

Abstract
By employing a pharmacological approach, we have shown that phospholipase C (PLC) activity is involved in the regulation of gene expression of transcription factors such as c-Fos and c-Jun in cardiomyocytes in response to norepinephrine (NE). However, there is no information available regarding the identity of specific PLC isozymes involved in the regulation of c-Fos and c-Jun or on the involvement of these transcription factors in PLC isozyme gene expression in adult cardiomyocytes. In this study, transfection of cardiomyocytes with PLC isozyme specific siRNA was found to prevent the NE-mediated increases in the corresponding PLC isozyme gene expression, protein content and activity. Unlike PLC γ1 gene, silencing of PLC β1, β3 and δ1 genes with si RNA prevented the increases in c-Fos and c-Jun gene expression in response to NE. On the other hand, transfection with c-Jun si RNA suppressed the NE-induced increase in c-Jun as well as PLC β1, β3 and δ1 gene expression, but had no effect on PLC γ1 gene expression. Although transfection of cardiomyocytes with c-Fos si RNA prevented NE-induced expression of c-Fos, PLC β1 and PLC β3 genes, it did not affect the increases in PLC δ1 and PLC γ1 gene expression. Silencing of either c-Fos or c-Jun also depressed the NE-mediated increases in PLC β1, β3 and γ1 protein content and activity in an isozyme specific manner. Furthermore, silencing of all PLC isozymes as well as of c-Fos and c-Jun resulted in prevention of the NE-mediated increase in atrial natriuretic factor gene expression. These findings, by employing gene silencing techniques, demonstrate that there occurs a reciprocal regulation of transcription factors and specific PLC isozyme gene expression in cardiomyocytes.
Keywords: adult rat cardiomyocytes, c-Fos, c-Jun, phospholipase C isozymes, gene expression, gene silencing, hypertrophic response
Introduction
Hypertrophic growth is an adaptive response of the heart to diverse pathological stimuli and is characterized by cardiomyocyte enlargement, protein synthesis, and activation of a foetal programme of cardiac gene expression [1–4]. Cardiomyocyte enlargement is also known to occur without upregulation of foetal genes during, for example, exercise- and thyroid-induced cardiac hypertrophy [5, 6]. The development of the hypertrophic phenotype is manifested by a coordinated series of events, where several transcription factors are activated in response to different hypertrophic agents [7–9]. Cardiac hypertrophy is also associated with hyperactivity of the sympathetic nervous system as well as elevated levels of plasma norepinephrine (NE) [10]. In fact, NE is known to produce cardiac hypertrophy [11–13], which may, in part, be mediated via the activation of α1-adrenoceptors (α1-AR) [14–17]. Furthermore, an early transient increase in the transcription factors such as c-Fos and c-Jun has been observed in response to sympathetic stimulation [18–20]. Mechanical overload has also been shown to increase c-Jun expression transiently [21]. Transfection of cardiac myocytes with a dominant negative c-Jun has been reported to inhibit cardiomyocyte hypertrophy in response to phenylephrine (PhE), an α1-AR agonist [22]. Moreover, activated c-Fos and c-Jun complexes are capable of inducing the hypertrophic phenotype, in cultured cardiomyocytes [23].
Earlier we have shown that the hypertrophic response of cardiomyocytes to NE, as demonstrated by an increase in atrial natriuretic factor (ANF) expression and protein synthesis, was due to activation of phospholipase C (PLC) via the α1-AR [15]. We have also reported that activity of PLC isozymes regulate their own gene expression [14], which might represent a cycle of events that perpetuates the hypertrophic response to NE. Furthermore, by using different pharmacological agents, we have demonstrated that c-Fos and c-Jun gene expression is regulated by PLC activity in adult cardiomyocytes through a protein kinase C (PKC)- and extracellular regulated kinases (ERK) 1/2-dependent pathway in response to (NE) [24]. However, the identity of the specific PLC isozymes involved in the regulation of c-Fos and c-Jun gene expression as well as the role of these transcription factors in the regulation of PLC isozyme gene expression in adult cardiomyocytes is not known. Therefore, by employing a gene silencing approach, this study was undertaken to determine which PLC isozyme regulates the expression of c-Fos and c-Jun and to determine if c-Fos and c-Jun are involved in the regulation of PLC isozyme gene expression in response to NE in adult cardiomyocytes.
Materials and methods
Cardiomyocyte isolation and stimulation
All experimental protocols for animal studies were approved by the Animal Care Committee of the University of Manitoba, in accordance with the guidelines established by the Canadian Council on Animal Care. Left ventricle (LV) cardiomyocytes were isolated from male Sprague-Dawley rats (250–300 g) as previously described [14, 15]. The cells were incubated with 5 μM NE for 2 hrs for gene expression; this concentration and time of incubation were previously determined to be optimal [14].
Silencing of c-Fos, c-Jun and PLC isozyme genes by small interfering (si) RNA
Pre-designed siRNAs were purchased from Ambion. The sequences of primers used to generate siRNA were as follows: c-Fos siRNA: 5′-CCUGUCUGGUUCCUUCUAUtt-3′ (sense), 5′-AUAGAAGGAACCAGACAGGtc-3′ (antisense); c-Jun siRNA: 5′-CGAUGGACUUUUCGUUAACtt-3′ (sense), 5′-GUUAACGAAAAGUCCAUCGtt-3′ (antisense); PLC β1 siRNA: 5′-CGAUGACUGUAAAAAGUCUtt-3′ (sense), 5′-AGACUUUUUACAGUCAUCGtc-3′ (antisense); PLC β3 siRNA: 5′-GCAGCUUAGUCGCAUCUACtt-3′ (sense), 5′-GUAGAUGCGACUAAGCUGCtg-3′ (antisense); PLC γ1 siRNA: 5′-GCCAUUAAUGAGAUGUACUtt-3′ (sense), 5′-AGUACAUCUCAUUAAUGGCtt-3′ (antisense) and PLC γ1 siRNA: 5′-GGAUAAACUAAAGCUGGUGtt-3′ (sense), 5′-CACCAGCUUUAGUUUAUCCtc-3′ (antisense). The negative control siRNA comprised of a 19bp scrambled sequence with 3′dt overhangs. The sequence had no homology to any known gene sequences. Optimal results were obtained by transfection of 5 nM siRNA and 5 nM non-silencer siRNA (scrambled sequence, negative control) with the Hi PerFect transfection reagent for 24 hrs, according to the manufacturer’s instructions (Qiagen). After 24 hr incubation, cells were incubated with 5 μM NE for 2 hrs for gene expression and 24 hrs for protein levels and activity measurements.
RNA isolation and semi-quantitative PCR
Total RNA was isolated from LV myocytes using RNA isolation kit (Life Technologies, ON, Canada), as described [14, 15]. Primers used for amplification were synthesized as follows: PLC β1: 5′-ATTCGGCCAGGCTATCACTA-3′ (forward), 5′-TGCATACGTGTCTGGGACAT-3′ (reverse); PLC β3: 5′-TTGGAAATCTTCGAGCGGTT-3′ (forward), 5′-AGGAACTGTTTGTTCGGCTCAT-3′ (reverse); PLC γ1: 5′-AGCCAAGGACCTGAAGAACA-3′ (forward), 5′-GCAAACTGCCCATAGGTGAT-3′ (reverse); PLC γ1: 5′-ACACAAGCCCAAGGAGGATA-3′ (forward), 5′-ACGGACAAAACCATTTCCTG-3′ (reverse); ANF: 5′-AGATCTGCCCTCTTGAAAAGCA-3′ (forward) and 5′-TCGAGCAGATTTGGCTGTTATC-3′ (reverse); c-Fos: 5′- GGAGCCGGTCAAGAACATTA-3′ (forward), 5′-ATGATGCCGGAAACAAGAAG-3′ (reverse); c-Jun: 5′-TGACTGCAAAGATGGAAACGA-3′ (forward), 5′-CAGGTTCAAGGTCATGCTCTGT-3′ (reverse). For the purpose of normalization of the data, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) primers: 5′-CATGACAACTTTGGCATCGT-3′ (forward) and 5′-GGATGCAGGGATGATGTTCT-3′ (reverse) were used to amplify GAPDH gene as a multiplex with the target genes. The PCR products were analysed by electrophoresis in 2% agarose gels. The intensity of each band was photographed and quantified using a Molecular Dynamics STORM scanning system (Amersham Biosciences Corp., PQ, Canada) as a ratio of a target gene over GAPDH.
Protein synthesis
Cardiomyocyte protein synthesis was assessed as incorporation of [3H] phenylalanine into cells as previously described [15]. Briefly, after a 10 min pre-incubation of PLC isozyme, c-Fos and c-Jun si RNA transfected cardiomyocytes with 0.5 μCi of [3H] phenylalanine, cells were treated with NE (5 μM) for 6 hrs. The reaction was terminated by addition of 5 ml ice-cold 10% w/v trichloroacetic acid (TCA). Cells were harvested and the suspension kept on ice for 20 min followed by centrifugation at 3838 ×g for 5 min at 4 °C. The cardiomyocyte pellet was washed with 1ml ice-cold 10% w/v TCA and centrifuged again at 3838 ×g for 5 min at 4 °C; this step was repeated once more. The precipitated protein was then suspended in 0.5 ml of 0.5 N NaOH and radioactivity in the neutralized aliquots was determined by liquid scintillation counting in 5 ml of CytoScint™, ICN.
Western blot analysis
Total membrane proteins (20 μg) and high molecular weight marker (Bio-Rad, Hercules, CA, USA) were separated on SDS-PAGE as described previously [14, 24–26]. Proteins were transferred onto 0.45-μm polyvinylidene difluoride (PVDF) membrane. PVDF membrane was blocked overnight at 4 °C in Tris-buffered saline (TBS) containing 5% skim milk and probed with monoclonal antibodies for PLC isozymes β1, γ1 and γ1 (Upstate, NY, USA) and polyclonal antibodies for PLC β3 (Santa Cruz Biotechnology, CA, USA). Primary antibodies were diluted in TBS with 0.1% (vol/vol) Tween 20 (TBS-T) (1:200 for PLC β1 and β3; 1:2000 for PLC γ1 and 1:10,000 for PLC γ1) according to the manufacturer’s instructions. Horseradish peroxidase-labelled antimouse IgG (Bio-Rad) was diluted 1:3000 in TBS-T and used as secondary antibody for PLC β1, γ1 and γ1 and 1:2000 in TBS-T for PLC β3 isozyme. Protein bands were visualized by enhanced chemiluminescence according to the manufacturer’s instructions (Boehringer Mannheim, Laval, PQ, Canada). Band intensities of the Western blot analysis were quantified using a CCD camera imaging densitometer (Bio-Rad GS 800).
Determination of PLC isozyme activities
The PLC isozyme activities were determined by measuring the hydrolysis of [3H]-PIP2, following immunoprecipitation (IP) as described previously [25, 26]. The IP was conducted overnight at 4 °C with monoclonal antibodies to PLC β1, and γ1, and polyclonal antibodies to β3 (5 μg of antibody to 350 μg membrane extract). For control experiments, IP and subsequent activity measurements were conducted with non-immune mouse IgG. It is pointed out that the IP of the specific PLC isozymes is complete under the condition described [25, 26].
Statistical analysis
All values are expressed as mean ± S.E. The differences between two groups were evaluated by Student’s t-test. The data from more than two groups were evaluated by one-way ANOVA followed by Duncan’s multiple comparison tests. A probability of 95% or more (P < 0.05) was considered significant.
Results
Prevention of the NE-induced increases in PLC isozyme gene expression, protein content and activity in cardiomyocytes transfected with PLC isozyme siRNA
Cardiomyocytes were transfected with specific PLC β1, β3, γ1 and δ1 si RNA in a dose dependent manner (1, 5, 10 and 25 nM). Our preliminary data showed that highly efficient transfection and silencing was obtained using a low concentration (5 nM) of siRNA, without resulting in any off target effects as demonstrated by the absence of mRNA degradation. In addition, cardiomyocytes remained viable and there was no loss of cardiomyocytes following transfection (data not shown). The prevention of NE-induced increases in PLC isozyme gene expression (Fig. 1), protein content and activity (Fig. 2) in PLC isozyme siRNA transfected cardiomyocytes indicated successful siRNA transfection.
Fig 1.
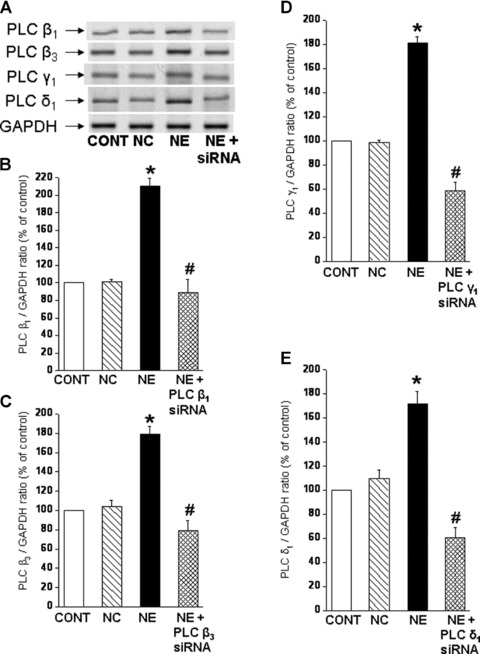
Prevention of the norepinephrine-induced increases in PLC isozyme mRNA levels by siRNA transfection of cardiomyocytes. Representative blots (A) showing PLC isozyme β1 (114bp), β3 (230bp), γ1 (123bp) and δ;1 (190bp) mRNA levels relative to GAPDH (138bp) mRNA level in LV cardiomyocytes. Quantified data shows PLC β1 (B), β3 (C), γ1 (D) and δ;1 (E) mRNA levels relative to GAPDH mRNA level in cardiomyocytes transfected with 5 nM PLC β1, β3, γ1 and γ1 siRNA and treated with NE (5 μM) for 2 hrs. Control cardiomyocytes were transfected with 5 nM negative control siRNA as described in ‘materials and methods’ section. Values are mean ± S.E. of five experiments performed with five different cardiomyocyte preparations. *Significantly different (P < 0.05) versus control; #significantly different (P < 0.05) versus NE. GAPDH, Glyceraldehyde 3-phosphate dehydrogenase; CONT, control; NC, negative control; NE, norepinephrine; siRNA, small interfering RNA.
Fig 2.
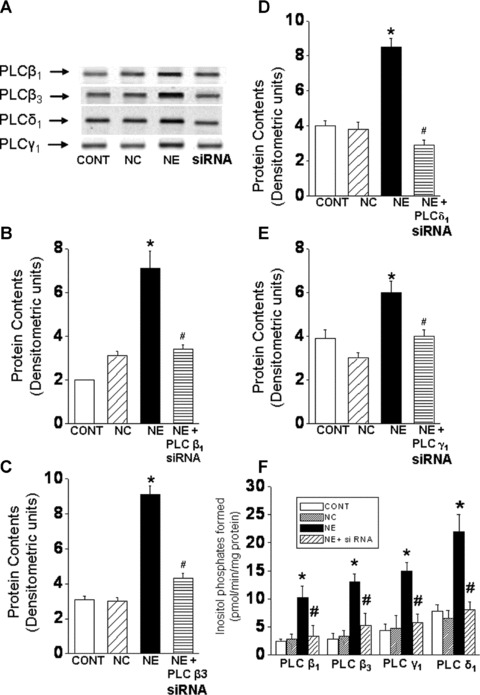
Prevention of the norepinephrine-induced increases in PLC isozyme protein content and activity in siRNA transfection of cardiomyocytes. Representative blots (A) showing PLC isozyme β1 (150 kD), β3 (150 kD), γ1 (145 kD) and δ1 (87 kD) protein levels in cardiomyocytes. Quantified data shows PLC β1 (B), β3 (C), δ1 (D) and γ1 (E) protein content in cardiomyocytes transfected with 5 nM PLC β1, β3, γ1 and δ1 siRNA and treated with NE (5 μM) for 24 hrs. The PLC isozyme activities (F) were determined in siRNA transfected cardiomyocytes following immunoprecipitation and hydrolysis of 3[H]-PIP2. preparations. Control cardiomyocytes were transfected with 5 nM negative control siRNA as described in ‘materials and methods’ section. Values are mean ± S.E. of five experiments performed with five different cardiomyocyte preparations. *Significantly different (P < 0.05) versus control; #significantly different (P < 0.05) versus. NE. GAPDH, Glyceraldehyde 3-phosphate dehydrogenase; CONT, control; NC, negative control; NE, norepinephrine; siRNA, small interfering RNA.
Regulation of c-Fos and c-Jun gene expression in response to NE in cardiomyocytes transfected with PLC isozyme siRNA
To examine the involvement of specific PLC isozymes in the NE-induced increases in c-Fos, c-Jun gene expression, cardiomyocytes were genetically manipulated to silence PLC isozyme genes with si RNA. Transfection of cardiomyocytes with PLC β1, β3 and δ1 si RNA inhibited the NE-induced increases in c-Fos and c-Jun gene expression, whereas PLC γ1 gene silencing did not affect the activation of c-Fos and c-Jun gene expression in response to NE (Fig. 3).
Fig 3.
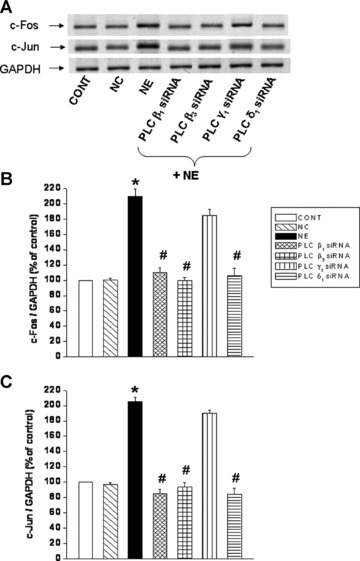
Inhibition of norepinephrine-induced increases in c-Fos and c-Jun mRNA levels in cardiomyocytes transfected with PLC isozymes siRNA. Representative blots (A) and quantified data (B) and (C) showing c-Fos (74 bp) and c-Jun (163 bp) mRNA levels respectively relative to GAPDH (138 bp) mRNA level in LV cardiomyocytes transfected with 5 nM PLC isozyme β1, β3, γ1 and δ;1 siRNA and treated with NE (5 μM) for 2 hrs. Control cardiomyocytes were transfected with 5 nM negative control siRNA as described in ‘Materials and methods’ section. Values are mean ± S.E. of five experiments performed with five different cardiomyocyte preparations. *Significantly different (P < 0.05) versus control; #significantly different (P < 0.05) versus NE. GAPDH, Glyceraldehyde 3-phosphate dehydrogenase; CONT, control; NC, negative control; NE, norepinephrine; siRNA, small interfering RNA.
Regulation of PLC isozyme gene expression, protein content and activity by c-Fos and c-Jun siRNA
While a pharmacological approach was previously used to examine the role of c-Fos and c-Jun in regulating PLC isozyme gene expression in response to NE [24], the involvement of transcription factors, c-Fos and c-Jun was further examined in cardiomyocytes genetically manipulated to silence c-Fos and c-Jun gene. Again, transfection of cardiomyocytes with c-Fos and c-Jun siRNA revealed that a highly efficient transfection and silencing was obtained using 5 nM siRNA without resulting in any off target effects and loss of cardiomyocytes following transfection (data not shown). The prevention of the NE-induced increases in c-Fos and c-Jun gene expression in siRNA treated cells also indicated successful siRNA transfection (Fig. 4A and B). It should be noted that a significant basal expression of c-fos and c-Jun in the isolated cardiomyocytes was observed (Fig. 4C). We consider this not to be an artifact due to the isolation procedure as it was determined that there is a substantial expression of c-Fos and c-Jun in control LV tissue. Indeed the amounts of c-Fos and c-Jun mRNA (as demonstrated by conventional-PCR) in the isolated control cardiomyocyte preparations were 75% and 78%, respectively, of that expressed in the LV tissue (Fig. 4C). Transfection of a cardiomyocytes with c-Fos si RNA inhibited the NE-induced increase in PLC β1 and β3 isozyme gene expression, but did not affect the increase in PLC γ1 and δ1 isozyme gene expression in response to NE (Fig. 5). On the other hand, transfection of cardiomyocytes with c-Jun siRNA prevented the NE-induced increase in PLC β1, β3 and δ1 isozyme whereas the increase in PLC γ1 isozyme mRNA level due to NE was unaffected (Fig. 5). These results suggest that both c-Fos and c-Jun are the transcription factors involved in the regulation of PLC β1 and β3 gene expression in response to NE, and that c-Jun specifically may also be involved in the regulation of PLC δ1 isozyme gene expression. Furthermore, it was revealed that the attenuation of the PLC β1, β3 and γ1 gene expression in response to NE in c-Fos and c-Jun si RNA transfected cardiomyocytes was also associated with an attenuation of the protein content (Fig. 6) as well as activity of these PLC isozymes (Fig. 7).
Fig 4.
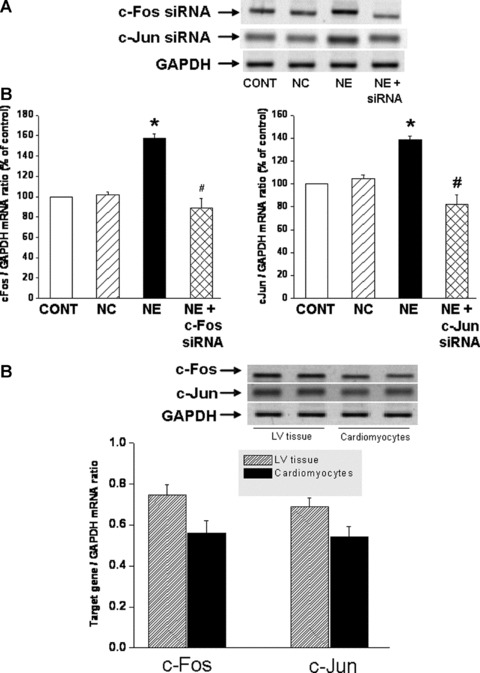
Prevention of the norepinephrine-induced increases in c-Fos and c-Jun mRNA levels by siRNA transfection of cardiomyocytes. Representative blots (A) and quantified data showing c-Fos (74 bp) and c-Jun (163 bp) mRNA levels relative to GAPDH (138 bp) mRNA level in cardiomyocytes transfected with 5 nM c-Fos or c-Jun siRNA and treated with NE (5 μM) for 2 hrs. Control cardiomyocytes were transfected with 5 nM negative control siRNA as described in ‘materials and methods’ section. Representative blots and quantified data (B) showing basal c-Fos and c-Jun expression in LV tissue and cardiomyocytes. Values are mean ± S.E. of five experiments performed with five different cardiomyocyte preparations. *Significantly different (P < 0.05) versus control; #significantly different (P < 0.05) versus NE. GAPDH, Glyceraldehyde 3-phosphate dehydrogenase; CONT, control; NC, negative control; NE, norepinephrine; siRNA, small interfering RNA.
Fig 5.
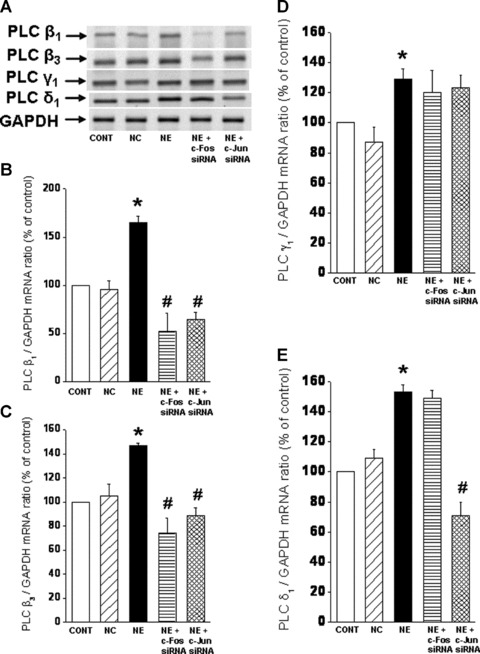
Norepinephrine-induced PLC isozyme mRNA expression in cardiomyocytes transfected with c-Fos and c-Jun siRNA. Representative blots (A) of PLC isozyme β1 (114 bp), β3 (230 bp), γ1 (123 bp) and δ1 (190 bp) mRNA levels relative to GAPDH (138 bp) mRNA level and quantified data showing PLC β1 (B), β3 (C), δ1 (D) and γ1 (E) mRNA levels in 5 nM c-Fos and c-Jun siRNA transfected cardiomyocytes treated with NE (5 μM) for 2 hrs. Control cardiomyocytes were treated with 5 nM negative control siRNA as described in ‘materials and methods’ section. Values are mean ± S.E. of five experiments performed with five different cardiomyocyte preparations. *Significantly different (P < 0.05) versus control; #significantly different (P < 0.05) versus NE. GAPDH, Glyceraldehyde 3-phosphate dehydrogenase; CONT, control; NC, negative control; NE, norepinephrine; siRNA, small interfering RNA; LV left ventricle.
Fig 6.
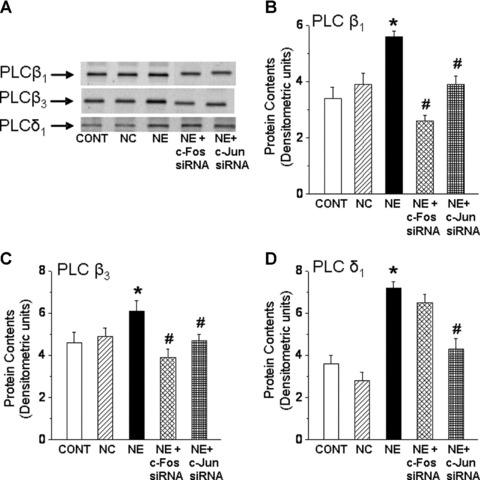
Norepinephrine-induced PLC isozyme protein content in cardiomyocytes transfected with c-Fos and c-Jun siRNA. Representative blots (A) of PLC isozyme β1 (150 kD), β3 (150 kD) and δ1 (85 kD) protein content and quantified data showing PLC β1 (B), β3 (C) and δ1 (D) protein content (arbitrary densitometric units) in c-Fos and c-Jun siRNA (5 nM) transfected cardiomyocytes treated with NE (5 μM) for 24 hrs. Control cardiomyocytes were treated with 5 nM negative control siRNA as described in ‘materials and methods’ section. Values are mean ± S.E. of five experiments performed with five different cardiomyocyte preparations. *Significantly different (P < 0.05) versus control; #significantly different (P < 0.05) versus NE. CONT, control; NC, negative control; NE, norepinephrine; siRNA, small interfering RNA.
Fig 7.
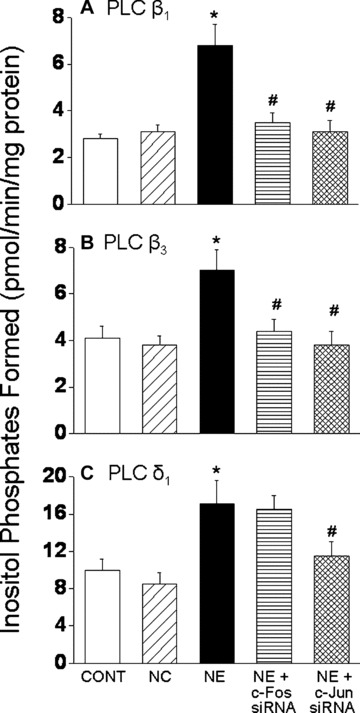
PLC isozyme activities in response to norepinephrine in cardiomyocytes transfected with c-Fos and c-Jun siRNA. PLC β1,β3 and δ1 isozyme activities were determined in c-Fos and c-Jun transfected cardiomyocytes treated with 5 μM NE by measuring hydrolysis of [3H]-PIP2 as described in ‘materials and methods’ section. Values are mean ± S.E. of five experiments performed with five different cardiomyocyte preparations. *Significantly different (P < 0.05) versus control; #significantly different (P < 0.05) versus NE. CONT, control; NC, negative control; NE, norepinephrine; siRNA, small interfering RNA.
Hypertrophic response to NE in cardiomyocytes transfected with siRNA
To understand the relationship of the prevention of increases in PLC isozymes and c-Fos and c-Jun gene expression with the hypertrophic response to NE, ANF mRNA levels and protein synthesis (markers of the cardiomyocytes hypertrophic response) were measured in cardiomyocytes transfected with siRNA to silence PLC isozyme and c-Fos and c-Jun genes. It can be seen in Figure 8 that transfection of cardiomyocytes with PLC β1, β3, δ1 and γ1 si RNA prevented the NE-induced increase in ANF mRNA levels and protein synthesis (as demonstrated by the incorporation of [3H] phenylalanine) (Fig. 8). Furthermore, it was revealed that the NE-induced increase in the ANF mRNA level (Fig. 9A–C) and protein synthesis (Fig. 9D) were prevented in c-Fos and c-Jun transfected cardiomyocytes.
Fig 8.
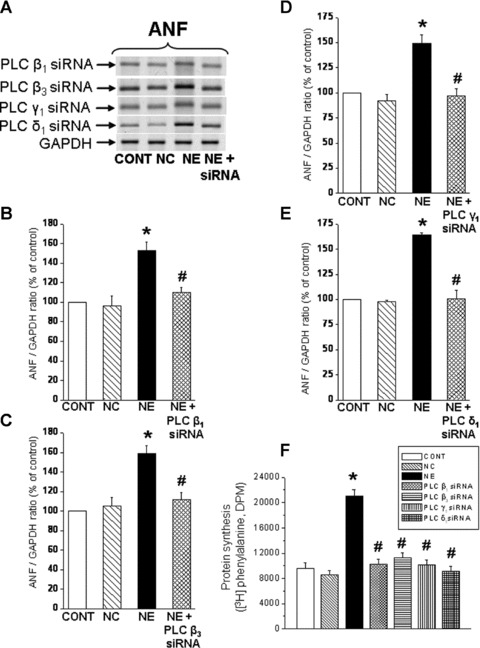
Inhibition of norepinephrine-induced increases in ANF mRNA levels and protein synthesis in cardiomyocytes transfected with PLC isozyme siRNA. Representative blots (A) and quantified data showing ANF (161bp) mRNA levels relative to GAPDH (138bp) mRNA level in cardiomyocytes transfected with 5 nM PLC isozyme (B) β1, (C) β3, (D) γ1 and (E) δ1 siRNA and treated with NE (5 μM) for 2 hrs. Protein synthesis (F) was determined as incorporation of [3H] phenylalanine (DPM) into newly synthesized protein. Control cardiomyocytes were transfected with 5 nM negative control siRNA as described in ‘materials and methods’ section. Values are mean ± S.E. of five experiments performed with five different cardiomyocyte preparations. *Significantly different (P < 0.05) versus control; #significantly different (P < 0.05) versus NE. GAPDH, Glyceraldehyde 3-phosphate dehydrogenase; CONT, control; NC, negative control; NE, norepinephrine; siRNA, small interfering RNA.
Fig 9.
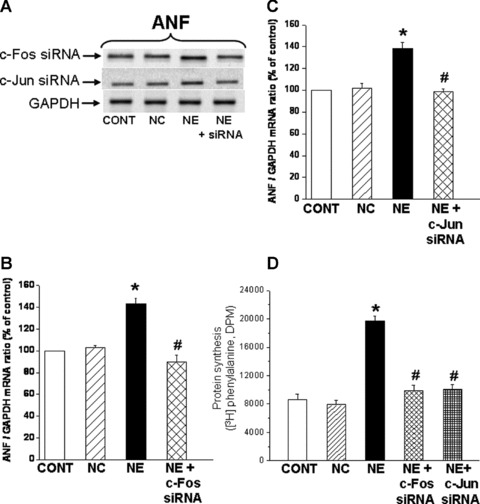
Inhibition of norepinephrine-induced increases in ANF mRNA levels and protein synthesis in cardiomyocytes transfected with c-Fos and c-Jun siRNA. Representative blots (A) and quantified data showing ANF (161bp) mRNA levels relative to GAPDH (138 bp) mRNA level in cardiomyocytes transfected with 5 nM c-Fos (B) and c-Jun (C) siRNA and treated with NE (5 μM) for 2 hrs. Protein synthesis (D) was determined by measurement of [3H] phenylalanine incorporation into newly synthesized cardiomyocyte protein. Control cardiomyocytes were transfected with 5 nM negative control siRNA as described in ‘materials and methods’ section. Values are mean ± S.E. of five experiments performed with five different cardiomyocyte preparations. *Significantly different (P < 0.05) versus control; #significantly different (P < 0.05) versus NE. GAPDH, Glyceraldehyde 3-phosphate dehydrogenase; CONT, control; NC, negative control; NE, norepinephrine; siRNA, small interfering RNA.
Discussion
Although by using different pharmacological agents we have reported earlier that PLC activity is involved in the regulation of transcription factor, c-Fos and c-Jun gene expression [24], we have now shown in this study that specific PLC isozyme activity regulate c-Fos and c-Jun gene expression in response to NE in adult cardiomyocytes. Because concomitant increases in PLC isozyme, c-Jun and c-Fos mRNA levels have been observed in response to NE, the relationship between these transcription factors and PLC isozyme gene expression was further investigated. The specific knockdown of PLC β1, β3, and δ1 with siRNA and subsequent attenuation of their protein content and activity prevented the NE-induced increases in c-Fos and c-Jun gene expression, indicating the involvement of PLC β1, β3 and γ1 isozyme activity in the regulation of c-Fos and c-Jun gene expression. It is interesting to note that knockdown of PLC γ1 did not prevent the NE-induced increase in the expression of c-Fos and c-Jun genes, although it prevented the increase in ANF gene expression and protein synthesis in response to NE. It could be inferred that PLC γ1 may be involved in a signal transduction pathway, independent of c-Fos and c-Jun, which regulates ANF gene expression as well as protein synthesis. A possible candidate in this pathway could be the early growth response factor-1 (Egr-1), the expression of which is known to be induced in response to α1-AR stimulation [27] and is reported to regulate ANF gene expression [28]. Thus, it is possible that PLC isozymes induce a distinct set of immediate early genes, but all induce ANF expression and protein synthesis in adult cardiomyocytes. In addition, Egr-1 could be the transcription factor involved in the NE-induced increase in PLC γ1. Furthermore, PLC γ1, unlike the G-protein mediated activation of PLC β and PLC δ isozymes [29–31], is activated primarily through tyrosine phosphorylation [29]. However, it is interesting to note that a link between Gq and tyrosine kinase provides a mechanism for the α1-AR mediated activation of PLC γ[4, 32]. Thus, while NE was seen to activate all PLC isozymes in this study, the α1-AR may display a discrete requirement for PLC γ1 that does not involve the regulation of c-Fos and c-Jun expression. It is possible that PLC γ1 may be involved in the regulation of Egr-1 gene expression.
To understand the significance of the prevention of the NE-induced increases in PLC isozyme gene expression in c-Fos and c-Jun siRNA transfected cardiomyocytes, the protein content as well as activities of PLC β1, β3 and δ1 isozymes were also determined. Consistent to the changes in their gene expression, silencing of c-Jun also resulted in a prevention of the NE-induced increases in PLC β1, β3 and δ1 isozyme activity and protein levels, whereas silencing of c-Fos specifically inhibited the increase in PLC β1 and β3 isozyme protein content and activity. Therefore, our data would seem to indicate that both these transcription factors are differentially involved in the regulation of PLC β1, β3 and γ1 isozyme gene and protein expression as well as activity in response to NE. Silencing of c-Fos and c-Jun abolished the NE-induced increase in ANF gene expression indicating that c-Fos and c-Jun play an important role in the regulation of cardiac hypertrophic response. It has been suggested that the in vitro binding of c-Fos/c-Jun heterodimers to ANF gene sequences plays a role in the regulation of ANF gene transcription [33] and continued increase in ANF is related to increases in c-Fos and c-Jun expression [28]. It is also interesting to note that the overexpression of c-Jun has been reported to result in an increase in the transcription of ANF [34, 35]. Thus, we consider that the NE-induced increase in c-Fos/c-Jun, which is involved in mediating NE-induced cardiac hypertrophic response, is also characterized by increases in the gene expression of specific PLC isozymes and concomitant increases in protein levels as well as activity. Furthermore, while it is already known that PLC is upstream of c-Fos/c-Jun and ANF, our data also seem to suggest that in the signaling pathway mediated by NE, activation of c-Fos and c-Jun is upstream of ANF.
The c-Fos and c-Jun proteins form the activator protein-1 (AP-1), which is a transcription factor considered to be involved in cardiac hypertrophy [16, 36, 37]. Taimor et al.[16] reported that PhE promotes the formation of c-Fos/c-Jun AP-1 transcription complex in cardiomyocytes and that the functional involvement of AP-1 in hypertrophic growth could only be demonstrated for α-adrenergic stimulation in adult cardiomyocytes. Accordingly, it is possible that the AP-1 mediated hypertrophy could involve the activation of PLC isozyme gene expression. It should also be mentioned that the hypertrophic phenotype in dominant negative c-Jun transfected cardiomyocytes has been reported to be inhibited in response to PhE [22]. On the basis of our earlier [11, 12] and present data, it is likely that this inhibitory response to PhE is attributed to an attenuation of specific PLC isozymes. It should be pointed out that studies in neonatal cardiomyocytes have shown that preincubation with PLC β3 antisense oligonucleotides abolished insulin like growth factor-1 (IGF-1)-induced upregulation of c-Fos and c-Jun genes [38], indicating that PLC β3 expression seems to be required for the induction of immediate early genes by IGF-1. The present data demonstrate a reciprocal regulation of specific PLC isozymes and c-Fos/c-Jun gene expression may exist in adult cardiomyocytes in response to NE, which may constitute not only an early event, but also the progression of cardiac hypertrophy. Indeed, we consider the activation of PLC isozymes as a primary signal transduction event in response to α1-AR stimulation due to NE, involving PKC and ERK 1/2 as downstream signalling mediators. This is borne out from the fact that PLC activity is involved in the (a) NE-mediated increases in c-Fos and c-Jun mRNA and protein levels via PKC and ERK 1/2 [24], (b) NE-induced increase in the expression of ANF [15], (c) NE induces increases in PLC isozyme gene expression, protein content and activity in a PKC- and ERK 1/2-dependent manner [14] and (d) inhibition of PLC activity [14] silencing of PLC isozymes attenuates the hypertrophic response to NE. Thus it is evident that activation of PLC isozymes by α1-AR mediated mechanisms increases the expression of c-Fos and c-Jun for inducing the hypertrophic response. Because silencing of c-Fos and c-Jun was observed to depress the NE-mediated increases in PLC isozyme (with the exception of PLCγ1), it is likely that the increased expression of c-Fos and c-Jun may further augment the expression of specific PLC isozymes, which then leads to the progression of the hypertrophic response to NE.
Prazosin, an α1-AR blocker has been observed to attenuate the progression of cardiac hypertrophy to heart failure [16, 29, 39, 40], it is likely that the mechanism of action of prazosin would include prevention of the activation of PLC and subsequent signal transduction events regulating the early response genes, associated with cardiac hypertrophy. It should be mentioned that in addition to the activation of the sympathetic nervous system, activation of the renin–angiotensin system is also known to occur during the development of cardiac hypertrophy [41]. Angiotensin II (ANG II) can initiate cardiac hypertrophy and upregulate PLC β3[42, 43] as well as increase the expression of c-Fos and c-Jun [44]. Losartan, an ANG II type 1 receptor blocker has also been reported to regress cardiac hypertrophy [40, 45, 46]; an effect that may, in part, be due to an inhibition of the upregulation of PLC isozymes [43, 47]. It should also be noted that the activation of specific PLC isozymes occur in pressure [48] and volume [25] overloaded hypertrophied heart. Taken together, this study supports the view that the activation of PLC isozymes is considered to be an important step in cardiac hypertrophy and thus may be a target for its prevention.
Acknowledgments
This study was supported by a grant from the Heart & Stroke Foundation of Manitoba. TS was a recipient of a Manitoba Health Research Council Ph.D. Studentship. Infrastructural support for this project was provided by the St. Boniface Hospital Research Foundation.
References
- 1.Brown JH, Del Re DP, Sussman MA. The rac and Rho hall of fame: a decade of hypertrophic signaling hits. Circ Res. 2006;98:730–42. doi: 10.1161/01.RES.0000216039.75913.9e. [DOI] [PubMed] [Google Scholar]
- 2.Clerk A, Cullingford TE, Fuller SJ, et al. Signaling pathways mediating cardiac myocyte gene expression in physiological and stress response. J Cell Physiol. 2007;212:311–22. doi: 10.1002/jcp.21094. [DOI] [PubMed] [Google Scholar]
- 3.Diwan A, Dorn GW., 2nd Decompensation of cardiac hypertrophy: cellular mechanisms and novel therapeutic targets. Physiology. 2007;22:56–64. doi: 10.1152/physiol.00033.2006. [DOI] [PubMed] [Google Scholar]
- 4.Hefti MA, Harder BA, Eppenberger HM, et al. Signaling pathways in cardiac myocyte hypertrophy. J Mol cell Cardiol. 1997;29:2873–92. doi: 10.1006/jmcc.1997.0523. [DOI] [PubMed] [Google Scholar]
- 5.McMullen JR, Jennings GL. Differences between pathological and physiological cardiac hypertrophy: novel therapeutic strategies to treat heart failure. Clin Exp Pharmacol Physiol. 2007;34:255–62. doi: 10.1111/j.1440-1681.2007.04585.x. [DOI] [PubMed] [Google Scholar]
- 6.Wang B, Ouyang J, Xia Z. Effects of triiodo-thyronine on angiotensin-induced cardiomyocyte hypertrophy: reversal of increased beta-myosin heavy chain gene expression. Can J Physiol Pharmacol. 2006;84:935–41. doi: 10.1139/y06-043. [DOI] [PubMed] [Google Scholar]
- 7.Li HL, She ZG, Li TB, et al. Overexpression of myofibrillogenesis regulator-1 aggravates cardiac hypertrophy induced by angiotensin II in mice. Hypertension. 2007;49:1399–1408. doi: 10.1161/HYPERTENSIONAHA.106.085399. [DOI] [PubMed] [Google Scholar]
- 8.Passier R, Zeng H, Frey N, et al. CaM kinase signaling induces cardiac hypertrophy and activates the MEF2 transcription factor in vivo. J Clin Invest. 2000;105:1395–1406. doi: 10.1172/JCI8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanna B, Bueno OF, Dai YS, et al. Direct and indirect interactions between calcineurin-NFAT and MEK1-extracellular signal-regulated kinase 1/2 signaling pathways regulate cardiac gene expression and cellular growth. Mol Cell Biol. 2005;25:865–78. doi: 10.1128/MCB.25.3.865-878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grassi G. Sympathetic overdrive as an independent predictor of left ventricular hypertrophy: prospective evidence. J Hypertens. 2006;24:815–7. doi: 10.1097/01.hjh.0000222748.37078.2d. [DOI] [PubMed] [Google Scholar]
- 11.Luodonpää M, Leskinen H, Ilves M, et al. Adrenomedullin modulates hemodynamic and cardiac effects of angiotensin II in conscious rats. Am J Physiol Regul Integr Comp Physiol. 2004;286:R1085–92. doi: 10.1152/ajpregu.00726.2003. [DOI] [PubMed] [Google Scholar]
- 12.Rapacciuolo A, Esposito G, Caron K, et al. Important role of endogenous norepinephrine and epinephrine in the development of in vivo pressure-overload cardiac hypertrophy. J Am Coll Cardiol. 2001;38:876–82. doi: 10.1016/s0735-1097(01)01433-4. [DOI] [PubMed] [Google Scholar]
- 13.Vecchione C, Fratta L, Rizzoni D, et al. Cardiovascular influences of α1b-adrenergic receptor defect in mice. Circulation. 2002;105:1700–7. doi: 10.1161/01.cir.0000012750.08480.55. [DOI] [PubMed] [Google Scholar]
- 14.Singal T, Dhalla NS, Tappia PS. Norepinephrine-induced changes in gene expression of phospholipase C in cardiomyocytes. J Mol Cell Cardiol. 2006;41:126–37. doi: 10.1016/j.yjmcc.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 15.Singal T, Dhalla NS, Tappia PS. Phospholipase C may be involved in norepinephrine-induced cardiac hypertrophy. Biochem Biophys Res Commun. 2004;320:1015–9. doi: 10.1016/j.bbrc.2004.06.052. [DOI] [PubMed] [Google Scholar]
- 16.Taimor G, Schlüter KD, Best P, et al. Transcription activator protein 1 mediates α- but not β-adrenergic hypertrophic growth responses in adult cardiomyocytes. Am J Physiol Heart Circ Physiol. 2004;286:H2369–75. doi: 10.1152/ajpheart.00741.2003. [DOI] [PubMed] [Google Scholar]
- 17.Yamazaki T, Yazaki Y. Molecular basis of cardiac hypertrophy. Z Kardiol. 2000;89:1–6. doi: 10.1007/s003920050001. [DOI] [PubMed] [Google Scholar]
- 18.Hannan RD, Stennard FA, West AK. Expression of c-fos and related genes in the rat heart in response to norepinephrine. J Mol Cell Cardiol. 1993;25:1137–48. doi: 10.1006/jmcc.1993.1127. [DOI] [PubMed] [Google Scholar]
- 19.Hannan RD, West AK. Adrenergic agents, but not triiodo-L-thyronine induce c-fos and c-myc expression in the rat heart. Basic Res Cardiol. 1991;86:154–64. doi: 10.1007/BF02190548. [DOI] [PubMed] [Google Scholar]
- 20.Saadane N, Alpert L, Chalifour LE. Expression of immediate early genes, GATA-4, and Nkx-2.5 in adrenergic-induced cardiac hypertrophy and during regression in adult mice. Br J Pharmacol. 1999;127:1165–76. doi: 10.1038/sj.bjp.0702676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schunkert H, Jahn L, Izumo S, et al. Localization and regulation of c-fos and c-jun protooncogene induction by systolic wall stress in normal and hypertrophied rat hearts. Proc Natl Acad Sci USA. 1991;88:11480–4. doi: 10.1073/pnas.88.24.11480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Omura T, Yoshiyama M, Yoshida K, et al. Dominant negative mutant of c-Jun inhibits cardiomyocyte hypertrophy induced by endothelin 1 and phenylephrine. Hypertension. 2002;39:81–6. doi: 10.1161/hy0102.100783. [DOI] [PubMed] [Google Scholar]
- 23.Bishopric NH, Jayasena V, Webster KA. Positive regulation of the skeletal α-actin gene by Fos and Jun in cardiac myocytes. J Biol Chem. 1992;267:25535–40. [PubMed] [Google Scholar]
- 24.Singal T, Dhalla NS, Tappia PS. Regulation of c-Fos and c-Jun gene expression by phospholipase C activity in adult cardiomyocytes. Mol Cell Biochem. 2009;327:229–39. doi: 10.1007/s11010-009-0061-1. [DOI] [PubMed] [Google Scholar]
- 25.Dent MR, Dhalla NS, Tappia PS. Phospholipase C gene expression, protein content, and activities in cardiac hypertrophy and heart failure due to volume overload. Am J Physiol Heart Circ Physiol. 2004;287:H719–27. doi: 10.1152/ajpheart.01107.2003. [DOI] [PubMed] [Google Scholar]
- 26.Ziegelhoffer A, Tappia PS, Mesaeli N, et al. Low level of sarcolemmal phosphatidylinositol 4,5-bisphosphate in cardiomyopathic hamster (UM-X7.1) heart. Cardiovasc Res. 2001;49:118–26. doi: 10.1016/s0008-6363(00)00209-1. [DOI] [PubMed] [Google Scholar]
- 27.Brand T, Sharam HS, Schaper W. Expression of nuclear proto-oncogenes in isoproterenol-induced cardiac hypertrophy. J Mol Cell Cardiol. 1993;25:1325–37. doi: 10.1006/jmcc.1993.1145. [DOI] [PubMed] [Google Scholar]
- 28.Saadane N, Alpert L, Chalifour LE. Expression of immediate early genes, GATA-4, and Nkx-2.5 in adrenergic-induced cardiac hypertrophy and during regression in adult mice. Br J Pharmacol. 1999;127:1165–76. doi: 10.1038/sj.bjp.0702676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tappia PS, Singal PK. Regulation of phospholipase C in cardiac hypertrophy. Clin Lipidol. 2009;4:79–90. [Google Scholar]
- 30.Feng JF, Gray CD, Im MJ. α1B-adrenoceptor interacts with multiple sites of transglutaminase II: characteristics of the interaction in binding and activation. Biochemistry. 1999;38:2224–32. doi: 10.1021/bi9823176. [DOI] [PubMed] [Google Scholar]
- 31.Park H, Park ES, Lee HS, et al. Distinct characteristic of Gαh (transglutaminase II) by compartment: GTPase and transglutaminase activities. Biochem Biophys Res Commun. 2001;284:496–500. doi: 10.1006/bbrc.2001.4997. [DOI] [PubMed] [Google Scholar]
- 32.Tappia PS, Padua RR, Panagia V, et al. Fibroblast growth factor-2 stimulates phospholipase Cβ in adult cardiomyocytes. Biochem Cell Biol. 1999;77:569–75. [PubMed] [Google Scholar]
- 33.Rosenzweig A, Halazonetis TD, Seidman JG, et al. Proximal regulatory domains of rat atrial natriuretic factor gene. Circulation. 1991;84:1256–65. doi: 10.1161/01.cir.84.3.1256. [DOI] [PubMed] [Google Scholar]
- 34.Kovacic-Milivojevic B, Gardner DG. Regulation of the human atrial natriuretic peptide gene in atrial cardiocytes by the transcription factor AP-1. Am J Hypertens. 1993;6:258–63. doi: 10.1093/ajh/6.4.258. [DOI] [PubMed] [Google Scholar]
- 35.Kovacic-Milivojevic B, Wong VS, Gardner DG. Selective regulation of the atrial natriuretic peptide gene by individual components of the activator protein-1 complex. Endocrinology. 1996;137:1108–17. doi: 10.1210/endo.137.3.8603581. [DOI] [PubMed] [Google Scholar]
- 36.Curran T, Franza BR., Jr Fos and Jun: the AP-1 connection. Cell. 1998;55:395–97. doi: 10.1016/0092-8674(88)90024-4. [DOI] [PubMed] [Google Scholar]
- 37.Kaminska B, Pyrzynska B, Ciechomska I, et al. Acta Neurobiol Exp. Vol. 60. (Wars): 2000. Modulation of the composition of AP-1 complex and its impact on transcriptional activity; pp. 395–402. [DOI] [PubMed] [Google Scholar]
- 38.Schnabel P, Mies F, Nohr T, et al. Differential regulation of phospholipase C-β isozymes in cardiomyocyte hypertrophy. Biochem Biophys Res Commun. 2000;275:1–6. doi: 10.1006/bbrc.2000.3255. [DOI] [PubMed] [Google Scholar]
- 39.Giles TD, Sander GE, Thomas MG, et al. α-adrenergic mechanisms in the pathophysiology of left ventricular heart failure-An analysis of their role in systolic and diastolic dysfunction. J Mol Cell Cardiol. 1986;18:33–43. doi: 10.1016/s0022-2828(86)80459-x. [DOI] [PubMed] [Google Scholar]
- 40.Okin PM, Devereux RB, Gerdts E, et al. Impact of diabetes mellitus on regression of electrocardiographic left ventricular hypertrophy and the prediction of outcome during antihypertensive therapy: the Losartan Intervention For Endpoint (LIFE) Reduction in Hypertension Study. Circulation. 2006;113:1588–96. doi: 10.1161/CIRCULATIONAHA.105.574822. [DOI] [PubMed] [Google Scholar]
- 41.Dent MR, Singal T, Tappia PS. Involvement of renin-angiotensin system in the pathogenesis of cardiovascular disease. In: Ray A, Gulati K, et al., editors. Current trends in pharmacology. New Delhi: IK International Publishing House Pvt. Ltd; 2007. pp. 137–160. [Google Scholar]
- 42.Aoki H, Richmond M, Izumo S, et al. Specific role of the extracellular signal-regulated kinase pathway in angiotensin Ii-induced cardiac hypertrophy in vitro. Biochem J. 2000;347:275–84. [PMC free article] [PubMed] [Google Scholar]
- 43.Bai H, Wu LL, Xing DQ, et al. Angiotensin II induced upregulation of G αq/11, phospholipase C β3 and extracellular signal-regulated kinase 1/2 via angiotensin II type 1 receptor. Chin Med J. 2004;117:88–93. [PubMed] [Google Scholar]
- 44.Lijnen P, Petrov V. Renin-angiotensin system, hypertrophy and gene expression in cardiac myocytes. J Mol Cell Cardiol. 1999;31:949–70. doi: 10.1006/jmcc.1999.0934. [DOI] [PubMed] [Google Scholar]
- 45.Moen MD, Wagstaff AJ. Losartan: a review of its use in stroke risk reduction in patients with hypertension and left ventricular hypertrophy. Drugs. 2005;65:2657–74. doi: 10.2165/00003495-200565180-00012. [DOI] [PubMed] [Google Scholar]
- 46.Wachtell K, Okin PM, Olsen MH, et al. Regression of electrocardiographic left ventricular hypertrophy during antihypertensive therapy and reduction in sudden cardiac death: the LIFE Study. Circulation. 2007;116:700–5. doi: 10.1161/CIRCULATIONAHA.106.666594. [DOI] [PubMed] [Google Scholar]
- 47.Dent MR, Aroutiounova N, Dhalla NS, et al. Losartan attenuates phospholipase C isozyme gene expression in hypertrophied hearts due to volume overload. J Cell Mol Med. 2006;10:470–9. doi: 10.1111/j.1582-4934.2006.tb00412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jalili T, Takeishi Y, Song G, et al. PKC translocation without changes in Gαq and PLC-β protein abundance in cardiac hypertrophy and failure. Am J Physiol. 1999;277:H2298–304. doi: 10.1152/ajpheart.1999.277.6.H2298. [DOI] [PubMed] [Google Scholar]