

Background: Recently, an immunosuppressive motif was identified in the HIV-1 envelope glycoprotein complex.
Results: Both d- and l-stereoisomers of the motif inhibit T-cell receptor activation and preferentially bind T-cells over B-cells.
Conclusion: The motif immunomodulates T-cells through interactions occurring within the membrane milieu.
Significance: This study provides new insights into the immunosuppressive activity of the envelope glycoprotein complex and the molecular recognition within the membrane.
Keywords: Autoimmune Diseases, Immunosuppression, Membrane Fusion, Protein-Protein Interactions, T-cell, Viral Fusion Protein, Membrane Recognition
Abstract
An immunosuppressive motif was recently found within the HIV-1 gp41 fusion protein (termed immunosuppressive loop-associated determinant core motif (ISLAD CM)). Peptides containing the motif interact with the T-cell receptor (TCR) complex; however, the mechanism by which the motif exerts its immunosuppressive activity is yet to be determined. Recent studies showed that interactions between protein domains in the membrane milieu are not always sterically controlled. Therefore, we utilized the unique membrane leniency toward association between d- and l-stereoisomers to investigate the detailed mechanism by which ISLAD CM inhibits T-cell activation. We show that a d-enantiomer of ISLAD CM (termed ISLAD d-CM) inhibited the proliferation of murine myelin oligodendrocyte glycoprotein (MOG)-(35–55)-specific line T-cells to the same extent as the l-motif form. Moreover, the d- and l-forms preferentially bound spleen-derived T-cells over B-cells by 13-fold. Furthermore, both forms of ISLAD CM co-localized with the TCR on activated T-cells and interacted with the transmembrane domain of the TCR. FRET experiments revealed the importance of basic residues for the interaction between ISLAD CM forms and the TCR transmembrane domain. Ex vivo studies demonstrated that ISLAD d-CM administration inhibited the proliferation (72%) and proinflammatory cytokine secretion of pathogenic MOG(35–55)-specific T-cells. This study provides insights into the immunosuppressive mechanism of gp41 and demonstrates that chirality-independent interactions in the membrane can take place in diverse biological systems. Apart from HIV pathogenesis, the d-peptide reported herein may serve as a potential tool for treating T-cell-mediated pathologies.
Introduction
HIV-1 is the etiological agent for AIDS, which infects millions of people worldwide (1–3). HIV-1 possesses several strategies to modify cellular pathways and to evade the immune response to infect and persist in the host (4–6). One example is the ability of HIV-1 to alter immune receptor signaling cascades (6, 7). During the life cycle of the virus, it is able to both hyperactivate and down-regulate the T-cell response at different stages. This is accomplished by several virulent proteins, such as Nef and Tat, which enable HIV-1 to evade the immune response (8, 9).
Other than mediating virus entry by membrane fusion (10, 11), the HIV-1 envelop glycoprotein complex Env is also capable of impairing the cross-talk between antigen-presenting cells (APCs)3 and T-cells, which is required for T-cell activation (12, 13). T-cell inactivation was shown as early as the stage of HIV-1 entry into T-cells (14, 15). Furthermore, CD4 T-cells exposed to noninfectious HIV expressing functional Env failed to properly activate APCs, which, in turn, damaged their capability to stimulate naive T-cells (14). Overall, Env acts as an additional virulent factor that assists the virus to escape from the immune response.
HIV-1 Env is composed of surface and transmembrane subunits. The surface subunit, gp120, is responsible for the recognition of CD4 and co-receptors. The transmembrane subunit, the gp41 fusion protein, is responsible for merging the viral membrane with that of its target T-cell (16). The ectodomain of gp41 consists of several regions, including a transmembrane domain (TMD), a C-terminal heptad repeat, a loop, an N-terminal heptad repeat, and a fusion peptide. The ability of gp41 to mediate fusion is facilitated by the interaction between the C- and N-terminal heptad repeats, which form a six-helix bundle structure (17, 18). The two hydrophobic portions of gp41, the fusion peptide and the TMD, also contribute to membrane fusion (19–21). These two regions possess a second function and are able to interact with the T-cell receptor (TCR) complex (22–25). The TCR complex is important for T-cell activation (12) and is found in proximity to the HIV fusion site (26). Emerging studies showed that the hydrophobic portions of gp41 are able not only to interact with the TCR but also to inhibit activation of the complex (22–25).
The gp41 loop region is also of membranotropic nature and takes part in the membrane fusion process (27–29). Recently, we showed that a peptide derived from this region is a potent inhibitor of T-cell activation both in vitro and in vivo (30). This peptide, termed immunosuppressive loop-associated determinant (ISLAD), contains a highly conserved core motif (CM) of Trp repeat and acidic (Asp and Glu) residues and interacts with the TCR complex (30). However, the mechanism by which ISLAD exerts its immunosuppressive effect on T-cells is yet to be determined.
We hypothesized that the interaction of ISLAD with the TCR complex takes place in the membrane, taking into account the membrane-binding capacity of the motif (30). Membrane interactions between the TMDs of the TCR complex are fundamental for the initiation of T-cell activation signals (31, 32). Importantly, recent studies suggested that interactions within the membrane can be chirality-independent for several cell membrane proteins (33–35). Therefore, we investigated the immunosuppressive mechanism of the ISLAD motif (ISLAD l-CM) and its d-enantiomer form (ISLAD d-CM) with an opposite chirality (see Table 1). Our results demonstrate that the d-enantiomer of ISLAD CM interacts with the TCR complex in the membrane and inhibits T-cell activation in vitro and ex vivo. The results are highlighted in the context of the immunosuppressive mechanism of HIV-1 gp41.
TABLE 1.
Designation, sequence, origin, and fluorescent labeling of the peptides used in the study
The boldface amino acids in the sequence represent d-amino acids. AMP, antimicrobial peptide.
| Designation | Protein of origin | Fluorescent labeling | Sequence |
|---|---|---|---|
| ISLAD | HIV-1 gp41 | SNKSLEQIWNHTTWMEWD | |
| ISLAD-Rho | HIV-1 gp41 | Rho | Rho-SNKSLEQIWNHTTWMEWD |
| ISLAD l-CM | HIV-1 gp41 | WNHTTWMEWD | |
| ISLAD l-CM-Rho | HIV-1 gp41 | Rho | Rho-WNHTTWMEWD |
| ISLAD d-CM | HIV-1 gp41 | WNHTTWMEWD | |
| ISLAD d-CM-Rho | HIV-1 gp41 | Rho | Rho-WNHTTWMEWD |
| Scrambled ISLAD l-CM | HIV-1 gp41 | WTHWMENWTD | |
| Control peptide | HIV-1 gp41 | TTAVPWNASW | |
| TCR l-CP-NBD | Mouse TCRα | NBD | NBD-GLRILLLKV |
| TCR l-CP2G-NBD | Mouse TCRα | NBD | NBD-GLGILLLGV |
| TCR d-CP2G-Rho | Mouse TCRα | Rho | Rho-GLGILLLGV |
| AMP-Rho | Synthetic antimicrobial | Rho | Rho-KLKLLKLLKLLKLLK |
| LL37-Rho | Human cathelicidin | Rho | Rho-LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES |
| MOG35–55 | Mouse myelin | MEVGWYRSPFSRVVHLYRNGK |
EXPERIMENTAL PROCEDURES
Mice
C57BL/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME). All mice were 2–3 months old when used in the experiments. The Institutional Animal Care and Use Committee of the Weizmann Institute approved the experiments (permit number 03530710-3), which were performed in accordance with the relevant guidelines and regulations.
Cell Lines
Antigen-specific T-cell lines were selected in vitro (36) from primed lymph node cells derived from C57BL/6J mice that had been immunized 9 days before with antigen (100 μg of myelin oligodendrocyte glycoprotein (MOG)-(35–55) peptide) emulsified in complete Freund's adjuvant containing 150 μg of Mycobacterium tuberculosis H37Ra (Difco, Detroit, MI). All T-cell lines were maintained in vitro in medium containing IL-2 with alternate stimulation with the antigen every 10–14 days. The human T-cell line Jurkat E6-1 was obtained Dr. Arthur Weiss through the AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, National Institutes of Health (37).
Peptide Synthesis and Fluorescent Labeling
Peptides were synthesized on Rink amide 4-methylbenzhydrylamine resin (Calbiochem-Novabiochem AG) using the Fmoc (N-(9-fluorenyl)methoxycarbonyl) strategy (38). All peptides were purified by reverse phase HPLC to >95% homogeneity. The molecular weight of the peptides was confirmed by mass spectrometry. The addition of 4-chloro-7-nitrobenzo-2-oxa-1,3-diazole (NBD) fluoride (Molecular Probes) to the N terminus of selected peptides was performed in dimethylformamide for 1 h. The addition of rhodamine N-hydroxysuccinimide (Rho; Molecular Probes) to the N terminus of selected peptides was performed in dimethylformamide containing 2% N,N-diisopropylethylamine for 24 h.
In Vitro T-cell Proliferative Responses
Murine MOG(35–55)-specific line T-cells (referred to hereafter as mMOG(35–55) T-cells) were plated onto round 96-well plates in RPMI 1640 medium supplemented with 2.5% FCS, 100 units/ml penicillin, 100 μg/ml streptomycin, 50 μm β-mercaptoethanol, and 2 mm l-glutamine. Each of the 96 wells had a final volume of 200 μl and contained 20 × 103 T-cells and 5 × 105 irradiated (25 gray) syngeneic spleen cells as APCs, without or with 1–5 μg/ml MOG(35–55). In addition, the relevant HIV peptide was added. To exclude interaction between the examined peptides and the MOG(35–55) antigen, we initially added the MOG(35–55) antigen to the APCs in a test tube, and in a second test tube, we added the examined peptides to the T-cells. After 1 h, the APCs were mixed with the T-cells and co-incubated for 48 h in a 96-well flat bottom plate. The T-cells were pulsed with 1 μCi [3H]thymidine (specific activity of 5.0 Ci/mmol), and after overnight incubation, [3H]thymidine incorporation was measured using a Matrix 96 direct β-counter (Packard Instrument Co.).
Ex Vivo Studies in Experimental Autoimmune Encephalomyelitis (EAE) Mice
Mice were subcutaneously immunized with 150 μg of MOG(35–55) peptide emulsified in complete Freund's adjuvant containing 150 μg of M. tuberculosis H37Ra with or without 0.5 mg/kg HIV peptides. Ten days post-immunization, draining lymph nodes were removed and cultured in triplicate in the absence or presence of antigen as described previously (39). The cultures were incubated for 72 h at 37 °C. [3H]Thymidine (1 mCi/well) was added for an additional 16 h of incubation, and the cultures were then harvested and counted using the β-counter. Proinflammatory cytokine analysis of IFNγ and IL-17 was performed by ELISA 24 h after cell activation according to standard protocols from Pharmingen as described previously (40).
Peptide Binding to Mouse Spleen Cells Detected by FACS
Splenocytes derived from C57BL mice were treated for red blood cell lysis, washed, and incubated for 20 min (room temperature) with 0.15 μm rhodamine-conjugated peptides. Thereafter, the splenocytes were washed and stained with antibodies according to the BioLegend protocols. Fluorochrome-labeled monoclonal antibodies (phycoerythrin-conjugated anti-mouse CD3 and B220) were purchased from BioLegend. Cells were analyzed on Cytomics FC 500 system (Beckman Coulter) and analyzed using Beckman Coulter software.
Co-localization of Peptides with TCR Molecules
Activated mMOG(35–55) T-cells (5 × 104) were fixed with 3% paraformaldehyde for 20 min and washed with PBS. The cells were then treated with 10% FCS in PBS at room temperature to block unspecific binding. After 30 min, the cells were washed, and rabbit anti-TCRα polyclonal antibody (Santa Cruz Biotechnology) was added (1:100) in 2% FCS in PBS for 1 h at room temperature. This was followed by the addition of FITC-labeled rat anti-rabbit antibody (1:100; Santa Cruz Biotechnology) for 40 min at room temperature. The rhodamine-labeled fluorescent peptide was added during the last 10 min of incubation at a final concentration 1 μm. The cells were then washed with PBS and deposited onto a glass slide. The labeled cell samples were observed under an Olympus FV1000 fluorescence confocal microscope, and confocal slices were obtained.
Detection of Binding of Fluorescently Labeled Peptide to T-cell Proteins by SDS-PAGE
Jurkat T-cells (4 × 106) were incubated overnight at 37 °C in the presence of 1 μm rhodamine-labeled peptides. The cells were washed then with PBS and cross-linked with 1% formaldehyde, and after 10 min, a 125 mm glycine solution was added to stop the reaction. Formaldehyde is a well known cross-linking agent used in the characterization of protein-protein interactions. It was used to increase specificity because only closely associated proteins can be cross-linked due to the small size of formaldehyde. The cells were lysed for 30 min on ice in 300 μl of lysis buffer, followed by a sonication step. Insoluble material was removed by centrifugation at 3000 × g for 4 min at 4 °C. The proteins were resolved by 12% SDS-PAGE, and proteins bound to the fluorescently labeled peptide were detected using the Typhoon 9400 variable mode imager (Amersham Biosciences). Excitation was set at 532 nm, and fluorescence emission was collected using a TAMRA filter (580 ± 30 nm) at 100 μm resolution and 600 V. The protein loading amount was confirmed by Western blotting for actin.
Immunoprecipitation of Fluorescently Labeled Peptides with TCR
Jurkat T-cells (4 × 106) were incubated overnight at 37 °C in the presence of 10 μm rhodamine-labeled peptides, cross-linked with 1% formaldehyde, and lysed. The lysate was then incubated overnight with anti-TCRα antibody (1 μg) following 2 h of incubation with Protein G Plus-agarose beads (Santa Cruz Biotechnology). The beads were washed then with lysis buffer and boiled for 10 min; the protein supernatant was subjected to 12% SDS-PAGE. The presence of co-immunoprecipitated peptide was detected using the Typhoon 9400 variable mode imager.
XTT Cytotoxicity Assay
Aliquots of 2.5 × 104 mMOG(35–55) T-cells were distributed onto a 96-well plate in the presence of the peptides (40 μm) for 4 h. Wells in the last two columns served as a blank (medium only) and a 100% survival control (cells and medium only). After incubation, the XTT (2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide) reaction solution (50:1 benzenesulfonic acid hydrate and N-methyl dibenzopyrazine methyl sulfate) was added for an additional 2 h. Optical density was read at a 450-nm wavelength in an ELISA plate reader. The percentage of toxicity was calculated relative to the control (2.5 × 104 cells in medium with no peptide added).
Secondary Structure Determination
CD spectroscopy measurements were performed using an Applied Photophysics spectropolarimeter. The spectra were scanned using a thermostatic quartz cuvette with a path length of 1 mm. Wavelength scans were performed at 25 °C; the average recording time was 15 s in 1-nm steps at a wavelength range of 190–260 nm. Peptides were scanned at a concentration of 100 μm in a membrane mimetic environment of 1% lysophosphatidylcholine in HEPES solution (Sigma).
Fluorescence Measurements of Peptide-Membrane Interactions
Peptides interacting with membranes were analyzed and quantified using the fluorescence anisotropy of their intrinsic Trp residue in the presence of large unilamellar vesicle (LUV) model membranes. The LUVs were composed of phosphatidylcholine and cholesterol (9:1) as described previously (41). Excitation and emission wavelengths were set to 280 and 350 nm, respectively, and 1 μm peptide (in 400 μl of PBS) was titrated successively with 13.3 mm membrane solution. Because Trp is known to change its emission in a hydrophobic environment, a change in its emission represents the amount of peptide bound to membranes. The system reached binding equilibrium (Fmax) at a certain lipid/peptide ratio, allowing us to calculate the affinity constant from the relations between the equilibrium level of Trp emission and the lipid concentration using a steady-state affinity model. The affinity constants were then determined by nonlinear least-squares analysis. The nonlinear least-squares fitting was done using Equation 1,
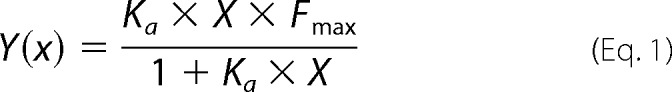 |
where X is the lipid concentration, Y(x) is the fluorescence emission, Fmax is the maximum difference in the emission of Trp-containing peptide before and after the addition of lipids (it represents the maximum amount of peptides bound to a lipid), and Ka is the affinity constant.
FRET Measurements
The FRET experiments were performed using the NBD- and Rho-labeled peptides as energy donors and energy acceptors, respectively. Fluorescence spectra were obtained at room temperature, with excitation set at 467 nm (10-nm slit) and emission scan at 500–600 nm (10-nm slits). An NBD-labeled peptide was added first from a stock solution in dimethyl sulfoxide at a final concentration of 0.1 μm and a maximum of 0.25% (v/v) dimethyl sulfoxide to a dispersion of phosphatidylcholine:cholesterol (9:1) LUVs (200 μm) in PBS. This was followed by the addition of the Rho-labeled peptide (stock solution in dimethyl sulfoxide) in several sequential doses. Fluorescence spectra were obtained before and after the addition of the Rho-labeled peptide. The fluorescence values were corrected by subtracting the corresponding blank (buffer with the same vesicle concentration).
RESULTS
Structural Characterization of ISLAD l-CM and ISLAD d-CM
ISLAD CM is conserved within all clades of HIV and simian immunodeficiency virus fusion proteins (30). CM-derived 10-mer peptides were prepared with opposite chirality of both the all-l-form (ISLAD l-CM) and the all-d-form (ISLAD d-CM) (see the peptide sequences in Table 1). The peptides were examined for their secondary structure by CD in a lipid environment. The opposite chirality of the peptides was verified by the opposite millidegrees exhibiting a mirror image (Fig. 1). As a control, a scrambled peptide was prepared from ISLAD l-CM. This peptide exhibited a different spectrum compared with wild-type ISLAD l-CM. The peptide structure was further examined by CDNN analysis (Gerald Böhm) of ISLAD l-CM and its scrambled analog (Table 2). Although ISLAD l-CM was found to possess mainly a helical structure in a lipid environment, the scrambled analog was much less helical. This suggests that the location of the amino acids within the motif facilitates its structure within the membrane.
FIGURE 1.
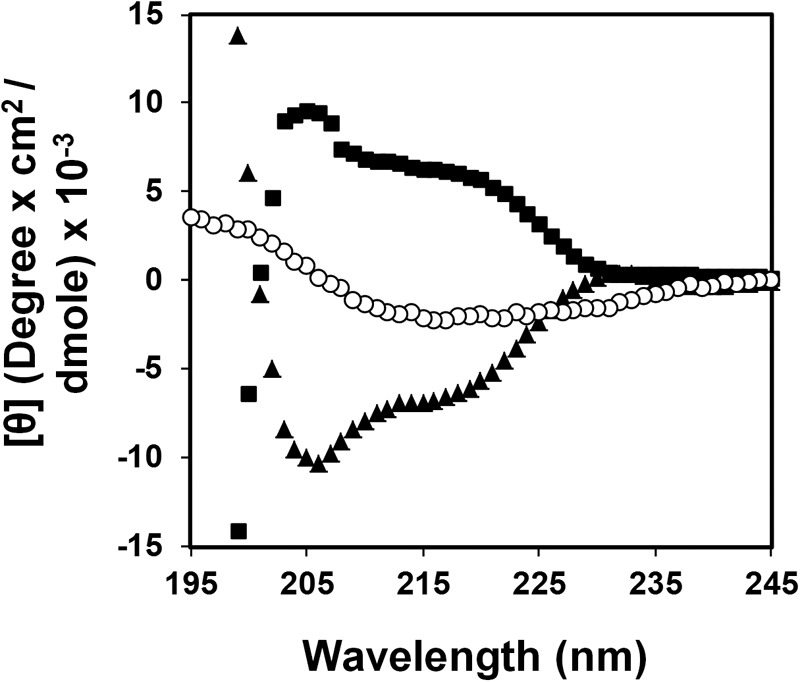
The ISLAD l and d CM peptides have mirror image structures. The CD spectra of ISLAD l-CM (▴), ISLAD d-CM CP (■), and scrambled ISLAD l-CM (○) were collected in a membrane mimetic environment (1% lysophosphatidylcholine). The y axis represents θ after subtracting the background spectrum of 1% lysophosphatidylcholine alone.
TABLE 2.
Analysis of the CD spectra of ISLAD CM and its scrambled analog by the CDNN secondary structure analysis program
| Structure | Fraction |
|
|---|---|---|
| Wild-type ISLAD CM | Scrambled ISLAD CM | |
| % | ||
| α-Helix | 68.9 | 15.3 |
| Antiparallel β-sheet | 1.7 | 17.1 |
| Parallel β-sheet | 3.3 | 12.0 |
| β-Turn | 12.7 | 16.3 |
| Random coil | 13.5 | 39.3 |
ISLAD d-CM and ISLAD l-CM Inhibit Proliferation of MOG(35–55) T-cells in Vitro
The ability of the ISLAD l-CM and ISLAD d-CM peptides to inhibit T-cell proliferation was investigated in C57BL/6J mMOG(35–55)-specific line T cells. Splenocytes, together with the MOG(35–55) antigen, were cultured with mMOG(35–55) T-cells with and without HIV-1 peptides. Fig. 2 shows the proliferative response of the T-cells measured by their incorporation of radioactive thymidine. Fig. 2A shows that both ISLAD CM forms exhibited dose-dependent inhibition of T-cell proliferation that was comparable to ISLAD. As a control, a 10-mer peptide derived from the gp41 loop adjacent to ISLAD was examined (the sequence is provided in Table 1). The control peptide was significantly less active than the CM peptides. Moreover, the peptides were not toxic to T-cells up to 40 μm, the maximum concentration examined and 4–16-fold more than the concentration range used for investigating biological activity (Fig. 2B). Additionally, the MOG(35–55) T-cell proliferative response was significantly higher in the presence of the scrambled analog of ISLAD CM compared with the wild-type sequence (Fig. 2C). These results suggest a chirality-independent mode of action, as ISLAD d-CM is capable of inhibiting T-cell proliferation with a similar potency to ISLAD l-CM.
FIGURE 2.
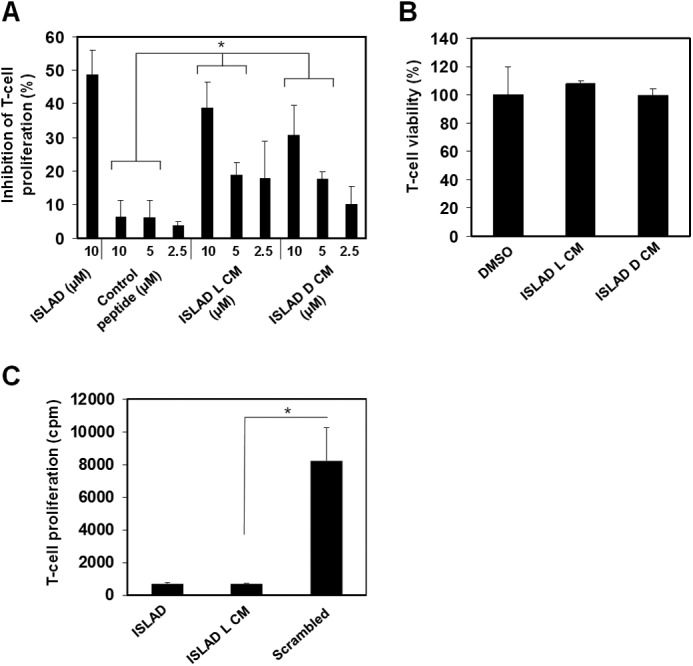
Inhibition of mMOG(35–55) T-cells by ISLAD CM and its d-enantiomer. mMOG(35–55) T-cells were cultured in microtiter plates with irradiated syngeneic splenocytes as APCs and the MOG(35–55) peptide in the presence or absence of several gp41-derived peptides. Their proliferative response was measured in a [3H]thymidine proliferation assay. A, inhibition of proliferation by ISLAD, ISLAD l-CM, ISLAD d-CM, and a control gp41-derived peptide. The results presented are the mean percent inhibition ± S.E. of the proliferative response to the MOG(35–55) peptide relative to the control (in the absence of gp41 peptides) from three experiments. B, ISLAD CM is not toxic to T-cells. mMOG(35–55) T-cells were incubated with 40 μm ISLAD l-CM and ISLAD d-CM. The viability of the cells was then analyzed by an XTT cytotoxicity assay. Results are the mean percent viability ± S.D. from the control (cells with no peptide added; n = 3). DMSO, dimethyl sulfoxide. C, the proliferative response of MOG(35–55) T-cells is higher in the presence of scrambled ISLAD l-CM. MOG(35–55) T-cells were cultured in the presence of MOG(35–55) and the gp41-derived peptides (10 μm), and proliferation was determined by [3H]thymidine uptake (cpm). The results presented are the mean cpm ± S.E. of the proliferative response. The proliferative response value without the gp41-derived peptides was 20716 cpm. *, p < 0.05 in same concentrations between groups.
ISLAD CM Peptides Preferentially Bind T-cells over B-cells in Mouse Spleen
Once the ability of the ISLAD CM peptides to inhibit T-cell proliferation was validated, the affinity of the peptides for lymphocyte populations was examined. Different Rho-labeled peptides were introduced to cells isolated from a naive mouse C57BL/6J spleen. The ISLAD l-CM and ISLAD d-CM peptides were compared with the control peptide LL37, which is an unrelated antimicrobial peptide. Using flow cytometry, we gated only on the lymphocytes that were labeled with rhodamine (Fig. 3, A and B); we then examined the percentage of the labeled cells that were T-cells or B-cells utilizing antibodies for the CD3 and B220 markers, respectively (Fig. 3C). Both ISLAD l-CM and ISLAD d-CM demonstrated much higher affinity for T-cells compared with B-cells (24- and 13-fold, respectively). However, the LL37 control peptide demonstrated similar affinity for both lymphocyte populations. This interesting result shows that the short 10-mer peptides have the ability to specifically interact with particular cell types.
FIGURE 3.

ISLAD CM peptides preferentially bind T-cells over B-cells. Splenocytes derived from C57BL/6J mice were incubated with rhodamine-labeled peptides. Thereafter, the cells were stained with either phycoerythrin-conjugated anti-CD3 or anti-B220 antibody and analyzed by flow cytometry. A, defining a lymphocyte gate using forward (FS) and side (SS) scatter analysis on splenocytes. B, defining a rhodamine-labeled lymphocyte gate. C, the percentage of T-cells and B-cells in the rhodamine-labeled cells was analyzed using anti-CD3 and anti-B220 antibody, respectively.
ISLAD l-CM and ISLAD d-CM Co-localize with TCR
The ability of the ISLAD CM peptides to inhibit T-cell proliferation was suggested to stem from their interaction with the TCR (30). Given that the ISLAD l-CM and ISLAD d-CM peptides are derived from ISLAD, we suspected that they interact with the TCR as well. Confocal microscopy was utilized to assess whether the peptides co-localized with the TCR (Fig. 4). Activated mMOG(35–55) T-cells were incubated with Rho-labeled peptides (denoted in red) and thereafter stained with anti-TCRα primary antibody, followed by FITC-labeled secondary antibody (denoted in green). Co-localization is shown in the merged panels, indicating the proximity of the two fluorophores on the cell membrane. Fig. 4 shows that both ISLAD CM forms co-localized with the TCR in a cap shape, which is often found in activated T-cells, compared with a control non-related antimicrobial peptide, which was all over the cell membrane.
FIGURE 4.
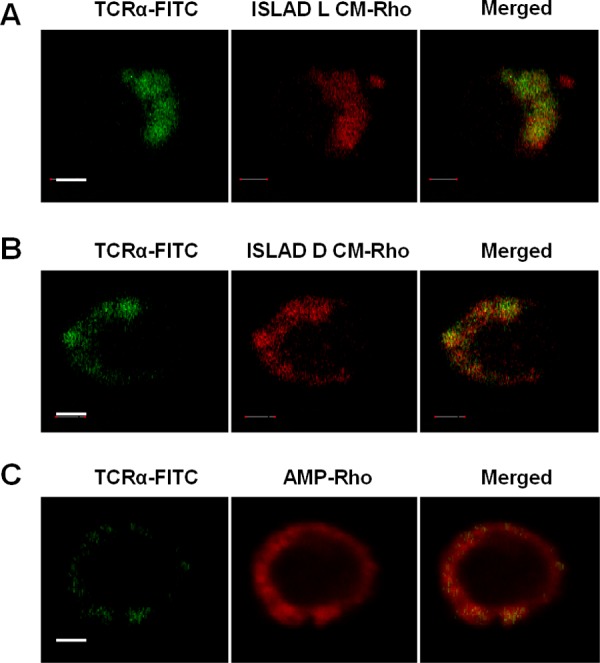
Both ISLAD CM forms co-localize with the TCR. Activated mMOG(35–55) T-cells were probed with anti-TCRα antibody, followed by staining with FITC-labeled secondary antibody (left panels, green) and with rhodamine-labeled fluorescent peptides (ISLAD l-CM-Rho, ISLAD d-CM-Rho, and a control antimicrobial peptide (AMP-Rho); middle panels, red). The merged images of the molecules are shown (right panels). Scale bars = 2 μm.
ISLAD CM Peptides Interact with TCRα TMD
The ability of ISLAD CM peptides to inhibit T-cell activation could be attributed to their interaction with one or more cellular counterparts. To characterize the interaction of the peptides with T-cell proteins, T-cells were incubated with peptides that were fluorescently labeled with Rho and then cross-linked. The cross-linked cells were lysed, and the proteins were subjected to SDS-PAGE (Fig. 5A). The protein-peptide complexes are evident by the presence of fluorescent bands on the gel. Remarkably, full-length ISLAD, ISLAD l-CM, and ISLAD d-CM all presented one main fluorescent band, which is about the same size of the TCR heterodimer (∼70 kDa). This band was not detected without the addition of any peptide or with the addition of a d-enantiomer of CP2G, an established negative control for binding studies with the TCR (42).
FIGURE 5.

ISLAD CM and its d-enantiomer interact with the TCR. A, biochemical analysis of peptide interaction with T-cell proteins. Jurkat T-cells were incubated with fluorescently labeled peptides (ISLAD l-CM-Rho, ISLAD d-CM-Rho, ISLAD-Rho, and a TCR d-CP2G-Rho control peptide), cross-linked, and lysed. The T-cell proteins were resolved by SDS-PAGE, and proteins bound to Rho-labeled peptides were detected by the fluorescence of rhodamine (the ladder of protein sizes is indicated in kDa). Subsequently, the gel was transferred to a membrane and subjected to Western blotting for actin. B, Jurkat T-cells were incubated with ISLAD l-CM-Rho, ISLAD d-CM-Rho, and TCR d-CP2G-Rho. The cells were then cross-linked, lysed, and immunoprecipitated with anti-TCRα antibody. Bound proteins were separated by SDS-PAGE and analyzed for the presence of the fluorescently labeled peptides. The results are presented as the mean -fold of fluorescence intensity relative to the control (TCR d-CP2G-Rho) ± S.D. (n = 2). *, p < 0.05. C, fluorescence measurements of peptide-membrane interactions. ISLAD l-CM (□) and ISLAD d-CM (■) were titrated with increasing concentrations of phosphatidylcholine:cholesterol (9:1) LUVs, and changes in the fluorescence anisotropy (arbitrary units (a.u)) of their intrinsic Trp residue were measured. The fitting curve from the nonlinear least-squares model is presented (Equation 1), which indicates the membrane binding affinity constant. D, d-enantiomer CM interaction with the TCRα TMD. FRET experiments used (i) TCR l-CP-NBD and its mutant TCR l-CP2G-NBD as fluorescent donors and (ii) ISLAD l-CM-Rho and ISLAD d-CM-Rho as acceptors. Spectra were obtained with excitation set at 467 nm and an emission scan at 500–600 nm. An NBD-labeled peptide was added to phosphatidylcholine:cholesterol (9:1) LUVs in PBS to a final concentration of 0.1 μm. This was followed by the addition of Rho-labeled peptides in several sequential doses. A nonlinear regression plot of FRET efficiency (arbitrary units) is presented by the relative percentage of emission at 530 nm between the NBD-labeled peptides alone and the peptides at different Rho/NBD-labeled ratios. ▴, ISLAD l-CM-Rho with TCR l-CP-NBD; ■, ISLAD d-CM-Rho with TCR l-CP-NBD; ▵, ISLAD l-CM-Rho with TCR l-CP2G-NBD; □, ISLAD d-CM-Rho with TCR l-CP2G-NBD.
TCR d-CP2G is a mutant of the d-enantiomer core peptide (d-CP) with Lys/Gly and Arg/Gly mutations. The CP is derived from the TCRα TMD and is known to interact with the TMD members of the TCR complex and to inhibit T-cell activation (35, 43). Due to the charge mutation, CP2G is unable to interact and inhibit the TCR and is therefore used as a negative control (35). SDS-PAGE was followed by an immunoprecipitation experiment using rhodamine-labeled peptides with the TCR, which validated that ISLAD l-CM and ISLAD d-CM bind the TCR (Fig. 5B).
The signaling cascade of the TCR complex is mediated by the interaction of the basic amino acids in its helical TMD with the acidic amino acids of the CD3 TMD helices (32, 44). ISLAD CM peptides are helical in the membrane and possess acidic residues as well as Trp residues that facilitate membrane binding. Thus, we investigated their interaction with the TCRα TMD (represented by the established TCR l-CP sequence) (Table 1). First, we examined the ability of the CM peptides to interact with the membrane utilizing the fluorescence anisotropy of their intrinsic Trp residue in the presence of LUV model membranes. ISLAD CM forms were titrated with increasing concentrations of LUVs, and the change in the fluorescence of their intrinsic Trp residue was measured. Fig. 5C demonstrates that both of the peptides bound the membrane with similar affinity (affinity constants of (1.7 ± 0.3) × 103 m−1 and (1.3 ± 0.07) × 103 m−1 for ISLAD l-CM and ISLAD d-CM, respectively; n = 2).
Once the peptides were shown to equally bind the membrane, the ability of the ISLAD CM peptides to interact with the TCRα CP was assessed by a FRET experiment in the membrane. The energy transfer between the donor NBD-labeled wild-type or mutant CP and the acceptor Rho-labeled ISLAD CM peptides was measured in phosphatidylcholine:cholesterol (9:1) LUVs. Fig. 5D shows similar FRET between both ISLAD CM forms and the CP. Curiously, FRET between the CM peptides and the mutant TCR TMD (CP2G) was considerably weaker compared with the wild-type CP. This indicates that the Arg and Lys residues of the TCRα TMD are involved in the interaction with ISLAD l-CM and ISLAD d-CM.
Administration of ISLAD d-CM to MOG(35–55)-immunized C57BL/6J Mice Down-regulates Pathogenic MOG(35–55) T-cells
Previously, ISLAD was shown to inhibit pathogenic T-cell activation ex vivo (30). The present results showed that the d-enantiomer form of ISLAD CM inhibited T-cell proliferation in vitro. Thus, we investigated whether the corresponding effect could take place ex vivo. The EAE model of multiple sclerosis is well established for investigating T-cell responses (45). PBS or ISLAD d-CM (0.5 mg/kg) was administrated to MOG(35–55)-immunized C57BL/6J mice. Ten days following immunization, the lymph node cells (LNCs) were analyzed ex vivo for their recall proliferative response to the immunizing MOG(35–55) peptide. LNCs derived from mice that were treated with ISLAD d-CM exhibited a significant reduction in T-cell proliferation (72% inhibition) compared with PBS-treated mice (Fig. 6A). Moreover, treatment with ISLAD d-CM resulted in a decrease in proinflammatory cytokine secretion. Fig. 6 (B and C) shows that compared with LNCs from PBS-treated mice, LNCs from mice treated with ISLAD d-CM showed a significant reduction in INFγ secretion (65% reduction) (Fig. 6B) and a 60% reduction in IL-17 secretion (Fig. 6C).
FIGURE 6.
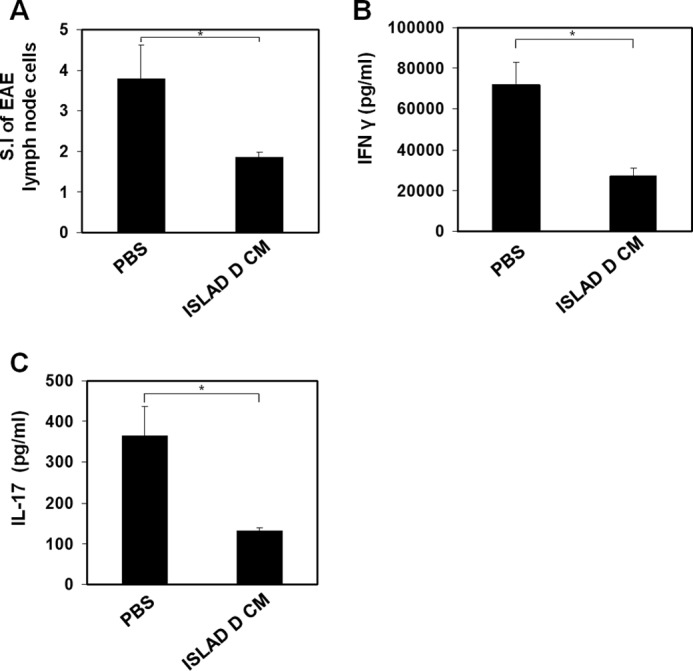
Administering ISLAD d CM to EAE mice down-regulates pathogenic T-cells. A, ex vivo down-regulation of encephalitogenic MOG(35–55) T-cells following ISLAD d-CM administration. C57BL/6J mice were injected for EAE induction with the MOG(35–55) peptide. Ten days after immunization, draining LNCs from each treatment group (n = four mice per group) were pooled and analyzed for their ex vivo recall proliferative response to MOG(35–55). The LNCs from each treatment group (ISLAD d-CM or PBS) were cultured for 72 h in microtiter wells in triplicate (0.5 × 106/well) in the absence or presence of MOG(35–55) (5 μg/ml). [3H]Thymidine was added for the last 18 h. The results are expressed as the stimulation index (S.I; mean cpm of antigen-containing cultures/mean cpm of medium-containing cultures). B and C, secretion of proinflammatory cytokines from cultured LNCs derived from PBS- or ISLAD d-CM-administered EAE mice. LNCs from each treatment group were cultured (5 × 106/ml) in the absence or presence of MOG(35–55) (5 μg/ml) for 24 h, and the supernatants were collected to detect the secreted IFNγ (B) and IL-17 (C) by ELISA. The results are the mean ± S.D. from a representative experiment. *, p < 0.05.
DISCUSSION
One of the ways in which HIV-1 evades the immune response is by T-cell inactivation during the membrane fusion event, mediated by the Env gp41 fusion protein. Recently, an immunosuppressive motif was found in the hydrophobic portion of gp41 associated with the loop region (ISLAD CM) (30). Here, we describe new insights into the mode of action of ISLAD CM in living cells. We show that ISLAD CM preferentially binds T-cells over B-cells (by 13-fold), interacts with the TCR complex within the membrane milieu, and thereby inhibits T-cell activation in vitro and ex vivo.
Intermolecular interactions between helices of soluble proteins are sterically dependent. However, recent studies show that the surrounding membrane is capable of leniency toward interactions between the d- and l-helices (34). Therefore, peptides composed of d-amino acids are being used as a tool to study TMD-TMD interactions of membrane-embedded receptors, as in the case of the glycophorin A and the bacterial aspartate receptor (33, 46). Furthermore, exogenously added TMD peptides can be used to investigate the assembly of membrane proteins in vivo by interacting with their corresponding segments within the membrane (43, 47–50).
The interactions between the TCR complex TMD members are crucial for the activation of the complex. These interactions are mediated by hydrogen bonds between conserved acidic and basic amino acids incorporated in the TMDs of CD3 and the TCR, respectively (31, 32). In this study, we have shown that in hydrophobic environment, ISLAD d-CM creates a mirror image helical structure of ISLAD l-CM, both of which can interact with the TCR TMD. Furthermore, both ISLAD CM forms interact with the wild-type TCRα TMD but not with the mutant TMD comprising Gly substitutions for its basic residues (Arg and Lys). Moreover, in support of our results here, we previously showed that Gly substitutions for ISLAD acidic residues prevent interaction with the wild-type TCRα TMD (30). This suggests sequence specificity, which involves the interaction between basic residues of the TCRα TMD and acidic residues of ISLAD CM. Another parameter that facilitates the immunosuppressive activity of ISLAD is the ability to adopt an α-helical structure in the membrane. The formation of a helical structure in the membrane is governed by the location of the amino acids within the motif, as scrambling them reduced both the helical content and immunosuppressive activity of the peptide.
Multiple regions within gp41 act together to enhance membrane binding and to facilitate structural changes within the protein that are required for membrane fusion (10, 51, 52). Therefore, the local concentration of the motif needed to exert an immunosuppressive effect might be much lower in the context of intact Env than that achieved with isolated peptides.
The gp41 ISLAD-containing loop region has the potential to bind and insert into the membrane when gp41 forms its hairpin conformation (28, 53, 54). Hence, it is reasonable to assume that the immunosuppressive activity of ISLAD takes place during the late fusion steps. In addition to ISLAD, other hydrophobic gp41 motifs located in the TMD and the fusion peptide bind the TMDs of the TCR complex and inhibit T-cell activation via different mechanisms (22–24). The fusion peptide presumably initiates the immunosuppressive activity of gp41. This happens when the fusion peptide is inserted into the cell membrane in the gp41 pre-hairpin conformation during the early membrane fusion steps (25). During the late fusion steps, when the gp41 hairpin conformation is formed, the gp41 TMD and ISLAD can further interfere with the TCR complex assembly, as postulated in an emerging model (25). This proposed immunosuppressive action of gp41 is carried out in addition to other immunomodulating segments of Env located in gp120 (55, 56) and in the gp41 immunosuppressive unit (57).
It has been shown that a d-enantiomer of the TCRα TMD is capable of interacting with the TMDs of the TCR complex and thereby interferes with the complex activation (35). Hence, chirality-independent interaction in the membrane is a more general phenomenon that is also extended to the TMDs of the TCR complex (34, 35). These observations reinforce our findings here that the ISLAD CM mode of action is interacting with the TMDs of the TCR complex within the membrane milieu.
In the clinical aspect, the efficacy of ISLAD d-CM may have an important application for inhibiting undesired T-cell activation in vivo. This short d-peptide is not cleaved by endogenous proteases and is efficiently incorporated into the membrane vicinity, which may provide a prolonged half-life to exert a desirable therapeutic effect.
Acknowledgments
We thank Batya Zarmi for valuable help with peptide purification and Vladimir Kiss for assistance with confocal imaging.
This work was supported by the Israel Science Foundation.
- APC
- antigen-presenting cell
- TMD
- transmembrane domain
- TCR
- T-cell receptor
- ISLAD
- immunosuppressive loop-associated determinant
- CM
- core motif
- MOG
- myelin oligodendrocyte glycoprotein
- mMOG
- murine MOG
- NBD
- 4-chloro-7-nitrobenzo-2-oxa-1,3-diazole
- Rho
- rhodamine N-hydroxysuccinimide
- EAE
- experimental autoimmune encephalomyelitis
- LUV
- large unilamellar vesicle
- CP
- core peptide
- LNC
- lymph node cell.
REFERENCES
- 1. Barré-Sinoussi F., Chermann J. C., Rey F., Nugeyre M. T., Chamaret S., Gruest J., Dauguet C., Axler-Blin C., Vézinet-Brun F., Rouzioux C., Rozenbaum W., Montagnier L. (1983) Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 220, 868–871 [DOI] [PubMed] [Google Scholar]
- 2. Gallo R. C., Salahuddin S. Z., Popovic M., Shearer G. M., Kaplan M., Haynes B. F., Palker T. J., Redfield R., Oleske J., Safai B. (1984) Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS. Science 224, 500–503 [DOI] [PubMed] [Google Scholar]
- 3. Maltêz F., Doroana M., Branco T., Valente C. (2011) Recent advances in antiretroviral treatment and prevention in HIV-infected patients. Curr. Opin. HIV AIDS 6, S21–S30 [DOI] [PubMed] [Google Scholar]
- 4. Douek D. C., Picker L. J., Koup R. A. (2003) T cell dynamics in HIV-1 infection. Annu. Rev. Immunol. 21, 265–304 [DOI] [PubMed] [Google Scholar]
- 5. Grossman Z., Meier-Schellersheim M., Sousa A. E., Victorino R. M., Paul W. E. (2002) CD4+ T-cell depletion in HIV infection: are we closer to understanding the cause? Nat. Med. 8, 319–323 [DOI] [PubMed] [Google Scholar]
- 6. Stevenson M. (2003) HIV-1 pathogenesis. Nat. Med. 9, 853–860 [DOI] [PubMed] [Google Scholar]
- 7. Baur A. S., Sawai E. T., Dazin P., Fantl W. J., Cheng-Mayer C., Peterlin B. M. (1994) HIV-1 Nef leads to inhibition or activation of T cells depending on its intracellular localization. Immunity 1, 373–384 [DOI] [PubMed] [Google Scholar]
- 8. Wu Y., Marsh J. W. (2001) Selective transcription and modulation of resting T cell activity by preintegrated HIV DNA. Science 293, 1503–1506 [DOI] [PubMed] [Google Scholar]
- 9. Kirchhoff F. (2010) Immune evasion and counteraction of restriction factors by HIV-1 and other primate lentiviruses. Cell Host Microbe 8, 55–67 [DOI] [PubMed] [Google Scholar]
- 10. Eckert D. M., Kim P. S. (2001) Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 70, 777–810 [DOI] [PubMed] [Google Scholar]
- 11. Harrison S. C. (2008) Viral membrane fusion. Nat. Struct. Mol. Biol. 15, 690–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weiss A., Imboden J., Hardy K., Manger B., Terhorst C., Stobo J. (1986) The role of the T3/antigen receptor complex in T-cell activation. Annu. Rev. Immunol. 4, 593–619 [DOI] [PubMed] [Google Scholar]
- 13. Huppa J. B., Davis M. M. (2003) T-cell-antigen recognition and the immunological synapse. Nat. Rev. Immunol. 3, 973–983 [DOI] [PubMed] [Google Scholar]
- 14. Zhang R., Lifson J. D., Chougnet C. (2006) Failure of HIV-exposed CD4+ T cells to activate dendritic cells is reversed by restoration of CD40/CD154 interactions. Blood 107, 1989–1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fernando K., Hu H., Ni H., Hoxie J. A., Weissman D. (2007) Vaccine-delivered HIV envelope inhibits CD4+ T-cell activation, a mechanism for poor HIV vaccine responses. Blood 109, 2538–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Finzi A., Xiang S. H., Pacheco B., Wang L., Haight J., Kassa A., Danek B., Pancera M., Kwong P. D., Sodroski J. (2010) Topological layers in the HIV-1 gp120 inner domain regulate gp41 interaction and CD4-triggered conformational transitions. Mol. Cell 37, 656–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chan D. C., Fass D., Berger J. M., Kim P. S. (1997) Core structure of gp41 from the HIV envelope glycoprotein. Cell 89, 263–273 [DOI] [PubMed] [Google Scholar]
- 18. Weissenhorn W., Dessen A., Harrison S. C., Skehel J. J., Wiley D. C. (1997) Atomic structure of the ectodomain from HIV-1 gp41. Nature 387, 426–430 [DOI] [PubMed] [Google Scholar]
- 19. Gallaher W. R., Ball J. M., Garry R. F., Griffin M. C., Montelaro R. C. (1989) A general model for the transmembrane proteins of HIV and other retroviruses. AIDS Res.. Hum Retroviruses 5, 431–440 [DOI] [PubMed] [Google Scholar]
- 20. Freed E. O., Myers D. J., Risser R. (1990) Characterization of the fusion domain of the human immunodeficiency virus type 1 envelope glycoprotein gp41. Proc. Natl. Acad. Sci. U.S.A. 87, 4650–4654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shang L., Yue L., Hunter E. (2008) Role of the membrane-spanning domain of human immunodeficiency virus type 1 envelope glycoprotein in cell-cell fusion and virus infection. J. Virol. 82, 5417–5428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Quintana F. J., Gerber D., Kent S. C., Cohen I. R., Shai Y. (2005) HIV-1 fusion peptide targets the TCR and inhibits antigen-specific T cell activation. J. Clin. Invest. 115, 2149–2158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bloch I., Quintana F. J., Gerber D., Cohen T., Cohen I. R., Shai Y. (2007) T-cell inactivation and immunosuppressive activity induced by HIV gp41 via novel interacting motif. FASEB J. 21, 393–401 [DOI] [PubMed] [Google Scholar]
- 24. Cohen T., Cohen S. J., Antonovsky N., Cohen I. R., Shai Y. (2010) HIV-1 gp41 and TCRα transmembrane domains share a motif exploited by the HIV virus to modulate T-cell proliferation. PLoS Pathog. 6, e1001085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ashkenazi A., Faingold O., Shai Y. (2013) HIV-1 fusion protein exerts complex immunosuppressive effects. Trends Biochem. Sci. 38, 345–349 [DOI] [PubMed] [Google Scholar]
- 26. McDonald D., Wu L., Bohks S. M., KewalRamani V. N., Unutmaz D., Hope T. J. (2003) Recruitment of HIV and its receptors to dendritic cell-T cell junctions. Science 300, 1295–1297 [DOI] [PubMed] [Google Scholar]
- 27. Caffrey M., Cai M., Kaufman J., Stahl S. J., Wingfield P. T., Covell D. G., Gronenborn A. M., Clore G. M. (1998) Three-dimensional solution structure of the 44 kDa ectodomain of SIV gp41. EMBO J. 17, 4572–4584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bär S., Alizon M. (2004) Role of the ectodomain of the gp41 transmembrane envelope protein of human immunodeficiency virus type 1 in late steps of the membrane fusion process. J. Virol. 78, 811–820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ashkenazi A., Viard M., Wexler-Cohen Y., Blumenthal R., Shai Y. (2011) Viral envelope protein folding and membrane hemifusion are enhanced by the conserved loop region of HIV-1 gp41. FASEB J. 25, 2156–2166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ashkenazi A., Faingold O., Kaushansky N., Ben-Nun A., Shai Y. (2013) A highly conserved sequence associated with the HIV gp41 loop region is an immunomodulator of antigen-specific T cells in mice. Blood 121, 2244–2252 [DOI] [PubMed] [Google Scholar]
- 31. Manolios N., Bonifacino J. S., Klausner R. D. (1990) Transmembrane helical interactions and the assembly of the T cell receptor complex. Science 249, 274–277 [DOI] [PubMed] [Google Scholar]
- 32. Call M. E., Pyrdol J., Wiedmann M., Wucherpfennig K. W. (2002) The organizing principle in the formation of the T cell receptor-CD3 complex. Cell 111, 967–979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gerber D., Shai Y. (2002) Chirality-independent protein-protein recognition between transmembrane domains in vivo. J. Mol. Biol. 322, 491–495 [DOI] [PubMed] [Google Scholar]
- 34. Fink A., Sal-Man N., Gerber D., Shai Y. (2012) Transmembrane domains interactions within the membrane milieu: principles, advances and challenges. Biochim. Biophys. Acta 1818, 974–983 [DOI] [PubMed] [Google Scholar]
- 35. Gerber D., Quintana F. J., Bloch I., Cohen I. R., Shai Y. (2005) d-Enantiomer peptide of the TCRα transmembrane domain inhibits T-cell activation in vitro and in vivo. FASEB J. 19, 1190–1192 [DOI] [PubMed] [Google Scholar]
- 36. Ben-Nun A., Lando Z. (1983) Detection of autoimmune cells proliferating to myelin basic protein and selection of T cell lines that mediate experimental autoimmune encephalomyelitis (EAE) in mice. J. Immunol. 130, 1205–1209 [PubMed] [Google Scholar]
- 37. Weiss A., Wiskocil R. L., Stobo J. D. (1984) The role of T3 surface molecules in the activation of human T cells: a two-stimulus requirement for IL 2 production reflects events occurring at a pre-translational level. J. Immunol. 133, 123–128 [PubMed] [Google Scholar]
- 38. Merrifield R. B., Vizioli L. D., Boman H. G. (1982) Synthesis of the antibacterial peptide cecropin A(1–33). Biochemistry 21, 5020–5031 [DOI] [PubMed] [Google Scholar]
- 39. Ben-Nun A., Kerlero de Rosbo N., Kaushansky N., Eisenstein M., Cohen L., Kaye J. F., Mendel I. (2006) Anatomy of T cell autoimmunity to myelin oligodendrocyte glycoprotein (MOG): prime role of MOG44F in selection and control of MOG-reactive T cells in H-2b mice. Eur. J. Immunol. 36, 478–493 [DOI] [PubMed] [Google Scholar]
- 40. Kaushansky N., Zilkha-Falb R., Hemo R., Lassman H., Eisenstein M., Sas A., Ben-Nun A. (2007) Pathogenic T cells in MOBP-induced murine EAE are predominantly focused to recognition of MOBP21F and MOBP27P epitopic residues. Eur. J. Immunol. 37, 3281–3292 [DOI] [PubMed] [Google Scholar]
- 41. Cohen T., Pevsner-Fischer M., Cohen N., Cohen I. R., Shai Y. (2008) Characterization of the interacting domain of the HIV-1 fusion peptide with the transmembrane domain of the T-cell receptor. Biochemistry 47, 4826–4833 [DOI] [PubMed] [Google Scholar]
- 42. Quintana F. J., Gerber D., Bloch I., Cohen I. R., Shai Y. (2007) A structurally altered d,l-amino acid TCRα transmembrane peptide interacts with the TCRα and inhibits T-cell activation in vitro and in an animal model. Biochemistry 46, 2317–2325 [DOI] [PubMed] [Google Scholar]
- 43. Manolios N., Collier S., Taylor J., Pollard J., Harrison L. C., Bender V. (1997) T-cell antigen receptor transmembrane peptides modulate T-cell function and T cell-mediated disease. Nat. Med. 3, 84–88 [DOI] [PubMed] [Google Scholar]
- 44. Cosson P., Lankford S. P., Bonifacino J. S., Klausner R. D. (1991) Membrane protein association by potential intramembrane charge pairs. Nature 351, 414–416 [DOI] [PubMed] [Google Scholar]
- 45. Sospedra M., Martin R. (2005) Immunology of multiple sclerosis. Annu. Rev. Immunol. 23, 683–747 [DOI] [PubMed] [Google Scholar]
- 46. Sal-Man N., Gerber D., Shai Y. (2004) Hetero-assembly between all-l- and all-d-amino acid transmembrane domains: forces involved and implication for inactivation of membrane proteins. J. Mol. Biol. 344, 855–864 [DOI] [PubMed] [Google Scholar]
- 47. Fink A., Reuven E. M., Arnusch C. J., Shmuel-Galia L., Antonovsky N., Shai Y. (2013) Assembly of the TLR2/6 transmembrane domains is essential for activation and is a target for prevention of sepsis. J. Immunol. 190, 6410–6422 [DOI] [PubMed] [Google Scholar]
- 48. Rath A., Johnson R. M., Deber C. M. (2007) Peptides as transmembrane segments: decrypting the determinants for helix-helix interactions in membrane proteins. Biopolymers 88, 217–232 [DOI] [PubMed] [Google Scholar]
- 49. Bennasroune A., Fickova M., Gardin A., Dirrig-Grosch S., Aunis D., Crémel G., Hubert P. (2004) Transmembrane peptides as inhibitors of ErbB receptor signaling. Mol. Biol. Cell 15, 3464–3474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chen L., Merzlyakov M., Cohen T., Shai Y., Hristova K. (2009) Energetics of ErbB1 transmembrane domain dimerization in lipid bilayers. Biophys. J. 96, 4622–4630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sackett K., Shai Y. (2003) How structure correlates to function for membrane associated HIV-1 gp41 constructs corresponding to the N-terminal half of the ectodomain. J. Mol. Biol. 333, 47–58 [DOI] [PubMed] [Google Scholar]
- 52. Melikyan G. B., Markosyan R. M., Hemmati H., Delmedico M. K., Lambert D. M., Cohen F. S. (2000) Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J. Cell Biol. 151, 413–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ashkenazi A., Merklinger E., Shai Y. (2012) Intramolecular interactions within the human immunodeficiency virus-1 gp41 loop region and their involvement in lipid merging. Biochemistry 51, 6981–6989 [DOI] [PubMed] [Google Scholar]
- 54. Pascual R., Moreno M. R., Villalaín J. (2005) A peptide pertaining to the loop segment of human immunodeficiency virus gp41 binds and interacts with model biomembranes: implications for the fusion mechanism. J. Virol. 79, 5142–5152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Masci A. M., Galgani M., Cassano S., De Simone S., Gallo A., De Rosa V., Zappacosta S., Racioppi L. (2003) HIV-1 gp120 induces anergy in naive T lymphocytes through CD4-independent protein kinase-A-mediated signaling. J. Leukoc. Biol. 74, 1117–1124 [DOI] [PubMed] [Google Scholar]
- 56. Chirmule N., McCloskey T. W., Hu R., Kalyanaraman V. S., Pahwa S. (1995) HIV gp120 inhibits T cell activation by interfering with expression of costimulatory molecules CD40 ligand and CD80 (B71). J. Immunol. 155, 917–924 [PubMed] [Google Scholar]
- 57. Ruegg C. L., Monell C. R., Strand M. (1989) Inhibition of lymphoproliferation by a synthetic peptide with sequence identity to gp41 of human immunodeficiency virus type 1. J. Virol. 63, 3257–3260 [DOI] [PMC free article] [PubMed] [Google Scholar]