

Background: The increase of PPARγ stability could contribute to lower blood glucose levels.
Results: PPARγ stability is increased by the deubiquitinating activity of HAUSP.
Conclusion: HAUSP overexpression could decrease blood glucose and triglyceride levels at least in part by deubiquitinating and stabilizing PPARγ in the liver.
Significance: Identification of a novel enzyme (HAUSP) that deubiquitinates and stabilizes PPARγ and its potential role in liver glucose and lipid metabolism are significant.
Keywords: Deubiquitination, Diabetes, Glucose Metabolism, Metabolism, Peroxisome Proliferator-activated receptor (PPAR)
Abstract
The peroxisome proliferator-activated receptor γ (PPARγ) is a central regulator of adipogenesis and modulates glucose and lipid metabolism. In this study, herpesvirus-associated ubiquitin-specific protease (HAUSP) was isolated as a binding partner of PPARγ. Both endogenous and exogenous PPARγ associated with HAUSP in co-immunoprecipitation analysis. HAUSP, but not the catalytically inactive HAUSP C223S mutant, increased the stability of both endogenous and exogenous PPARγ through its deubiquitinating activity. Site-directed mutagenesis experiments showed that the Lys462 residue of PPARγ is critical for ubiquitination. HBX 41,108, a specific inhibitor of HAUSP, abolished the increase in PPARγ stability induced by HAUSP. In addition, knockdown of endogenous HAUSP using siRNA decreased PPARγ protein levels. HAUSP enhanced the transcriptional activity of both exogenous and endogenous PPARγ in luciferase activity assays. Quantitative RT-PCR analysis showed that HAUSP increased the transcript levels of PPARγ target genes in HepG2 cells, resulting in the enhanced uptake of glucose and fatty acids, and vice versa, upon siRNA knockdown of HAUSP. In vivo analysis using adenoviruses confirmed that HAUSP, but not the HAUSP C223S mutant, decreased blood glucose and triglyceride levels, which are associated with the increased expression of endogenous PPARγ and lipid accumulation in the liver. Our results demonstrate that the stability and activity of PPARγ are modulated by the deubiquitinating activity of HAUSP, which may be a target for the development of anti-diabetic drugs.
Introduction
PPARγ3 functions as a central regulator of various physiological processes, including adipogenesis, glucose homeostasis, and lipid metabolism; it is a member of the nuclear hormone receptor superfamily and a ligand-activated transcription factor (1). PPARγ is composed of an N-terminal activation function-1 (AF-1) domain, a DNA-binding domain (DBD), a hinge domain, and a C-terminal ligand-binding domain (LBD). The AF-1 domain has a ligand-independent transcriptional activation function, whereas the DBD domain recognizes a PPAR-response element (PPRE) in the promoters of target genes. The LBD domain is necessary for heterodimerization with the 9-cis-retinoic X receptor (RXR) and contains a ligand-dependent transcriptional activation function (AF-2) domain at the C terminus. PPARγ is activated by its ligands, including hydroxyoctadeca-9Z,11E-dienoic acid, 13-hydroxyoctadecadienoic acid, 15-deoxy-Δ12,14 prostaglandin J2, nitrated fatty acids, and thiazolidinediones (2). Ligand binding to PPARγ induces the formation of a heterodimer between PPARγ and retinoic X receptor α, which then binds to the PPRE to increase the transcription of target genes.
PPARγ activity is modulated by various post-translational modifications, such as phosphorylation, sumoylation, and ubiquitination (3). The activation of MAPK and cyclin-dependent kinases promotes the phosphorylation of PPARγ at Ser112, leading to the increase or decrease of its transcriptional activity (4–9). The phosphorylation of PPARγ Ser273 by CDK5 does not affect its adipogenic activity, but it does down-regulate the expression of adiponectin (10). Additionally, PPARγ is regulated by sumoylation, whereas sumoylation inhibits its activity, although desumoylation by SUMO1/sentrin/SMT3-specific peptidase 2 (SENP2) increases its transcriptional activity (11, 12). Furthermore, PPARγ is regulated by polyubiquitination, which leads to proteasomal degradation (13, 14). Post-translational modifications of PPARγ have been studied in detail. However, to date, no enzymes have been identified that can deubiquitinate or dephosphorylate PPARγ. In this study, the ubiquitin-specific protease USP7, also called herpesvirus-associated ubiquitin-specific protease (HAUSP), was identified as an enzyme capable of deubiquitinating PPARγ. HAUSP increases the stability of p53 protein via deubiquitination (15). In addition, HAUSP binds and regulates the stability and function of a variety of molecules largely related to cancer progression, including mouse double minute 2 (Mdm2), phosphatase and tensin homolog, forkhead box O (FOXO4), p16/INK4a, histone 2B, and Epstein-Barr nuclear antigen 1 (EBNA1) (16–20).
In this study, HAUSP was identified as a binding partner of PPARγ in a GST pulldown assay using GST-PPARγ. The interaction between PPARγ and HAUSP was examined by exogenous and endogenous co-immunoprecipitation, and deletion mapping was carried out using different PPARγ deletion mutants. Additionally, site-directed mutagenesis was performed to identify the critical residue responsible for the ubiquitination of PPARγ. The functional relationship between PPARγ and HAUSP was investigated by performing assays specific for the deubiquitination, stability, and transcriptional activity of PPARγ, as well as quantitative RT-PCR of PPARγ target genes and in vitro fatty acid and glucose uptake assays. In addition, the effect of adenovirus-mediated HAUSP overexpression on endogenous PPARγ levels in the liver was investigated.
EXPERIMENTAL PROCEDURES
Cell Culture
COS7 cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 50 μg/ml penicillin and streptomycin in a 5% CO2 incubator. HepG2 cells were cultured in minimum Eagle's medium supplemented with 10% FBS and 50 μg/ml penicillin and streptomycin. The 3T3-L1 cells were maintained in DMEM containing 10% calf serum and 50 μg/ml penicillin and streptomycin in a 5% CO2 incubator. Adipocyte differentiation was induced by addition of 10% FBS-supplemented DMEM containing 0.5 nm 3-isobutyl-1-methylxanthine, 0.25 μm dexamethasone, and 5 μg/ml insulin for 2 days. The cells were then maintained in DMEM with 10% FBS and 1 μg/ml insulin for the following 2 days and further maintained in DMEM with 10% FBS for the following 4 days.
Construction of Plasmids, Adenoviruses, and Antibodies
Expression vectors containing each domain of mouse PPARγ2 were constructed by subcloning the corresponding cDNAs into N-terminal HA-tagged pcDNA3.1. The deletion mutants of mouse PPARγ2 were constructed into HA-tagged pcDNA3.1 as follows. The cDNAs encoding the activation function-1 (AF-1) domain from amino acids 1 to 138, DBD, the hinge domain from amino acids 139 to 279, ligand binding domain (LBD/AF2) from 279 to 505 amino acids, AF1/DBD from 1 to 280 amino acids, and DBD/LBD from 137 to 505 amino acids of mouse PPARγ type 2 were generated using polymerase chain reaction (PCR), and then ligated into HA-tagged pcDNA3.1 using the KpnI and XbaI sites. Site-directed mutagenesis of PPARγ was performed using the QuikChange site-directed mutagenesis kit (Agilent Technologies, Palo Alto, CA). Primers are summarized in Table 1. The FLAG-HAUSP and His-ubiquitin expression vectors were kindly provided by C. H. Chung (Seoul National University, South Korea). The PPRE reporter plasmid (PPRE-pk-Luc), control reporter plasmid (pk-Luc), and β-galactosidase expression vector (β-Gal) were kindly provided by S. H. Koo (Sung Kyun Kwan University, South Korea). Adenoviruses encoding human HAUSP (Ad-GFP/HAUSP) were generated by insertion of the HAUSP ORF into pAdTrack-CMV expressing GFP (Addgene, MA). Adenoviruses were prepared as described previously (21). Antibodies against HAUSP, PPARγ, HA, γ-tubulin, and ubiquitin were purchased from Santa Cruz Biotechnology. Anti-FLAG antibodies were purchased from Sigma.
TABLE 1.
Primers used for site-directed mutagenesis
F means forward, and R means reverse.
| Primers | Sequences |
|---|---|
| mPPARγ2-K382R | F, 5′-GGATTCATGACCAGGGAGTTCCTCAGAAACCTGCGG-3′ |
| R, 5′-CCGCAGGTTTCTGAGGAACTCCCTGGTCATGAATCC-3′ | |
| mPPARγ2-K386R | F, 5′-GTTCCTCAAAAACCTGCGGAGACCCTTCGGTGACTTTATGG-3′ |
| R, 5′-CCATAAAGTCACCGAAGGGTCTCCGCAGGTTTTTGAGGAAC-3′ | |
| mPPARγ2-K395R | F, 5′-CCTTTGGTGACTTTATGGAGCCTAGATTTGAGTTTGCTGTG-3′ |
| R, 5′-CACAGCAAACTCAAATCTAGGCTCCATAAAGTCACCAAAGG-3′ | |
| mPPARγ2-K462R | F, 5′-CAGCTGTTCGCCAGGGTGCTCCAGAAGATGAC-3′ |
| R, 5′-GTCATCTTCTGGAGCACCCTGGCGAACAGCTG-3′ |
Isolation of PPARγ-binding Protein
Bacterial expression of GST or GST-PPARγ was induced by 0.1 mm isopropyl 1-thio-β-d-galactopyranoside at 25 °C for 8 h. The GST or GST-PPARγ proteins were purified by glutathione affinity chromatography according to the manufacturer's instructions. HeLa cells were lysed in lysis buffer (50 mm Tris-HCl, pH 7.4, 50 mm NaCl, 0.5 mm EDTA, 1 mm PMSF, 5 μg/ml aprotinin, 1% Triton X-100). 10 mg of HeLa extracts were incubated with GST or GST-PPARγ bound to agarose beads at 4 °C for 4 h. After washing three times, proteins were separated by SDS-PAGE and visualized with a silver staining kit (Bio-Rad). Bands of interest were in-gel digested with trypsin (Promega). For MALDI-TOF MS analysis, peptides were loaded onto the MALDI plate (Opti-TOFTM 384-well insert, Applied Biosystems). MALDI-TOF MS was performed on a 4800 MALDI-TOF/TOFTM analyzer (Applied Biosytems) equipped with a 355-nm Nd:YAG laser. Data were obtained in the reflectron mode with an accelerating voltage of 20 kV and sum from 500 laser pulses and calibrated with the 4700 calibration mixture (Applied Biosystems). Database search criteria were as follows: taxonomy, Homo sapiens, fixed modification; carboxyamidomethylated (+57) at cysteine residues; variable modification; oxidized (+16) at methionine residues, maximum allowed missed cleavage, 1, MS tolerance, 100 ppm. Typical contaminants such as trypsin (used for proteolysis) and keratin were excluded.
Co-immunoprecipitation and Western Blot Analysis
To assess the interaction of PPARγ with HAUSP, COS7 cells were transfected with following vectors using Lipofectamine Plus reagent (Invitrogen): HA-PPARγ WT, HA-AF1, HA-DBD, HA-LBD, HA-AF1/DBD, and HA-DBD/LBD with FLAG-HAUSP. Cell lysates were prepared with RIPA buffer containing 20 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS, 10 mm NaF, 0.1 mm Na3VO4, 12 mm β-glycerophosphate, 7 μg/ml aprotinin, 7 μg/ml leupeptin, and 1 mm PMSF. 300 μg of cell lysates were used for immunoprecipitation with anti-HA antibody coupled to agarose beads (Roche Applied Science) for 4 h at 4 °C. To evaluate the endogenous interaction of PPARγ with HAUSP, differentiated 3T3-L1 cells were lysed in RIPA buffer as described above. Then 300 μg of cell lysates were incubated with 2 μg of normal IgG or PPARγ-specific antibody overnight at 4 °C and further incubated with protein G-Sepharose (Roche Applied Science) for 1 h. To investigate the HAUSP-dependent deubiquitination of PPARγ, 60-mm dishes of COS7 cells (1.25 × 106) were transfected with pHA-PPARγ (0.5 μg), and pHis-ubiquitin (0.1 μg), in the presence or absence of pFLAG-HAUSP (0.5 μg) expression vectors. After 24 h, the cells were harvested and lysed with lysis buffer containing 20 mm HEPES, pH 7.4, 150 mm NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 1 mm EDTA, and 10 mm Na4P2O7, 100 mm NaF, 2 mm Na3VO4, 7 μg/ml aprotinin, 7 μg/ml leupeptin, and 1 mm PMSF. Then 500 μg of the cell lysates were incubated with anti-HA antibody bound to the agarose beads for 4 h at 4 °C. The precipitates were washed three times, subjected to 9% SDS-PAGE, and then blotted with their specific antibodies. All blots were developed using the enhanced chemiluminescence kit (Pierce).
Transient Transfection and Stability Assay
COS7 cells (5 × 105) were seeded into 12-well plates and transfected with HA-PPARγ and FLAG-HAUSP using Lipofectamine 2000 (Invitrogen) or infected with HAUSP-expressing adenovirus for 36 h. To assess the effect of HAUSP on PPARγ stability, COS7 or HepG2 cells were transfected with FLAG-HAUSP or HAUSP-specific siRNAs for 24 or 48 h. For HepG2 cells, polyethyleneimine (Mr 43.07, Sigma) was used for transfection. The cells were then treated with cycloheximide (5 μm) for the indicated times and analyzed for PPARγ expression by Western blot analysis.
Luciferase Activity Assay
HepG2 cells (5 × 105) were seeded into 12-well plates and transfected with luciferase reporter vectors containing PPRE (0.3 μg) and β-gal (0.1 μg) in the presence or absence of FLAG-HAUSP (0.1 and 0.3 μg) and cultured for 24 h. To assess the knockdown effect of HAUSP, HepG2 cells were transfected with luciferase reporter vectors containing PPRE (0.3 μg) and β-gal (0.1 μg) in the presence of control or HAUSP-specific siRNAs (20 and 50 nm) for 48 h. Then cells were treated with DMSO or rosiglitazone (10 μm) for 24 h and harvested using reporter lysis buffer. Luciferase activity was determined using the luciferase assay system kit (Promega) and quantified using GLOMAX (Promega) according to the manufacturer's instructions. Luciferase activity was normalized by β-galactosidase activity.
RT-PCR
Total RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. cDNA was prepared by reverse transcription with 1 μg of total RNA, and each gene transcript was amplified by PCR with its specific primers (Table 2). Real time PCR was performed with AccuPower Greenstar qPCR premix (SYBR Green pre-mix, Bioneer) and ExicyclerTM 96 real time PCR systems (Bioneer). GAPDH was used as an endogenous control.
TABLE 2.
Primers used for qRT-PCR
F means forward, and R means reverse.
| Primers | Sequences | Species |
|---|---|---|
| ADRP | F, 5′-TTGCAGTTGCCAATACCTATGC-3′ | Human |
| R, 5′-CCAGTCACAGTAGTCGTCACA-3′ | ||
| GK | F, 5′-CTGGGACAAGATAACTGGAGAGC-3′ | |
| R, 5′-TCAACGGTAGACTGGGTTCTTA-3′ | ||
| FABP1 | F, 5′-GTGTCGGAAATCGTGCAGAAT-3′ | |
| R, 5′-GACTTTCTCCCCTGTCATTGTC-3′ | ||
| GLUT2 | F, 5′-CCATCTTCCTCTTTGTCAGCTT-3′ | |
| R, 5′-AAATTGCAGGTCCAATTGCT-3′ | ||
| CD36 | F, 5′-AAGCCAGGTATTGCAGTTCTTT-3′ | |
| R, 5′-GCATTTGCTGATGTCTAGCACA-3′ | ||
| PPARγ | F, 5′-TACTGTCGGTTTCAGAAATGCC-3′ | |
| R, 5′-GTCAGCGGACTCTGGATTCAG-3′ | ||
| HAUSP | F, 5′-CCCTCCGTGTTTTGTGCGA-3′ | |
| R, 5′-AGACCATGACGTGGAATCAGA-3′ | ||
| GAPDH | F, 5′-AAGGTGAAGGTCGGAGTCAAC-3′ | |
| R, 5′-GGGGTCATTGATGGCAACAATA-3′ | ||
| PPARγ | F, 5′-TACTGTCGGTTTCAGAAATGCC-3′ | Monkey |
| R, 5′-GTCAGCGGACTCTGGGTTCAG-3′ | ||
| GAPDH | F, 5′-CTGGGCTACACTGAGCACC-3′ | |
| R, 5′-AAGTGGTCGTTGAGGGCAATG-3′ | ||
| ADRP | F, 5′-GACCTTGTGTCCTCCGCTTAT-3′ | Mouse |
| R, 5′-CAACCGCAATTTGTGGCTC-3′ | ||
| GK | F, 5′-TGAACCTGAGGATTTGTCAGC-3′ | |
| R, 5′-CCATGTGGAGTAACGGATTTCG-3′ | ||
| FABP1 | F, 5′-ATGAACTTCTCCGGCAAGTACC-3′ | |
| R, 5′-CTGACACCCCCTTGATGTCC-3′ | ||
| GLUT2 | F, 5′-GCCTGTGTATGCAACCATTG-3′ | |
| R, 5′-GAAGATGGCAGTCATGCTCA-3′ | ||
| CD36 | F, 5′-AGATGACGTGGCAAAGAACAG-3′ | |
| R, 5′-CCTTGGCTAGATAACGAACTCTG-3′ | ||
| PPARγ | F, 5′-TCAGGGCTGCCAGTTTCG-3′ | |
| R, 5′-GTAATCAGCAACCATTGGGTCA-3′ | ||
| HAUSP | F, 5′-CCTTAGCCCTCCGTGTTTTGT-3′ | |
| R, 5′-CCAGTCGTTTTCCTTGTGGAAG-3′ | ||
| GAPDH | F, 5′-ACCCCAGCAAGGACACTGAGCAAG-3′ | |
| R, 5′-GGCTCCCTAGGCCCCTCCTGTTATT-3′ | ||
| GFP | F, 5′-ATGGTGAGCAAGGGCGAGG-3′ | |
| R, 5′-TTACTTGTACAGCTCGTCCATG-3′ |
Metabolite Uptake Assay
HepG2 cells were maintained in minimum Eagle's medium as described above. HepG2 cells (5 × 105) were seeded into 12-well plates and cultured for 12 h, transfected with the indicated plasmids or siRNAs for 12 or 24 h, and then treated with DMSO or rosiglitazone (10 μm) for 24 h. HepG2 cells were treated with [3H]BSA/palmitate (1 μci/well) for 1 h. The cells were then washed with Hanks' buffered saline and lysed with 1 n NaOH. Radioactivity was quantified using a liquid scintillation counter (LKB). In addition, deoxyglucose uptake was measured as described (22). Briefly, after transfection with the indicated plasmids or siRNAs, cells were treated with DMSO or rosiglitazone (10 μm) and treated with 2-d-[14C]deoxyglucose (1 μCi/well) for 1 h. The incorporation rate was quantified as described above.
In Vivo Experiment
C57BL/6 (male) mice were purchased from Orient Bio (Seoul, Korea) at 6 weeks of age and maintained until 8 weeks of age. The animals were housed in a temperature-controlled room (23 °C) on a 12-h dark/12-h light cycle and maintained with standard rodent chow. The CHA Animal Care and Use Committee approved all animal studies, and the investigation conformed to the Guide for the Care and Use of Laboratory Animals, published by the National Institutes of Health. Adenoviruses expressing GFP or GFP/HAUSP were diluted in sterile PBS and intravenously injected through the tail vein at a dose of 2 × 109 infectious units/200 μl/mouse. Five days after infection, mice were euthanized with isoflurane (Hana Pharm, Korea), and blood and liver samples were harvested for metabolite, total RNA, and protein analysis.
Biochemical Analysis
Levels of triglycerides (TG), glucose concentrations, alanine aminotransferase, and aspartate aminotransferase were determined by standard laboratory procedures. Free fatty acid levels were determined using the Nefa kit (Randox Lab, UK) according to the manufacturer's instructions.
Intraperitoneal Glucose Tolerance Test
Mice were fasted for 10 h, and blood glucose levels were measured with a glucose meter (Lifescan) at time 0. Immediately thereafter, a 20% sterile glucose solution was injected intraperitoneally to reach a concentration of 2 g/kg of body weight. Blood was collected at the indicated time points, and glucose levels were measured. Area under the curve was calculated by trapezoidal rule.
Oil Red O Staining
Liver tissue was immediately embedded in optimal cutting temperature (OCT) compound (Sakura Finetek, Japan) after isolation and frozen at −70 °C. Frozen sections (4 μm) were fixed with 4% paraformaldehyde for 15 min and washed with PBS three times. Sections were stained for 1 h in freshly diluted Oil Red O solution (60% stock solution and 40% distilled water; Oil Red O stock solution is 0.5% Oil Red O in isopropyl alcohol). Sections were washed with distilled water for 10 min, and then sections were mounted with glycerin jelly. Images were captured under the microscope (Olympus, Japan) at ×100 magnification.
Immunohistochemistry
Liver tissues were fixed with 10% formalin for 24 h and embedded in paraffin block. Sections were cut with a Minot microtome at a thickness of 4 μm and were dewaxed and rehydrated. The endogenous peroxidase activity was quenched with 0.3% H2O2. Antigens were retrieved by incubation at 0.01 m citrate buffer, pH 6.0. GFP, HAUSP, and PPARγ were detected with their specific antibodies (Santa Cruz Biotechnology). Next, HRP-conjugated anti-rabbit or anti-mouse IgG was used to detect primary antibodies (DAKO). Sections were developed by incubation with a solution of 3,3′-diaminobenzidine tetrahydrochloride substrate for 3 min (DAKO). Nuclei were counterstained with Mayer's hematoxylin for 5 min.
Statistics
SPSS, version 10.0 (SPSS Inc., Chicago, IL), was used for statistical analysis. The data are expressed as means ± S.E. The differences between the means were calculated using the Mann-Whitney U test. A p value less than 0.05 denoted the presence of a statistically significant difference.
RESULTS
Association of PPARγ with HAUSP
PPARγ is a substrate of polyubiquitination and is degraded by proteasome-dependent pathways (13, 14). In addition, PPARγ is known to be regulated by sumoylation (11, 23, 24). To identify the proteins that regulate PPARγ, we performed a GST pulldown assay using HeLa extracts with GST or GST-PPARγ. Mass analysis identified the isolated binding candidate of PPARγ as HAUSP (Fig. 1, A and B). The interaction between PPARγ and HAUSP was confirmed by co-immunoprecipitation (Fig. 1C). To further investigate the endogenous interaction between PPARγ and HAUSP, co-immunoprecipitation was performed using differentiated 3T3-L1 adipocytes as described under “Experimental Procedures.” This confirmed that PPARγ specifically interacts with HAUSP but not with C/EBPα (Fig. 1D). To examine the ligand dependence of the interaction of PPARγ with HAUSP, differentiated adipocytes or COS7 cells transfected with HAUSP and PPARγ were treated with the PPARγ ligand rosiglitazone in a time-dependent manner, and co-immunoprecipitation analysis was performed. However, there was no ligand dependence in the interaction of PPARγ with HAUSP (data not shown). In addition, an in vitro competition assay using the HAUSP-PPARγ complex and rosiglitazone did not show ligand dependence (data not shown). To identify the PPARγ domains responsible for the interaction with HAUSP, various deletion mutants of PPARγ were generated (Fig. 1E) and used in co-immunoprecipitation assays. HA-PPARγ WT, HA-PPARγ DBD, HA-PPARγ LBD, HA-PPARγ AF1/DBD, and HA-PPARγ DBD/LBD interacted with HAUSP, but HA-AF1 did not (Fig. 1F).
FIGURE 1.

HAUSP interacts with PPARγ. A, GST or GST-PPARγ was incubated with 10 mg of HeLa cell extract and separated by SDS-PAGE followed by visualization through silver staining. B, band was in-gel digested with trypsin and analyzed using a 4800 MALDI-TOF/TOFTM analyzer. The red letters indicate the regions identified by mass analysis. C, COS7 cells were transfected with HA-PPARγ and FLAG-HAUSP. Cell lysates were subjected to immunoprecipitation (IP) using an anti-HA antibody and immunoblotted with anti-FLAG or anti-HA antibodies. WCL, whole cell lysate. D, WCLs of differentiated 3T3-L1 cells were immunoprecipitated with normal IgG or an anti-PPARγ antibody and blotted with anti-HAUSP, anti-C/EBPα, and anti-PPARγ antibody. E, to generate HA-PPARγ fusion proteins, the HA tag was fused to AF1, DBD, LBD, AF1/DBD, or DBD/LBD. F, HA-PPARγ-WT, HA-PPARγ-AF1, HA-PPARγ-DBD, HA-PPARγ-LBD, HA-PPARγ-AF1/DBD, and HA-PPARγ-DBD/LBD were co-transfected with FLAG-HAUSP into COS7 cells, and 500 μg of WCL was subjected to immunoprecipitation with an anti-FLAG antibody followed by immunoblotting with anti-HA or anti-FLAG antibodies. EV, empty vector. Arrowheads indicate IgG heavy chain (upper) and IgG light chain (lower).
Deubiquitination and Stabilization of PPARγ by HAUSP
HAUSP increases the stability of target proteins, including p53 and MDM2, by deubiquitinating these targets and preventing their proteasome-dependent degradation (15, 16, 25). Therefore, the deubiquitination of PPARγ by HAUSP was examined in COS7 cells. Co-immunoprecipitation using an anti-HA antibody showed that HAUSP efficiently deubiquitinated the ubiquitin-PPARγ conjugates (Fig. 2Α). The overexpression of FLAG-HAUSP increased PPARγ protein levels in whole cell extracts. Because PPARγ is degraded upon ubiquitination (26), the increase of PPARγ in whole cell lysates might be due to the deubiquitinating activity of HAUSP. Recently, it was reported that a catalytic mutant of HAUSP (C223S) failed to stabilize p53 due to the lack of deubiquitinating activity (15). Thus, we further confirmed the specificity of HAUSP for the deubiquitination of PPARγ by transfecting the HAUSP C223S mutant into COS7 cells. Wild-type HAUSP, but not the HAUSP C223S mutant, efficiently removed ubiquitin from ubiquitin-PPARγ conjugates (Fig. 2B). The protein levels of PPARγ were increased by wild-type HAUSP but not by the HAUSP C223S mutant (Fig. 2B). In addition, the ubiquitination assay was repeated using various PPARγ deletion mutants, and in agreement with a previous report, we identified the LBD of PPARγ as a major ubiquitinated domain (Fig. 2C) (26). Using the deubiquitination assay, we were also able to show HAUSP efficiently removed ubiquitin from the Ub-LBD of PPARγ (Fig. 2D). In addition, site-directed mutagenesis was performed to find the critical residue for ubiquitination of PPARγ. To predict the ubiquitinated residues of PPARγ, four mutants of PPARγ (K382R, K386R, K395R, and K462R) were examined in the ubiquitination assay. Our results showed that the Lys462 mutant of PPARγ showed reduced ubiquitination (Fig. 2E).
FIGURE 2.
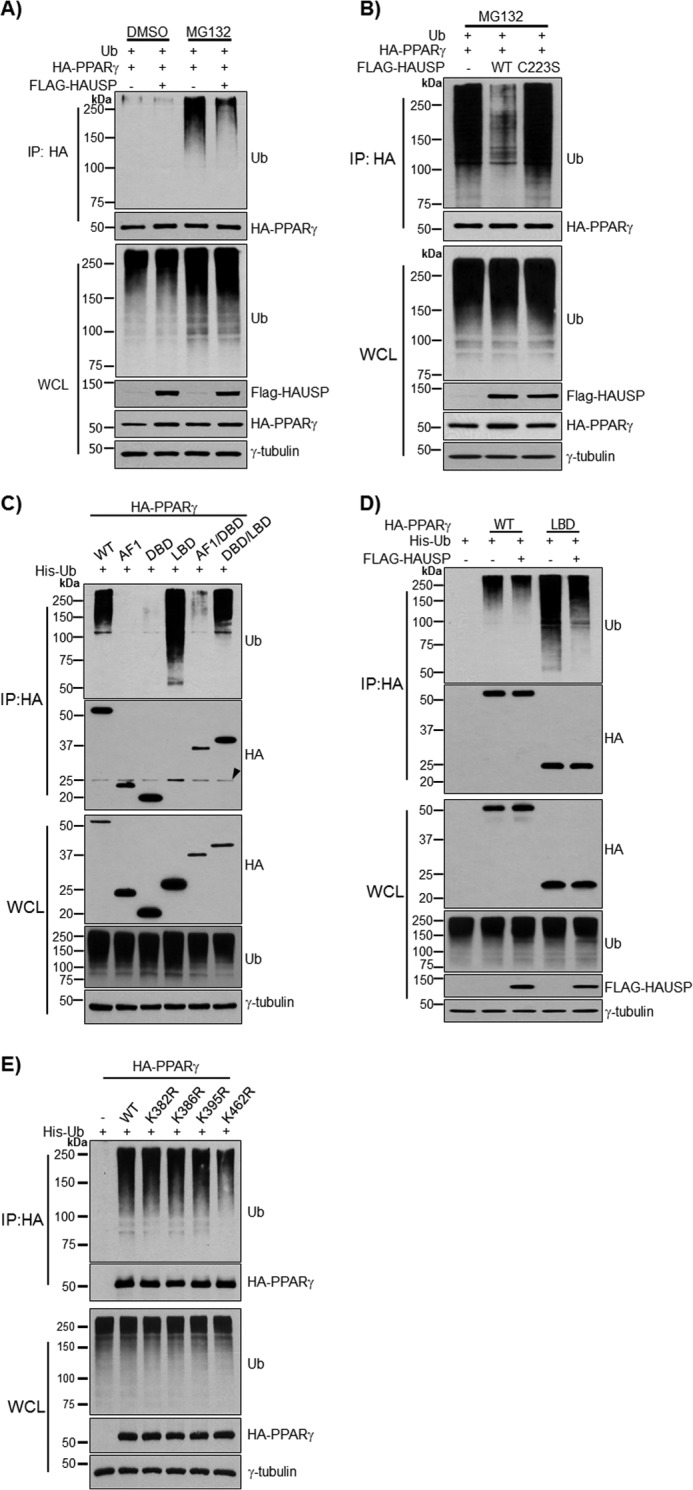
Deubiquitination of PPARγ by HAUSP. HA-PPARγ, His-Ub, and FLAG-HAUSP were co-transfected into COS7 cells for 24 h, and cells were treated with MG132 (10 μm), a proteasome inhibitor, for 5 h to inhibit degradation of ubiquitinated-PPARγ. A, PPARγ was precipitated with an anti-HA antibody and immunoblotted with the indicated antibodies. To confirm the specificity of HAUSP for PPARγ, HA-PPARγ and His-Ub were co-transfected with wild-type FLAG-HAUSP or mutant FLAG-HAUSP C223S for 24 h and treated with MG132 (10 μm) for 5 h. PPARγ was precipitated with an anti-HA antibody and immunoblotted with the indicated antibodies (B). C, HA-PPARγ-WT, HA-PPARγ-AF1, HA-PPARγ-DBD, HA-PPARγ-LBD, HA-PPARγ-AF1/DBD, and HA-PPARγ-DBD/LBD were co-transfected with His-Ub into COS7 cells, and 500 μg of WCL were subjected to immunoprecipitation (IP) with an anti-HA antibody followed by immunoblotting with anti-HA or anti-Ub antibody. Arrowhead indicates IgG light chain. D, HA-PPARγ-WT and HA-PPARγ-LBD were co-transfected with His-Ub into COS7 cells in the presence or absence of FLAG-HAUSP. PPARγ was precipitated with an anti-HA antibody and immunoblotted with anti-HA, anti-Ub, or anti-FLAG antibody. E, HA-PPARγ (wild type, K382R, K386R, K395R, and K462R) was co-transfected with His-Ub into COS7 cells and subjected to the ubiquitination assay. PPARγ was precipitated with an anti-HA antibody and immunoblotted with anti-HA or anti-Ub antibody. γ-tubulin was used as a loading control. The experiment was performed independently at least three times.
To further confirm the stabilization of PPARγ by HAUSP, FLAG-HAUSP and HA-PPARγ were transfected at different concentrations into COS7 cells. PPARγ protein expression was increased upon the expression of FLAG-HAUSP in a dose-dependent manner (Fig. 3A). Furthermore, endogenous PPARγ levels increased in a dose-dependent manner after infection with HAUSP-expressing adenoviruses (Ad-GFP/HAUSP) but not with control adenoviruses (Ad-GFP) (Fig. 3B). The endogenous PPARγ band detected in COS7 cells was further confirmed by Western blot and qRT-PCR after transfection of PPARγ siRNA (data not shown). qRT-PCR analysis showed that the transcript level of PPARγ was not affected by HAUSP overexpression (Fig. 3, B and C). To rule out the possibility that the regulation of PPARγ by HAUSP might be regulated at the translational level, cycloheximide treatment was used to inhibit de novo protein synthesis, and the stability of PPARγ was assessed. In this experiment, values were normalized to the basal intensity of the PPARγ signal to exclude density-dependent variation when calculating the time-dependent effects of HAUSP on the stability of PPARγ. In the control samples, PPARγ protein levels decreased by ∼50% within 2 h of cycloheximide treatment, and the protein was barely detected after 8 h, which is in agreement with previous reports (Fig. 3D) (3, 27, 28). The overexpression of HAUSP significantly prolonged the half-life of PPARγ for up to about 6 h after cycloheximide treatment (Fig. 3, D and E). However, the HAUSP C223S mutant had no effect on the half-life of PPARγ (Fig. 3, F and G). Recently, 7-chloro-9-oxo-9H-indeno[1,2-b]pyrazine-2,3-dicarbonitrile(HBX 41,108) was identified as an inhibitor of HAUSP (29). HBX 41,108 inhibited the increase of PPARγ stability induced by wild-type HAUSP, further confirming that PPARγ stability is regulated by HAUSP (Fig. 3, F and G).
FIGURE 3.

HAUSP increases the stability of PPARγ. A, HA-PPARγ and FLAG-HAUSP were transfected into COS7 cells for 24 h. Cell lysates (30 μg) were subjected to Western blot (W.B.) analysis. B and C, COS7 cells were infected with GFP or GFP/HAUSP adenoviruses as indicated, and cell lysates (70 μg) were subjected to immunoblotting. Total RNA was isolated, and qRT-PCR was performed to analyze the expression of PPARγ, HAUSP, and GFP mRNA. Data represent the mean ± S.E. of the averages of three independent experiments. moi, multiplicity of infection. D and E, empty vector (EV) or FLAG-HAUSP was co-transfected with HA-PPARγ to COS7 cells for 24 h, and cycloheximide (5 μm) was added for the indicated times. *, p < 0.01. F and G, wild-type FLAG-HAUSP or mutant FLAG-HAUSP C223S was co-transfected with HA-PPARγ to COS7 cells for 24 h, and cells were pretreated with HBX 41,108 (20 μm) for 2 h. Cells were treated with cycloheximide (5 μm) for the indicated times, and lysates were analyzed by Western blotting (*, versus HAUSP-C223S; †, versus HAUSP-WT +,HBX; ‡, versus HAUSP-C223S). The densities of the PPARγ bands at time point 0 were set to 100%, and the remaining densities were expressed as relative values. Data represent the mean ± S.D. of the averages of three independent experiments. *, p < 0.01; †, p < 0.05; ‡, p < 0.05.
PPARγ Transcriptional Activity Is Increased by HAUSP
To determine whether the increased stability of PPARγ could enhance its transcriptional activity, luciferase assays using PPRE were performed in COS7 or HepG2 cells. The results showed that the overexpression of HAUSP significantly increased the transcriptional activity of endogenous as well as overexpressed PPARγ by 2–4-fold in both the DMSO- and rosiglitazone-treated groups (Fig. 4, A and B). Transfection of the HAUSP C223S mutant or treatment of cells with HBX 41,108 did not increase the stability of endogenous PPARγ, eliminating the synergistic effect of HAUSP plus rosiglitazone (Fig. 4C). Additionally, we examined whether HBX 41,108 could decrease the transcriptional activity of endogenous PPARγ by inhibiting endogenous HAUSP. HBX 41,108 significantly reduced the basal transcriptional activity of PPARγ by up to 70% and completely abolished the transcriptional activity induced by rosiglitazone treatment (Fig. 4D). Furthermore, the inhibition of endogenous HAUSP by HBX 41,108 decreased the expression of endogenous PPARγ, further confirming that HAUSP stabilizes PPARγ protein (Fig. 4D). The specific siRNA-mediated knockdown of HAUSP in HepG2 cells significantly decreased the transcriptional activity of PPARγ as well as the expression of endogenous PPARγ (Fig. 4E). PPARγ target gene analyses using quantitative RT-PCR in HepG2 cells showed that HAUSP increased the transcription of adipose differentiation-related protein (ADRP), fatty acid-binding protein 1 (FABP1), glycerol kinase, glucose transporter 2 (GLUT2), and CD36 by about 2-fold even in the absence of rosiglitazone (Fig. 4F). Rosiglitazone further increased the expression of ADRP, FABP1, GLUT2, and CD36. Knockdown of HAUSP by siRNA in HepG2 cells decreased the transcript levels of ADRP, FABP1, glycerol kinase, GLUT2, and CD36 in the presence and absence of rosiglitazone, whereas the PPARγ transcript levels were unchanged (Fig. 4G). Overexpression of HAUSP in HepG2 cells significantly increased glucose uptake by ∼20% and fatty acid uptake by 30% compared with the control, in both the presence and absence of rosiglitazone (Fig. 4, H and I). Conversely, down-regulation of HAUSP by siRNA significantly decreased the uptake of glucose and fatty acids in the presence and absence of rosiglitazone (Fig. 4, J and K). Taken together, these results suggest that HAUSP increases the stability and transcriptional activity of PPARγ at the post-translational level.
FIGURE 4.

HAUSP increases the transcriptional activity of PPARγ. A, COS7 cells were transfected with HA-PPARγ, luciferase reporter vectors containing PPRE, and β-gal, in the presence or absence of FLAG-HAUSP, and DMSO or rosiglitazone (10 μm) was applied for 24 h. Luciferase activity was determined as described under “Experimental Procedures.” Cell lysates were blotted with anti-HA or anti-FLAG antibody. HepG2 cells were co-transfected with a luciferase reporter construct containing the PPRE (PPRE-pk-luc) and the β-gal reporter in the presence or absence of FLAG-HAUSP (B) and in the presence of wild-type FLAG-HAUSP or the mutant FLAG-HAUSP C223S. Cells were pretreated with HBX 41,108 (5 μm) for 2 h and then treated with DMSO or rosiglitazone (10 μm) for 24 h. W, wild type; M, mutant (C). HepG2 cells were co-transfected with a luciferase reporter construct containing the PPRE (PPRE-pk-luc) and the β-gal reporter for 24 h, pretreated with HBX 41,108 (5 μm) for 2 h, and then treated with DMSO or rosiglitazone (10 μm) for 24 h (D). HepG2 cells were transfected with control siRNA or HAUSP siRNA (20 and 50 nm) for 24 h and then with the luciferase reporter vectors (pk-luc or PPRE-pk-luc) with β-gal for 24 h (E). Luciferase activity assays were performed as described under “Experimental Procedures.” Data represent the mean ± S.D. of the averages of four independent experiments. In addition, Western blot analysis was performed using anti-PPARγ, anti-HAUSP, and anti-FLAG antibody. γ-Tubulin was used as loading control. To examine the expression of PPARγ target genes in response to HAUSP, empty vector (EV), or FLAG-HAUSP (100 ng) (F) and control siRNA or HAUSP siRNA (20 nm) (Invitrogen) (G) were transfected into HepG2 cells for 24 and 48 h, respectively. Total RNAs were isolated and synthesized into cDNA by qRT-PCR using specific primers as described in Table 2. H–K, HepG2 cells were transfected with the indicated plasmids (100 ng) or siRNAs (20 nm), and treated with DMSO or rosiglitazone (10 μm). The uptake of [3H]BSA/palmitate or 2- d-[14C]deoxyglucose was analyzed as described under “Experimental Procedures.” Data represent the mean ± S.D. of the averages of three independent experiments. *, p < 0.05; **, p < 0.01.
HAUSP Overexpression Stabilizes Endogenous PPARγ in the Liver
To examine the effect of HAUSP on endogenous PPARγ expression in vivo, adenoviruses expressing GFP (Ad-GFP), HAUSP C223S (Ad-GFP/HAUSP C223S), or HAUSP (Ad-GFP/HAUSP) were injected intravenously into the tail vein of C57BL/6 mice. The adenoviruses used in this study did not cause changes in body weight, and there were no differences in the levels of aspartate aminotransferase and alanine aminotransferase between the Ad-GFP-, Ad-GFP/HAUSP C223S-, and Ad-GFP/HAUSP-injected groups (data not shown). Wild-type HAUSP induced the stabilization of endogenous PPARγ in the liver, but Ad-GFP and Ad-GFP/HAUSP C223S had no effect (Fig. 5A). Immunohistochemical analysis showed that wild-type HAUSP increased the levels of endogenous PPARγ, whereas the HAUSP C223S mutant had no effect, further confirming that HAUSP stabilizes endogenous PPARγ in vivo as well as in vitro (Fig. 5B). H&E and Oil Red O staining showed that wild-type HAUSP (Ad-GFP/HAUSP) caused TG accumulation in the liver compared with the control (Ad-GFP) and the HAUSP C223S mutant (Ad-GFP/HAUSP C223S) (Fig. 5B), which was confirmed by quantification of hepatic TG (Fig. 5C). In addition, the analyses of PPARγ target genes by qRT-PCR showed that the expression levels of ADRP, glycerol kinase, FABP1, GLUT2, and CD36 were significantly increased by Ad-GFP/HAUSP but not by Ad-GFP or Ad-GFP/HAUSP C223S (Fig. 5, D–F). The effect of increased PPARγ levels caused by HAUSP overexpression on various metabolites was examined. Adenoviruses expressing wild-type HAUSP (Ad-GFP/HAUSP) significantly decreased glucose, free fatty acid, and TG levels in the blood compared with the GFP control (Ad-GFP) and the HAUSP C223S mutant (Ad-GFP/HAUSP C223S) (Fig. 5, G–I). In addition, an intraperitoneal glucose tolerance test showed that the blood glucose of mice injected with Ad-GFP/HAUSP was more rapidly decreased than that of the control group (Ad-GFP) and the HAUSP C223S mutant group (Ad-GFP/HAUSP C223S) (Fig. 5J). Analysis of the area under the curve showed that blood glucose levels in the Ad-GFP/HAUSP group were significantly lower than those in the control group (Ad-GFP) and the HAUSP C223S mutant group (Ad-GFP/HAUSP C223S) (Fig. 5J, bottom panel). These results suggest that HAUSP overexpression could regulate glucose and fatty acid metabolism via stabilization of endogenous PPARγ.
FIGURE 5.

HAUSP induced the expression of PPARγ in vivo. C57BL/6 mice (n = 5 in each group) were intravenously injected with Ad-GFP, Ad-GFP/HAUSP C223S, or Ad-GFP/HAUSP adenoviruses through the tail vein and sacrificed 5 days later. A, liver samples from each group were immunoblotted with the indicated antibodies. B, expression of HAUSP, PPARγ, and GFP was examined by immunohistochemical staining. H&E staining showed TG accumulation in mice injected with Ad-GFP/HAUSP. TG accumulation was further confirmed by Oil Red O staining. Liver samples were harvested for the quantification of TG content (C) and the analysis of PPARγ target genes (D), GFP (E), and HAUSP (F) (n = 5 in each group). Blood glucose level (G), blood TG levels (H), and blood free fatty acid (FFA) (I) were quantified using specific assay kits. J, intraperitoneal glucose tolerance test was performed, and the area under the curve (AUC) was calculated (n = 5 in each group). The data represent the mean ± S.D. *, p < 0.05; **, p < 0.001.
DISCUSSION
PPARγ plays a critical role in a variety of physiological processes, including adipogenesis, glucose homeostasis, lipid metabolism, and osteogenesis (1). PPARγ is polyubiquitinated in response to ligand binding or phosphorylation and then degraded by the proteasome (13, 14). However, to date, no enzymes have been identified that can deubiquitinate or dephosphorylate PPARγ. HAUSP was isolated as a novel PPARγ regulatory protein that stabilizes its target protein by deubiquitination (15, 16, 30). The DBD and LBD of PPARγ were responsible for its interaction with HAUSP (Fig. 1). A recent report showed that only the LBD of PPARγ is polyubiquitinated to target the protein for degradation (26), suggesting that HAUSP stabilizes the PPARγ protein by removing the ubiquitin conjugated to the LBD. In this study, the K462R mutant of PPARγ showed a reduced ubiquitination pattern. In addition, we found that PPARγ was still ubiquitinated even when Lys462 was substituted with arginine, suggesting that PPARγ is ubiquitinated at multiple residues.
Analysis of the stability of PPARγ showed that it has a half-life of ∼2 h (Fig. 3E). This result is consistent with previous reports (3, 27). Surprisingly, HAUSP significantly increased the half-life of PPARγ by up to 4–6 h. Whereas PPARγ was hardly detected 8 h after cycloheximide treatment in the controls, ∼30% of the protein was still detectable in COS7 cells overexpressing HAUSP. This was further confirmed by transfection with the HAUSP C223S mutant, which did not increase PPARγ stability. Furthermore, treatment with HBX 41,108, a specific inhibitor of HAUSP, abolished the increase of PPARγ stability induced by the ectopic expression of wild-type HAUSP (Fig. 3E). In addition, knockdown of HAUSP using a specific siRNA destabilized endogenous PPARγ (Fig. 4E). A luciferase assay using the PPRE showed that HAUSP increased the transcriptional activity of PPARγ. However, there were no changes in the specific activity of PPARγ, as shown by normalization of luciferase activity using PPARγ (data not shown). This suggests that the HAUSP-mediated increase in PPARγ was due to an increase in PPARγ protein, rather than an effect on its specific activity. The functional relationship between PPARγ and HAUSP appears to be most evident in liver cells, as the altered PPARγ stability affected by HAUSP overexpression or siRNA knockdown was not observed in differentiated 3T3-L1 adipocytes. Although HAUSP has deubiquitinating activity against PPARγ in differentiating adipocytes, increased PPARγ deubiquitination may be obscured by the high PPARγ expression levels in this cell type. However, as the expression of PPARγ in liver is relatively minimal compared with adipocytes, its up-regulation by HAUSP in liver might have a more significant effect upon metabolic regulation than in adipocytes. Additionally, because PPARγ expression is not abundant in liver, the physiological function of PPARγ stabilized by HAUSP might be underestimated in the regulation of blood glucose and TG. However, recent reports suggest that hepatic PPARγ is up-regulated in obese animal models (31), and increased hepatic PPARγ is an initial event leading to lipid accumulation and fatty liver (32). Liver-specific disruption of PPARγ improved fatty liver but aggravated systemic insulin resistance in ob/ob mice. This suggests that hepatic PPARγ plays a critical role in the regulation of TG content and in the homeostasis of blood glucose and insulin resistance in steatotic diabetic mice (33). In addition, we could not rule out the possibility that HAUSP has additional targets that might affect metabolism in hepatocytes. Based on our data and previous studies on the role of PPARγ in hepatic steatosis and insulin sensitivity, we suggest that HAUSP overexpression could decrease blood glucose and TG concentration at least in part by stabilizing PPARγ in liver.
An increase in PPARγ expression in the liver induces TG accumulation (34, 35). The overexpression of HAUSP, but not the HAUSP C223S mutant, also promoted lipid accumulation in the liver while decreasing TG levels in the blood (Fig. 5, B, C, and H), which suggests that VLDL secretion from liver might be decreased by HAUSP overexpression. Investigation of VLDL secretion using an ELISA showed that HAUSP overexpression decreased VLDL secretion from HepG2 cells (data not shown). The chronic treatment of pioglitazone, a PPARγ agonist, has been known to reduce VLDL TG levels by increasing the expression of lipoprotein lipase (36, 37). In this study, however, the expression level of lipoprotein lipase was not changed by HAUSP (data not shown). Therefore, the decreased secretion of VLDL may be due to an lipoprotein lipase-independent regulatory mechanism brought about by HAUSP-stabilized PPARγ.
Wild-type HAUSP decreased blood glucose and TG levels, which are associated with the increased expression of endogenous PPARγ and lipid accumulation in the liver (Figs. 4 and 5). These results suggest that HAUSP could be a critical regulator of metabolism, in addition to its tumor-promoting activity mediated by p53 and MDM2 (15, 16, 25, 30, 38). PPARγ is overexpressed in a variety of cancers, including those of the colon (39), breast (40), and prostate (41), because cancer cells require large amounts of metabolites to maintain their highly proliferative status. However, the molecular mechanism underlying the increased expression of PPARγ in cancer is not well understood. We used immunohistochemical staining to investigate whether PPARγ was increased in human hepatocellular carcinoma (HCC) and found that HAUSP and PPARγ levels were higher in tumorous areas than in adjacent nontumorous tissues (data not shown). Increased levels of PPARγ in HCC could be, at least in part, due to the overexpression of HAUSP.
PPARγ is suggested to play a tumor-suppressing role in carcinogenesis (42). Treatment with thiazolidinediones, which are PPARγ ligands, induced cancer regression by triggering apoptosis (43, 44). However, recent studies using PPARγ−/− cells demonstrated that the anticancer effect of TZDs is independent of PPARγ (45, 46). Of note, some reports suggest that PPARγ functions as an oncogene in certain cancers (47, 48). The role of PPARγ as an oncogene or tumor suppressor is still controversial. However, the increase in PPARγ levels induced by HAUSP may be important in cancer metabolism, and further investigation is needed to determine the inter-relationship between the multiple functions of PPARγ, its many modifications, and cancer.
A recent report indicates that CDK5-mediated phosphorylation of PPARγ may be involved in the pathogenesis of insulin resistance, suggesting a possible target for the development of novel anti-diabetic drugs (10). Similarly, the effect of HAUSP on increasing the stability of PPARγ suggests that HAUSP could be a target for the development of anti-diabetic drugs, which could then be used in combination with insulin or TZDs to decrease blood glucose levels.
This work was supported by the Bio and Medical Technology Development Program of the National Research Foundation funded by Korean Government Grant 2012M3A9C6049719 (to S. G. P.), Basic Science Research Program through the National Research Foundation of Korea funded by Ministry of Education, Science, and Technology Grant 2012R1A1A2005546 (to K. S. P.), and the Basic Research Program through the National Research Foundation of Korea funded by Ministry of Education, Science, and Technology Grant 2010-0008279 (K.-H. B.).
- PPARγ
- peroxisome proliferator-activated receptor γ
- HAUSP
- herpesvirus-associated ubiquitin-specific protease
- TG
- triglyceride
- PPRE
- PPAR-response element
- DBD
- DNA-binding domain
- LBD
- ligand-binding domain
- HAUSP
- herpesvirus-associated ubiquitin-specific protease
- TG
- triglyceride
- AF
- activation function
- ADRP
- adipose differentiation-related protein
- WCL
- whole cell lysate
- Ub
- ubiquitin
- qRT
- quantitative RT.
REFERENCES
- 1. Desvergne B., Wahli W. (1999) Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr. Rev. 20, 649–688 [DOI] [PubMed] [Google Scholar]
- 2. Lehrke M., Lazar M. A. (2005) The many faces of PPARγ. Cell 123, 993–999 [DOI] [PubMed] [Google Scholar]
- 3. van Beekum O., Fleskens V., Kalkhoven E. (2009) Post-translational modifications of PPAR-γ: fine-tuning the metabolic master regulator. Obesity 17, 213–219 [DOI] [PubMed] [Google Scholar]
- 4. Adams M., Reginato M. J., Shao D., Lazar M. A., Chatterjee V. K. (1997) Transcriptional activation by peroxisome proliferator-activated receptor γ is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. J. Biol. Chem. 272, 5128–5132 [DOI] [PubMed] [Google Scholar]
- 5. Camp H. S., Tafuri S. R. (1997) Regulation of peroxisome proliferator-activated receptor γ activity by mitogen-activated protein kinase. J. Biol. Chem. 272, 10811–10816 [DOI] [PubMed] [Google Scholar]
- 6. Camp H. S., Tafuri S. R., Leff T. (1999) c-Jun N-terminal kinase phosphorylates peroxisome proliferator-activated receptor-γ1 and negatively regulates its transcriptional activity. Endocrinology 140, 392–397 [DOI] [PubMed] [Google Scholar]
- 7. Hu E., Kim J. B., Sarraf P., Spiegelman B. M. (1996) Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARγ. Science 274, 2100–2103 [DOI] [PubMed] [Google Scholar]
- 8. Zhang B., Berger J., Zhou G., Elbrecht A., Biswas S., White-Carrington S., Szalkowski D., Moller D. E. (1996) Insulin- and mitogen-activated protein kinase-mediated phosphorylation and activation of peroxisome proliferator-activated receptor γ. J. Biol. Chem. 271, 31771–31774 [DOI] [PubMed] [Google Scholar]
- 9. Iankova I., Petersen R. K., Annicotte J. S., Chavey C., Hansen J. B., Kratchmarova I., Sarruf D., Benkirane M., Kristiansen K., Fajas L. (2006) Peroxisome proliferator-activated receptor γ recruits the positive transcription elongation factor b complex to activate transcription and promote adipogenesis. Mol. Endocrinol. 20, 1494–1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Choi J. H., Banks A. S., Estall J. L., Kajimura S., Boström P., Laznik D., Ruas J. L., Chalmers M. J., Kamenecka T. M., Blüher M., Griffin P. R., Spiegelman B. M. (2010) Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARγ by Cdk5. Nature 466, 451–456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ohshima T., Koga H., Shimotohno K. (2004) Transcriptional activity of peroxisome proliferator-activated receptor γ is modulated by SUMO-1 modification. J. Biol. Chem. 279, 29551–29557 [DOI] [PubMed] [Google Scholar]
- 12. Chung S. S., Ahn B. Y., Kim M., Kho J. H., Jung H. S., Park K. S. (2011) SUMO modification selectively regulates transcriptional activity of peroxisome-proliferator-activated receptor γ in C2C12 myotubes. Biochem. J. 433, 155–161 [DOI] [PubMed] [Google Scholar]
- 13. Hauser S., Adelmant G., Sarraf P., Wright H. M., Mueller E., Spiegelman B. M. (2000) Degradation of the peroxisome proliferator-activated receptor γ is linked to ligand-dependent activation. J. Biol. Chem. 275, 18527–18533 [DOI] [PubMed] [Google Scholar]
- 14. Floyd Z. E., Stephens J. M. (2002) Interferon-γ-mediated activation and ubiquitin-proteasome-dependent degradation of PPARγ in adipocytes. J. Biol. Chem. 277, 4062–4068 [DOI] [PubMed] [Google Scholar]
- 15. Li M., Chen D., Shiloh A., Luo J., Nikolaev A. Y., Qin J., Gu W. (2002) Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature 416, 648–653 [DOI] [PubMed] [Google Scholar]
- 16. Cummins J. M., Rago C., Kohli M., Kinzler K. W., Lengauer C., Vogelstein B. (2004) Tumour suppression: disruption of HAUSP gene stabilizes p53. Nature 428, 1 page following 486 [DOI] [PubMed] [Google Scholar]
- 17. Song M. S., Salmena L., Carracedo A., Egia A., Lo-Coco F., Teruya-Feldstein J., Pandolfi P. P. (2008) The deubiquitinylation and localization of PTEN are regulated by a HAUSP-PML network. Nature 455, 813–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van der Horst A., de Vries-Smits A. M., Brenkman A. B., van Triest M. H., van den Broek N., Colland F., Maurice M. M., Burgering B. M. (2006) FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat. Cell Biol. 8, 1064–1073 [DOI] [PubMed] [Google Scholar]
- 19. van der Knaap J. A., Kumar B. R., Moshkin Y. M., Langenberg K., Krijgsveld J., Heck A. J., Karch F., Verrijzer C. P. (2005) GMP synthetase stimulates histone H2B deubiquitylation by the epigenetic silencer USP7. Mol. Cell 17, 695–707 [DOI] [PubMed] [Google Scholar]
- 20. Saridakis V., Sheng Y., Sarkari F., Holowaty M. N., Shire K., Nguyen T., Zhang R. G., Liao J., Lee W., Edwards A. M., Arrowsmith C. H., Frappier L. (2005) Structure of the p53 binding domain of HAUSP/USP7 bound to Epstein-Barr nuclear antigen 1 implications for EBV-mediated immortalization. Mol. Cell 18, 25–36 [DOI] [PubMed] [Google Scholar]
- 21. Chung S. S., Kim M., Youn B. S., Lee N. S., Park J. W., Lee I. K., Lee Y. S., Kim J. B., Cho Y. M., Lee H. K., Park K. S. (2009) Glutathione peroxidase 3 mediates the antioxidant effect of peroxisome proliferator-activated receptor γ in human skeletal muscle cells. Mol. Cell. Biol. 29, 20–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kase E. T., Wensaas A. J., Aas V., Højlund K., Levin K., Thoresen G. H., Beck-Nielsen H., Rustan A. C., Gaster M. (2005) Skeletal muscle lipid accumulation in type 2 diabetes may involve the liver X receptor pathway. Diabetes 54, 1108–1115 [DOI] [PubMed] [Google Scholar]
- 23. Lim S., Ahn B. Y., Chung S. S., Park H. S., Cho B. J., Kim M., Choi S. H., Lee I. K., Lee S. W., Choi S. J., Chung C. H., Cho Y. M., Lee H. K., Park K. S. (2009) Effect of a peroxisome proliferator-activated receptor γ sumoylation mutant on neointimal formation after balloon injury in rats. Atherosclerosis 206, 411–417 [DOI] [PubMed] [Google Scholar]
- 24. Jennewein C., Kuhn A. M., Schmidt M. V., Meilladec-Jullig V., von Knethen A., Gonzalez F. J., Brüne B. (2008) Sumoylation of peroxisome proliferator-activated receptor γ by apoptotic cells prevents lipopolysaccharide-induced NCoR removal from κB-binding sites mediating transrepression of proinflammatory cytokines. J. Immunol. 181, 5646–5652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li M., Brooks C. L., Kon N., Gu W. (2004) A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol. Cell 13, 879–886 [DOI] [PubMed] [Google Scholar]
- 26. Kilroy G. E., Zhang X., Floyd Z. E. (2009) PPAR-γ AF-2 domain functions as a component of a ubiquitin-dependent degradation signal. Obesity 17, 665–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Christianson J. L., Nicoloro S., Straubhaar J., Czech M. P. (2008) Stearoyl-CoA desaturase 2 is required for peroxisome proliferator-activated receptor γ expression and adipogenesis in cultured 3T3-L1 cells. J. Biol. Chem. 283, 2906–2916 [DOI] [PubMed] [Google Scholar]
- 28. Waite K. J., Floyd Z. E., Arbour-Reily P., Stephens J. M. (2001) Interferon-γ-induced regulation of peroxisome proliferator-activated receptor γ and STATs in adipocytes. J. Biol. Chem. 276, 7062–7068 [DOI] [PubMed] [Google Scholar]
- 29. Colland F., Formstecher E., Jacq X., Reverdy C., Planquette C., Conrath S., Trouplin V., Bianchi J., Aushev V. N., Camonis J., Calabrese A., Borg-Capra C., Sippl W., Collura V., Boissy G., Rain J. C., Guedat P., Delansorne R., Daviet L. (2009) Small-molecule inhibitor of USP7/HAUSP ubiquitin protease stabilizes and activates p53 in cells. Mol. Cancer Ther. 8, 2286–2295 [DOI] [PubMed] [Google Scholar]
- 30. Cummins J. M., Vogelstein B. (2004) HAUSP is required for p53 destabilization. Cell Cycle 3, 689–692 [PubMed] [Google Scholar]
- 31. Rahimian R., Masih-Khan E., Lo M., van Breemen C., McManus B. M., Dubé G. P. (2001) Hepatic overexpression of peroxisome proliferator-activated receptor γ2 in the ob/ob mouse model of non-insulin dependent diabetes mellitus. Mol. Cell. Biochem. 224, 29–37 [DOI] [PubMed] [Google Scholar]
- 32. Yamazaki T., Shiraishi S., Kishimoto K., Miura S., Ezaki O. (2011) An increase in liver PPARγ2 is an initial event to induce fatty liver in response to a diet high in butter: PPARγ2 knockdown improves fatty liver induced by high-saturated fat. J. Nutr. Biochem. 22, 543–553 [DOI] [PubMed] [Google Scholar]
- 33. Matsusue K., Haluzik M., Lambert G., Yim S. H., Gavrilova O., Ward J. M., Brewer B., Jr., Reitman M. L., Gonzalez F. J. (2003) Liver-specific disruption of PPARγ in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J. Clin. Invest. 111, 737–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Memon R. A., Tecott L. H., Nonogaki K., Beigneux A., Moser A. H., Grunfeld C., Feingold K. R. (2000) Up-regulation of peroxisome proliferator-activated receptors (PPAR-α) and PPAR-γ messenger ribonucleic acid expression in the liver in murine obesity: troglitazone induces expression of PPAR-γ-responsive adipose tissue-specific genes in the liver of obese diabetic mice. Endocrinology 141, 4021–4031 [DOI] [PubMed] [Google Scholar]
- 35. Bedoucha M., Atzpodien E., Boelsterli U. A. (2001) Diabetic KKAy mice exhibit increased hepatic PPARγ1 gene expression and develop hepatic steatosis upon chronic treatment with antidiabetic thiazolidinediones. J. Hepatol. 35, 17–23 [DOI] [PubMed] [Google Scholar]
- 36. Laplante M., Sell H., MacNaul K. L., Richard D., Berger J. P., Deshaies Y. (2003) PPAR-γ activation mediates adipose depot-specific effects on gene expression and lipoprotein lipase activity: mechanisms for modulation of postprandial lipemia and differential adipose accretion. Diabetes 52, 291–299 [DOI] [PubMed] [Google Scholar]
- 37. Nagashima K., Lopez C., Donovan D., Ngai C., Fontanez N., Bensadoun A., Fruchart-Najib J., Holleran S., Cohn J. S., Ramakrishnan R., Ginsberg H. N. (2005) Effects of the PPARγ agonist pioglitazone on lipoprotein metabolism in patients with type 2 diabetes mellitus. J. Clin. Invest. 115, 1323–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hu M., Gu L., Li M., Jeffrey P. D., Gu W., Shi Y. (2006) Structural basis of competitive recognition of p53 and MDM2 by HAUSP/USP7: implications for the regulation of the p53-MDM2 pathway. PLoS Biol. 4, e27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. DuBois R. N., Gupta R., Brockman J., Reddy B. S., Krakow S. L., Lazar M. A. (1998) The nuclear eicosanoid receptor, PPARγ, is aberrantly expressed in colonic cancers. Carcinogenesis 19, 49–53 [DOI] [PubMed] [Google Scholar]
- 40. Mueller E., Sarraf P., Tontonoz P., Evans R. M., Martin K. J., Zhang M., Fletcher C., Singer S., Spiegelman B. M. (1998) Terminal differentiation of human breast cancer through PPARγ. Mol. Cell 1, 465–470 [DOI] [PubMed] [Google Scholar]
- 41. Mueller E., Smith M., Sarraf P., Kroll T., Aiyer A., Kaufman D. S., Oh W., Demetri G., Figg W. D., Zhou X. P., Eng C., Spiegelman B. M., Kantoff P. W. (2000) Effects of ligand activation of peroxisome proliferator-activated receptor γ in human prostate cancer. Proc. Natl. Acad. Sci. U.S.A. 97, 10990–10995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kinzler K. W., Vogelstein B. (1996) Lessons from hereditary colorectal cancer. Cell 87, 159–170 [DOI] [PubMed] [Google Scholar]
- 43. Núñez N. P., Liu H., Meadows G. G. (2006) PPAR-γ ligands and amino acid deprivation promote apoptosis of melanoma, prostate, and breast cancer cells. Cancer Lett. 236, 133–141 [DOI] [PubMed] [Google Scholar]
- 44. Keshamouni V. G., Reddy R. C., Arenberg D. A., Joel B., Thannickal V. J., Kalemkerian G. P., Standiford T. J. (2004) Peroxisome proliferator-activated receptor-γ activation inhibits tumor progression in non-small-cell lung cancer. Oncogene 23, 100–108 [DOI] [PubMed] [Google Scholar]
- 45. Chawla A., Barak Y., Nagy L., Liao D., Tontonoz P., Evans R. M. (2001) PPAR-γ dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat. Med. 7, 48–52 [DOI] [PubMed] [Google Scholar]
- 46. Palakurthi S. S., Aktas H., Grubissich L. M., Mortensen R. M., Halperin J. A. (2001) Anticancer effects of thiazolidinediones are independent of peroxisome proliferator-activated receptor γ and mediated by inhibition of translation initiation. Cancer Res. 61, 6213–6218 [PubMed] [Google Scholar]
- 47. Saez E., Tontonoz P., Nelson M. C., Alvarez J. G., Ming U. T., Baird S. M., Thomazy V. A., Evans R. M. (1998) Activators of the nuclear receptor PPARγ enhance colon polyp formation. Nat. Med. 4, 1058–1061 [DOI] [PubMed] [Google Scholar]
- 48. Lefebvre A. M., Chen I., Desreumaux P., Najib J., Fruchart J. C., Geboes K., Briggs M., Heyman R., Auwerx J. (1998) Activation of the peroxisome proliferator-activated receptor γ promotes the development of colon tumors in C57BL/6J-APCMin/+ mice. Nat. Med. 4, 1053–1057 [DOI] [PubMed] [Google Scholar]