

Background: LILRA3 is a soluble receptor with unknown functions that is abundantly present in human serum.
Results: Optimally glycosylated recombinant LILRA3 protein produced only in a mammalian system binds potential ligands and suppresses monocyte function.
Conclusion: LILRA3 suppresses LPS-mediated TNF production, suggesting that it is a new anti-inflammatory protein.
Significance: This work provides first insight into the biochemical characteristics and functions of LILRA3.
Keywords: Glycoprotein, Glycosylation, Leukocyte, Mass Spectrometry (MS), Monocytes, Protein Deamidation, Protein Expression, Protein Purification, Recombinant Protein Expression, Tumor Necrosis Factor (TNF)
Abstract
The leukocyte immunoglobulin-like receptor (LILR) A3 is a member of the highly homologous activating and inhibitory receptors expressed on leukocytes. LILRA3 is a soluble receptor of unknown functions but is predicted to act as a broad antagonist to other membrane-bound LILRs. Functions of LILRA3 are unclear primarily because of the lack of high quality functional recombinant protein and insufficient knowledge regarding its ligand(s). Here, we expressed and characterized recombinant LILRA3 (rLILRA3) proteins produced in 293T cells, Escherichia coli, and Pichia pastoris. We found that the purified rLILRA3 produced in the mammalian system was the same size as a 70-kDa native macrophage LILRA3. This is 20 kDa larger than the calculated size, suggesting significant post-translational modifications. In contrast, rLILRA3 produced in E. coli was similar in size to the unprocessed protein, but yeast-produced protein was 2–4 times larger than the unprocessed protein. Treatment with peptide-N-glycosidase F reduced the size of the mammalian cell- and yeast-produced rLILRA3 to 50 kDa, suggesting that most modifications are due to glycosylation. Consistent with this, mass spectrometric analysis of the mammalian rLILRA3 revealed canonical N-glycosylation at the predicted Asn140, Asn281, Asn302, Asn341, and Asn431 sites. Functionally, only mammalian cell-expressed rLILRA3 bound onto the surface of monocytes with high affinity, and importantly, only this significantly abrogated LPS-induced TNFα production by monocytes. Binding to monocytes was partially blocked by β-lactose, indicating that optimally glycosylated LILRA3 might be critical for ligand binding and function. Overall, our data demonstrated for the first time that LILRA3 is a potential new anti-inflammatory protein, and optimal glycosylation is required for its functions.
Introduction
Leukocyte immunoglobulin-like receptor (LILR)4 A3 belongs to a family of highly homologous activating and inhibitory receptors that are primarily co-expressed on monomyeloid leukocytes and are increasingly recognized to regulate innate immune responses (1). Activating LILRs (LILRAs) have a short cytoplasmic tail that links to the intracellular immunoreceptor tyrosine-based activation motifs of the common γ chain of Fc receptors and transduces activating signals via protein tyrosine kinases (2). Inhibitory LILRs (LILRB) have a long cytoplasmic tail that contains two to four immunoreceptor tyrosine-based inhibitory motifs that recruit SH2-containing inhibitory phosphatases such as SHP-1 (3). Co-engagement by shared ligands may regulate the threshold and amplitude of leukocyte activation. LILRA3 is a soluble protein with unknown function but bears close sequence homology to the extracellular domains of activating LILRA1 and LILRA2 and thus may act as a soluble antagonist (4, 5). We showed previously that LILRA3 is abundantly present in normal serum and is significantly increased in sera of patients with rheumatoid arthritis (6). We proposed that LILRA3 may antagonize the effects of LILRA2, which is increased in highly inflamed synovial tissue (7). Interestingly, absence of a functional allele due to a natural gene deletion (4) has been shown to be strongly associated with increased incidence of multiple sclerosis (8, 9) and Sjögren syndrome (10), both characterized by excessive inflammation, suggesting that LILRA3 may play a key role in the pathogenesis of chronic inflammatory diseases. Consistent with this, LILRA3 is strongly up-regulated by the anti-inflammatory cytokine IL-10 and down-regulated by proinflammatory cytokine TNFα (6). However, the exact in vivo and in vitro functions of LILRA3 are poorly established primarily due to inadequate knowledge about its ligand(s). The major impediments for comprehensive identification of high affinity LILR ligand(s) and understanding functions in vivo are the lack of properly folded and post-translationally modified, full-length recombinant LILR proteins and absence of robust protocols capable of simultaneously identifying ligands and/or co-ligands. The former is particularly relevant because LILRs are predicted to be highly glycosylated and have multiple disulfide bonds; thus, recombinant LILRs produced in non-eukaryotic cells are likely to be unsuitable for functional assays.
In this study, we produced high quality, properly folded, full-length rLILRA3 protein with or without C-terminal a placental alkaline phosphatase tag in a mammalian system using 293T cells. More importantly, rLILRA3 protein produced in 293T cells was successfully used to screen specific binding of this protein to various cell types. We show for the first time that LILRA3 strongly and specifically bound onto the surface of the monocytic cell line U937 and primary peripheral blood monocytes, suggesting expression of LILRA3 ligand(s) on these cells. Moreover, treatment of primary monocytes with purified mammalian recombinant LILRA3 significantly suppressed LPS-mediated TNFα production, indicating functional interaction of LILRA3 with its yet uncharacterized ligand. By contrast, full-length rLILRA3 produced in bacteria or yeast had poor binding and failed to suppress LPS-induced activation of monocytes. This variability in binding and function might be due to the optimal post-translational modification of the rLILRA3 produced in 293T cells. Indeed, rLILRA3 protein produced in 293T cells showed five N-glycosylation sites that likely have contributed to its superior ability to functionally bind to its potential ligand(s). Consistent with this finding, pretreatment of cells with β-lactose partially abrogated binding of rLILRA3 to the surface of monocytes. Taken together, recombinant LILRA3 that can be used for screening and identification of its ligand(s) and characterization of functions is best produced in higher eukaryote expression systems.
EXPERIMENTAL PROCEDURES
Cloning of LILRA3 cDNA into Mammalian, Bacterial, and Yeast Expression Vectors
Full-length LILRA3 cDNA was amplified from PBMC mRNA and inserted into pCR2.1 vector (Invitrogen). This was used for further subcloning into mammalian (pAPtag-5, GenHunter), bacterial (pET30/LIC, Novagen), and yeast (pPICZβ, Invitrogen) expression vectors. In brief, a full-length LILRA3 without signal peptide was reamplified using 5′-AAGCTTTAAGGACCCACGTGCAGGCAGG-3′ forward primer and 5′-AAGCTTCCCACTCACCAGCCTTGGAGTC-3′ reverse primer containing HindIII restriction sites. This was inserted into HindIII-digested pAPtag-5 (GenHunter) vector to generate mammalian rLILRA3 protein with heat-resistant placental alkaline phosphatase and His6 tags on its C terminus (rLILRA3-APtag-His). Mammalian rLILRA3-His protein without APtag was generated by introduction of a new His6 sequence and a stop codon using 5′-CCGAAGCTTTAAGGACCCACGT-3′ forward primer and 5′-GGCCTCGAGTCAATGATGATGATGATGATGCTCACCAGCCTTGGAG-3′ reverse primer and directionally subcloned to pAPtag-5 vector, linearized with HindIII and XhoI. To generate rLILRA3-His protein in Escherichia coli, rLILRA3 without signal peptide was amplified using 5′-GACGACGACAAGACCAGGACCCACGTG-3′ forward primer and 5′-GAGGAGAAGCCCGGTCACCAGCCTTGG-3′ reverse primer and ligated into pET30 EK/LIC expression vector (Novagen). rLILRA3-His was produced in yeast after subcloning LILRA3 without signal peptide from pPIC9k vector using 5′-CCGCTCGAGAAAAGAGGGCCCCTCCCCAAGCCC-3′ forward primer and 5′-CCACTCGTAGTAGTAGTAGTAGTAACTGAGCTCCGG-3′ reverse primer and inserted into pPICZβ expression vector at the XhoI cloning site. See supplemental Fig. 1 for illustration of the recombinant proteins.
Production of Secreted Recombinant LILRA3 and Placental Alkaline Phosphatase in a Mammalian System
The LILRA3 with or without APtag in pAPtag-5 vector alone was stably transfected into the 293T cell line using Lipofectamine LTX reagent (Invitrogen), cultured in DMEM +10% FBS, and selected with 300 μg/ml Zeocin (Invitrogen). During each weekly passage, secreted recombinant LILRA3-APtag-His and APtag-His alone were detected in culture supernatants using a simple alkaline phosphatase activity assay as described (11). Secreted fusion proteins were further confirmed by Western blotting 15 μl of culture supernatants using mouse anti-human placental alkaline phosphatase mAb (GenHunter). Production of rLILRA3-His without APtag protein was detected in culture supernatants by Western blotting using mouse anti-human LILRA3 mAb (Abcam). Cells that produced a high level of recombinant LILRA3 proteins were gradually adapted to DMEM containing 1% FBS, and selection was maintained with 30 μg/ml Zeocin. Cells were then grown to confluence in 1 liter of the serum-minimized medium, recombinant protein-containing culture supernatants were collected, and debris was removed by high speed centrifugation at 330 × g for 30 min at 4 °C followed by filtration with 0.22-μm filters. Culture supernatants were then buffer-exchanged and concentrated to 150 ml in binding buffer (20 mm Tris, pH 7.4, 150 mm NaCl, 5 mm imidazole) using an Amicon ultrafiltration system with a 30-kDa-cutoff membrane (Millipore). Buffer-exchanged proteins were loaded onto 1 ml of cobalt-immobilized metal affinity resin (Clontech) and connected to a BioLogic DuoFlow FPLC (Bio-Rad). The column was then stringently washed at a flow rate of 2 ml/min with 20 bed volumes of 20 mm Tris, pH 7.4, 150 mm NaCl (wash buffer) containing 10 mm imidazole for the APtag-His column or wash buffer containing 20 mm imidazole for rLILRA3-APtag-His and rLILRA3-His columns. Finally proteins were stepwise eluted with 5 × 2-ml fractions of 20 mm Tris, pH 7.4, 300 mm NaCl elution buffers containing 50, 150, and 300 mm imidazole. rLILRA3-APtag-His and rAPtag-His proteins in each eluted fraction were quantitated by comparing placental alkaline phosphatase (AP) activity using AP standards (Sigma), and the purified rLILRA3 without APtag was quantitated using a standard BCA assay (Pierce). Proteins were further quality-controlled by silver staining of SDS-PAGE and Western blots, and their identities were verified by mass spectrometry. Fractions that contained a high concentration of high quality proteins were pooled, dialyzed into sterile LPS-minimized TBS (20 mm Tris, 150 mm NaCl, pH 7.4), and requantitated. The resulting estimates of specific activity for the dialyzed rLILRA3-APtag-His and rAPtag-His proteins were 960 and 1500 units/mg, respectively. The concentration of the dialyzed rLILRA3 protein without APtag-His was 0.4 μg/ml. Totals of 750, 1000, and 400 μg of rLILRA3-APtag-His, rAPtag-His, and rLILRA3-His, respectively, were produced from 1 liter of culture supernatants. These proteins were stable at 4 °C for several months.
Production of Recombinant LILRA3 in E. coli
LILRA3 in pET30 EK/LIC in BL21-DE3 with 50 μg/ml kanamycin selection was induced with 0.1 mm isopropyl 1-thio-β-d-galactopyranoside when it reached optimal growth (A600 of 0.7–1.0). After overnight culture, the solubilization and refolding of the recombinant protein from E. coli inclusion bodies were custom optimized by Protein'eXpert (Grenoble, France). In brief, the bacterial cell pellet from 1 liter of culture was lysed by sonication and washed twice with cold TBS, and the inclusion body was solubilized in 40 ml of 50 mm Tris, pH 8.5, 500 mm NaCl, 6 m guanidine, 10 mm β-mercaptoethanol overnight at 4 °C. Solubilized protein was separated by centrifugation at 21,000 × g for 30 min at 4 °C and dialyzed three times against 1 liter of buffer A (50 mm Tris, pH 8.5, 500 mm NaCl, 8 m urea, 1 mm glycine, 10 mm β-mercaptoethanol) each time using a 10,000-dalton-cutoff membrane (Pierce). After dialysis, Sarkosyl was added to a final concentration of 0.3%, and the solution was incubated for 4 h at 4 °C, sonicated five times, and centrifuged at 21,000 × g for 20 min. The soluble fraction was then loaded overnight at 4 °C onto 1-ml cobalt-containing metal affinity resin (Clontech) connected to an FPLC (Bio-Rad). The column was washed with buffer A containing 0.3% Sarkosyl and eluted with TBS buffer containing 150 mm imidazole (5 × 5 ml). Fractions of the eluted proteins with high purity and concentration were pooled (20 ml), dialyzed three times over 12 h against 1 liter of cold buffer B (50 mm Tris, pH 8.5, 500 mm NaCl, 1 m urea, 25 mm CaCl2, 10 mm β-mercaptoethanol, 0.1% Brij35), and finally centrifuged at 21,000 × g for 20 min, and the soluble fraction was collected. This was then refolded by gradual dialysis into four changes (1 liter each) of cold 50 mm Tris, pH 8.5, 200 mm NaCl, 1 mm DTT over a period of 18 h and centrifuged at 21,000 × g for 45 min to remove aggregated protein. The refolded soluble protein was quality-controlled by HPLC and quantitated using the BCA assay. Up to 15 mg of refolded protein was produced from 1 liter of bacterial culture. To prevent oxidation and aggregation, protein was stored under argon gas at −80 °C in small aliquots in silicon-coated sealed vials at 1.3 mg/ml.
Production of Recombinant LILRA3 in Pichia pastoris
LILRA3 in pPICZα-B vector was linearized with the restriction enzyme PmeI and transfected to P. pastoris (X33) by electroporation. Yeast strains showing single crossover recombination were selected to grow on minimal dextrose medium, and transformants showing a Mut+ phenotype were picked and cultured on minimal methanol agar plates. An rLILRA3-expressing colony was selected and grown in BMGY agar (100 mm potassium phosphate, pH 6.0, 1% (w/v) yeast extract, 2% (w/v) peptone, 1.34% (w/v) yeast nitrogen base, 4 × 10−5% (w/v) biotin, 1% (v/v) glycerol, 200 μg/ml Zeocin) at 30 °C until a 1:10 diluted culture had an A600 of 1.6–2.0. The culture medium was changed to BMMY broth (1% (w/v) yeast extract, 2% (w/v) peptone, 100 mm potassium phosphate, pH 6.0, 1.34% (w/v) yeast nitrogen base, 4 × 10−5% (w/v) biotin) and cultured at 30 °C for 24 h. Cells were then induced with 0.5% (v/v) methanol every 24 h until a total of 96 h. The induced culture supernatant was harvested by removing cells with centrifugation at 3000 × g for 20 min at 4 °C and filtration using 0.22-μm-cutoff filter. The filtrate was then diluted 1:1 with binding buffer containing 50 mm sodium phosphate, pH 8.0, 300 mm sodium chloride, 10 mm imidazole and loaded onto a nickel-MAC cartridge column (Novagen) at a flow rate of 1 ml/min (ÄKTA purifier, GE Healthcare). The column was washed with 40 bed volume of 50 mm sodium phosphate, pH 8.0, 300 mm sodium chloride, 20 mm imidazole and eluted with 10 ml of elusion buffer (50 mm sodium phosphate, pH 8.0, 300 mm sodium chloride, 250 mm imidazole). Approximately 250 μg of protein was produced from 1 liter of yeast culture. The protein was stored at 4 °C and used for up to 1 month.
Deglycosylation of Recombinant LILRA3 Produced in 293T and P. pastoris
To determine the extent of LILRA3 glycosylation, the mammalian cell- and yeast-produced recombinant proteins were treated with PNGase F according to the manufacturer's instructions (New England Biolabs). In brief, 2 μl of 10× glycoprotein denaturing buffer was added to 2.5 μg of protein in 20 μl of buffer and incubated at 95 °C for 10 min. 3 μl of G7 reaction buffer, 3 μl of Nonidet P-40, and 1 μl (500 units/μl) of PNGase F were then added to each reaction mixture, and samples were incubated at 37 °C for 1 h. Changes in the deglycosylated rLILRA3 size and isoelectric focusing were determined by Western blotting of membranes from one- and two dimensional SDS-PAGE gels, respectively, and mass spectrometry.
Generation of Primary Human Macrophages in Vitro
Peripheral blood mononuclear cells from two healthy subjects were suspended at 5 × 106/ml in RPMI 1640 medium containing 2 mm l-glutamine, 10 units/ml penicillin, 100 mg/ml streptomycin (all from Invitrogen), and 10% autologous sera and seeded onto 6-well Costar® plates. Cells were incubated at 37 °C in a humidified atmosphere of 95% air and 5% CO2 for 2 h, and non-adherent cells were removed by two washes with phosphate-buffered saline (PBS). The adherent cells were then cultured in 3 ml of medium supplemented with 25 ng/ml GM-CSF (Invitrogen) for 3 days before washing twice in PBS and culturing for another 3 days in culture medium without GM-SCF but containing a 1:1000 dilution of brefeldin A solution (BioLegend, San Diego, CA). Cells were then washed twice with PBS, lysed in radioimmune precipitation assay buffer (1% Nonidet P-40, 0.5% sodium deoxycholate, 50 mm Tris, pH 8, 2 mm EDTA, 0.5 mm sodium orthovanadate, 5 mm sodium fluoride, protease inhibitors), and stored at −80 °C until used for Western blotting to detect native LILRA3.
Western Blotting and Silver Staining of One- and Two-dimensional Gels
For one-dimensional gels, 10 μg of purified recombinant proteins or in vitro derived primary human macrophage (∼1 × 104 cells) lysates were resolved by 10% SDS-PAGE under reducing and non-reducing conditions before and after treatment with PNGase. Gels were either silver-stained or transferred onto PVDF membranes for Western blotting. In brief, PVDF membranes were rinsed with TBS and blocked in 5% skim milk in TBS for 1 h at room temperature. Membranes were then probed with a 1 μg/ml concentration of either mouse anti-LILRA3 mAb (clone 2E9, Abnova) or rabbit anti-AP antibody (GenHunter) for 2 h at room temperature and washed 4 × 5 min with TBS + 0.1% Tween 20. This was followed by incubation of membranes with horseradish peroxidase (HRP)-conjugated anti-mouse or anti-rabbit antibody (Bio-Rad) for 1.5 h at room temperature, 3 × 5-min washes, development with ECL reagent (PerkinElmer Life Sciences), and imaging with ImageQuant LAS 4000 (GE Healthcare). For two-dimensional gel electrophoresis, PNGase F-treated and -untreated rLILRA3 proteins were precipitated with 20% acetone for 1 h at −20 °C. The precipitated proteins were air-dried for 10 min and rehydrated overnight at room temperature with IPG buffer (8 m urea, 2% (w/v) CHAPS, 10 mm DTT). The next day, 2% ampholyte was added to the rehydrated mixture. Isoelectric focusing of the denatured samples was determined by running a 13-cm pH 3–10 Immobiline dry strip (GE Healthcare) for a total of 9350 V. After focusing, the strips were equilibrated in 50 mm Tris, pH 6.8, 6 m urea, 30% glycerol, 2% SDS containing 2% DTT for 10 min. The reduced proteins were alkylated by incubation in the same buffer containing 2.5% iodoacetamide and electrophoretically separated by layering the strips onto 10% polyacrylamide gels, and resolved proteins were visualized by silver staining.
Identification of Glycosylation Sites on the Recombinant Mammalian LILRA3 by Nano-liquid Chromatography Tandem Mass Spectrometry (Nano-LC-MS/MS)
In brief, 10 μg of purified rLILRA3-His from 293T cells was deglycosylated, separated under reducing conditions by 10% SDS-PAGE, and silver-stained. Specific bands were then excised, reduced, alkylated with iodoacetamide, and dehydrated with acetonitrile. In-gel proteins were digested with 2 ng/μl trypsin, 6 ng/μl chymotrypsin, or 6 ng/μl Glu-C. Treated gel bands were incubated in 1% formic acid and 3 × 100 μl of acetonitrile at room temperature. Pooled supernatants from each digest were dried. The dried residues were resuspended in 15 μl of 0.05% heptafluorobutyric anhydride and 1% formic acid and injected into a fritless Nano column (75 μm × 10 cm) of an Ultimate 3000 HPLC and autosampler system (Dionex, Amsterdam, Netherlands). Peptides were then eluted using a linear gradient from mobile phase A (0.1% formic acid in H2O) to mobile phase B (0.1% formic acid in 80% acetonitrile) over 35 min at a flow rate of 0.3 μl/min. Positive ions of tryptic digests were generated by electrospray, and a survey scan (m/z 350–1750) was acquired in the Fourier transform ion cyclotron resonance cell of an LTQ-FT Ultra mass analyzer (Thermo Electron, Bremen, Germany). For chymotrypsin and Glu-C digests, an Orbitrap mass analyzer (Thermo Electron, Bremen, Germany) was used. Peak lists of MS/MS data were generated using Mascot Daemon/extract_msn (Matrix Science, London, UK), interrogated using Mascot version 2.1 (Matrix Science), and searched against Homo sapiens proteins in the Swiss-Prot protein database (version 80). Precursor tolerances were 4.0 ppm, and product ion tolerances were ±0.4 Da. Modifications accounted for were acrylamide (Cys), carbamidomethyl (Cys), deamidation (Asn or Gln), and oxidation (Met) with a maximum of one missed cleavage permitted. Enzyme specificity was trypsin and semitrypsin for tryptic digests, V8-DE and no enzyme for Glu-C digests, and chymotrypsin and no enzyme for chymotryptic digests. Acceptable cutoff scores for individual MS/MS spectra were set to 20. Comparisons of experimental and theoretical tandem mass spectra were automatically performed by Mascot and verified manually. PNGase F-treated and -untreated E. coli-produced rLILRA3 and PNGase F-untreated 293T rLILRA3-His proteins were used as negative controls.
Prediction of N-Glycosylation Sites in LILRA3
The N-glycosylation analysis tool NetNGlyc-1.0 retrieved April 17, 20135 set at a threshold of 0.25–0.5 was used to predict N-linked glycosylation sites in LILRA3.
Quantitative Assay for rLILRA3-APtag-His Binding to Potential Ligand(s) on Cell Surface
To assay for rLILRA3-APtag-His binding, over 25 cell lines from different lineage and peripheral blood leukocyte subsets were initially screened using 100 nm purified protein or control rAPtag-His. We consistently found specific high affinity binding to U937 cells and peripheral blood monocytes. The U937 cells were used for subsequent kinetic studies, competition experiments, and in situ staining. In brief, cell lines were cultured in their suitable media containing 10% FBS, and primary mononuclear leukocytes were purified by density gradient centrifugation followed by negative selection of monocytes, T cells, B cells, and NK cells using magnetic beads (Miltenyi Biotech, Germany) (6). Primary polymorphonuclear cells (>80% neutrophils) were enriched from whole blood by 4.5% dextran in PBS precipitation (Sigma) followed by density gradient centrifugation (13). Cells were washed twice with PBS and once with HBHA buffer (Hanks' balanced salt solution (Sigma) containing 0.5 mg/ml BSA, 0.1% NaN3, 20 mm HEPES, pH 7.0) and transferred to 1.5-ml Eppendorf tubes at 5 × 106/ml of HBHA containing 100 nm rLILRA3-APtag-His or rAPtag-His control. Cells were incubated for 90 min at room temperature followed by four washes with 1.5 ml of cold HBHA and centrifugation at 200 × g at 4 °C for 5 min and aspiration of supernatant. Cell pellets were lysed with 400 μl of 1% Triton X-100 in 10 mm Tris-HCl, pH 8.5. Lysates were then vortexed vigorously, the nuclei were spun out, and soluble lysates were collected. After heat inactivation of endogenous phosphatases at ∼65 °C for 15 min, the soluble lysates were assayed in duplicate for alkaline phosphatase activity as described (11).
For competition assays, 5 × 106 U937 cells in 1 ml of HBHA buffer were preincubated with increasing concentrations (0–300 nm) of untagged purified recombinant LILRA3 proteins for 1 h at room temperature. Cells were spun at 200 × g for 5 min, unbound protein was aspirated, and cells were washed one time with cold HBHA buffer. Each cell pellet was then resuspended in 1 ml of HBHA buffer, and binding to an optimal concentration of rLILRA3-APtag-His (30 nm) was performed as described above. To determine whether glycosylation contributes for ligand binding, U937 cells were preincubated with 0.1–0.2 m β-lactose or control sucrose or NaCl (Sigma) for 1 h at 37 °C followed by a brief wash with cold HBHA buffer and binding to 30 nm rLILRA3-APtag-His or rAPtag-His control performed as described (14, 15).
In Situ Staining of U937 Cell with rLILRA3-APtag-His
U937 cells were washed twice with PBS and once with HBHA buffer. Aliquots of 3 × 106 cells in 300 μl of HBHA buffer were then dispensed into 1.5-ml Eppendorf tubes and incubated with 100 nm purified rLILRA3-APtag-His or rAPtag-His control proteins in HBHA buffer for 90 min at room temperature. Then rLILRA3-APtag-His and rAPtag-His control treated cells were washed four times with 1.5 ml of cold HBHA buffer with gentle vortexing and centrifugation at 200 × g at 4 °C for 5 min and resuspended in HBHA buffer at 5 × 104/ml. 200 μl (1 × 104) of cells from each treatment were then cytospun at 800 rpm for 5 min onto silanized Superfrost® glass slides; fixed in 60% acetone, 3% formaldehyde, 20 mm HEPES for 30 s; and washed twice in 150 mm NaCl, 20 mm HEPES. Endogenous phosphatases were inactivated by heating slides in 150 mm NaCl, 20 mm HEPES at 65 °C for 10 min. This was followed by rinsing slides with 100 mm Tris-HCl, pH 9.5, 100 mm NaCl, 5 mm MgCl2 and incubation for 15 min with alkaline phosphatase substrate containing 10 mm l-homoarginine, 0.17 mg/ml 5-bromo-4-chloro-3-indolyl phosphate, and 0.33 mg/ml nitro blue tetrazolium in the same buffer (BCIP/NBT Substrate Kit IV, Vector Laboratories, Burlingame, CA).
Inhibition of LPS-induced Monocyte Activation by Recombinant LILRA3
PBMCs from three healthy donors were seeded in duplicate in 96-well flat bottom plates at a concentration of 4 × 105 monocytes/well in 200 μl of RPMI 1640 medium containing 10% fetal calf serum. Cells were treated with or without 100 ng/ml LPS and in the presence of 0–280 nm purified recombinant LILRA3 produced in 293T cells or E. coli for a dose dependence study. PBMCs from a further five donors were used for repeat studies using the optimal concentration of rLILRA3 (70 nm) and 100 ng/ml LPS. Culture supernatants were collected 24 h post-treatment, and levels of secreted TNF-α were measured by sandwich ELISA according the manufacturer's instructions (DuoSet, R&D Systems).
RESULTS
High Quality, Full-length Recombinant LILRA3 Was Successfully Produced in Mammalian Cells, E. coli, and Yeast
Stable transfected 293T cell lines constitutively secreted rLILRA3 with or without APtag-His proteins to culture supernatants. These were purified by cobalt column affinity chromatography to 95–98% purity (Fig. 1, A and C). Similarly, 85–90% pure, full-length rLILRA3 was produced in methanol-induced yeast culture supernatants (data not shown) and in inclusion bodies of transformed E. coli (Fig. 1E) using one step cobalt column affinity chromatography. Most of the latter protein remained soluble during sequential dialysis and refolding. A typical yield of purified rLILRA3-APtag-His and rLILRA3-His from 1 liter of 293T culture supernatant was 0.75 and 0.4 mg, respectively. Similarly, the amount of rLILRA3 produced from 1 liter of yeast culture was a modest 0.2 mg. In contrast, up to 15 mg of correctly refolded rLILRA3 protein was purified from a liter of isopropyl 1-thio-β-d-galactopyranoside-induced E. coli culture. The apparent molecular masses of rLILRA3 from 293T cells with (Fig. 1, A and B) or without (Fig. 1, C and D) rAPtag-His, rLILRA3 from E. coli (Fig. 1, E and F), and rLILRA3 from yeast (data not shown) on SDS-PAGE under reducing conditions were ∼130, 70, 50, and 65+ kDa, respectively. The sizes of the rLILRA3 proteins produced in the 293T cells and yeast were substantially larger than their respective calculated mass of 110 kDa for rLILRA3-APtag-His and 46.61 kDa for the rLILRA3s without tag, whereas the size of the rLILRA3 produced in E. coli was similar to the calculated mass of 52.36 kDa, suggesting significant post-translational modification in recombinant proteins produced by the mammalian and yeast cells. Interestingly, under non-reducing conditions, rLILRA3 proteins produced in all systems exist as both monomeric and dimeric forms (Fig. 1). This is not surprising as LILRA3 contains 12 cysteine residues that potentially can form homodimers through multiple disulfide bonds.
FIGURE 1.
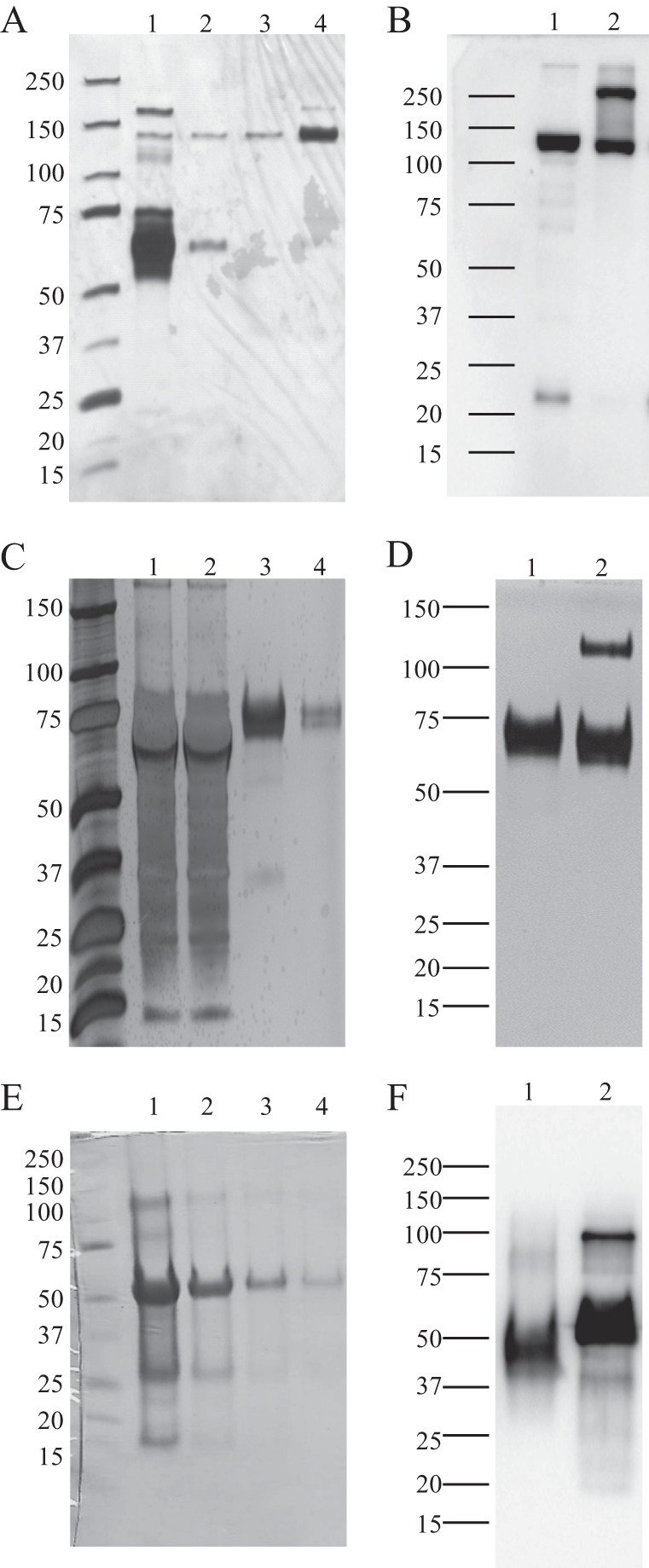
Representative silver staining and Western blotting of rLILRA3 from 293T cells and E. coli showing high quality protein production. A, silver staining of culture supernatant from rLILRA3-APtag-His-overexpressing 293T cells showing abundant expression of protein (lane 1) and sequential fractions of cobalt column-purified proteins from the same culture supernatant eluted using 50 mm imidazole (lanes 2 and 3) and 150 mm imidazole (lane 4) showing highly purified protein. B, Western blotting of cobalt column-purified rLILRA3-APtag-His proteins using anti-AP antibody showing a single band under reduced conditions with DTT (lane 1) but some dimerization under non-reduced conditions (lane 2). C, silver staining of culture supernatant from rLILRA3-His-overexpressing 293T cells showing abundant expression of protein (lane 1), unbound protein after cobalt column affinity binding (lane 2), and sequential fractions of cobalt column-purified proteins from the same culture supernatant eluted using 50 mm imidazole (lane 3) and 150 mm imidazole (lane 4) showing highly purified protein. D, Western blotting of cobalt column-purified rLILRA3-His proteins using anti-LILRA3 mAb showing a single band under reduced conditions (lane 1) but some dimerization under non-reduced conditions (lane 2). E, Coomassie staining of rLILRA3 in inclusion body of E. coli after solubilization with 8 m urea (lane 1) and after purification with a cobalt affinity column (lane 2) followed by dialysis in buffer B (lane 3) and refolding in buffer A (lane 4). F, Western blotting of refolded rLILRA3 from E. coli using anti-LILRA3 mAb showing a single band under reduced conditions (lane 1) and some dimerization under non-reduced conditions (lane 2).
The Increase in the Molecular Mass of LILRA3 Produced in Mammalian and Yeast Expression Systems Is Due to Extensive Glycosylation
PNGase F cleaves between N-acetylglucosamine (GlcNAc) and asparagine (Asn) residues on N-linked high mannose glycans, hybrid glycans, and complex glycans (16). Treatment of recombinant LILRA3 produced in 293T cells and P. pastoris with this enzyme reduced their molecular mass to a size similar to the non-glycosylated rLILRA3 produced in E. coli (Fig. 2A). This strongly indicates that carbohydrates added to rLILRA3 during post-translational modifications are responsible for the marked increase in mass by more than 15 kDa (Fig. 2A). As expected, the size of native LILRA3 in primary macrophages was similar to the recombinant LILRA3 produced in 293T cells but larger than the protein produced in E. coli (Fig. 2A). However, the protein produced in yeast was significantly larger than the native LILRA3 (Fig. 2A). This suggests that different expression systems produce highly varied rLILRA3 proteins that may potentially alter LILRA3 characteristics such as its ability to bind its potential ligands and the ability to modulate cell functions in vitro. Because rLILRA3 produced in E. coli lacks post-translational modifications, whereas rLILRA3 produced in the lower eukaryote (P. pastoris) is possibly hypermannosylated, these recombinant proteins are likely to be functionally inferior to rLILRA3 produced in the higher eukaryotic 293T cells with complex N-glycosylation similar to the native protein.
FIGURE 2.

N-Glycosylation altered the molecular mass and biochemical properties of LILRA3. A, Western blotting of PNGase F-treated (P+) and PNGase F-untreated (P−) rLILRA3 from 293T cells and P. pastoris using anti-LILRA3 mAb showed a substantial reduction in molecular mass of both eukaryotic cell-produced recombinant proteins following deglycosylation. Recombinant LILRA3 produced in E. coli served as a control for non-glycosylated protein. Non-PNGase F-treated LILRA3 from native macrophages (Macs) of two individual donors (lanes 1 and 2) is shown as positive references to optimally glycosylated protein. B, silver staining of two-dimensional gel of non-deglycosylated purified rLILRA3 from 293T cells showed a spectrum of isoelectric focusing with pI ranging from 6 to 9 (upper panel), but upon deglycosylation using PNGase F, it was reduced to a single focus with a pI of 7 (lower panel).
Glycosylation Contributes to the Properties of LLRA3
The glycosylated rLILRA3 protein was focused on a spectrum of pH ranging from 6 to 9 with the majority focusing at around 7 despite having the same molecular weight (Fig. 2B, upper panel). Treatment with PNGase F reduced the wide pI spectra of the untreated protein to a single focusing at pI 7 (Fig. 2B, lower panel). This suggests that the addition of highly charged N-glycan sugar moieties led to the alteration in the charge of the unprocessed protein that has a calculated pI of 8.43. The wide range of isoelectric focusing found on the glycosylated recombinant LILRA3 from 293T cells (Fig. 2B, upper panel) indicates the presence of a mixture of proteins with varying degrees of glycosylation.
Identification of N-Glycosylation Sites to the Predicted Canonical N-Glycans in LILRA3
The N-glycosylation analysis tool NetNGlyc-1.0 predicted five N-linked glycosylation sites in LILRA3 at Asn140, Asn281, Asn341, and Asn431, whereas Asn302 was predicted at a lower threshold value of 0.25 (supplemental Fig. 2). Sites at Asn140, Asn281, Asn302, and Asn341 were predicted with an analysis threshold of 0.5, whereas Asn431 was predicted at a lower threshold value of 0.25 (supplemental Fig. 2). Experimentally, PNGase F-treated recombinant LILRA3 from 293T cells was digested with trypsin, chymotrypsin, or Glu-C, and peptides were analyzed using tandem nano-LC-MS/MS (Fig. 3). Digestion with Glu-C identified 3 deamidated residues at Asn140 (Fig. 3Ai), Asn281 (Fig. 3Aii), and Asn431 (Fig. 3Aiii), trypsin digestion identified 2 deamidated asparagine residues at Asn281 (Fig. 3Bi) and Asn431 (Fig. 3Bii), and chymotrypsin identified 2 deamidated residues at Asn281 (Fig. 3Ci) and Asn341 (Fig. 3Cii). It is noteworthy that some sites were identified by more than one enzyme (Fig. 3). Deamidation of the predicted Asn302 site was not found in the initial experiment (Fig. 3). In two subsequent experiments, in addition to confirming the findings on the Glu-C- and chymotrypsin-digested peptides, digestion of peptides with trypsin detected all the predicted sites including Asn302 (Fig. 4) (Table 1). Importantly, we found <2% deamidation (1 in 54 occurrences) of any sites in E. coli-produced rLILRA3 control with or without PNGase F treatment followed by peptide digest (Table 1) (supplemental Fig. 3). As expected, rLILRA3 produced in the mammalian 293T cells not treated with PNGase F did not generate matching peptides to the PNGase F-treated protein due to the presence of the large N-glycans (Table 1).
FIGURE 3.

Representative nano-LC-MS/MS of PNGase F-deglycosylated tryptic-digested peptides of mammalian rLILRA3 confirmed four predicted N-glycosylation sites. A, in-gel peptide digestion of deglycosylated rLILRA3 with Glu-C showed deamidation of asparagine to aspartic acid at Asn140 (panel i), Asn281 (panel ii), and Asn431 (panel iii) indicating N-linked glycosylation of these sites. B, digestion with chymotrypsin showed deamidation at Asn281 (panel i) and Asn341 (panel ii). C, digestion with trypsin detected Asn281 (panel i) and Asn431 (panel ii). It is noteworthy that some sites were detected in peptides digested by more than one enzyme, and none of the enzymes provided full peptide coverage. The predicted Asn302 was not detected. The sequence of the peptide, the fragmentation pattern, and the detected fragment ions are shown at the top right in each panel. b ions contain the N-terminal region of the peptide; y ions contain the C-terminal region of the peptide. Deamidation of asparagine to aspartic acid is designated as “N” with an underscore.
FIGURE 4.

Repeat nano-LC-MS/MS of PNGase F-deglycosylated trypsin-digested mammalian rLILRA3 confirmed all five predicted N-glycosylation sites. In-gel peptide digestion of deglycosylated rLILRA3 with trypsin showed deamidation of asparagine to aspartic acid at Asn140 (A), Asn281 (B), Asn302 (C), Asn341 (D), and Asn431 (E) indicating N-linked glycosylation of these sites. The sequence of the peptide, the fragmentation pattern, and the detected fragment ions are shown at the top right in each panel. b ions contain the N-terminal region of the peptide; y ions contain the C-terminal region of the peptide. Deamidation of asparagine to aspartic acid is designated as “N” with an underscore.
TABLE 1.
LC-MS/MS summary of N-linked glycosylation sites on PNGase F-treated, trypsin-digested recombinant LILRA3 produced in 293T cells showing deamidation of all five predicted sites as compared with <2% spontaneous deamidation (false positive) in E. coli-produced protein
Non-PNGase F-treated, trypsin-digested recombinant LILRA3 produced in both mammalian cells and E. coli was used as relevant negative controls. The peptide fragments containing Asn were given an ion score and analyzed for deamidation. Positive deamidation is designated as “Yes,” and no deamidation is designated as “No.” If no peptide was detected, it is designated as “ND.”
| Asn site | PNGase F-treated |
Non-PNGase F-treated |
||||||
|---|---|---|---|---|---|---|---|---|
| Mammalian |
E. coli |
Mammalian |
E. coli |
|||||
| Ion score | Deamidation (yes/no) | Ion score | Deamidation (yes/no) | Ion score | Deamidation (yes/no) | Ion score | Deamidation (yes/no) | |
| 140 | 51 | Yes | 66 | No | ND | ND | 47 | No |
| 25 | Yes | 68 | No | 45 | No | |||
| 80 | No | |||||||
| 281 | 26 | Yes | 52 | No | 60 | No | 41 | No |
| 28 | Yes | 55 | No | 85 | No | |||
| 109 | Yes | 90 | No | 34 | No | |||
| 32 | Yes | 29 | No | 30 | No | |||
| 110 | Yes | 42 | No | 65 | No | |||
| 76 | Yes | 34 | No | 23 | No | |||
| 30 | Yes | 57 | No | 32 | No | |||
| 91 | Yes | 59 | No | 39 | No | |||
| 53 | Yes | 64 | No | 78 | No | |||
| 20 | Yes | 42 | No | 51 | No | |||
| 77 | Yes | 59 | No | 26 | No | |||
| 57 | Yes | 54 | No | 58 | No | |||
| 56 | No | 29 | No | |||||
| 48 | No | 71 | No | |||||
| 32 | No | 43 | No | |||||
| 30 | No | 73 | No | |||||
| 24 | No | |||||||
| 91 | No | |||||||
| 39 | No | |||||||
| 33 | No | |||||||
| 302 | 49 | Yes | 44 | No | ND | ND | 45 | No |
| 95 | Yes | 81 | No | 79 | No | |||
| 55 | No | |||||||
| 341 | 64 | Yes | 20 | No | ND | ND | 30 | No |
| 28 | Yes | 49 | No | 39 | No | |||
| 33 | Yes | 21 | No | 24 | No | |||
| 42 | Yes | 28 | No | 50 | No | |||
| 53 | Yes | 60 | No | 23 | No | |||
| 65 | Yes | 57 | No | 44 | No | |||
| 72 | Yes | 43 | No | 34 | No | |||
| 38 | Yes | 36 | No | 5 | No | |||
| 39 | Yes | 53 | No | 56 | No | |||
| 60 | No | 53 | No | |||||
| 84 | No | 56 | No | |||||
| 61 | No | 64 | No | |||||
| 55 | No | 40 | No | |||||
| 39 | No | 63 | No | |||||
| 27 | No | 46 | No | |||||
| 33 | No | 24 | No | |||||
| 47 | No | |||||||
| 56 | No | |||||||
| 42 | No | |||||||
| 39 | No | |||||||
| 71 | No | |||||||
| 61 | No | |||||||
| 35 | No | |||||||
| 431 | 51 | No | 53 | No | 31 | No | 25 | No |
| 54 | Yes | 31 | No | 49 | No | |||
| 38 | Yes | 36 | No | 34 | No | |||
| 52 | Yes | 30 | No | 25 | No | |||
| 31 | Yes | 23 | No | 31 | No | |||
| 30 | Yes | 38 | No | 48 | No | |||
| 22 | Yes | 43 | No | 45 | No | |||
| 29 | Yes | 49 | No | 38 | No | |||
| 21 | Yes | 30 | No | 67 | No | |||
| 44 | Yes | 29 | Yes | 100 | No | |||
| 30 | No | |||||||
| 36 | No | |||||||
Screening for LILRA3 Binding on Cell Surface Using Placental Alkaline Phosphatase-tagged Recombinant LILRA3
Surface binding of rLILRA3-APtag-His to primary leukocytes subsets and cell lines was screened using a simple and highly sensitive method (11). The U937 monocytic cell line showed the highest binding to rLILRA3-APtag-His with ∼80 microunits of specific alkaline phosphatase activity followed by Raji B cells (∼50 microunits), PBMCs (∼37 microunits), and HL-60 myeloid cells (∼14 microunits) (Fig. 5A). There was little or no binding of rLILRA3-APtag to THP-1 cells, Jurkat T cells, and NK-92 cells (Fig. 5A). Among the primary leukocytes, purified primary monocytes showed the strongest binding to rLILRA3-APtag-His with net alkaline phosphatase activity of ∼90 microunits as compared with polymorphonuclear leukocytes that showed moderate binding (∼24 microunits). In contrast T, B, and NK cells showed minimal binding to rLILRA3-APtag-His (Fig. 5B). None of the 20 cell lines of epithelial or mesenchymal lineage tested showed significant binding to rLILRA3-APtag-His except for a neuronal cell line (SKNSH) that showed substantial binding (data not shown). In situ staining of U937 cytospins using rLILRA3-APtag-His or rAPtag-His control showed cell surface staining only on cells incubated with rLILRA3-APtag-His but not rAPtag-His (Fig. 5C), suggesting expression of LILRA3 ligand(s) on the surface of these cells. Binding of rLILRA3-APtag-His to U937 cells was competitively blocked by preincubation of cells with untagged rLILRA3 but not buffer control in a dose-dependent manner (Fig. 5D), confirming specific binding. Interestingly, preincubation of U937 cells with a 10-fold excess of recombinant LILRA3 produced in yeast poorly blocked rLILRA3-APtag-His binding by only 20% (data not shown), and E. coli-produced rLILRA3 totally failed to inhibit binding in contrast to >90% blocking by mammalian cell-produced untagged protein (Fig. 5E). These results suggest that optimal post-translational modification in the mammalian expression system is required for binding of LILRA3 to its potential cell surface ligand(s).
FIGURE 5.

High affinity binding of rLILRA3 to the surface of monocytes and partial blocking of binding by β-lactose. A, screening of leukocytic cell lines and PBMCs showing strong surface binding of purified rLILRA3-rAPtag-His on the surface of PBMCs, U937 monocytic cell line, and Raji B cell line. There was minimal binding to rAPtag-His control protein (n = 6). B, binding assay using purified primary leukocyte subsets showing significant binding of purified rLILRA3-APtag-His on the surface of monocytes (Mono) and neutrophils (polymorphonuclear leukocytes (PMN)) but limited binding to T cells, B cells, or NK cells (n = 3). C, representative in situ staining of U937 cells using purified rLILRA3-APtag-His or rAPtag-His alone showed strong surface staining/binding of rLILRA3-APtag-His (left) but not rAPtag-His alone (right) as detected by 5-bromo-4-chloro-3-indolyl phosphate/nitro blue tetrazolium alkaline phosphatase substrate stained in blue and neutral red nuclear counterstain (250× magnification; n = 5). D, binding of 30 nm purified rLILRA3-rAPtag-His to U937 cells was competitively blocked with preincubation of cells with untagged rLILRA3 in a dose-dependent manner, confirming binding specificity (n = 3) (one-way ANOVA; *, p < 0.05; **, p < 0.01 as compared with corresponding buffer control). E, binding of 30 nm purified rLILRA3-APtag to U937 cells was blocked by a 10-fold excess of untagged purified rLILRA3 from 293T cells but not E. coli recombinant LILRA3 (n = 3) (one-way ANOVA; *, p < 0.05 compared with rLILRA3-APtag alone; NS, not significant). F, binding of 30 nm purified rLILRA3-APtag to U937 cells was partially blocked by β-lactose in a dose-dependent manner, suggesting that the sugar moiety components may be required for ligand binding (n = 3) (one-way ANOVA; *, p < 0.05 compared with buffer (PBS) control). G, 0.2 m β-lactose but not 0.2 m sucrose or 0.2 n NaCl in PBS blocked rLILRA3-APtag (30 nm) binding to the surface of U937 cells, confirming specificity (n = 5) (one-way ANOVA; *, p < 0.05; **, p < 0.01 compared with PBS (buffer) control). Error bars represent S.E.
LILRA3 Glycosylation Contributes to Effective Ligand Binding
Given that glycosylation was the major post-translational modification that altered the physical properties of the mammalian rLILRA3 (Figs. 2–4), the effects of these modifications on its ability to bind U937 cells were assessed by pretreatment of cells with β-lactose. Preincubation of cells with 0.1 or 0.2 m β-lactose blocked rLILRA3 binding by an average of 10 ± 0.3 and 40 ± 0.5%, respectively (Fig. 5F), but control sucrose or NaCl did not (Fig. 5G), indicating that N-glycosylation partially played a role in its ability to bind ligand(s). These results strongly complement our observation that E. coli-produced non-glycosylated rLILRA3 (Fig. 5E) and yeast-produced “inappropriately” glycosylated rLILRA3 (data not shown) do not competitively block binding.
Mammalian but Not E. coli-derived Recombinant LILRA3 Down-regulates LPS-mediated TNFα Production in PBMCs
Incubation of freshly isolated normal PBMCs with 100 pg/ml LPS in vitro induced high levels of TNFα production ranging between 5500 and 9000 pg/ml (Fig. 6). Simultaneous treatment of cells with 17–280 nm recombinant mammalian LILRA3 significantly abrogated LPS-induced TNFα production in a dose-dependent manner (Fig. 6A). Treatment of cells with the optimal dose of rLILRA3 consistently suppressed TNF production by up to 60% in a dose-dependent manner (Fig. 6A) in multiple donors (Fig. 6B). However, equivalent amounts of recombinant LILRA3 produced in E. coli did not suppress LPS-induced TNF production in all donors (data not shown). This is consistent with the ability of mammalian cell- but not E. coli-derived rLILRA3 to bind to the surface of PBMCs and modulate function.
FIGURE 6.
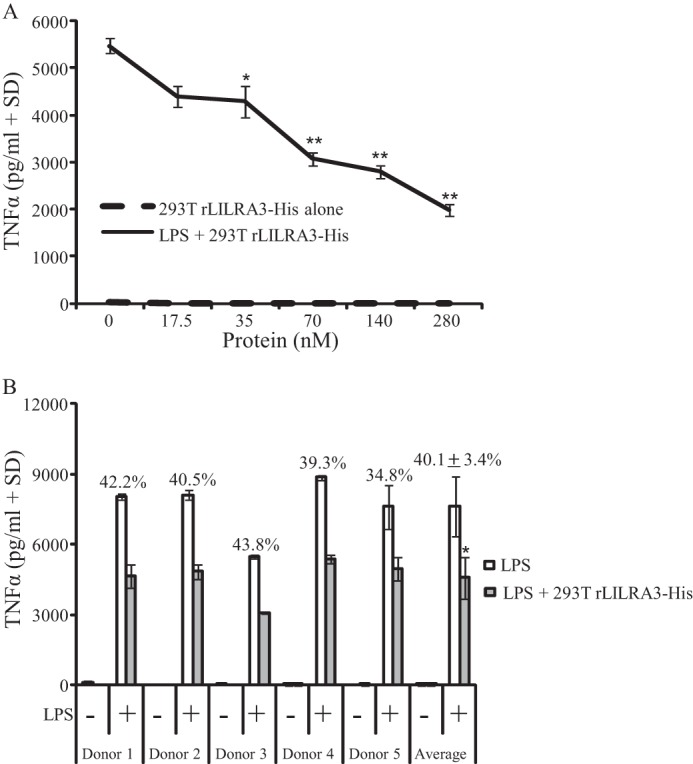
Recombinant LILRA3 produced in 293T cells suppressed LPS-mediated TNFα production by PBMCs. A, simultaneous treatment of PBMCs with increasing concentrations of purified rLILRA3 and 100 ng/ml LPS for 24 h caused dose-dependent suppression of TNFα production. Cells treated with rLILRA3 alone produced minimal TNFα (n = 3) (one-way ANOVA; *, p < 0.05; **, p < 0.01 compared with no rLILRA3 control). B, treatment of PBMCs from five healthy subjects with 100 ng/ml LPS and the optimal concentration of rLILRA3 (70 nm) for 24 h consistently showed 35–45% suppression of TNFα production. Cells treated with rLILRA3 but not LPS showed minimal TNF production (one-way ANOVA; *, p < 0.05 compared no rLILRA3 control). Error bars represent S.D.
DISCUSSION
In this study, we have successfully generated recombinant LILRA3 proteins in E. coli, yeast, and 293T cells and assessed their yield, properties, and biological functions. We found that P. pastoris and 293T cells but not E. coli produced rLILRA3 of a substantially larger size than the calculated mass of the unmodified protein, indicating marked post-translational modifications (Figs. 1 and 2). Treatment with PNGase F reduced yeast- and mammalian cell-produced proteins to a size equivalent to the expected unmodified protein, suggesting that both proteins underwent N-glycan modifications but with varied outcomes. N-Glycan modification in all eukaryotic cells starts as a common Man8GlcNAc2 precursor produced after some initial processing, but in higher eukaryotes, the α-1,2-mannose residues are removed, and complex N-linked glycosylated proteins are formed (17). By contrast, Man8GlcNAc2 in yeast are further elongated by several mannosyltransferases, resulting in the formation of large hyperglycosylated or hypermannosylated products of native (18) or complex foreign proteins (19). Hence, the significantly larger molecular mass of rLILRA3 produced in P. pastoris as compared with native LILRA3 is likely due to hyperglycosylation or hypermannosylation (Fig. 2A), whereas the mammalian cell-produced protein may have undergone appropriate glycosylation and is thus structurally and functionally closer to native LILRA3. Consistent with the latter, we showed that native LILRA3 from primary macrophages was the same size as recombinant protein produced in mammalian cells but not in yeast or E. coli (Fig. 2).
LILRA3 produced in the mammalian system showed multiple isoelectric focusing but upon treatment with PNGase F reduced to a single pI (Fig. 2), indicating that changes in the biochemical property of this protein were due to addition of highly charged N-glycans. This is similar to previous data showing that glycosylation significantly alters the pI of other similar glycoproteins (20, 21). Moreover, glycosylation can provide conformational and structural stability (22, 23) and may facilitate correct folding (23, 24) and importantly modulate ligand binding and functions (20, 25, 26). We found that rLILRA3 in its non-glycosylated form required extensive refolding steps, was highly susceptible to aggregation/oxidation, and was non-functional despite being of high purity and high yield (up to 15 mg/liter). Similarly, production of hyperglycosylated rLILRA3 in yeast may have altered its biochemical property that contributed to its low production efficiency (0.25 mg/liter) and poor function. By contrast, optimal glycosylation of the mammalian cell-produced rLILRA3 protein may have facilitated its efficient folding, sufficient production (0.4–0.8 mg/liter), high stability, and excellent biological functions (Figs. 5 and 6). Our successful blocking experiment using β-lactose further supports the suggestion that ligand binding and possibly suppression of TNFα production were at least partially dependent on optimal glycosylation (Fig. 5).Therefore, we were compelled to identify the specific N-glycosylation sites as a prelude for future functional characterization using site-targeted mutagenesis.
LILRA3 is predicted to contain five N-glycosylation sites on specific asparagine residues within NX(S/T) consensus sequences (X is any amino acid other than Pro) (27). We utilized a combination of electrophoresis and nano-LC-MS/MS and identified all five N-glycosylated sites at Asn140, Asn281, Asn302, Asn341, and Asn431. This is the first study that has experimentally mapped specific glycosylation sites on any LILR protein. Recently it has been reported that deglycosylation with PNGase F can lead to false positive assignments of N-glycosylation sites (28). False positive deamidation commonly occurs in small and hydrophilic amino acids such as glycine and serine at position X of the NX(S/T/C) consensus sequence (16). Although LILRA3 does not contain such consensus sequences, we performed stringent control experiments using E. coli-produced rLILRA3 with or without PNGase treatment and PNGase-untreated mammalian rLILRA3-His. As expected, E. coli-produced rLILRA3 with or without PNGase F treatment or untreated mammalian cell-derived rLILRA3 showed negligible spontaneous deamidation with a false discovery rate of <2%, indicating specificity. Initially, three different peptide digestion enzymes were required to map four of the five sites: Asn281 was detected with all three enzymes, Asn140 and Asn341 were apparent with Glu-C or chymotrypsin digest, and Asn431 was detected in both trypsin- and Glu-C-digested peptides (Fig. 3). This is in agreement with reports showing that bottom-up proteomics would generate different peptide coverage depending on the choice of enzyme (29, 30). Interestingly, in subsequent experiments, digestion with trypsin alone was sufficient for full peptide coverage and detected all five N-glycosylation sites including Asn302 (Fig. 4). The Asn302 site could not be detected initially possibly due to the inability to generate enough peptides for full coverage as a result of the use of an insufficient amount of rLILRA3.
It is noteworthy that LILRA3 is also predicted to have up to eight potential O-linked glycosylated sites (NetOGlyc 4.0 analysis tool) (31) that may contribute to its structure and/or functions. This will require future investigation.
Our data demonstrate that production of optimally glycosylated LILRA3 in the mammalian system was necessary for its high affinity binding to its potential ligand(s) (Fig. 5). However, most studies of the LILR family to date did not consider the likely importance of appropriate glycosylation to high affinity ligand binding and function. To date, LILRB1, LILRB2, LILRA1, and LILRA3 have been shown to bind various classical and non-classical MHC class I molecules (32–34) and a viral homolog, UL-18 (32). However, these are mostly low affinity interactions with varied dissociation constants ranging from 2 to 12 μm (35, 36). These studies also lack robust functional data possibly due to the use of truncated extracellular domains of LILRs produced in E. coli that are inefficiently folded and not appropriately post-translationally modified. Alternatively, LILRs may have hierarchical tissue-specific interactions with multiple ligands in vivo, or MHC class I molecules might be co-ligands, an issue grossly overlooked so far. Indeed, recently several LILRs were also shown to functionally bind non-MHC class I molecules with much higher affinity than binding to MHC class I molecules. These interactions include binding of Nogo-66 (37) and ANGPTL5 (38) to LILRB2 and binding of BST-2 to LILRA4 (39).
In this study, we presented a simple robust approach for rapid high throughput screening and in situ localization of LILRA3 cell surface binding. The additional advantages for the use of mammalian LILRA3 protein tagged to placental alkaline phosphatase include the high specific activity of the mammalian enzyme, its high stability including placental isoenzyme stability to heat, the availability of isoenzyme-specific inhibitors, the availability of a variety of indicator substrates for alkaline phosphatase (11), and the availability of high quality anti-placental alkaline phosphatase antibodies (GenHunter). This provided us with key tools for future simultaneous identification of LILR ligands and co-ligands using selected LILRA3-binding cells. Moreover, the rapid screening of rLILRA3-APtag-His-binding proteins allowed us to objectively select suitable cells for our functional studies.
We showed that LILRA3 preferably binds on the surface of monocytes and the monocytic cell line U937. This suggests that LILRA3 may predominantly regulate monomyeloid cells. We showed for the first time that LILRA3 abrogated LPS-mediated TNFα production, suggesting a direct inhibitory effect transduced through a yet unknown surface ligand(s). LILRA3 may also exert its effects by competitively antagonizing closely related cell surface activating LILRs. A recent study showed that LILRA3 and LILRA1 (88% homology) may share common MHC class I ligand(s) (34), although the functional consequence of this needs to be defined. We propose that LILRA3 is a novel anti-inflammatory protein that directly suppresses excessive leukocyte activation and/or acts as a soluble antagonist to activating LILRs akin to the soluble TNFα receptor and IL-6 receptor (40, 41). This is consistent with our recent finding of the presence of abundant LILRA3 protein in sera of healthy individuals and its significant up-regulation by the anti-inflammatory cytokine IL-10 (6). This is further supported by recent reports showing an association of a lack of LILRA3 with an increased incidence of multiple sclerosis (8, 9) and Sjögren syndrome (10), diseases characterized by chronic inflammation. Interestingly, we found a significant increase in LILRA3 in sera of patients with active rheumatoid arthritis (6) together with increased expression of activating and inhibitory LILRs in synovial tissue (7). This may suggest that an increase in LILRA3 opposes the ongoing inflammation or that a proportion of the high level of LILRA3 in patient sera might be aberrantly glycosylated, leading to poor function. Abnormal ligand binding and functions due to altered glycosylation of endogenous proteins such as IgG have been reported in rheumatoid arthritis (12, 42). Whether LILRA3, which has structural similarities to IgG, also displays abnormal glycan modifications that alter its function requires further investigation.

This article contains supplemental Figs. 1–3.
R. Gupta, E. Jung, and S. Brunak (2004) Prediction of N-glycosylation sites in human proteins, unpublished data.
- LILR
- leukocyte immunoglobulin-like receptor
- rLILRA3
- recombinant LILRA3
- PNGase
- peptide-N-glycosidase
- LILRA
- activating LILR
- LILRB
- inhibitory LILR
- APtag
- alkaline phosphatase tag
- rAPtag
- recombinant APtag
- PBMC
- peripheral blood mononuclear cell
- NK
- natural killer
- ANOVA
- analysis of variance.
REFERENCES
- 1. Brown D., Trowsdale J., Allen R. (2004) The LILR family: modulators of innate and adaptive immune pathways in health and disease. Tissue Antigens 64, 215–225 [DOI] [PubMed] [Google Scholar]
- 2. Nakajima H., Samaridis J., Angman L., Colonna M. (1999) Human myeloid cells express an activating ILT receptor (ILT1) that associates with Fc receptor γ-chain. J. Immunol. 162, 5–8 [PubMed] [Google Scholar]
- 3. Cosman D., Fanger N., Borges L., Kubin M., Chin W., Peterson L., Hsu M. L. (1997) A novel immunoglobulin superfamily receptor for cellular and viral MHC class I molecules. Immunity 7, 273–282 [DOI] [PubMed] [Google Scholar]
- 4. Torkar M., Haude A., Milne S., Beck S., Trowsdale J., Wilson M. J. (2000) Arrangement of the ILT gene cluster: a common null allele of the ILT6 gene results from a 6.7-kbp deletion. Eur. J. Immunol. 30, 3655–3662 [DOI] [PubMed] [Google Scholar]
- 5. Wilson M. J., Torkar M., Haude A., Milne S., Jones T., Sheer D., Beck S., Trowsdale J. (2000) Plasticity in the organization and sequences of human KIR/ILT gene families. Proc. Natl. Acad. Sci. U.S.A. 97, 4778–4783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. An H., Chandra V., Piraino B., Borges L., Geczy C., McNeil H. P., Bryant K., Tedla N. (2010) Soluble LILRA3, a potential natural antiinflammatory protein, is increased in patients with rheumatoid arthritis and is tightly regulated by interleukin 10, tumor necrosis factor-α, and interferon-γ. J. Rheumatol. 37, 1596–1606 [DOI] [PubMed] [Google Scholar]
- 7. Tedla N., An H., Borges L., Vollmer-Conna U., Bryant K., Geczy C., McNeil H. P. (2011) Expression of activating and inhibitory leukocyte immunoglobulin-like receptors in rheumatoid synovium: correlations to disease activity. Tissue Antigens 77, 305–316 [DOI] [PubMed] [Google Scholar]
- 8. Koch S., Goedde R., Nigmatova V., Epplen J. T., Müller N., de Seze J., Vermersch P., Momot T., Schmidt R. E., Witte T. (2005) Association of multiple sclerosis with ILT6 deficiency. Genes Immun. 6, 445–447 [DOI] [PubMed] [Google Scholar]
- 9. Ordóñez D., Sánchez A. J., Martínez-Rodríguez J. E., Cisneros E., Ramil E., Romo N., Moraru M., Munteis E., López-Botet M., Roquer J., García-Merino A., Vilches C. (2009) Multiple sclerosis associates with LILRA3 deletion in Spanish patients. Genes Immun. 10, 579–585 [DOI] [PubMed] [Google Scholar]
- 10. Kabalak G., Dobberstein S. B., Matthias T., Reuter S., The Y. H., Dörner T., Schmidt R. E., Witte T. (2009) Association of immunoglobulin-like transcript 6 deficiency with Sjögren's syndrome. Arthritis Rheum. 60, 2923–2925 [DOI] [PubMed] [Google Scholar]
- 11. Flanagan J. G., Leder P. (1990) The kit ligand: a cell surface molecule altered in steel mutant fibroblasts. Cell 63, 185–194 [DOI] [PubMed] [Google Scholar]
- 12. Alavi A., Arden N., Spector T. D., Axford J. S. (2000) Immunoglobulin G glycosylation and clinical outcome in rheumatoid arthritis during pregnancy. J. Rheumatol. 27, 1379–1385 [PubMed] [Google Scholar]
- 13. Tedla N., Bandeira-Melo C., Tassinari P., Sloane D. E., Samplaski M., Cosman D., Borges L., Weller P. F., Arm J. P. (2003) Activation of human eosinophils through leukocyte immunoglobulin-like receptor 7. Proc. Natl. Acad. Sci. U.S.A. 100, 1174–1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fermino M. L., Polli C. D., Toledo K. A., Liu F. T., Hsu D. K., Roque-Barreira M. C., Pereira-da-Silva G., Bernardes E. S., Halbwachs-Mecarelli L. (2011) LPS-induced galectin-3 oligomerization results in enhancement of neutrophil activation. PLoS One 6, e26004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stillman B. N., Hsu D. K., Pang M., Brewer C. F., Johnson P., Liu F. T., Baum L. G. (2006) Galectin-3 and galectin-1 bind distinct cell surface glycoprotein receptors to induce T cell death. J. Immunol. 176, 778–789 [DOI] [PubMed] [Google Scholar]
- 16. Martinez-Donato G., Acosta-Rivero N., Morales-Grillo J., Musacchio A., Vina A., Alvarez C., Figueroa N., Guerra I., Garcia J., Varas L., Muzio V., Dueñas-Carrera S. (2006) Expression and processing of hepatitis C virus structural proteins in Pichia pastoris yeast. Biochem. Biophys. Res. Commun. 342, 625–631 [DOI] [PubMed] [Google Scholar]
- 17. Helenius A., Aebi M. (2004) Roles of N-linked glycans in the endoplasmic reticulum. Annu. Rev. Biochem. 73, 1019–1049 [DOI] [PubMed] [Google Scholar]
- 18. Vervecken W., Kaigorodov V., Callewaert N., Geysens S., De Vusser K., Contreras R. (2004) In vivo synthesis of mammalian-like, hybrid-type N-glycans in Pichia pastoris. Appl. Environ. Microbiol. 70, 2639–2646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guo M., Hang H., Zhu T., Zhuang Y., Chu J., Zhang S. (2008) Effect of glycosylation on biochemical characterization of recombinant phytase expressed in Pichia pastoris. Enzyme Microb. Technol. 42, 340–345 [Google Scholar]
- 20. Margraf-Schönfeld S., Böhm C., Watzl C. (2011) Glycosylation affects ligand binding and function of the activating natural killer cell receptor 2B4 (CD244) protein. J. Biol. Chem. 286, 24142–24149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kunicki T. J., Cheli Y., Moroi M., Furihata K. (2005) The influence of N-linked glycosylation on the function of platelet glycoprotein VI. Blood 106, 2744–2749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zou S., Huang S., Kaleem I., Li C. (2013) N-Glycosylation enhances functional and structural stability of recombinant β-glucuronidase expressed in Pichia pastoris. J. Biotechnol. 164, 75–81 [DOI] [PubMed] [Google Scholar]
- 23. Wormald M. R., Dwek R. A. (1999) Glycoproteins: glycan presentation and protein-fold stability. Structure 7, R155–160 [DOI] [PubMed] [Google Scholar]
- 24. Shental-Bechor D., Levy Y. (2008) Effect of glycosylation on protein folding: a close look at thermodynamic stabilization. Proc. Natl. Acad. Sci. U.S.A. 105, 8256–8261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Guseva N. V., Fullenkamp C. A., Naumann P. W., Shey M. R., Ballas Z. K., Houtman J. C., Forbes C. A., Scalzo A. A., Heusel J. W. (2010) Glycosylation contributes to variability in expression of murine cytomegalovirus m157 and enhances stability of interaction with the NK-cell receptor Ly49H. Eur. J. Immunol. 40, 2618–2631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Uchibori-Iwaki H., Yoneda A., Oda-Tamai S., Kato S., Akamatsu N., Otsuka M., Murase K., Kojima K., Suzuki R., Maeya Y., Tanabe M., Ogawa H. (2000) The changes in glycosylation after partial hepatectomy enhance collagen binding of vitronectin in plasma. Glycobiology 10, 865–874 [DOI] [PubMed] [Google Scholar]
- 27. Geetha-Habib M., Park H. R., Lennarz W. J. (1990) In vivo N-glycosylation and fate of Asn-X-Ser/Thr tripeptides. J. Biol. Chem. 265, 13655–13660 [PubMed] [Google Scholar]
- 28. Palmisano G., Melo-Braga M. N., Engholm-Keller K., Parker B. L., Larsen M. R. (2012) Chemical deamidation: a common pitfall in large-scale N-linked glycoproteomic mass spectrometry-based analyses. J. Proteome Res. 11, 1949–1957 [DOI] [PubMed] [Google Scholar]
- 29. Kalli A., Håkansson K. (2008) Comparison of the electron capture dissociation fragmentation behavior of doubly and triply protonated peptides from trypsin, Glu-C, and chymotrypsin digestion. J. Proteome Res. 7, 2834–2844 [DOI] [PubMed] [Google Scholar]
- 30. Zielinska D. F., Gnad F., Wiśniewski J. R., Mann M. (2010) Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell 141, 897–907 [DOI] [PubMed] [Google Scholar]
- 31. Steentoft C., Vakhrushev S. Y., Joshi H. J., Kong Y., Vester-Christensen M. B., Schjoldager K. T., Lavrsen K., Dabelsteen S., Pedersen N. B., Marcos-Silva L., Gupta R., Bennett E. P., Mandel U., Brunak S., Wandall H. H., Levery S. B., Clausen H. (2013) Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J. 32, 1478–1488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vitale M., Castriconi R., Parolini S., Pende D., Hsu M. L., Moretta L., Cosman D., Moretta A. (1999) The leukocyte Ig-like receptor (LIR)-1 for the cytomegalovirus UL18 protein displays a broad specificity for different HLA class I alleles: analysis of LIR-1 + NK cell clones. Int. Immunol. 11, 29–35 [DOI] [PubMed] [Google Scholar]
- 33. Chapman T. L., Heikeman A. P., Bjorkman P. J. (1999) The inhibitory receptor LIR-1 uses a common binding interaction to recognize class I MHC molecules and the viral homolog UL18. Immunity 11, 603–613 [DOI] [PubMed] [Google Scholar]
- 34. Jones D. C., Kosmoliaptsis V., Apps R., Lapaque N., Smith I., Kono A., Chang C., Boyle L. H., Taylor C. J., Trowsdale J., Allen R. L. (2011) HLA class I allelic sequence and conformation regulate leukocyte Ig-like receptor binding. J. Immunol. 186, 2990–2997 [DOI] [PubMed] [Google Scholar]
- 35. Shiroishi M., Kajikawa M., Kuroki K., Ose T., Kohda D., Maenaka K. (2006) Crystal structure of the human monocyte-activating receptor, “Group 2” leukocyte Ig-like receptor A5 (LILRA5/LIR9/ILT11). J. Biol. Chem. 281, 19536–19544 [DOI] [PubMed] [Google Scholar]
- 36. Chapman T. L., Heikema A. P., West A. P., Jr., Bjorkman P. J. (2000) Crystal structure and ligand binding properties of the D1D2 region of the inhibitory receptor LIR-1 (ILT2). Immunity 13, 727–736 [DOI] [PubMed] [Google Scholar]
- 37. Atwal J. K., Pinkston-Gosse J., Syken J., Stawicki S., Wu Y., Shatz C., Tessier-Lavigne M. (2008) PirB is a functional receptor for myelin inhibitors of axonal regeneration. Science 322, 967–970 [DOI] [PubMed] [Google Scholar]
- 38. Zheng J., Umikawa M., Cui C., Li J., Chen X., Zhang C., Huynh H., Kang X., Silvany R., Wan X., Ye J., Cantó A. P., Chen S. H., Wang H. Y., Ward E. S., Zhang C. C. (2012) Inhibitory receptors bind ANGPTLs and support blood stem cells and leukaemia development. Nature 485, 656–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cao W., Bover L., Cho M., Wen X., Hanabuchi S., Bao M., Rosen D. B., Wang Y. H., Shaw J. L., Du Q., Li C., Arai N., Yao Z., Lanier L. L., Liu Y. J. (2009) Regulation of TLR7/9 responses in plasmacytoid dendritic cells by BST2 and ILT7 receptor interaction. J. Exp. Med. 206, 1603–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jones S. A., Horiuchi S., Topley N., Yamamoto N., Fuller G. M. (2001) The soluble interleukin 6 receptor: mechanisms of production and implications in disease. FASEB J. 15, 43–58 [DOI] [PubMed] [Google Scholar]
- 41. Arck P. C., Troutt A. B., Clark D. A. (1997) Soluble receptors neutralizing TNF-α and IL-1 block stress-triggered murine abortion. Am. J. Reprod. Immunol. 37, 262–266 [DOI] [PubMed] [Google Scholar]
- 42. Parekh R. B., Dwek R. A., Sutton B. J., Fernandes D. L., Leung A., Stanworth D., Rademacher T. W., Mizuochi T., Taniguchi T., Matsuta K., Takeuchi F., Nagano Y., Miyamoto T., Kobata A. (1985) Association of rheumatoid arthritis and primary osteoarthritis with changes in the glycosylation pattern of total serum IgG. Nature 316, 452–457 [DOI] [PubMed] [Google Scholar]