

Background: Activated TRAIL receptors accumulate in lipid raft membrane domains to trigger cell death.
Results: Ack1 is required for ligand-induced TRAIL receptor recruitment to lipid rafts and induction of cell death.
Conclusion: Ack1 is a novel key regulator of TRAIL receptor membrane dynamics.
Significance: Insights gained into the spatiotemporal control of TRAIL receptors may help to predict or modulate therapeutic responsiveness to TRAIL.
Keywords: Apoptosis, Cancer, Cell Signaling, TRAIL, Tyrosine-Protein Kinase (Tyrosine Kinase), Lipid Rafts
Abstract
TNF-related apoptosis-inducing ligand (TRAIL) holds promise for treatment of cancer due to its ability to selectively kill cancer cells while sparing normal cells. Ligand-induced translocation of TRAIL receptors (TRAIL-R) 1 and 2 (also called DR4 and DR5, respectively) into lipid raft membrane microdomains is required for TRAIL-induced cell death by facilitating receptor clustering and formation of the death-inducing signaling complex, yet the underlying regulatory mechanisms remain largely unknown. We show here that the non-receptor tyrosine kinase Ack1, previously implicated in the spatiotemporal regulation of the EGF receptor, is required for TRAIL-induced cell death in multiple epithelial cell lines. TRAIL triggered a transient up-regulation of Ack1 and its recruitment to lipid rafts along with TRAIL-R1/2. siRNA-mediated depletion of Ack1 disrupted TRAIL-induced accumulation of TRAIL-R1/2 in lipid rafts and efficient recruitment of caspase-8 to the death-inducing signaling complex. Pharmacological inhibition of Ack1 did not affect TRAIL-induced cell death, indicating that Ack1 acts in a kinase-independent manner to promote TRAIL-R1/2 accumulation in lipid rafts. These findings identify Ack1 as an essential player in the spatial regulation of TRAIL-R1/2.
Introduction
Apoptosis is triggered either by intrinsic signaling pathways that depend on release of pro-apoptotic factors from the mitochondria in response to various cellular stresses or through binding of extracellular ligands to death receptors on the cell surface. These ligands include the TNF-related apoptosis-inducing ligand (TRAIL),4 a member of the TNF superfamily that initiates apoptosis by activating two cognate receptors, TRAIL-R1 (DR4) and TRAIL-R2 (DR5). Due to the unique ability of TRAIL to trigger apoptosis in various types of cancer cells while sparing normal cells, therapeutic agonistic agents are being evaluated in clinical trials. Innate or acquired resistance has, however, hampered clinical success; hence, understanding what determines TRAIL receptor signaling outcome and mechanisms of TRAIL resistance is of vital importance for developing effective therapeutics. One proposed mechanism of TRAIL resistance is constitutive endocytosis and subsequent reduced surface expression of TRAIL-R1 and TRAIL-R2, which has been reported to reduce TRAIL sensitivity in breast and colon cancers (1, 2).
Ligand-bound TRAIL receptors oligomerize, which facilitates recruitment of the adaptor protein Fas-associated death domain (FADD) and procaspase-8 to form the so-called death-inducing signaling complex (DISC) (1, 3, 4). This promotes autoprocessing and release of active caspase-8, which in turn cleaves and activates the effector caspase-3 directly or indirectly through cleavage of Bcl-2 inhibitory BH3 domain protein (5) and activation of the intrinsic mitochondrial apoptotic pathway. Recent evidence suggests that accumulation of TRAIL receptors within cholesterol- and sphingolipid-enriched membrane compartments commonly referred to as lipid rafts is essential for efficient receptor clustering and DISC formation in several epithelial cancer cells (6–9). Agents that disrupt lipid rafts, such as methyl β-cyclodextrin, have been shown to inhibit TRAIL-induced DISC formation and subsequent TRAIL sensitivity (6, 10–12). Furthermore, several chemotherapeutic drugs have been reported to promote TRAIL sensitivity by driving accumulation and clustering of TRAIL receptors in lipid rafts, whereas disrupting the lipid rafts represses the sensitizing effect of these drugs (6, 10, 11, 13). The molecular mechanisms regulating TRAIL receptor clustering and association with lipid rafts remain largely unclear, although glycosylation (14, 15) of TRAIL-R1/2 or palmitoylation of TRAIL-R1 (9) is essential for receptor clustering and translocation to lipid rafts. Another proposed level of regulation involves TRAIL-induced translocation of acid sphingomyelinase to the plasma membrane, where it synthesizes ceramide, a key component of lipid rafts (6, 7).
Ack1 (activated Cdc42-associated kinase 1), also known as Tnk2, is a ubiquitously expressed non-receptor tyrosine kinase that carries a number of protein interaction domains, including a CRIB (Cdc42/Rac-interactive binding) domain, a SAM (sterile alpha-motif) domain, an SH3 domain, a clathrin-binding region, and a C-terminal region homologous to the EGF receptor (EGFR) interaction region found in Mig6 (mitogen-inducible gene 6). Ack1 was first identified as an effector of Cdc42 (16) that is activated by several growth factors or integrins (17–21). Ack1 has been implicated in the spatiotemporal regulation of EGFR, which it binds via its Mig6 homology region (22). Ack1 has been reported to either promote clathrin-mediated endocytosis of EGFR and target it for degradation (23, 24) or, conversely, to stabilize cell surface expression of EGFR (25), suggesting different functions for Ack1 in different cell types. Ack1 has also been reported to regulate the receptor tyrosine kinase Axl and the androgen receptor (22, 26) and to interact with a number of molecules involved in vesicular trafficking, including clathrin, AP2 (adaptor protein 2), and SNX9 (sorting nexin 9) (27–29).
Recent findings also implicate Ack1 in carcinogenesis. Amplification or activating mutations of Ack1 in prostate, lung, ovarian, and pancreatic cancers correlate with increased invasiveness (30–34). The cancer-promoting properties of Ack1 may be mediated through its regulation of p130cas (17) or the tumor suppressor Wwox (35). We here show for the first time that the Ack1 tyrosine kinase is a key regulator of TRAIL receptor membrane compartmentalization that promotes TRAIL-induced receptor accumulation in lipid rafts and activation of pro-apoptotic signaling.
EXPERIMENTAL PROCEDURES
Cell Lines and Cell Culture
MCF10A cells were cultured in DMEM/F-12 supplemented with 5% horse serum (Invitrogen), 20 ng/ml EGF, 0.5 mg/ml hydrocortisone, 100 ng/ml cholera toxin and 10 μg/ml insulin (Sigma). HeLa and HEK-293 cells were cultured in DMEM with 10% FBS. NCI-H460 cells were cultured in RPMI 1460 medium with 10% FBS.
Reagents and Antibodies
EGF and FITC-conjugated annexin V (Miltenyi Biotech), nickel-nitrilotriacetic acid-agarose beads (Qiagen), HybondTM-P PVDF (Millipore), HaltTM phosphatase/protease inhibitor and Streptavidin Plus UltraLink resin (Pierce), and lapatinib (LC Laboratories) were obtained from the indicated manufacturers. All other reagents were from Sigma. The following primary antibodies were used: anti-Ack1 (clone A11, sc-28336, Santa Cruz Biotechnology); anti-phospho-Ack1 (Tyr-284, 09-142, Millipore); anti-caspase-8 (clone 1-1-37, MAB4708, Millipore); anti-caveolin-1 (610406, BD Transduction Laboratories); anti-α-tubulin (homemade); and anti-caspase-3 (9662), anti-cleaved caspase-8 (Asp-391, 9496), anti-cleaved caspase-3 (Asp-175, 9661), anti-FADD (2782), anti-GAPDH, and anti-phospho-EGFR (Tyr-1068, 2234) (Cell Signaling Technology). Anti-TRAIL-R1 (1139) and anti-TRAIL-R2 (2019) (ProSci Inc.); phycoerythrin (PE)-conjugated anti-TRAIL-R1 (clone DJR1), PE-conjugated anti-TRAIL-R2 (clone DJR2), and PE-conjugated anti-mouse IgG1 isotype control (eBioscience); and HRP-conjugated goat anti-mouse or goat anti-rabbit (The Jackson Laboratory) secondary antibodies were used. Alexa 594-conjugated anti-rabbit antibody was used for immunofluorescence (The Jackson Laboratory).
siRNA Transfection
Cells were transfected with RNAiMAX (Invitrogen) at a final concentration of 12 nm siRNA for all cell lines except NCI-H460, which was transfected twice with 30 nm siRNA. Ack1a (siRNA ID 103419), Ack1b (siRNA ID s19850), and non-targeting control (siRNA ID AM4636) siRNAs were obtained from Applied Biosciences. The Ack1b oligonucleotide was used for Ack1 knockdown unless stated otherwise. Experiments were performed 48–72 h post-transfection.
Plasmid DNA Transfection
H460 cells were transfected with full-length Ack1 (WT) or kinase-dead Ack1 (mutant K158A) cloned in a pcDNA-DEST40 expression vector using JetPRIME transfection reagent (Polyplus Transfection) according to the supplier's recommendations.
Site-directed Mutagenesis
Site-directed mutagenesis was performed using the QuikChange® XL site-directed mutagenesis kit (Stratagene) according to the supplier's recommendations. A kinase-dead Ack1 mutant (K158A, a472g;a473c) was generated using primer 3′-GGTGAGTGTGGCTGTGGCGTGCCTGAAGC-5′.
Generation and Purification of Recombinant TRAIL
The construct for generating recombinant human TRAIL was a kind gift from Dr. Marion MacFarlane (University of Leicester) and generated as described previously (36). Briefly, TRAIL residues 95–281 was cloned into the expression vector pET28b containing in-frame N-terminal His6 and T7 tags. The pET28b-TRAIL construct was transformed into BL21(DE3)RIL cells for propagation. Expression of recombinant TRAIL was induced by 1 mm isopropyl β-d-thiogalactopyranoside for 3 h at 27 °C. Induced bacterial pellets were lysed in Triton lysis buffer A (20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 10% glycerol, 1% Triton X-100, and 1 mm PMSF) for 30 min on ice and sonicated. Lysates were cleared by centrifugation at 3220 × g for 45 min at 4 °C and supplemented with 20 mm imidazole. His-TRAIL was batch-purified by absorption onto nickel-nitrilotriacetic acid-agarose beads for 1.5 h at 4 °C. After washing, TRAIL was eluted from the beads by the addition of elution buffer (300 mm NaCl and 100 mm imidazole dissolved in PBS).
Western Blotting
Cells were lysed in SDS sample buffer (120 mm Tris-HCl (pH 6.8), 3% SDS, 15% glycerol, 0.03% bromphenol blue, and 75 mm DTT) and run on a 7.5–12.5% polyacrylamide gel at 100 V. Proteins were transferred onto HybondTM-P PVDF membranes at 100 V for 60–90 min. Membranes were blocked in 4% milk or 5% BSA dissolved in TBS/Tween. The protein bands were visualized using ECLTM or ECL PlusTM reagent and Hyperfilm® ECL (GE Healthcare).
FACS Analysis of Apoptosis with Annexin V/Propidium Iodide Labeling
MCF10A cells were treated with 30 units/ml TRAIL for 2 h. Cells were trypsinized and washed twice with annexin V binding buffer (10 mm HEPES (pH 7.4), 150 mm NaCl, 5 mm KCl, 1 mm MgCl2, and 1.8 mm CaCl2). 10 μl of FITC-conjugated annexin V was added and incubated for 15 min in the dark at room temperature. Cells were washed twice with annexin V binding buffer, and 1 mg/ml propidium iodide solution was added immediately prior to analysis by flow cytometry. Summit software was used to analyze the data. 10,000 events/sample were collected, and three independent experiments were performed.
FACS Analysis of TRAIL Receptor Cell Surface Expression
Cell surface expression of TRAIL-R1 and TRAIL-R2 was analyzed as described previously (36). Briefly, MCF10A cells were trypsinized, washed once with cell culture medium, and left to recover for 30 min at 37 °C. Cells (2.5 × 105) were pelleted and blocked in 40–45 μl of normal goat serum on ice for 5 min. 10 μl of PE-conjugated TRAIL-R1 (clone DJR1), 5 μl of PE-conjugated TRAIL-R2 (clone DJR2), or 10 μl of PE-conjugated mouse IgG1 isotype control was added to the cells for 1 h on ice in the dark. Cells were washed with PBS and resuspended in 1 ml of PBS. The mean fluorescence intensity was measured by flow cytometry with excitation at 488 nm and emission at 575 nm. 10,000 events/sample were collected, and three independent experiments were performed.
Lipid Raft Isolation
Lipid rafts were isolated by sucrose gradient centrifugation and ultracentrifugation. One 15-cm dish with cells was used per sample. Cells were treated with 30 units/ml TRAIL for 1 h, washed once with PBS, and lysed in 1 ml of lysis buffer (10 mm Tris-HCl (pH 7.5), 150 mm NaCl, 5 mm EDTA, and 1% Triton X-100 supplemented with 1 mm vanadate, 1 mm NaF, and HaltTM phosphatase/protease inhibitor). 50-μl aliquots of the lysate were taken as the input control. The lysates were incubated on ice for 30 min, homogenized with 10 strokes of a tissue grinder, and mixed with 1 ml of 85% (w/v) sucrose (dissolved in lysis buffer without Triton X-100). The lysate and sucrose mixture was transferred to the bottom of a precooled 14-ml open top thin-wall ultracentrifuge tube (Beckman Instruments) and carefully overlaid with 7.5 ml of 35% sucrose and 3.5 ml of 5% sucrose. The samples were centrifuged at 38,000 rpm for 18 h at 4 °C in a swing-out SW 40 rotor in an Beckman Optima L-100 XP centrifuge. 1-ml fractions were carefully collected from the top to bottom of the tube and resuspended 1:1 with 3× SDS sample buffer. Samples were then subjected to Western blotting.
Subcellular Fractionation
Fractionation was performed with the subcellular proteome extraction kit (Calbiochem) according to the supplier's protocol.
TRAIL Receptor Clustering Assay
TRAIL receptor clustering was analyzed by SDS-PAGE with lysates without reducing agent. Cells were treated with 30 units/ml TRAIL for 15 min or 1 h, washed with PBS, and lysed in cell lysis buffer (20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 10% glycerol, and 1% Triton X-100 supplemented with HaltTM phosphatase/protease inhibitor, 1 mm vanadate, and 1 mm NaF). The lysates were incubated on ice for 30 min and then centrifuged at 16,000 × g at 4 °C. The cleared lysate was divided in two aliquots and diluted 1:1 with 3× SDS sample buffer with or without 150 mm DTT. The samples with DTT were incubated at 95 °C for 3 min, whereas the nonreduced samples were kept on ice before loading.
Confocal Imaging
NCI-H460 cells were fixed with ice-cold methanol for 10 s and then blocked in 5% goat serum in TBS/Tween. Primary and secondary antibodies and FITC-conjugated cholera toxin were diluted in blocking buffer. Images were taken with Zeiss LSM 710 confocal microscope and processed using Zeiss ZEN 2011 software.
DISC Formation Assay
Cells was treated with 30 units/ml biotinylated TRAIL for 30 min at 37 °C, collected, and lysed for 30 min on ice in Triton lysis buffer B (20 mm Tris-HCl (pH 7.5), 150 mm NaCl, 1 mm EDTA, 10% glycerol, 1% Triton X-100, 1 mm vanadate, Complete protease inhibitor mixture, and 1 mm NaF). 50-μl aliquots of the lysates were taken as input samples. Lysates were cleared by centrifugation at 16,000 × g for 30 min at 4 °C. To analyze total TRAIL receptor levels, 3 units/ml biotinylated TRAIL was instead added to the cleared lysate, allowing TRAIL to bind all receptors in the cells. One sample without any addition of TRAIL was used as a negative control for the immunoprecipitation. TRAIL receptors with bound biotinylated TRAIL were precipitated with streptavidin-Sepharose beads overnight at 4 °C. Beads were washed three times with lysis buffer, resuspended in 60 μl of SDS sample buffer, incubated at 95 °C for 3 min, and centrifuged at 16,000 × g for 20 s. The supernatant was subject to Western blotting and blotted for proteins of interest.
Inhibitor Treatment of Cultured Cells
Clathrin-mediated endocytosis was inhibited in MCF10A cells by pretreatment with 10 μg/ml chlorpromazine hydrochloride (dissolved in Me2SO) for 1 h prior to treatment with 30 units/ml TRAIL for 2 h. Me2SO alone was added to the non-treated samples. H460 cells were treated with the lysosomal inhibitor chloroquine for 16 h at a final concentration of 50 or 100 μm prior to TRAIL treatment for 90 min. MCF10A cells were pretreated with 1 μm lapatinib and incubated for 1 h prior to treatment with 30 units/ml TRAIL. MCF10A cells were pretreated for 6 h with 50 mg/ml methyl β-cyclodextrin or 2 μg/ml nystatin (Sigma) in starvation medium (no serum or EGF) and then treated for 3 h with 3 units/ml TRAIL. MCF10A or HEK-293T cells were pretreated with 10 μm AIM-100 for 2 h to inhibit Ack1.
RESULTS
Ack1 Is Required for TRAIL-induced Apoptosis
We (37) and others (38) have recently found a pro-apoptotic role for the Ack1-related protein Mig6. We therefore asked whether Ack1 also plays a role in apoptotic signaling in response to different cellular stresses or pro-apoptotic ligands. Strikingly, siRNA-mediated depletion of Ack1 in MCF10A breast epithelial cells largely abolished TRAIL-induced apoptosis as shown by cell surface binding of annexin V (Fig. 1A). Quantification of annexin V-positive cells following 3 h of TRAIL treatment revealed a 72% decrease in the number of apoptotic cells after RNAi-mediated knockdown of Ack1 (Fig. 1, B and B′). To confirm the specificity of the siRNA, we transfected the MCF10A cells with two independent siRNA oligonucleotides that effectively reduced Ack1 expression. Both siRNAs prevented TRAIL-induced cleavage of caspase-8 and caspase-3 at different doses (30 and 60 units/ml) up to 3 h after adding TRAIL as revealed by immunoblotting (Fig. 1C). We next asked whether Ack1 is required for TRAIL-induced apoptosis in other TRAIL-sensitive epithelial cell lines. Indeed, depletion of Ack1 largely abolished TRAIL-induced caspase-8 or caspase-3 cleavage in the other epithelial cell lines examined: human non-small cell lung cancer NCI-H460, HeLa, and colorectal adenocarcinoma SW480 cells (Fig. 1, D and E) (data not shown), consistent with a general requirement for Ack1 in TRAIL-induced epithelial cell death. However, RNAi-mediated knockdown of Ack1 (unlike knockdown of Mig6) did not affect cell death induced by growth factor withdrawal (Fig. 1F), arguing against a general role for Ack1 in mediating cell death or promoting growth factor-independent cell survival. Furthermore, the impaired cleavage of caspase-8 and caspase-3 was not due to a reduction in expression of these caspases because RNAi-mediated depletion of Ack1 did not alter the expression levels of procaspase-8 and procaspase 3 (Fig. 1G).
FIGURE 1.

Ack1 is required for TRAIL-induced apoptosis. A and B, flow cytometry analysis with the apoptosis markers annexin V and propidium iodide of MCF10A cells transfected with non-targeting (siCtrl) or Ack1 (siAck1) siRNA and mock-treated or treated with 30 or 60 units/ml TRAIL for 3 h. A representative dot blot is shown in A, and the percentage of annexin V-positive cells is quantified in B (n = three independent experiments; p < 0.006, Student's t test). Error bars represent S.D. B′, Western blot analysis confirming RNAi-mediated knockdown of Ack1 in MCF10A cells. C–E, Western blot analysis with Ack1 and the indicated markers of apoptosis of MCF10A (C), NCI-H460 (D), and HeLa (E) cells transfected with non-targeting or Ack1 siRNA and treated with TRAIL (C and D) for the indicated times or with 30 or 60 units/ml TRAIL for 3 h (E). Note the reduced cleavage of caspase-8 and caspase-3 in Ack1 knockdown cells compared with control cells. F, Western blot analysis of MCF10A cells subjected to non-targeting or Ack1 siRNA transfection and cultured without serum and growth factors (GF) for 48–72 h. G, quantification of uncleaved caspase-8 and caspase-3 levels by immunoblotting of MCF10A cells 48 h after transfection with non-targeting or Ack1 siRNA (n = 3). Error bars represent S.E. ns, not significant.
Given the reported role of Ack1 as a negative regulator of EGFR signaling, we addressed whether gain of EGFR-mediated mitogenic/prosurvival signaling could underlie the observed impairment of TRAIL-induced cell death in Ack1 knockdown cells. However, this does not appear to be the case because pretreatment of MCF10A cells with the EGFR/ErbB2 dual-specificity inhibitor lapatinib failed to restore TRAIL-induced cleavage of caspase-8 and caspase 3 in Ack1-depleted cells (Fig. 2). Furthermore, siRNA-mediated knockdown of Ack1 in MCF10A cells did not increase activation of EGFR or the canonical mitogenic/survival signaling molecules Erk1/2 and Akt under normal growth conditions as evident from immunoblotting with phosphospecific antibodies (Fig. 2) (data not shown). Taken together, these results show that Ack1 is required for TRAIL-induced apoptosis in different epithelial cell lines, independent of its reported role as a negative regulator of EGFR signaling.
FIGURE 2.
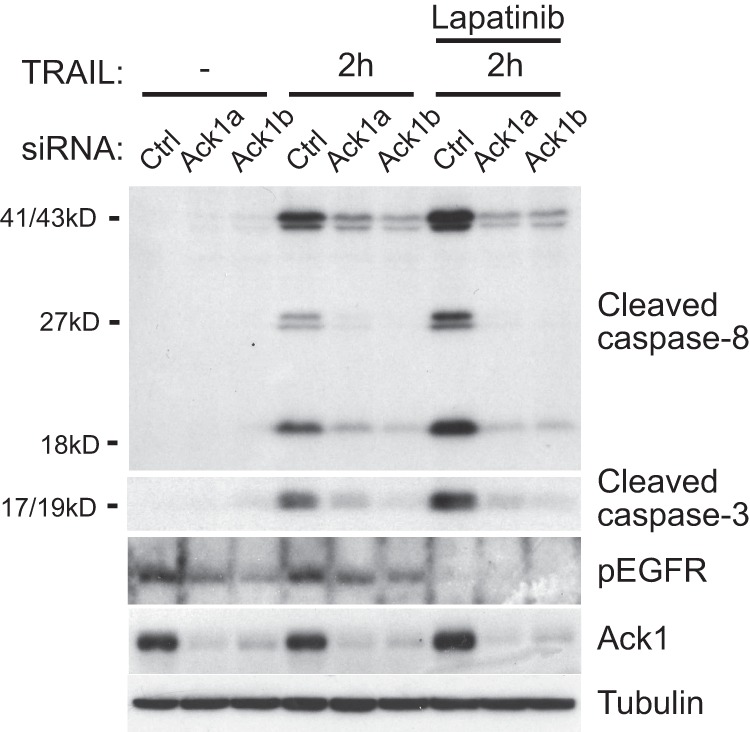
Ack1 regulates TRAIL-induced apoptosis independent of EGFR. Shown are the results from Western blot analysis of MCF10A cells treated with EGFR and the ErbB2 dual-specificity inhibitor lapatinib for 1 h, as indicated, followed by treatment with TRAIL for 2 h. Note that inhibition of ErbB signaling did not restore the reduced cleaved caspase-8 and caspase-3 levels in Ack1 knockdown cells. Ctrl, control; pEGFR, phospho-EGFR.
Ack1 Is Transiently Induced upon TRAIL Treatment
We next asked whether Ack1 expression or activity is regulated by TRAIL. We found that TRAIL treatment of MCF10A cells led to a rapid and transient increase in Ack1 protein levels that was evident after 30 min and that peaked (3.2-fold increase) after 1 h (Fig. 3, A and B). At later time points (2–3 h), Ack1 was degraded (Figs. 1C and 3B). Similarly, TRAIL-induced up-regulation of Ack1 was also observed in NCI-H460 and HeLa cells, typically peaking 30–60 min after the addition of TRAIL (Fig. 1, D and E) (data not shown). To address whether the TRAIL-mediated increase in Ack1 protein levels is due to stabilization or de novo synthesis, we pretreated the MCF10A cells with the protein synthesis inhibitor cycloheximide prior to TRAIL treatment. Cycloheximide completely abolished the TRAIL-induced increase in Ack1 expression levels, indicating that de novo synthesis contributes to the up-regulation of Ack1 (data not shown). We next asked whether the ensuing degradation of Ack1 observed after 2–3 h of TRAIL stimulation occurs by proteasomal or lysosomal degradation. Treatment of the MCF10A cells with the lysosomal inhibitor chloroquine (100 μm) for 16 h led to a 3.7-fold increase in Ack1 expression levels (Fig. 3, C and D), whereas inhibition of the proteasome with MG132 had a limited effect (Fig. 3E). Chloroquine treatment also prevented the down-regulation of Ack1 observed after prolonged TRAIL treatment (Fig. 3D). These results indicate that Ack1 is targeted for degradation primarily via the lysosomes in these cells both in non-treated cells and upon TRAIL treatment. Notably, TRAIL-R1 also accumulated upon treatment with either chloroquine or MG132 in wild-type but not Ack1-depleted cells, suggesting that Ack1 promotes basal turnover of TRAIL-R1 (Fig. 3, C and E).
FIGURE 3.

TRAIL transiently increases Ack1 levels. Ack1 levels were analyzed by Western blotting following TRAIL treatment of MCF10A for the times indicated (A), and band intensities were quantified (B). Error bars represent S.D. (n = three independent experiments). C, Western blot analysis of NCI-H460 cells subjected to Ack1 or non-targeting siRNA transfection followed by treatment with the lysosomal inhibitor chloroquine. D, quantification of Ack1 by densitometry of Western blots of NCI-H460 cells pretreated with chloroquine followed by treatment with TRAIL for the indicated times (n = 5). E, Western blot analysis of NCI-H460 cells subjected to Ack1 or non-targeting siRNA transfection followed by treatment with the proteasomal inhibitor MG132 for 16 h (representative of three independent experiments). F, TRAIL-induced cleavage of caspase-8 levels determined by Western blot analysis after a 2-h preincubation with the selective TRAIL inhibitor AIM-100 (10 μm) or vehicle alone (mock). G, Western blot analysis of Tyr-284 phosphorylation of ectopically expressed Ack1 (pAck1) treated with AIM-100 as described for F.
We then asked whether the kinase activity of Ack1 is required for its role in promoting TRAIL-induced cell death. However, this does not appear to be the case because pretreating MCF10A cells with the Ack1 inhibitor AIM-100 prior to treatment with TRAIL did not significantly alter cleavage of caspase-8 (Fig. 3F). The effectiveness of AIM-100 in inhibiting Ack1 autophosphorylation at the dose used was confirmed by immunoblotting (Fig. 3G).
Loss of Ack1 Prevents TRAIL Receptor Recruitment to Lipid Rafts
To address whether Ack1 acts proximal to the TRAIL receptors, we first investigated whether Ack1 regulates receptor localization to lipid rafts, known to be essential for efficient pro-apoptotic signaling. Cells were subjected to siRNA-mediated silencing of Ack1 prior to stimulation with TRAIL. Lipid rafts were then isolated from cell lysates as nonionic detergent-resistant low-density membrane fractions resolved by sucrose gradient centrifugation (Fig. 4A). Western blot analysis of the gradient fractions revealed that TRAIL-R1 and, to a lesser extent, TRAIL-R2 accumulated in the lipid raft fractions positive for the raft marker caveolin-1 upon TRAIL stimulation in MCF10A cells (Fig. 4A). RNAi-mediated silencing of Ack1 completely abolished the association of TRAIL-R1 and TRAIL-R2 with lipid rafts upon TRAIL treatment (Fig. 4, A and B). Interestingly, Ack1 was also recruited to lipid rafts in response to TRAIL, consistent with a role for Ack1 in regulating TRAIL-R1 and TRAIL-R2 recruitment to lipid rafts (Fig. 4A).
FIGURE 4.
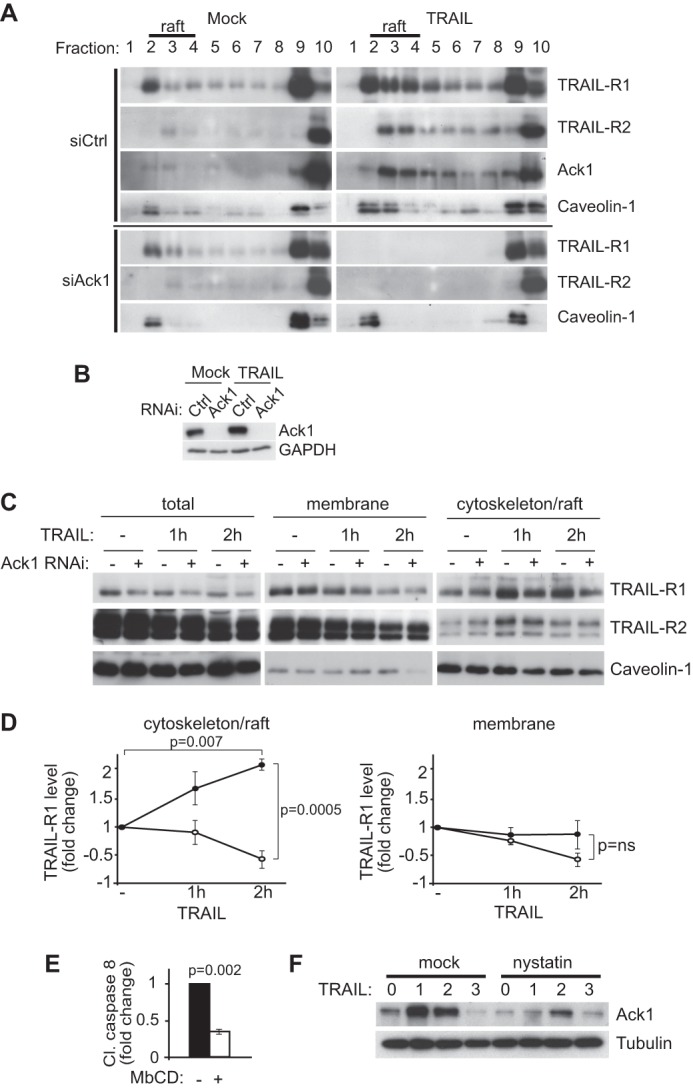
Ack1 is required for translocation of TRAIL receptors to lipid rafts. A, lipid raft-containing proteins were analyzed by Western blotting of sucrose gradient fractions of MCF10A cells transfected with non-targeting control (siCtrl) or Ack1 (siAck1) siRNA and subjected to TRAIL or mock treatment. Note that lipid rafts containing caveolin-1 were enriched in low-density fractions 1–3. B, Western blot confirming efficient Ack1 knockdown in the cell lysates used in A. C and D, subcellular fractionation by sequential detergent extraction of MCF10A cells transfected with non-targeting or Ack1 siRNA followed by treatment with TRAIL for the indicated time periods. D, quantification of TRAIL-R1 levels in the soluble membrane versus insoluble cytoskeleton/raft fractions as described for C by densitometry analysis of Western blots (n = 3). Note the Ack1-dependent translocation of TRAIL-R1 to the cytoskeleton/raft fraction following TRAIL treatment. E, quantification of the effect of the lipid raft-disrupting compound methyl β-cyclodextrin (MbCD) on TRAIL-induced cell death by densitometry analysis of Western blots against cleaved caspase-8 (n = 3). F, Western blot showing the effect of pretreating cells with the raft-disrupting drug nystatin on TRAIL-induced Ack1 and cleaved (Cl) caspase-8 (representative of three independent experiments). Note that nystatin prevented TRAIL-induced accumulation of Ack1. Error bars indicate S.E. p values were obtained with Student's t test.
To confirm the sucrose gradient results by an alternative method, we performed a detergent-based cell fractionation assay that separates detergent-soluble (non-raft) from detergent-resistant (raft/cytoskeleton-associated) membrane fractions (Fig. 4, C and D). TRAIL stimulation for 2 h led to a 2-fold increase in TRAIL-R1 association with the lipid raft/cytoskeleton and a comparable decrease in the proportion of Ack1 found in the soluble membrane fraction (Fig. 4D). In contrast, TRAIL-R2 remained largely in the soluble non-raft membrane fraction upon TRAIL treatment (Fig. 4C). siRNA silencing of Ack1 abolished TRAIL-induced TRAIL-R1 accumulation in the detergent-insoluble membrane fraction (Fig. 4, C and D).
To confirm the previously reported finding that lipid rafts are required for TRAIL-induced cell death, we pretreated the epithelial cells with the lipid raft-disrupting drug methyl β-cyclodextrin prior to TRAIL stimulation. Methyl β-cyclodextrin treatment led to a 67% decrease in TRAIL-induced cleavage of caspase-8 (Fig. 4E). Interestingly, we also found that disruption of lipid rafts with nystatin reduced the protein levels of Ack1 following TRAIL treatment (Fig. 4F), suggesting that the observed induction of Ack1 is due to stabilization of Ack1 within lipid rafts.
We then conducted confocal immunofluorescence imaging to assess co-localization of TRAIL-R1 with fluorophore-conjugated cholera toxin, a marker of lipid rafts. We found that a 1-h TRAIL treatment led to increased plasma membrane localization of TRAIL-R1 and extensive co-localization with FITC-conjugated cholera toxin (Fig. 5A). Strikingly, RNAi-mediated depletion of Ack1 largely abolished TRAIL-induced co-localization with FITC-conjugated cholera toxin (Fig. 5, A and B). Instead, TRAIL-R1 appears to be internalized in the absence of Ack1 upon TRAIL treatment. Taken together, these results show that Ack1 accumulates in lipid raft membranes upon TRAIL treatment and is required for recruitment of TRAIL-R1 to lipid rafts.
FIGURE 5.
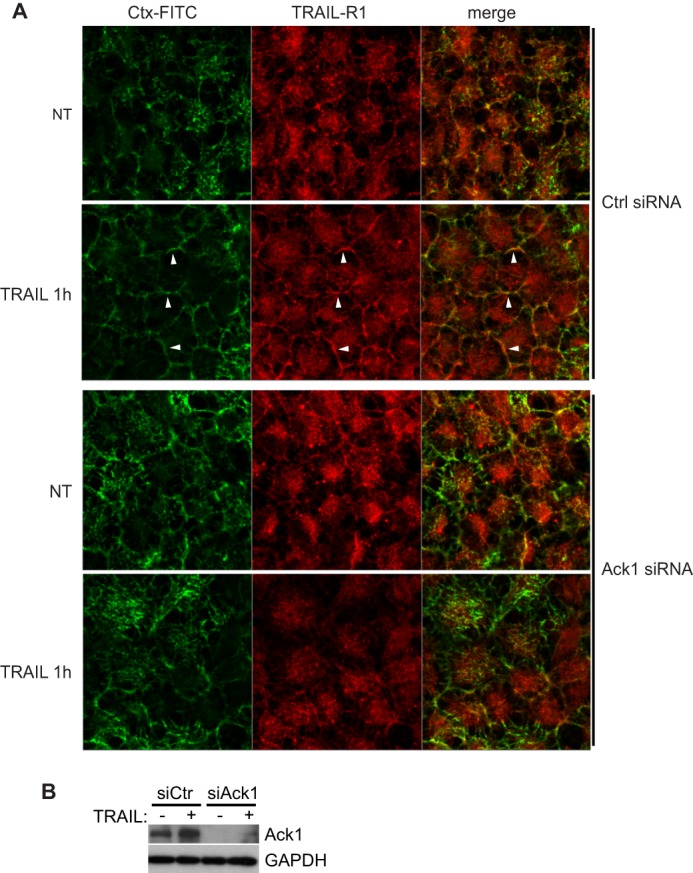
TRAIL-R1 co-localization with lipid rafts upon TRAIL treatment is disrupted in Ack1-depleted cells. A, confocal imaging with FITC-conjugated cholera toxin (Ctx) and anti-TRAIL-R1 antibody of NCI-H460 cells subjected to non-targeting control (siCtrl) or Ack1 (siAck1) siRNA transfection prior to treatment with TRAIL. White arrowheads highlight examples of plasma membrane with co-localization in wild-type cells. Note the absence of co-localization in TRAIL-treated Ack1 knockdown cells. B, confirmation of RNAi-mediated knockdown of Ack1 in A by Western blotting. Images are single confocal planes representative of four independent experiments. NT, non-treated.
Ack1 Is Required for Formation of a Functional TRAIL Receptor-DISC Complex
Previous studies indicate that efficient oligomerization and clustering of the TRAIL receptors are facilitated by their recruitment to lipid rafts (Ref. 3 and references therein). We therefore addressed whether receptor oligomerization also requires Ack1. siRNA-mediated knockdown of Ack1 in MCF10A cells led to impaired TRAIL-induced oligomerization of TRAIL-R1, but not TRAIL-R2, as detected by nonreduced SDS-PAGE followed by immunoblotting (Fig. 6A). However, this was not the case for NCI-H460 cells, in which silencing of Ack1 failed to significantly alter TRAIL-R1 oligomerization (Fig. 6B). Failed receptor oligomerization is therefore unlikely to be the primary cause of the impaired cell death observed in the absence of Ack1.
FIGURE 6.
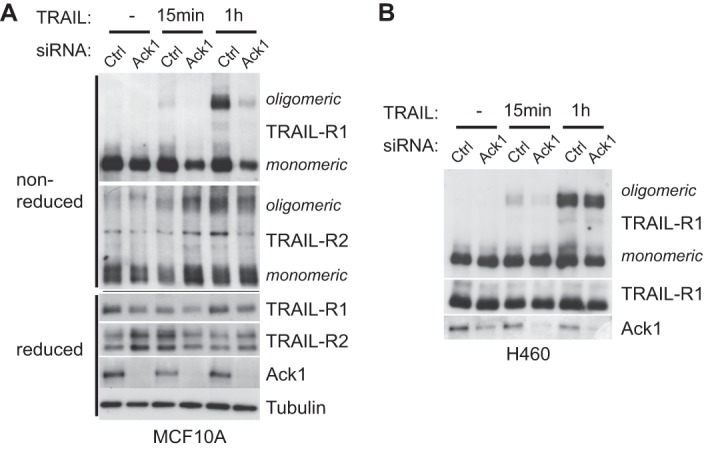
Ack1 is required for TRAIL-R1 clustering in a cell type-dependent manner. Monomeric and oligomeric forms of the TRAIL receptors were resolved by nonreduced SDS-PAGE followed by Western blot analysis of MCF10A (A) and NCI-H460 (B) cells after TRAIL treatment. Note that Ack1 knockdown prevented TRAIL-R1 oligomerization in MCF10A cells, but not in NCI-H460 cells. Ctrl, control.
Reduced surface expression of the TRAIL receptors due to their constitutive internalization has been described as a mechanism of acquired resistance to TRAIL-induced cell death (1, 2) and might therefore underlie the observed impairment of TRAIL-induced receptor localization to lipid rafts in the absence of Ack1. To address whether cell surface availability of the TRAIL receptors is reduced by the absence of Ack1, we labeled TRAIL-R1 and TRAIL-R2 using antibodies that recognize the extracellular part of the receptors and analyzed surface expression by flow cytometry. Fig. 7A shows that siRNA-mediated silencing of Ack1 (blue graphs) did not affect the cell surface expression of TRAIL-R1 or TRAIL-R2 compared with non-silencing siRNA control cells (green graphs). In agreement, inhibition of endocytosis with the clathrin inhibitor chlorpromazine (Fig. 7B) or the dynamin inhibitor Dynasore5 failed to restore TRAIL sensitivity in the absence of Ack1. Furthermore, we assessed the ability of biotinylated TRAIL to bind TRAIL-R1 and TRAIL-R2 and the associated DISC complex by performing streptavidin pulldown experiments (Fig. 7C). We found that TRAIL bound TRAIL-R1 at comparable levels in both the presence and absence of Ack1. Similarly, the death domain adaptor protein FADD co-purified with biotinylated TRAIL to the same extent with or without Ack1. Interestingly, procaspase-8 was not efficiently recruited to the DISC complex in the absence of Ack1. These results indicate that the impaired pro-apoptotic response in the absence of Ack1 is not due to reduced availability of the TRAIL receptors for binding to TRAIL at the cell surface. Taken together, our results show that Ack1 promotes TRAIL-induced apoptosis by facilitating TRAIL-R1 translocation to lipid rafts and efficient formation of a functional pro-apoptotic DISC complex.
FIGURE 7.

Ack1 knockdown does not alter TRAIL receptor surface exposure. A, flow cytometry analysis of cell surface expression of TRAIL-R1 and TRAIL-R2 in MCF10A cells. Cells were incubated with PE-conjugated antibodies targeting the ligand-binding domains of TRAIL-R1 and TRAIL-R2 or with an IgG isotype control (ctrl). kd, kinase-dead. B, Western blot analysis of MCF10A cells subjected to non-targeting control or Ack1 siRNA transfection prior to treatment with the clathrin inhibitor chlorpromazine (CPZ) followed by TRAIL stimulation for 3 h as indicated. C, DISC was co-purified by treating MCF10A cells with biotinylated TRAIL followed by pulldown with streptavidin beads and analysis by Western blotting. In the first and second lanes, biotinylated TRAIL was added to the cleared lysate. In the third and fourth lanes, cells were treated with biotinylated TRAIL for 2 h prior to harvest. In the fifth lane, no TRAIL was added. Note that the recruitment of FADD was not affected, whereas the recruitment of procaspase-8 was impaired in Ack1 knockdown cells.
DISCUSSION
The ability of TRAIL receptor agonists to selectively kill many types of cancer cells while sparing normal cells has provoked great interest in their potential use in the treatment of cancer; hence, elucidating the underlying molecular mechanism of TRAIL signaling is of vital importance (1, 3, 4). TRAIL triggers the assembly of a receptor-DISC complex that leads to caspase-8 activation and execution of cell death. This process requires translocation of the TRAIL receptors into lipid raft membrane domains (6–9). We have shown here that the tyrosine kinase Ack1 is required for TRAIL-induced apoptosis by promoting translocation of TRAIL-R1 into lipid rafts, TRAIL-R1 clustering, and formation of an active DISC complex. Notably, we found that TRAIL-R2 did not efficiently associate with lipid rafts in MCF10A cells, whereas TRAIL-R1 accumulation in lipid rafts has been reported to be sufficient and necessary to mediate TRAIL-induced cell death (9). RNAi-mediated knockdown of Ack1 largely abolished the apoptotic response to TRAIL in four cell lines of different epithelial origin: breast, lung, colon, and cervix. This suggests a general requirement for Ack1 in TRAIL-induced death of TRAIL-sensitive epithelial cells.
We found that in addition to abolishing ligand-induced TRAIL-R1 accumulation in lipid rafts, depletion of Ack1 also prevented oligomerization of TRAIL-R1 in MCF10A cells, but not NCI-H460 cells, raising the question as to which of the two events are directly regulated by Ack1. Whether lipid rafts facilitate TRAIL receptor oligomerization or whether oligomerization constitutes a prerequisite for lipid raft association is an issue that remains to be conclusively addressed. However, the former possibility is strongly supported by the reported finding that palmitoylation of TRAIL-R1 promotes both lipid raft localization and oligomerization of the receptor (9). Palmitoylation of transmembrane receptors has emerged as a key mediator of raft affinity (39). Ack1 is therefore likely to directly regulate TRAIL receptor recruitment to lipid rafts, which in turn facilitate oligomerization of TRAIL-R1 in certain cell types. It should be noted that we sometimes observed moderately reduced TRAIL receptor levels upon Ack1 depletion, which could contribute to resistance to TRAIL. However, this seems unlikely because FACS data indicate that the surface availability of the receptors in resting cells is not altered. Furthermore, TRAIL recruits comparable levels of receptor-associated FADD to the DISC complex in the absence or presence of Ack1.
How does Ack1 regulate TRAIL receptor recruitment to lipid rafts? Previous reports have shown that TRAIL-R1 and TRAIL-R2 can be internalized via clathrin-dependent and clathrin-independent endocytosis and that constitutive internalization of the receptors reduces their availability at the cell surface and, as a consequence, the sensitivity to TRAIL (1, 2). Given that Ack1 can bind clathrin and has been shown to modulate EGFR endocytosis, a possible mechanism could be that Ack1 negatively regulates clathrin-mediated internalization of TRAIL-R1 and TRAIL-R2, thereby shifting the balance of TRAIL-R1 and TRAIL-R2 dynamics from clathrin-mediated internalization to lipid raft/cytoskeleton association. However, this possibility can be ruled out because pretreating Ack1-depleted cells with inhibitors of clathrin- or dynamin-mediated endocytosis failed to restore TRAIL sensitivity. In addition, RNAi-mediated depletion of Ack1 did not reduce cell surface expression of TRAIL-R1 and TRAIL-R2 in resting cells or the ability of biotinylated TRAIL to bind its receptors. It is therefore unlikely that Ack1 promotes raft association of TRAIL-R1 and TRAIL-R2 by preventing clathrin-mediated endocytosis of the TRAIL receptors.
Instead, we propose that Ack1 acts by directly targeting the receptors to lipid rafts. This is supported by the observation that Ack1 associated with lipid rafts along with TRAIL-R1 upon receptor activation. Confocal imaging revealed that, in the absence of Ack1, the spatiotemporal dynamics of TRAIL-R1 was shifted from ligand-induced accumulation in rafts to internalization. Notably, we were unable to detect a physical interaction between Ack1 and TRAIL-R1 and TRAIL-R2 by co-immunoprecipitation or in biotinylated TRAIL pulldown experiments,5 indicating that Ack1 may not bind to the TRAIL receptors directly.
Interestingly, Ack1 kinase activity is not required to promote TRAIL-induced cell death. Instead, pharmacological inhibition of Ack1 moderately accelerates TRAIL-induced cell death. We therefore speculate that there are two distinct pools of Ack1 in the cell: 1) a pool that is activated downstream of growth factor receptors to regulate clathrin-mediated endocytosis and to promote mitogenic signaling and 2) a kinase-inactive pool of Ack1 that associates with lipid rafts to promote TRAIL receptor association with the raft microdomains and induction of cell death.
Therapeutic agonistic agents for the TRAIL receptors are being evaluated in clinical trials. Understanding what determines TRAIL receptor signaling outcome and mechanisms of TRAIL resistance is of vital importance for developing better therapeutics and predicting benefit. Ack1 has been found to be amplified or activated by mutations in lung, prostate, pancreatic, and ovarian cancers (30–34). It will be interesting to investigate whether Ack1 deregulation correlates with sensitivity to TRAIL-induced cell death and whether inhibition of Ack1 could potentiate the therapeutic response to TRAIL.
Acknowledgments
We thank Marion MacFarlane for TRAIL ligand and receptor cDNAs, and Carl-Henrik Heldin (Ludwig Institute for Cancer Research) and Taija Makinen (Cancer Research UK, London) for critically reading the manuscript.
This work was supported by Cancerfonden Grant CAN 2012/581, BBSRC, and the Ludwig Institute for Cancer Research.
E. Linderoth, G. Pilia, N. Mahajan, and I. Ferby, unpublished data.
- TRAIL
- TNF-related apoptosis-inducing ligand
- TRAIL-R
- TRAIL receptor
- FADD
- Fas-associated death domain
- DISC
- death-inducing signaling complex
- EGFR
- EGF receptor
- PE
- phycoerythrin.
REFERENCES
- 1. Gonzalvez F., Ashkenazi A. (2010) New insights into apoptosis signaling by Apo2L/TRAIL. Oncogene 29, 4752–4765 [DOI] [PubMed] [Google Scholar]
- 2. Jin Z., McDonald E. R., 3rd, Dicker D. T., El-Deiry W. S. (2004) Deficient tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor transport to the cell surface in human colon cancer cells selected for resistance to TRAIL-induced apoptosis. J. Biol. Chem. 279, 35829–35839 [DOI] [PubMed] [Google Scholar]
- 3. Pennarun B., Meijer A., de Vries E. G., Kleibeuker J. H., Kruyt F., de Jong S. (2010) Playing the DISC: turning on TRAIL death receptor-mediated apoptosis in cancer. Biochim. Biophys. Acta 1805, 123–140 [DOI] [PubMed] [Google Scholar]
- 4. Yang A., Wilson N. S., Ashkenazi A. (2010) Proapoptotic DR4 and DR5 signaling in cancer cells: toward clinical translation. Curr. Opin. Cell Biol. 22, 837–844 [DOI] [PubMed] [Google Scholar]
- 5. Li H., Zhu H., Xu C. J., Yuan J. (1998) Cleavage of BID by caspase-8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell 94, 491–501 [DOI] [PubMed] [Google Scholar]
- 6. Dumitru C. A., Carpinteiro A., Trarbach T., Hengge U. R., Gulbins E. (2007) Doxorubicin enhances TRAIL-induced cell death via ceramide-enriched membrane platforms. Apoptosis 12, 1533–1541 [DOI] [PubMed] [Google Scholar]
- 7. Martin S., Phillips D. C., Szekely-Szucs K., Elghazi L., Desmots F., Houghton J. A. (2005) Cyclooxygenase-2 inhibition sensitizes human colon carcinoma cells to TRAIL-induced apoptosis through clustering of DR5 and concentrating death-inducing signaling complex components into ceramide-enriched caveolae. Cancer Res. 65, 11447–11458 [DOI] [PubMed] [Google Scholar]
- 8. Muppidi J. R., Siegel R. M. (2004) Ligand-independent redistribution of Fas (CD95) into lipid rafts mediates clonotypic T cell death. Nat. Immunol. 5, 182–189 [DOI] [PubMed] [Google Scholar]
- 9. Rossin A., Derouet M., Abdel-Sater F., Hueber A. O. (2009) Palmitoylation of the TRAIL receptor DR4 confers an efficient TRAIL-induced cell death signalling. Biochem. J. 419, 185–192 [DOI] [PubMed] [Google Scholar]
- 10. Delmas D., Rébé C., Micheau O., Athias A., Gambert P., Grazide S., Laurent G., Latruffe N., Solary E. (2004) Redistribution of CD95, DR4 and DR5 in rafts accounts for the synergistic toxicity of resveratrol and death receptor ligands in colon carcinoma cells. Oncogene 23, 8979–8986 [DOI] [PubMed] [Google Scholar]
- 11. Gajate C., Mollinedo F. (2007) Edelfosine and perifosine induce selective apoptosis in multiple myeloma by recruitment of death receptors and downstream signaling molecules into lipid rafts. Blood 109, 711–719 [DOI] [PubMed] [Google Scholar]
- 12. Song J. H., Tse M. C., Bellail A., Phuphanich S., Khuri F., Kneteman N. M., Hao C. (2007) Lipid rafts and nonrafts mediate tumor necrosis factor related apoptosis-inducing ligand induced apoptotic and nonapoptotic signals in non small cell lung carcinoma cells. Cancer Res. 67, 6946–6955 [DOI] [PubMed] [Google Scholar]
- 13. Vanoosten R. L., Moore J. M., Ludwig A. T., Griffith T. S. (2005) Depsipeptide (FR901228) enhances the cytotoxic activity of TRAIL by redistributing TRAIL receptor to membrane lipid rafts. Mol. Ther. 11, 542–552 [DOI] [PubMed] [Google Scholar]
- 14. Wagner K. W., Punnoose E. A., Januario T., Lawrence D. A., Pitti R. M., Lancaster K., Lee D., von Goetz M., Yee S. F., Totpal K., Huw L., Katta V., Cavet G., Hymowitz S. G., Amler L., Ashkenazi A. (2007) Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat. Med. 13, 1070–1077 [DOI] [PubMed] [Google Scholar]
- 15. Yoshida T., Shiraishi T., Horinaka M., Wakada M., Sakai T. (2007) Glycosylation modulates TRAIL-R1/death receptor 4 protein: different regulations of two pro-apoptotic receptors for TRAIL by tunicamycin. Oncol. Rep. 18, 1239–1242 [PubMed] [Google Scholar]
- 16. Manser E., Leung T., Salihuddin H., Tan L., Lim L. (1993) A non-receptor tyrosine kinase that inhibits the GTPase activity of p21cdc42. Nature 363, 364–367 [DOI] [PubMed] [Google Scholar]
- 17. Eisenmann K. M., McCarthy J. B., Simpson M. A., Keely P. J., Guan J. L., Tachibana K., Lim L., Manser E., Furcht L. T., Iida J. (1999) Melanoma chondroitin sulphate proteoglycan regulates cell spreading through Cdc42, Ack-1 and p130cas. Nat. Cell biology 1, 507–513 [DOI] [PubMed] [Google Scholar]
- 18. Linseman D. A., Heidenreich K. A., Fisher S. K. (2001) Stimulation of M3 muscarinic receptors induces phosphorylation of the Cdc42 effector activated Cdc42Hs-associated kinase-1 via a Fyn tyrosine kinase signaling pathway. J. Biol. Chem. 276, 5622–5628 [DOI] [PubMed] [Google Scholar]
- 19. Satoh T., Kato J., Nishida K., Kaziro Y. (1996) Tyrosine phosphorylation of ACK in response to temperature shift-down, hyperosmotic shock, and epidermal growth factor stimulation. FEBS Lett. 386, 230–234 [DOI] [PubMed] [Google Scholar]
- 20. Yang W., Cerione R. A. (1997) Cloning and characterization of a novel Cdc42-associated tyrosine kinase, ACK-2, from bovine brain. J. Biol. Chem. 272, 24819–24824 [DOI] [PubMed] [Google Scholar]
- 21. Yang W., Lin Q., Guan J. L., Cerione R. A. (1999) Activation of the Cdc42-associated tyrosine kinase-2 (ACK-2) by cell adhesion via integrin β1. J. Biol. Chem. 274, 8524–8530 [DOI] [PubMed] [Google Scholar]
- 22. Pao-Chun L., Chan P. M., Chan W., Manser E. (2009) Cytoplasmic ACK1 interaction with multiple receptor tyrosine kinases is mediated by Grb2: an analysis of ACK1 effects on Axl signaling. J. Biol. Chem. 284, 34954–34963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grøvdal L. M., Johannessen L. E., Rødland M. S., Madshus I. H., Stang E. (2008) Dysregulation of Ack1 inhibits down-regulation of the EGF receptor. Exp. Cell Res. 314, 1292–1300 [DOI] [PubMed] [Google Scholar]
- 24. Shen F., Lin Q., Gu Y., Childress C., Yang W. (2007) Activated Cdc42-associated kinase 1 is a component of EGF receptor signaling complex and regulates EGF receptor degradation. Mol. Biol. Cell 18, 732–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Howlin J., Rosenkvist J., Andersson T. (2008) TNK2 preserves epidermal growth factor receptor expression on the cell surface and enhances migration and invasion of human breast cancer cells. Breast Cancer Res. 10, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mahajan K., Challa S., Coppola D., Lawrence H., Luo Y., Gevariya H., Zhu W., Chen Y. A., Lawrence N. J., Mahajan N. P. (2010) Effect of Ack1 tyrosine kinase inhibitor on ligand-independent androgen receptor activity. Prostate 70, 1274–1285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Shen H., Ferguson S. M., Dephoure N., Park R., Yang Y., Volpicelli-Daley L., Gygi S., Schlessinger J., De Camilli P. (2011) Constitutive activated Cdc42-associated kinase (Ack) phosphorylation at arrested endocytic clathrin-coated pits of cells that lack dynamin. Mol. Biol. Cell 22, 493–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Teo M., Tan L., Lim L., Manser E. (2001) The tyrosine kinase ACK1 associates with clathrin-coated vesicles through a binding motif shared by arrestin and other adaptors. J. Biol. Chem. 276, 18392–18398 [DOI] [PubMed] [Google Scholar]
- 29. Yeow-Fong L., Lim L., Manser E. (2005) SNX9 as an adaptor for linking synaptojanin-1 to the Cdc42 effector ACK1. FEBS Lett. 579, 5040–5048 [DOI] [PubMed] [Google Scholar]
- 30. Chua B. T., Lim S. J., Tham S. C., Poh W. J., Ullrich A. (2010) Somatic mutation in the ACK1 ubiquitin association domain enhances oncogenic signaling through EGFR regulation in renal cancer derived cells. Mol. Oncol. 4, 323–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. van der Horst E. H., Degenhardt Y. Y., Strelow A., Slavin A., Chinn L., Orf J., Rong M., Li S., See L. H., Nguyen K. Q., Hoey T., Wesche H., Powers S. (2005) Metastatic properties and genomic amplification of the tyrosine kinase gene ACK1. Proc. Natl. Acad. Sci. U.S.A. 102, 15901–15906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Prieto-Echagüe V., Gucwa A., Craddock B. P., Brown D. A., Miller W. T. (2010) Cancer-associated mutations activate the nonreceptor tyrosine kinase Ack1. J. Biol. Chem. 285, 10605–10615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mahajan K., Coppola D., Chen Y. A., Zhu W., Lawrence H. R., Lawrence N. J., Mahajan N. P. (2012) Ack1 tyrosine kinase activation correlates with pancreatic cancer progression. Am. J. Pathol. 180, 1386–1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mahajan N. P., Liu Y., Majumder S., Warren M. R., Parker C. E., Mohler J. L., Earp H. S., Whang Y. E. (2007) Activated Cdc42-associated kinase Ack1 promotes prostate cancer progression via androgen receptor tyrosine phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 104, 8438–8443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mahajan N. P., Whang Y. E., Mohler J. L., Earp H. S. (2005) Activated tyrosine kinase Ack1 promotes prostate tumorigenesis: role of Ack1 in polyubiquitination of tumor suppressor Wwox. Cancer Res. 65, 10514–10523 [DOI] [PubMed] [Google Scholar]
- 36. Harper N., MacFarlane M. (2008) Recombinant TRAIL and TRAIL receptor analysis. Methods Enzymol. 446, 293–313 [DOI] [PubMed] [Google Scholar]
- 37. Hopkins S., Linderoth E., Hantschel O., Suarez-Henriques P., Pilia G., Kendrick H., Smalley M. J., Superti-Furga G., Ferby I. (2012) Mig6 is a sensor of EGF receptor inactivation that directly activates c-Abl to induce apoptosis during epithelial homeostasis. Dev. Cell 23, 547–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Colvin E. S., Ma H. Y., Chen Y. C., Hernandez A. M., Fueger P. T. (2013) Glucocorticoid-induced suppression of beta-cell proliferation is mediated by Mig6. Endocrinology 154, 1039–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Levental I., Lingwood D., Grzybek M., Coskun U., Simons K. (2010) Palmitoylation regulates raft affinity for the majority of integral raft proteins. Proc. Natl. Acad. Sci. U.S.A. 107, 22050–22054 [DOI] [PMC free article] [PubMed] [Google Scholar]