransfection with PPARδ siRNA, treatment with the anti-TGF-β1 antibody partially suppressed COL3A1 promoter activity. Combined treatment with PPARδ siRNA and the anti-TGF-β1 antibody completely abolished the COL3A1 promoter activity induced by GW501516 (Fig. 5D). These observations support the hypothesis that both PPARδ and TGF-β1 are involved in the regulation of type III collagen expression.
Smad3 and an SBE mediated the PPARδ-induced increase in COL3A1 promoter activity
To elucidate the mechanism involved in the TGF-β1-mediated increase of type III collagen level by PPARδ, we carried out a cotransfection assay using the COL3A1–luciferase reporter gene constructs and expression vectors for several different Smad isoforms (2, 3, 4 and 7), Sp-1 or c-Jun/c-Fos, which are known to mediate TGF-β signalling [24]. As shown in Fig. 6A, coexpression of Smad3 alone increased COL3A1 promoter activity, which was enhanced in the presence of GW501516. The effect of Smad3 was significantly attenuated by a dominant-negative form of Smad3 (SmadΔ3) in the presence or absence of GW501516 (Fig. 6B). Next, we investigated the effect of PPARδ activation on the activity of a promoter bearing a tandem array of six copies of the SBE consensus motif, which was placed upstream of the luciferase coding region (p6 × SBEwt/Luc; 25). As shown in Fig. 6C, there was a significant increase in SBE-driven luciferase activity in cells treated with GW501516 for 38 hrs. Of note, treatment of cells with either siRNA against PPARδ, anti-TGF-β1 antibody or TβR-I inhibitor attenuated the induction of the SBE-driven luciferase expression by GW501516. A reporter construct containing specific mutations in the SBE motifs (p6 × SBEmut/Luc; [25]) was unresponsive to GW501516. These results were in agreement with the phosphorylation of Smad3 in response to PPARδ activation and suggest that the PPARδ-dependent increase in COL3A1 promoter activity is brought about by a cis-activation mediated by the binding of Smad3 to SBE (Fig. 6C and D).
Fig 6.
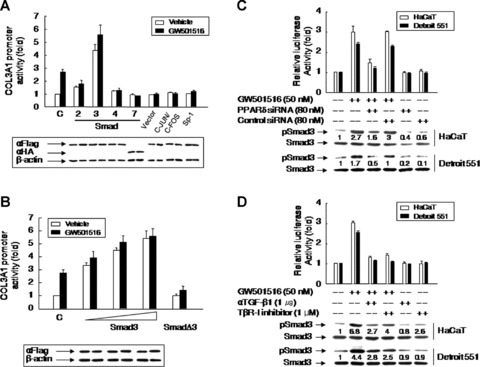
Smad3 is a key regulator of the up-regulation of PPARγ-induced COL3A1 promoter activity. (A and B) The COL3A1 promoter construct (–1685/+68; 1 μg) and the pSV β-Gal construct (0.5 μg) were cotransfected into HaCaT cells with the various expression plasmids indicated (1 μg) (A), or expression plasmids for Smad3 (0.1, 0.5 and 1 μg) or SmadΔ3 (1 μg) (B). The integrity and expression levels of the transfected genes were verified by Western blot analysis using anti-Flag (αFlag) and anti-haemagglutinin (αHA) antibodies to detect the indicated Flag- or HA-tagged proteins (A and B, lower panel). (C and D) A luciferase reporter construct (0.5 μg) containing six copies of wild-type (p6 × SBEwt/Luc) or mutant (p6 × SBEmt/Luc) SBE was transfected into cells pretreated for 72 hrs with PPARδ siRNA or control siRNA (C), or in the presence or absence of anti-TGF-β1 antibody or a TβR-1 inhibitor (D). After incubation for 24 hrs, cells were stimulated with 50 nM GW501516 for 38 hrs. Western blot analysis was performed using anti-Smad3 and anti-phospoSmad3 (phosphorylated form of Smad3) antibodies. Fold changes relative to untreated cells are indicated below each band. Luciferase activities were measured and data from 4 to 6 independent transfections normalized to β-galactosidase activity are expressed as means ± SE.
Both PPARδ and TGF-β1 are involved in the transcriptional regulation of type III collagen expression
To identify the promoter region responsible for the PPARδ-induced up-regulation of type III collagen, reporter assays were performed using a set of truncated constructs driven by the COL3A1 promoter (Fig. 7A). The response to GW501516 was markedly reduced after deletion of sequences between nt –1494 and –1030 (relative to the transcriptional start site at +1), or deletion of sequences up to nt –225 of the COL3A1 promoter (Fig. 7A). The anti-TGF-β1 antibody had no effect on the activity of these two constructs. Thus, the sequence region between nt –1494 and –1030 appears to be essential for the TGF-β1-mediated increase in COL3A1 promoter activity. Further truncation of the promoter up to nt –115 almost completely abolished its response to GW501516, which seems to indicate that this region is responsible for the increased promoter activity observed in response to GW501516.
Fig 7.
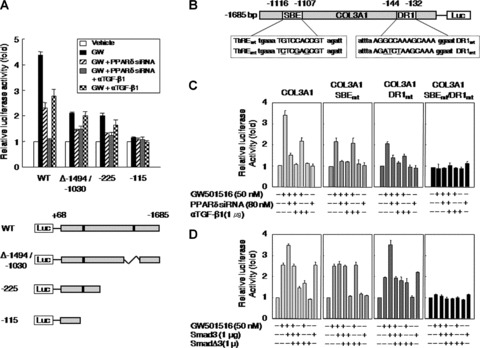
PPARδ and Smad3 increase COL3A1 promoter activity through PPRE and SBE, respectively. (A) HaCaT cells pretreated for 72 hrs with or without PPARδ siRNA were transfected with 1 μg of the deleted or truncated COL3A1 promoter constructs and 0.5 μg of the pSV β-Gal plasmid. Transfectants were grown for 24 hrs and then treated with GW501516 for 38 hrs in the presence or absence of anti-TGF-β1 antibody. (B) Schematic representation of the COL3A1 promoter. The putative response elements for Smad3 and PPARδ were mutated by site-directed mutagenesis (underlined). (C) HaCaT cells pretreated for 72 hrs in the presence or absence of PPARδ siRNA were transfected with the indicated luciferase reporter plasmids (1 μg) driven by the COL3A1 promoter, and with the pSV β-Gal construct (0.5 μg). Transfectants were grown for 24 hrs and then treated with 50 nM GW501516 for 38 hrs in the presence or absence of anti-TGF-β1 antibody (1 μg). (D) HaCaT cells were cotransfected with the indicated COL3A1-luciferase reporter constructs (1 μg) and the pSV β-Gal construct (0.5 μg), together with an expression plasmid for Smad3 (1 μg) or for SmadΔ3 (1 μg) for 24 hrs. These cells were subsequently incubated with 50 nM GW501516 for 38 hrs. Luciferase activity was normalized to β-galactosidase activity, and data (mean ± SE) from 4 to 6 independent transfections are expressed as fold induction.
PPARδ and Smad3 increase COL3A1 promoter activity through sites that contain a PPRE and an SBE
The results of our analysis of the COL3A1 promoter led us to speculate that the promoter elements responsible for mediating the effects of TGF-β1 and PPARδ were located between nt –1494 and –1030 and –225 and –115, respectively. We searched the sequence database (TFSEARCH) and identified a putative SBE at nt –1116 to –1107, and a putative PPRE containing a DR1 sequence at nt -144 to -132 of the COL3A1 promoter (Fig. 7B).
To determine whether the putative SBE and PPRE in the COL3A1 promoter were involved in PPARδ-mediated transcriptional activation, we introduced mutations into the SBE and DR1 site of the full-length COL3A1 promoter (1.68 kb). In the presence of the wild-type COL3A1 promoter, the activation of PPARδ by GW501516 resulted in a 3.5-fold increase in transcriptional activity. This effect was markedly attenuated in the presence of an siRNA against PPARδ, but was only partially affected by the anti-TGF-β1 antibody. Mutation in the putative SBE (COL3A1-SBEmt) resulted in a partial loss of promoter activity in response to GW501516. COL3A1-SBEmt activity was completely lost in the presence of an siRNA against PPARδ, whereas it was unaffected by the anti-TGF-β1 antibody. We next introduced mutations into the putative DR1 site of the COL3A1 promoter (COL3A1-DR1mt). Surprisingly, while the response of COL3A1-DR1mt to GW501516 was similar to that of COL3A1-SBEmt, transcriptional activity was almost completely lost in the presence of the anti-TGF-β1 antibody. These results indicate that the COL3A1 promoter is subject to dual regulation by PPARδ and TGF-β1, such that PPARδ binds directly to a DR1-type PPRE in the COL3A1 promoter, and also induces the expression of TGF-β1 to elicit full activation of COL3A1 expression. In support of this mechanism, simultaneous double mutation of the DR1 site and the SBE completely abolished the promoter activity of COL3A1 in response to GW501516 (Fig. 7C).
To gain further insight into the effector molecules involved in the TGF-β1-mediated induction of gene expression, we performed a cotransfection assay using wild-type and mutant COL3A1 promoter constructs, and expression vectors for wild-type and dominant-negative Smad3. The promoter activity of wild-type COL3A1 was dependent on Smad3, whereas COL3A1-SBEmt activity was unresponsive to the overexpression of Smad3 and/or SmadΔ3. The overexpression of Smad3 increased the promoter activity of COL3A1-DR1mt, which was suppressed by SmadΔ3. This indicates that Smad3 seems to play a primary role in the PPARδ/TGF-β1-mediated induction of COL3A1 promoter activity (Fig. 7D).
PPARδ and Smad3 bind to the PPRE and SBE of COL3A1 promoter
To determine whether Smad3 and PPARδ interact directly with the COL3A1 promoter to elicit transcriptional up-regulation, we carried out a ChIP assay. Cells were treated with GW501516 for various time periods and chromatin fragments were subjected to immunoprecipitation using anti-Smad3 or anti-PPARδ antibodies. Genomic DNA from the immunoprecipitates was amplified by PCR using primers that corresponded to the putative SBE or DR1 site. As shown in Fig. 8, PCR-amplified fragments were obtained from cells treated with GW501516, but not from untreated cells. The specificity of the assay was confirmed using a set of primers that amplified a non-specific region of the promoter (oligo #3). These results were consistent with the results of promoter assay and suggest that Smad3 and PPARδ bind to the SBE and DR1 site of the COL3A1 promoter, respectively.
Fig 8.
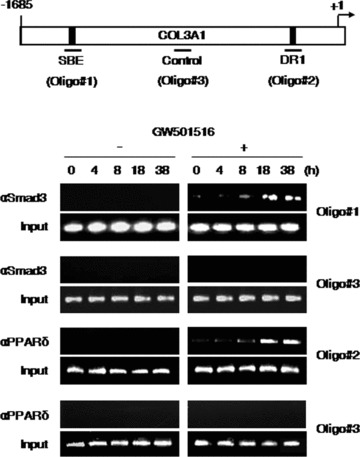
Smad3 and PPARγ associate physically with the SBE and DR1 site of the COL3A1 promoter, respectively. HaCaT cells were grown for the indicated times in the presence or absence of GW501516 (lane 0, untreated). Chromatin was subjected to immunoprecipitation using either anti-Smad3 or anti-PPARγ antibodies and the recovered genomic DNA was amplified by PCR using primers specific for the regions corresponding to oligo #1, oligo #2 or oligo #3 (indicated in the upper panel, which also depicts the location of the Smad3-binding and PPARγ-binding sites). Control amplifications were carried out using input chromatin obtained before immunoprecipitation. The results are representative of three independent experiments.
Discussion
A critical role for the nuclear receptor PPARδ in the process of tissue injury and wound repair has been suggested [7, 12]. While multiple factors associated with the activities of PPARδ participate in wound healing processes, little is known about the specific effector genes that transmit signals derived from the activation of PPARδ. In the present study, we demonstrated that the activation of PPARδ by a specific ligand, GW501516, induced the expression of collagen types I and III collagen and fibronectin, which are important factors for the process of wound healing [5]. The induction of these genes was, in part, mediated by TGF-β1, which is a master cytokine involved in the regulation of ECM homeostasis in the skin [4]. The effect of PPARδ on the expression of type III collagen was mediated by a dual mechanism involving the direct binding of PPARδ to a DR1-type PPRE in the COL3A1 promoter and the TGF-β1-mediated binding of Smad3 to an SBE in the COL3A1 promoter. Analysis of point mutations of the COL3A1 promoter demonstrated that the DR1 site and SBE are indispensable for the up-regulation of type III collagen by PPARδ. Furthermore, we showed that the activation of PPARδ promoted wound closure and was accompanied by the up-regulation of collagen types I and III, α-SMA, pSmad3 and TGF-β1 in the incisional wound sites, which seems to indicate that PPARδ-induced collagen expression via TGF-β1 had beneficial effects on wound repair.
Direct administration of GW501516 to wounded mice accelerated the wound repair process by enhancing expression of TGF-β1, collagen types I and III, α-SMA and pSmad3 at the wound sites. Although it is known that PPARδ protects keratinocytes against apoptosis and contributes to the process of skin wound closure [13, 14], our results suggest the possibility that PPARδ is also involved in wound healing processes through the regulation of the expression of ECM components, including collagens and α-SMA, to promote wound closure. In turn, TGF-β1, which is synthesized and secreted by PPARδ at the wound sites, promotes not only such cellular events as anti-inflammatory and anti-apoptotic actions [6], but also exerts pleiotropic effects in the wound healing process, including the stimulation of ECM production. Accordingly, TGF-β1-mediated up-regulation of collagen and ECM components, which has been demonstrated in the incisional wound model [4], is a key event in the evolution of wounds prone to healing. In this regard, the PPARδ-mediated induction of ECM proteins via the TGF-β1/Smad3 signalling-dependent or -independent pathway plays an important role in the regulation of wound healing. Accordingly, specific ligands for PPARδ are promising as future therapeutic interventions for wound healing.
The ligand-activated PPARδ increased the migration of human epidermal keratinocytes and dermal fibroblasts in an in vitro scrape wound assay. This observation was consistent with previous reports showing PPARδ-dependent promotion of cell migration in keratinocytes [26]. Most notably, the expression of ECM-related genes in human keratinocytes and fibroblasts was regulated by the expression of PPARδ-mediated TGF-β1. These results are consistent with the concept that both TGF-β1 and ECM components within the microenvironment of the wound stimulate migration of human keratinocytes [27, 28]. In addition to the migration of cells surrounding the edge of wounds, intercellular interactions are also essential to wound healing in which ECM molecules connect the neighbouring cells [5, 21, 29]. Thus, PPARδ-mediated induction of ECM-related genes, including collagen, may contribute to the cutaneous wound healing process, not only by accelerating the migration of keratinocytes but also by promoting intracellular interactions at the wound’s edge.
The TGF-β1-dependent activation of PPARδ, but not of PPARα or PPARγ, specifically induced the expression of collagen types I and III and fibronectin in human keratinocytes and skin fibroblasts. In a previous study, we showed that PPARδ induces the expression of TGF-β1 by binding to the DR1-type PPRE located on the promoter region of TGF-β1 gene [16]. Although the roles of all members of the PPAR family in wound healing processes have been well characterized using genetically modified mice [12], in the present study only PPARδ induced both TGF-β1 and ECM-related genes. Given that the interaction of PPARδ and TGF-β1 in mouse wounding models accelerates wound closure and induces the PPARδ expression in limited microenvironments [30], PPARγ- and/or TGF-β1-stimulated ECM biosynthesis may contribute to the promotion of wound healing. Therefore, it is possible to promote wound healing by increasing ECM components, including collagens, through the PPARδ-induced expression of TGF-β1 at the edge of wounds.
Two cis-acting elements identified in the COL3A1 promoter were responsible for the transcriptional up-regulation mediated by PPARδ activation. Other reports have demonstrated that multiple proteins, including lysyl oxidase, are involved in the formation of protein–DNA complexes within the COL3A1 proximal promoter region [23, 31]. In the present study, activation of PPARγ regulated the expression of COL3A1 at the transcriptional level through the direct binding of PPARγ to a DR1 site in the COL3A1 promoter, as well as via the direct interaction of Smad3 with an SBE by the induction of TGF-β1. The PPRE identified in this study contained a canonical DR1 motif separated by an adenine residue. The SBE was identified as an additional cis-acting element responsible for the PPARδ-induced transcriptional activation via TGF-β1, and Smad3 is suggested to incorporate into a TGF-β1-inducible complex formed after PPARγ stimulation. Thus, we have identified two sites, a DR1-type PPRE and an SBE in the COL3A1 promoter, that function as cis elements responsible for the PPARγ-induced activation of COL3A1 expression. These results establish a novel link between PPARδ-mediated collagen production and TGF-β1/Smad3 signalling.
Our findings suggest that PPARδ plays an important role in skin wound healing in vivo by accelerating ECM and/or α-SMA-mediated cellular interactions in a process mediated by the TGF-β1/Smad3 signalling-dependent or independent pathway. These results have important implications for the understanding of the molecular mechanisms underlying the wound healing that involve the PPARδ-mediated transcriptional regulation of ECM genes. Identification of PPARδ as an inducer of ECM protein expression may provide a therapeutically relevant key concept in the wound healing processes.
Acknowledgments
This work was supported in part by a grant of the Korea Healthcare technology R&D Project, Ministry of Health & Welfare (A080433), and by Korea Science and Engineering Foundation (KOSEF) grants funded by the Korea government (R13–2005-012–02001-0), Republic of Korea.
Supporting Information
Fig. S1 Effects of a small interfering (si)RNA on theexpression of PPARγ. Cells transfected with PPARγ siRNAor control siRNA for 72 hrs were incubated in the presence orabsence of GW501516 for 38 hrs. The levels of PPARγ weremarkedly reduced upon transfection with PPARγ siRNA, whereascontrol siRNA, consisting of a pool of nonspecific sequences, hadno effect on PPARγ levels. Fold changes are indicated below each band.
Fig. S2 Effects of actinomycin D and cycloheximide on GW501516-induced COL3A1 expression. Cells were incubated for 38 hrs with GW501516 in the presence or absence of cycloheximide or actinomycin. Total RNA was extracted and the levels of type III collagen mRNA (COL3A1) were assessed by Northern analysis using a cDNA probe for type III collagen. Fold changes are indicated below each band.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Santoro MM, Gaudino G. Cellular and molecular facets of keratinocyte reepithelization during wound healing. Exp Cell Res. 2005;304:274–86. doi: 10.1016/j.yexcr.2004.10.033. [DOI] [PubMed] [Google Scholar]
- 2.Verrecchia F, Mauviel A. Transforming growth factor-beta signaling through the Smad pathway: role in extracellular matrix gene expression and regulation. J Invest Dermatol. 2002;118:211–5. doi: 10.1046/j.1523-1747.2002.01641.x. [DOI] [PubMed] [Google Scholar]
- 3.Massague J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 2000;19:1745–54. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quaglino DJ, Nanney LB, Ditesheim JA, et al. Transforming growth factor-beta stimulates wound healing and modulates extracellular matrix gene expression in pig skin: incisional wound model. J Invest Dermatol. 1991;97:34–42. [PubMed] [Google Scholar]
- 5.Mutsaers SE, Bishop JE, McGrouther G, et al. Mechanisms of tissue repair: from wound healing to fibrosis. Int J Biochem Cell Biol. 1997;29:5–17. doi: 10.1016/s1357-2725(96)00115-x. [DOI] [PubMed] [Google Scholar]
- 6.O’Kane S, Ferguson MW. Transforming growth factor beta s and wound healing. Int J Biochem Cell Biol. 1997;29:63–78. doi: 10.1016/s1357-2725(96)00120-3. [DOI] [PubMed] [Google Scholar]
- 7.Tan NS, Michalik L, Di-Poï N, et al. Essential role of Smad3 in the inhibition of inflammation-induced PPARbeta/delta expression. EMBO J. 2004;23:4211–21. doi: 10.1038/sj.emboj.7600437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–50. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- 9.Kersten S, Desvergne B, Wahli W. Roles of PPARs in health and disease. Nature. 2000;405:421–4. doi: 10.1038/35013000. [DOI] [PubMed] [Google Scholar]
- 10.Tugwood JD, Issemann I, Anderson RG, et al. The mouse peroxisome proliferator activated receptor recognizes a response element in the 5’ flanking sequence of the rat acyl CoA oxidase gene. EMBO J. 1992;11:433–9. doi: 10.1002/j.1460-2075.1992.tb05072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649–88. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 12.Michalik L, Desvergne B, Tan NS, et al. Impaired skin wound healing in peroxisome proliferator-activated receptor (PPAR)alpha and PPARbeta mutant mice. J Cell Biol. 2001;154:799–814. doi: 10.1083/jcb.200011148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan NS, Michalik L, Noy N, et al. Critical roles of PPAR beta/delta in keratinocyte response to inflammation. Genes Dev. 2001;15:3263–77. doi: 10.1101/gad.207501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Di-Poï N, Tan NS, Michalik L, et al. Antiapoptotic role of PPARbeta in keratinocytes via transcriptional control of the Akt1 signaling pathway. Mol Cell. 2002;10:721–33. doi: 10.1016/s1097-2765(02)00646-9. [DOI] [PubMed] [Google Scholar]
- 15.Michalik L, Wahli W. Involvement of PPAR nuclear receptors in tissue injury and wound repair. J Clin Invest. 2006;116:598–606. doi: 10.1172/JCI27958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim HJ, Ham SA, Kim SU, et al. Transforming growth factor-beta1 is a molecular target for the peroxisome proliferator-activated receptor delta. Circ Res. 2008;102:193–200. doi: 10.1161/CIRCRESAHA.107.158477. [DOI] [PubMed] [Google Scholar]
- 17.Chang KC, Paek KS, Kim HJ, et al. Substrate induced up-regulation of aldose reductase by methylglyoxal, a reactive oxoaldehyde elevated in diabetes. Mol Pharmacol. 2002;61:1184–91. doi: 10.1124/mol.61.5.1184. [DOI] [PubMed] [Google Scholar]
- 18.Kang ES, Kim HJ, Paek KS, et al. Phorbol ester up-regulates aldose reductase expression in A549 cells: a potential role for aldose reductase in cell cycle modulation. Cell Mol Life Sci. 2005;62:1146–55. doi: 10.1007/s00018-005-5024-4. [DOI] [PubMed] [Google Scholar]
- 19.Oliver WR, Jr, Shenk JL, Snaith MR, et al. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc Natl Acad Sci. 2001;98:5306–11. doi: 10.1073/pnas.091021198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roberts AB, Heine UI, Flanders KC, et al. Transforming growth factor-beta. Major role in regulation of extracellular matrix. Ann N Y Acad Sci. 1990;580:225–32. doi: 10.1111/j.1749-6632.1990.tb17931.x. [DOI] [PubMed] [Google Scholar]
- 21.Broughton G, 2nd, Janis JE, Attinger CE. The basic science of wound healing. Plast Reconstr Surg. 2006;117:12S–34S. doi: 10.1097/01.prs.0000225430.42531.c2. [DOI] [PubMed] [Google Scholar]
- 22.Smola H, Thiekötter G, Fusenig NE. Mutual induction of growth factor gene expression by epidermal-dermal cell interaction. J Cell Biol. 1993;122:417–29. doi: 10.1083/jcb.122.2.417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yoshino T, Sumiyoshi H, Shin T, et al. Multiple proteins are involved in the protein-DNA complex in the proximal promoter of the human alpha1(III) collagen gene (COL3A1) Biochim Biophys Acta. 2005;1729:94–104. doi: 10.1016/j.bbaexp.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 24.Greenwel P, Inagaki Y, Hu W, et al. Sp1 is required for the early response of alpha2(I) collagen to transforming growth factor-beta1. J Biol Chem. 1997;272:19738–45. doi: 10.1074/jbc.272.32.19738. [DOI] [PubMed] [Google Scholar]
- 25.Qiu P, Ritchie RP, Fu Z, et al. Myocardin enhances Smad3-mediated transforming growth factor-beta1 signaling in a CArG box-independent manner: Smad-binding element is an important cis element for SM22alpha transcription in vivo. Circ Res. 2005;97:983–91. doi: 10.1161/01.RES.0000190604.90049.71. [DOI] [PubMed] [Google Scholar]
- 26.Tan NS, Icre G, Montagner A, et al. The nuclear hormone receptor peroxisome proliferator-activated receptor beta/delta potentiates cell chemotactism, polarization, and migration. Mol Cell Biol. 2007;27:7161–75. doi: 10.1128/MCB.00436-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sarret Y, Woodley DT, Grigsby K, et al. Human keratinocyte locomotion: the effect of selected cytokines. J Invest Dermatol. 1992;98:12–6. doi: 10.1111/1523-1747.ep12493517. [DOI] [PubMed] [Google Scholar]
- 28.Woodley DT. Re-epithelialization. In: Clark RAF, editor. The Molecular and Cellular Biology of Wound Repair. 2nd ed. New York: Plenum Publishing; 1996. pp. 339–50. [Google Scholar]
- 29.Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med. 1999;341:738–46. doi: 10.1056/NEJM199909023411006. [DOI] [PubMed] [Google Scholar]
- 30.Tan NS, Michalik L, Desvergne B, et al. Genetic- or transforming growth factor-beta 1-induced changes in epidermal peroxisome proliferator-activated receptor beta/delta expression dictate wound repair kinetics. J Biol Chem. 2005;280:18163–70. doi: 10.1074/jbc.M412829200. [DOI] [PubMed] [Google Scholar]
- 31.Giampuzzi M, Botti G, Di Duca M, et al. Lysyl oxidase activates the transcription activity of human collagene III promoter. Possible involvement of Ku antigen. J Biol Chem. 2000;275:36341–9. doi: 10.1074/jbc.M003362200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Effects of a small interfering (si)RNA on theexpression of PPARγ. Cells transfected with PPARγ siRNAor control siRNA for 72 hrs were incubated in the presence orabsence of GW501516 for 38 hrs. The levels of PPARγ weremarkedly reduced upon transfection with PPARγ siRNA, whereascontrol siRNA, consisting of a pool of nonspecific sequences, hadno effect on PPARγ levels. Fold changes are indicated below each band.
Fig. S2 Effects of actinomycin D and cycloheximide on GW501516-induced COL3A1 expression. Cells were incubated for 38 hrs with GW501516 in the presence or absence of cycloheximide or actinomycin. Total RNA was extracted and the levels of type III collagen mRNA (COL3A1) were assessed by Northern analysis using a cDNA probe for type III collagen. Fold changes are indicated below each band.