

Background: The role of the vascular IP3 receptor (IP3R) in hypertension is unknown.
Results: IP3R are up-regulated in vascular smooth muscle (VSM) in hypertension through the calcineurin-NFAT pathway.
Conclusion: Up-regulated IP3R in VSM sensitize Ca2+ release and enhance contraction.
Significance: Up-regulated vascular IP3R may contribute to vascular resistance in hypertension.
Keywords: Calcineurin, Calcium Signaling, Hypertension, NFAT Transcription Factor, Vascular Smooth Muscle Cells, A7r5, IP3 Receptor
Abstract
Resistance arteries show accentuated responsiveness to vasoconstrictor agonists in hypertension, and this abnormality relies partly on enhanced Ca2+ signaling in vascular smooth muscle (VSM). Although inositol 1,4,5-triphosphate receptors (IP3Rs) are abundant in VSM, their role in the molecular remodeling of the Ca2+ signaling machinery during hypertension has not been addressed. Therefore, we compared IP3R expression and function between mesenteric arteries of normotensive and hypertensive animals. Levels of IP3R transcript and protein were significantly increased in mesenteric arteries of hypertensive animals, and pharmacological inhibition of the IP3R revealed a higher contribution of IP3-dependent Ca2+ release to vascular contraction in these arteries. Subsequently, we established cultured aortic VSM A7r5 cells as a cellular model that replicates IP3R up-regulation during hypertension by depolarizing the VSM cell membrane. IP3R up-regulation requires Ca2+ influx through L-type Ca2+ channels, followed by activation of the calcineurin-NFAT axis, resulting in IP3R transcription. Functionally, IP3R up-regulation in VSM is associated with enhancement and sensitization of IP3-dependent Ca2+ release, resulting in increased VSM contraction in response to agonist stimulation.
Introduction
Hypertension is a complex disease that is the clinical manifestation of multiple genetic, environmental, nutritional, and hormonal pathologies that predispose individuals to cardiovascular disease, including heart failure and stroke (1, 2). In recent years, it has become clear that structural alterations in arteries, referred to as remodeling, increase their stiffness and contribute to the development of hypertension (3, 4). Furthermore, arterial wall injury leads to a phenotypic switch of VSM2 from a contractile to a proliferative phenotype, which is associated with alterations to the VSM Ca2+ signaling machinery (5).
The major drop in hydrostatic pressure in the vasculature occurs at the level of resistance arteries. Because blood flow is proportional to the vessel radius to the 4th power, as described by Poiseuille's law, small changes in the luminal diameter of resistance arteries will dramatically alter resistance to blood flow with a pronounced impact on blood pressure. Peripheral resistance depends on the diameter of resistance arteries, which hinges in part on the contractile state of the VSM cells (6, 7). The resting myogenic tone is set by the summed responses of the Ca2+ handling and contractile machineries to various inputs, including sympathetic neurotransmitters (norepinephrine) (8), endothelium-derived agents (NO, endothelin) (6), endocrine factors in the blood stream (angiotensin II) (2), and intraluminal pressure (9).
Several channels and transporters contribute to Ca2+ signaling and contraction in VSM, with two primary pathways being the agonist-activated PLC-IP3R pathway and the voltage-gated L-type Ca2+ channel (LTCC) (7). The pore-forming subunit of the LTCC channel in VSM is α1C (10). The fundamental role of these two Ca2+ signaling pathways in fueling vasoconstriction during hypertension is attested to clinically by the effective lowering of blood pressure following pharmacological block of the LTCC and α-adrenergic receptors (2). Consistently, norepinephrine produces greater vasoconstriction in hypertensive compared with normotensive individuals (11). However, the molecular mechanisms of this enhanced vasomotor response are unclear.
The role of the LTCC in modulating the myogenic response of VSM is well established through clinical, genetic, and physiological studies (7, 10, 12–15). In contrast, the contribution of the IP3R remains poorly characterized. However, several lines of evidence support a role for the PLC-IP3 axis in regulating the myogenic tone. The IP3R has been implicated in Ca2+-dependent contraction in response to vasoconstrictors central to blood pressure regulation, including norepinephrine and angiotensin II (16–18). In addition, G-protein-coupled receptor-dependent Ca2+ oscillations have been implicated in vasoconstriction (19), and activation of α1-adrenergic receptors increases the percentage of VSM exhibiting Ca2+ oscillations (20). Furthermore, PLC-IP3-dependent Ca2+ release plays an important role in regulating myogenic tone caused by increased intraluminal pressure in isolated arteries (21, 22), and stretch of isolated VSM cells results in enhanced IP3 levels, leading to Ca2+ release (23, 24). Finally, PLC inhibition or Ca2+ store depletion results in vasodilation or a decreased myogenic response (25–27). Collectively, these findings suggest that the activity of in situ IP3R is influenced by blood pressure and vasoactive agonists impinging on the VSM cells of resistance arteries.
Here, we report up-regulation of the IP3R in resistance-size mesenteric arteries in hypertensive mouse and rat models. IP3R up-regulation could be recapitulated in cultured VSM cells by depolarization-induced activation of the LTCC, leading to induction of the Ca2+-dependent calcineurin-NFAT signaling pathway. Importantly, IP3R up-regulation results in sensitization of both IP3-dependent Ca2+ release and VSM contraction. Hence, up-regulation of vascular IP3R is poised to contribute to enhanced Ca2+ signaling and vasoreactivity during hypertension.
EXPERIMENTAL PROCEDURES
Animals
All procedures involving animals were approved by the Institutional Animal Care and Use Committee at the University of Arkansas for Medical Sciences. C57BL/6 mice were obtained at 10 weeks of age from Harlan Laboratories (Indianapolis, IN) and maintained in a temperature-controlled room in a 12-h/12-h light/dark cycle with free access to food and water. Wistar Kyoto rats and spontaneously hypertensive rats (SHR) were obtained at 12–14 weeks of age from Taconic Farms (Germantown, NY). Sprague-Dawley rats were obtained from Harlan Laboratories (Madison, WI) at the same age.
Minipump Implantation and Blood Pressure Measurements
Mice were anesthetized by 2.5% isoflurane inhalation. Osmotic minipumps (Alzet 1002, Durect Corp.) loaded with angiotensin II (Ang II; Bachem) to accomplish an infusion dose of 2 ng/min/g for 2 weeks or an equal volume of vehicle (0.9% saline) were implanted subcutaneously. After recovery from anesthesia, mice were housed in individual cages and allowed free access to food and water (28). The systolic blood pressure (SBP) was recorded by tail cuff plethysmography (Kent Scientific) before (day 0) and on days 7 and 14 after osmotic minipump implantation. As described earlier (12, 13), blood pressure was recorded by intra-arterial catheter in anesthetized normotensive and hypertensive rats directly prior to studies.
Vascular Reactivity Assays
Second order mesenteric arteries (MA) were isolated from saline (SAL) and Ang II-infused hypertensive (AHT) mice. Arteries were cleaned of adhered fat and connective tissue in ice-cold physiological salt solution (PSS) of the following composition: 119 mm NaCl, 4.7 mm KCl, 1.17 mm MgSO4, 24 mm NaHCO3, 0.026 mm EDTA, 1.17 mm NaH2PO4, 5.5 mm glucose, and 1.6 mm CaCl2. Vessels were cannulated on both ends with tapered glass micropipettes in a microvessel perfusion system (Living Systems) containing PSS bubbled with a 95% O2, 5% CO2 gas mixture and maintained at 37 °C. Arteries were perfused with PSS at an intraluminal pressure of 60 mm Hg with no outflow. The PSS in the chamber was exchanged every 15 min during 1 h of equilibration. After equilibration, the viability of MA was verified by observing a contractile response to 60 mm KCl (28). Phenylephrine (PE) concentration-response curves (10−8 to 10−4 m; half-log increments) were obtained in the absence or presence of the IP3R blocker, 2-aminoethoxydiphenyl borate (2-APB) (50 μm). Changes in outer vessel diameter were recorded using an upright microscope/Spot RT camera and analyzed with automated edge detection and data acquisition software, DMTvas (Danish Myo Technology). The constrictor response to PE was calculated as percentage change in diameter from the initial resting diameter.
Cell Culture and Transfection
Embryonic rat aortic smooth muscle-derived A7r5 cells were purchased from the American Type Culture Collection (Manassas, VA). The cells were seeded onto a 100-mm dish (BD Biosciences) and grown in Dulbecco's modified Eagle's medium-high glucose supplemented with 4 mm glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, 1 mm sodium pyruvate, and 10% fetal bovine serum (Sigma). Cells were kept at 37 °C in a humidified atmosphere with 5% CO2, and fresh medium was added every 48 h until the start of studies.
The plasmid pEGFPC1-NFATc1 was a generous gift from G. Crabtree (Stanford University). A7r5 cells were transiently transfected with pEGFP-NFATc1 using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. At 3 days post-transfection, cells were washed with prewarmed Ca2+/Mg2+-free PBS and maintained for 30 min at 37 °C and 5% CO2 in HEPES-buffered saline (HBS): 145 mm NaCl, 4 mm KCl, 1 mm MgCl2, 10 mm Hepes, 2 mm CaCl2, 10 mm glucose, pH 7.4. Fluorescent confocal imaging was performed on a Zeiss LSM 710 confocal microscope (Carl Zeiss) using an EC Plan Neofluar ×40/1.3 numerical aperture oil differential interference contrast objective. GFP signal was excited with a 488-nm laser, and emissions were collected through a bandwidth of 492–558 nm. Images were analyzed using ZEN 2008 software (Carl Zeiss).
Western Blotting
Mesenteric Arteries
MA from SAL and AHT mice were carefully isolated and cleaned of adherent fat and connective tissue. Arteries from each mouse were homogenized on ice in radioimmune precipitation assay buffer (Thermo Scientific) containing protease inhibitor mixture (Roche Applied Science). Large tissue debris and cell fragments were removed by centrifugation at 12,000 × g at 4 °C for 10 min, and the protein-rich supernatant was carefully collected. Crude protein concentration was determined by the Bradford method using Bio-Rad protein assay dye reagent. Twenty-five μg of protein was loaded per well, and proteins were separated by electrophoresis on a 3–8% polyacrylamide gel (Invitrogen) under reducing conditions. After separation, the proteins were transferred to a PVDF membrane (Thermo Scientific). Membranes were first blocked with 10% nonfat dried milk in TBS-T (10 mm Tris, 100 mm NaCl, and 0.1% Tween 20, pH 7.5) for 1 h at room temperature, followed by overnight incubation at 4 °C with antibodies targeting specific epitopes in 5% nonfat dried milk in TBS-T. In a subset of studies, proteins were similarly prepared from rat MA (12, 13). In Western blots, monoclonal antibodies were directed against IP3R1 (1:1000; NeuroMab), STIM1 (1:1000; Abcam), PMCA1 (1:500; Santa Cruz Biotechnology, Inc.), and GAPDH (1:10,000; EMD Millipore). Polyclonal antibodies were directed against α1C and TRPC1 (1:1000; Alomone Labs), TRPC4 (1:2000; NeuroMab), or Orai1 (1:1000; ProSci Inc.). After exposure to primary antibody, membranes were incubated for 1 h at room temperature with horseradish peroxidase-labeled sheep anti-mouse IgG or donkey anti-rabbit IgG (GE Healthcare) in TBS-T containing 5% nonfat dried milk. The bands were identified by chemiluminescence and exposed to x-ray films. The density of immunoreactive bands was determined using ImageJ software (National Institutes of Health) and normalized to the GAPDH density for each lane (28). Similar procedures were used to detect rat arterial proteins on Western blot (12, 13).
A7r5 Cells
Total proteins were extracted from A7r5 cells (90–100% confluent) and subjected to Western blot analysis as described above. Forty μg of protein was loaded per well, and expression of Ca2+-handling proteins was detected using specific antibodies against IP3R1 (1:1000; Millipore), α1C (1:1000; Millipore), TRPC1 (1:200; Santa Cruz Biotechnology), TRPC4 (1:200; Novus Biologicals), STIM1 (1:1000; Cell Signaling Technology), Orai1 (1:1000; Sigma), and PMCA1 (1:500; Santa Cruz Biotechnology). The density of bands was determined using ImageJ and normalized to the α-smooth muscle actin density for each lane.
RT-PCR
Mesenteric Arteries
Total RNA was isolated from MA of SAL and AHT mice using the RNeasy minikit (Qiagen). First strand cDNA synthesis was performed with the iScriptTM cDNA synthesis kit (Bio-Rad). After reverse transcription, real-time PCR was performed using iQTM SYBR® Green Supermix (Bio-Rad) containing a hot start iTaqTM DNA polymerase and the following specific primers: IP3R1, 5′-GAGATGAGCCTGGCTGAGGTTCAA-3′ (forward) and 5′-TGTTGCCTCCTTCCAGAAGTGCGA-3′ (reverse); β-actin, 5′-ATCCTGTGGCATTCCATGAAACTAC-3′ (forward) and 5′-AGGAGCCAGGGCAGTAATCCTC-3′ (reverse).
Ten pmol of each primer pair and 1 μl of cDNA template were added to the 25-μl reaction mixture, and PCR was performed in an iCycler iQTM real-time PCR detection system (Bio-Rad) using the following conditions: 5 min at 94 °C for initial activation and 40 cycles of 30 s at 94 °C, 1 min at 65 °C, 2 min at 72 °C, and 10 min at 72 °C. A negative control with RNA instead of cDNA was carried out, and the integrity of PCR was verified by dissociation curve analysis. The relative gene expression was quantified from fluorescence cycle threshold (Ct) values using the ΔΔCt method.
For multicell PCR using populations of enzymatically isolated mouse mesenteric VSM cells, MA were enzymatically digested, and 60–80 VSM cells were aspirated into borosilicate glass micropipette filled with 1× phosphate-buffered saline under the microscope. Total RNA preparation and cDNA synthesis was performed using the Single Cell real-time RT-PCR assay kit (Signosis) per commercial instructions. cDNA products were amplified by nested PCR using gene-specific primer pairs. The following primer pairs were used for first round PCR amplification: IP3R1, 5′-CCCATCCTAACGGAACGAGC-3′ (forward) and 5′-GGCTTAGCAGCTTTCCAGAAC-3′ (reverse); α1C, 5′-TTACAACCAGCCTCACTGGC-3′ (forward) and 5′-GCGGGAAGTTATCGAACGTG-3′ (reverse); TRPC4, 5′-ACTGCCAATTCTCACCTGGG-3′ (forward) and 5′-GGGCGGAACCATTGCTTAGG-3′ (reverse); α-actin, 5′-TCTGCCTCTAGCACACAACTG-3′ (forward) and 5′-CTAGCTGTGAAGTCAGTGTCG-3′ (reverse); von Willebrand factor (vWF), 5′-GCTGTGGGAGATGCTTGCCT-3′ (forward) and 5′-TGGAGTACACAGCTTTGCTG-3′ (reverse).
The following gene-specific primers designed within a region of first round PCR products were used for second-round (nested) PCR amplification: IP3R1, 5′-CTGCTAACCCCTCTCCTGGT-3′ (forward) and 5′-TGGCTGTACAACACAACGGT-3′ (reverse); α1C, 5′-TGTCCCTCTTCAACCGCTTTGACT-3′ (forward) and 5′-GTTCAGCAAGGATGCCACAAGGTT-3′ (reverse); TRPC4, 5′-GGGCGGCGTGCTGCTGAT-3′ (forward) and 5′-CCGCGTTGGCTGACTGTATTGTAG-3′ (reverse); α-actin, 5′-TCTGCCTCTAGCACACAACTG-3′ (forward) and 5′-CTAGGCCAGGGCTACAAGTT-3′ (reverse); vWF, 5′-TTTGTGCAGGGTCAACGAGA-3′ (forward) and 5′-GTACTGCACCTTGGCTGTGA-3′ (reverse).
Reaction conditions for first round PCR and nested PCR were similar to those described above. PCR products were separated by electrophoresis on a 1.6% agarose gel, followed by ethidium bromide staining, UV transillumination, and documentation on Gel/Chemi Doc (Bio-Rad).
A7r5 Cells
Total RNA was prepared from A7r5 cells using TRIzol reagent (Invitrogen). cDNA was synthesized from total RNA using the SuperScript® III first strand synthesis kit (Invitrogen) with random hexamers as primers, according to the manufacturer's instructions. PCRs contained 1 μg of total RNA, 0.2 mm dNTP, Phusion GC Buffer (5×), and Phusion DNA Polymerase (New England Biolabs), and a 0.4 mm concentration of each primer in a total volume of 20 μl. Reactions were run at 94 °C for 2 min, followed by 25 cycles at 94 °C for 30 s, 56 °C for 30 s, and 72 °C for 90 s and a 10-min final extension using the following primers: IP3R1, 5′-CTCTGTTTGCTGCAAGGGTGATC-3′ (forward) and 5′-GTCCTTCACTTTCACCAGCACG-3′ (reverse); IP3R2, 5′-ATTGTCACCGTGCTGAACCAG-3′ (forward) and 5′-GCCACAGATGAAGCAGGTTGTC-3′ (reverse); IP3R3, 5′-CCTGAGATCCTAGAAGAGGACGAG-3′ (forward) and 5′-TCTCCAGACCACAGATGAAGCAC-3′ (reverse); α1C, 5′-CTTCAAACGTGGCCACAGAC-3′ (forward) and 5′-GCTGTGTGGAACTGACGGTA-3′ (reverse); β-actin, 5′-ATCCTGTGGCATTCCATGAAACTAC-3′ (forward) and 5′-AGGAGCCAGGGCAGTAATCTCCT-3′ (reverse).
The amplified products were separated by electrophoresis on 1.2% agarose gels and analyzed as described above. For real-time PCR, each PCR (25 μl) contained 12.5 μl of iQ SYBR Green Supermix (Bio-Rad), 400 nm primers, and an equal volume of cDNA template. Reactions were performed in triplicate on a 7500 Fast Real-Time PCR System (Applied Biosystems) at 95 °C for 10 min and 40 cycles of 20 s at 95 °C, 20 s at 65 °C, and 45 s at 72 °C.
Ca2+ Imaging
Cells were seeded onto 35-mm poly-d-lysine-coated glass coverslips (MatTek Corp.) and incubated at 37 °C and 5% CO2 in complete culture media for 4 days. Cells were incubated with the ratiometric Ca2+ dye Fura-2-AM (2 μm; Invitrogen) and placed in the incubator for 45 min in the dark. After incubation, cells were washed with prewarmed Ca2+/Mg2+-free PBS and allowed to rest in HBS for 10 min. The coverslips were then transferred to an inverted epifluorescence microscope (Olympus) equipped with a UPlanSapo ×40/0.95 numerical aperture oil objective and connected to a CCD camera (CoolSNAP HQ2). The fluorescence emission of individual cells was measured at 510 nm following excitation at 340 and 380 nm using an EasyRatioPro calcium imaging system (PTI). The fluorescence ratio (F340/F380) was recorded in real time at 0.5-s intervals as a relative indicator of intracellular Ca2+ concentration. The ratios were calibrated by determining Fmax and Fmin values in the presence of 0.1% saponin (Fmax) and 10 mm EGTA (Fmin).
Caged IP3
Ca2+ transients were analyzed in A7r5 cells loaded with the membrane-permeant caged IP3 analog ciIP3, purchased from SiChem. Briefly, cells were cultured on glass coverslips and kept in culture as described under “Ca2+ Imaging.” Prior to Ca2+ imaging, cells were washed with prewarmed Ca2+/Mg2+-free PBS and incubated in HBS containing 2 μm ciIP3 for 45 min in the dark, followed by incubation with a 2 μm concentration of the cell-permeant dye Ca-Green-1, AM (Invitrogen), which was added to the loading solution for a further 45 min before washing and allowing at least 30 min for de-esterification. Imaging of changes in [Ca2+]i was accomplished using an inverted epifluorescence microscope (Olympus) equipped with a UPlanSapo ×40/0.95 numerical aperture oil objective and connected to a CCD camera (CoolSNAP HQ2). IP3 was released by a photochemical destruction of the cage with brief exposures (100 ms every 1.5 s) of the cells to UV light (340 nm). The fluorescence of individual cells was collected simultaneously at 510 nm by excitation of Ca-Green-1 at 488 nm using an EasyRatioPro calcium imaging system (PTI). The fluorescence signals are expressed as ratios (F/F0) of the mean fluorescence F of each cell relative to the mean resting fluorescence F0.
Monitoring of A7r5 Contraction
Assessment of cell contraction was performed on A7r5 cells cultured on glass coverslips as described above. Attached cells were washed with prewarmed Ca2+/Mg2+-free PBS and allowed to settle in HBS for 10 min prior to live cell imaging. Cell contraction was visualized using an inverted microscope (Olympus) equipped with a LucPlan FLN ×40/0.60 numerical aperture objective. Images were acquired with a CCD camera (Olympus DP72) and processed using DP2-BSW software (Olympus Soft Imaging Solutions). Cell contraction was quantified by morphometric analysis using ImageJ. Briefly, the addition of PE induces the formation of contractile fibers, which appear as protruded edges on the surface of contracted cells. To quantify contractile responses to PE, contractile fibers were quantified using the edge detection function in ImageJ on thresholded time lapse images.
Statistics
Data represent means ± S.E. of the mean for the number (n) of animals or in vitro preparations indicated in parentheses. Student's t test was used to compare two data sets, and one-way/two-way analysis of variance and Bonferroni's post hoc test were used for multiple group comparisons. p ≤ 0.05 was considered significant.
RESULTS AND DISCUSSION
Hypertension Is Coupled to Increased Expression of Vascular IP3R
To determine the expression level of vascular IP3R during hypertension, male C57BL/6 mice were infused subcutaneously with Ang II (2 ng/g/min, AHT) for 2 weeks, resulting in average SBP values of 109 ± 2, 136 ± 3, and 152 ± 2 mm Hg at 0, 7, and 14 days, respectively (n = 12 each; Fig. 1A). In contrast, SBP values were unchanged from the preinfusion value of 105 ± 3 mm Hg in SAL-infused mice (n = 12 each; Fig. 1A). At 14 days, hypertension correlates with increased protein expression (2.39 ± 0.19-fold) of the type 1 IP3R (IP3R1) in MA (Fig. 1, B and C). Localization and functional studies in renal and mesenteric VSM and in A7r5 cells argue that IP3R1 is the primary isoform (29–31). Consistently, quantitative RT-PCR reveals a 1.83 ± 0.11-fold increase of IP3R1 transcript in arteries of AHT compared with SAL mice (Fig. 1D).
FIGURE 1.

Increased expression and functional contribution of vascular IP3Rs in experimental hypertension. A, measurement of SBP at base line and after infusion of SAL or Ang II to establish AHT. SBP was recorded by tail cuff plethysmography at base line (day 0) and at 7 and 14 days after infusion. SBP was significantly increased in AHT compared with SAL mice at 7 and 14 days (n = 12). B, immunoblot analysis of IP3R in lysates from mouse MA. Artery lysate from SAL (control) and AHT mice was added to each lane (3 animals/group). C, relative protein density after normalization to GAPDH shows that IP3R1 immunoreactivity was 2.4-fold higher in MA of AHT compared with SAL mice (n = 6). D, quantitative mRNA expression reveals a 1.8-fold increase of IP3R1 transcript in arteries of AHT compared with SAL mice (n = 4–6). Ct values were normalized to the Ct value of the amplification standard β-actin, which was similarly expressed between MA of SAL and AHT mice. E, representative traces showing changes in vessel diameter in response to increasing concentrations of PE (gray dots). Isolated second order MA from SAL and AHT mice were cannulated and pressurized to 60 mm Hg, and diameter was recorded on-line by edge detection. After washout (WO), PE-induced contractions were repeated in the presence of 2-APB (50 μm) (right). F, concentration-response curves to PE in MA from SAL and AHT mice in the presence (+2-APB) or absence of 2-APB. PE-induced contraction was enhanced in arteries of AHT mice (n = 8 and 6). PE-induced contraction was inhibited by 2-APB in MA from both SAL and AHT mice (n = 8 and 6). *, significant difference between SAL and AHT; @, significant difference between SAL and SAL+2-APB; #, significant difference between AHT and AHT+2-APB. G, arteries from AHT mice showed an increased 2-APB-sensitive component of PE-induced contraction compared with SAL mice (n = 6 and 8). The 2-APB-sensitive component was calculated by subtracting concentration-dependent contractions to PE obtained in the presence of 2-APB from those obtained in the absence of the blocker. Error bars, S.E. *, p ≤ 0.05.
PE-induced Contractions Are Enhanced in MA of AHT Mice
To assess the functional significance of IP3R up-regulation in arteries exposed to hypertension, we sought to determine the contribution of IP3-dependent Ca2+ release to vascular contractile responses to the vasoconstrictor agonist, PE. Isolated, pressurized (60 mm Hg) second order MA branches of AHT mice overexpressing IP3R1 showed accentuated contractions to PE, an α1-adrenoreceptor agonist that relies on IP3-dependent Ca2+ release for contraction (Fig. 1E) (32). The PE-induced contractions in MA from AHT mice averaged 38% higher at agonist concentrations of ≥10−6 m (Fig. 1F). The concentration of PE required for half-maximal contraction (ED50) did not differ significantly between MA of SAL (6.9 × 10−7 m) and AHT mice (6.7 × 10−7 m). Subsequently, we observed that the nonselective IP3R antagonist, 2-APB (33), suppressed contractions to PE in MA of SAL and AHT mice (Fig. 1E). Comparison of the amplitude of 2-APB-sensitive contraction between MA of SAL and AHT mice revealed a higher component in arteries from hypertensive animals (Fig. 1, F and G). These results argue that IP3-dependent Ca2+ release contributes more significantly to PE-induced vascular contractions in MA of hypertensive animals, supporting a functional role for the observed IP3R up-regulation in increasing vascular reactivity.
Specific Up-regulation of IP3R and α1C in Arteries of AHT Mice
We explored the possibility that Ang II-induced hypertension causes a nonspecific remodeling of Ca2+-handling proteins in VSM by comparing the expression of a panel of proteins implicated in VSM contraction between MA of SAL and AHT mice. It is well known that the α1C pore-forming subunit of the vascular LTCC up-regulates during hypertension (10, 12, 13). This event may partly account for the increased sensitivity of hypertensive subjects to the antihypertensive effect of clinical calcium channel antagonists; these drugs only mildly lower blood pressure in normotensive subjects (34–37). Indeed, Western blots confirmed that α1C up-regulation is coupled to increased IP3R1 expression in MA of AHT mice (Fig. 2A). We also evaluated the expression of two agonist-activated TRPC channels implicated in vascular reactivity, TRPC1 and TRPC4 (34–36, 38). TRPC1 expression was unchanged after Ang II infusion, but a pronounced up-regulation of TRPC4 was detected in MA of AHT mice (Fig. 2A). However, we failed to detect TRPC4 transcript in multicell PCRs limited to mouse mesenteric VSM cells. In this experiment, PCRs using cDNA derived from whole MA revealed transcripts coding for IP3R1, α1C, TRPC4, α-actin, and vWF; the latter two proteins are VSM and endothelium-specific markers, respectively (Fig. 2B, top). However, only IP3R1, α1C, and α-actin transcripts were detected in amplified products from isolated VSM cells (Fig. 2B, bottom). Thus, we ruled out TRPC4 as a direct contributor to VSM contraction, and our data suggest that this channel may only be expressed in the endothelium of small arteries, as suggested by others (34, 35).
FIGURE 2.
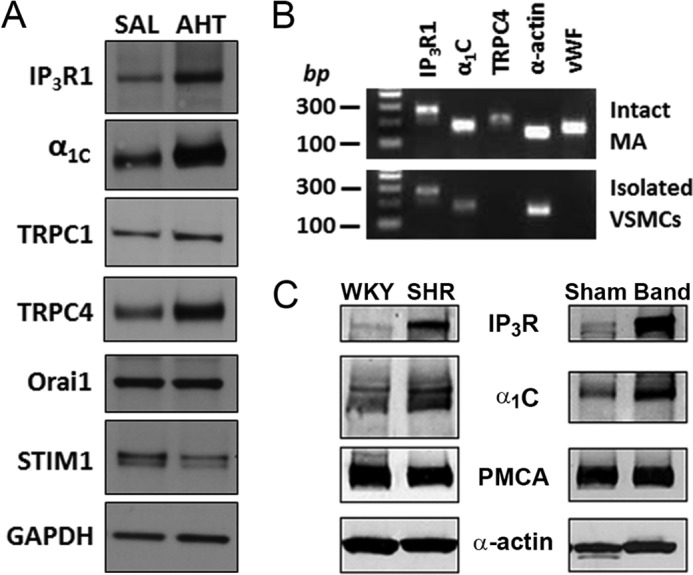
Screening of Ca2+-handling proteins reveals coupled up-regulation of the vascular IP3R and LTCC in experimental models of hypertension. A, immunoblot analysis of IP3R1, α1C, TRPC1, TRPC4, Orai1, and STIM1 in lysates from mouse MA. MA lysate pooled from three SAL or AHT mice was added to each lane. Increased expression levels of IP3R1, α1C, and TRPC4 are detected in MA of AHT mice, whereas levels of TRPC1, Orai1, and STIM1 were similar. B, agarose gel analysis of products obtained after PCRs for IP3R1 (274 bp), α1C (185 bp), and TRPC4 (226 bp) using specific primers. α-Actin (152 bp) and vWF (178 bp) were used as VSM and endothelial cell markers, respectively. IP3R, α1C, and α-actin were detected both in intact MA and isolated VSM cells as expected. In contrast, TRPC4 and vWF were detected in arteries but not isolated VSM cells, implying expression only in endothelium. C, immunoblots comparing the expression of IP3R, α1C, PMCA, and α-actin (as a loading control) between MA from normotensive Wistar Kyoto rats (WKY) and SHR and between MA from sham-operated and aortic-banded (Band) hypertensive rats. In each case, lysates were pooled from arteries of three or four rats. Only the IP3R and α1C proteins are up-regulated in MA from hypertensive SHR and aortic-banded rats. Blots are representative of three experiments.
Finally, we observed no difference in expression levels of STIM1 and Orai1 between MA of SAL and AHT mice. This finding is significant, because we presumed that the enhanced block of PE-induced contractions by 2-APB in arteries of AH mice reflected an increased contribution of IP3-dependent Ca2+ release to VSM reactivity in hypertension (Fig. 1, E–G), but 2-APB also can inhibit store-operated Ca2+ entry (SOCE) (33, 39, 40), which relies on the endoplasmic reticulum Ca2+ sensor STIM1 and the plasma membrane channel Orai1 (41). The comparable expression of STIM1 and Orai1 between MA of SAL and AHT mice is consistent with a relatively minor role of SOCE in VSM contraction and argues that the observed inhibition with 2-APB primarily relates to block of IP3Rs.
We also tested if IP3R up-regulation is specific to the AHT mouse model by evaluating IP3R1 expression in two rat models of hypertension, the SHR and aortic-banded rat (12, 13). The MA of the genetically hypertensive SHR were compared with those of the normotensive Wistar Kyoto rat. In the second model, the aorta of a Sprague-Dawley rat was partially banded between the left and right renal arteries, thus exposing the entire mesenteric circulation of banded rats to high blood pressure; MA of sham-operated rats exposed to normal blood pressure served as the control. In both rat models of hypertension, the IP3R1 and the α1C subunit of the LTCC were up-regulated in small MA (Fig. 2C). In contrast, expression of a membrane protein involved in Ca2+ extrusion, the plasma membrane Ca2+ pump (PMCA), was unchanged. Similarly, α-actin used as a loading control showed no change (Fig. 2C). We did observe some variability in IP3R up-regulation in different SHR animals, where in some instances no increase was observed. This may be a reflection of the genetic complexities underlying hypertension in the SHR model. Nonetheless, the fact that IP3R up-regulation was observed in three distinct models of hypertension with different genetic and neuroendocrine profiles and in two different species argues that it is a general phenomenon associated with hypertension.
Depolarization-induced Ca2+ Rise Up-regulates IP3R in A7r5 Cells
To identify mechanisms that up-regulate IP3R in VSM and thereby contribute to its overabundance in arteries exposed to hypertension, we used the embryonic rat aortic smooth muscle-derived A7r5 cell line. Notably, pressure-induced depolarization of small arteries and arterioles is recognized as a common abnormality of hypertension, which can induce the up-regulation of LTCCs and trigger enhanced vascular reactivity and vessel remodeling (12, 13). This loss of resting membrane potential (Em) extends to VSM cells of the renal, mesenteric, and skeletal muscle beds in experimental models of hypertension in which vessels are exposed to high intraluminal pressures (10, 12, 42–45). To mimic a key stimulus of vascular remodeling in hypertension, we exposed aortic A7r5 cells to incrementally higher concentrations of 20 (K20), 40 (K40), and 60 (K60) mm KCl for 24 h, to progressively depolarize and activate the SMCs as an in vitro surrogate for the hypertensive environment. These K+ concentrations would be predicted to shift EK by +43 mV (K20), +61.5 mV (K40), and +72.4 mV (K60) based on the Nernst potential. All three concentrations of KCl up-regulated the IP3R protein in A7r5 by 24 h, whereas expression levels of PMCA and smooth muscle α-actin were stable (Fig. 3A). Further studies used the lowest depolarizing stimulus of K20 that caused a 1.7-fold increase in IP3R expression (Fig. 3E). Importantly, up-regulation of IP3R was induced by depolarization rather than the osmotic challenge of K20, because enhanced IP3R was not observed in A7r5 aortic cells exposed to equiosmolar NaCl or sucrose (Sucr) (Fig. 3, A, B, and E). However, up-regulation of IP3R by 20K was prevented in A7r5 cells incubated in 10 μm nifedipine (Nif), a specific blocker of the LTCC (Fig. 3, A, B, and E). This finding argues that voltage-dependent Ca2+ influx through LTCC mediates IP3R up-regulation.
FIGURE 3.

Depolarization of A7r5 cells for 24 h replicates the induction of IP3R and α1C observed in VSM cells of hypertensive animals in vivo. A–E, immunoblot analyses of IP3R, PMCA, and α1C. Proteins were isolated from A7r5 cells and separated on SDS-PAGE, and the blots were probed for the appropriate antigens as indicated. A, IP3R1 expression is increased in A7r5 cells exposed to K20, K40, or K60 for 24 h but not in cells treated with NaCl or sucrose (Sucr) as osmolar controls. Depolarization-induced IP3R up-regulation is lost in the presence of the LTCC blocker, Nif (10 μm). Expression levels of PMCA are unchanged. B, the expression level of α1C is significantly increased in A7r5 cells incubated with K20 or K40 for 24 h but not in cells exposed to sucrose or 10 μm Nif (n = 3). C, expression levels of TRPC1, STIM1, and α-actin are not altered in A7r5 cells depolarized for 24 h by 20K. D, right blot shows that IP3R1 up-regulates in A7r5 cells exposed to 20K or treated with Thaps (1 μm) for 24 h. The up-regulation of IP3R1 by K20 is blocked by 10 μg/ml CHX or 10 μm ACD. Blots are representative of at least three independent experiments. Smooth muscle-specific α-actin was used as a loading control. E, relative immunodensity of the IP3R1 after normalization to α-actin. Cell depolarization with K20 or treatment with Thaps (1 μm) for 24 h increases IP3R1 expression, and this response is lost in the presence Nif (10 μm), CHX (10 μg/ml), ACD (10 μm), or CsA (10 μm) (n = 3–21; *, p < 0.05; **, p < 0.01). F, transcriptional up-regulation of IP3R1, IP3R3, and α1C in A7r5 cells depolarized by K20 for 24 h. Real-time PCR plots represent relative quantification. G, nuclear translocation of NFATc1 in A7r5 cells. Cells were transfected with plasmids expressing EGFP-NFATc1 and imaged using a confocal laser-scanning microscopy. At rest (Con), NFATc1 showed a basal cytoplasmic localization, but treatment with Thaps (1 μm) or K20 alone or in the presence of CHX (10 μg/ml), ACD (10 μm), or CsA (10 μm) promotes its nuclear translocation. In contrast, incubation of cells with 10 μm Nif or CsA prevents K20-induced nuclear translocation of NFATc1. Data are representative of four independent experiments. Scale bar, 20 μm. Error bars, S.E.
Similar to arteries exposed to hypertension, depolarization-induced IP3R up-regulation in A7r5 cells was coupled to an increased expression of LTCC α1C subunit (1.6 ± 0.07-fold; Fig. 3B), arguing that depolarization of A7r5 cells faithfully replicates the remodeling of Ca2+ signaling pathways observed in arteries from hypertensive animals.
Because SOCE has been previously implicated in VSM proliferation and migration (46), we tested the expression levels of STIM1 and Orai1 following depolarization in A7r5. Consistent with our findings in MA of AHT mice, STIM1 expression levels were stable (Fig. 3C), and we were unable to detect Orai1 expression on Western blots from A7r5 cells. Furthermore, no change in the expression levels of TRPC1 was detected in A7r5 cells exposed to KCl (Fig. 3C), whereas TRPC4 expression was not detected.
We then tested whether Ca2+ influx through LTCC is specifically required for IP3R up-regulation or whether a nonspecific cytoplasmic Ca2+ rise is sufficient to trigger and sustain IP3R1 enrichment in VSM cells. We raised intracellular Ca2+ levels by inhibiting the endoplasmic reticulum Ca2+-ATPase (SERCA) using the specific antagonist thapsigargin (Thaps) (47). Due to a continuous ill defined Ca2+ leak pathway at the endoplasmic reticulum membrane, Thaps results in a slow rise in cytoplasmic Ca2+ levels. This passive store depletion activates SOCE, thus accentuating the Ca2+ rise (43, 48). Indeed, Thaps treatment results in high basal Ca2+ levels (Fig. 4C) and was sufficient to induce significant IP3R up-regulation (1.5 + 0.14-fold; Fig. 3, D and E). Although depolarization-dependent Ca2+ influx through LTCC was more efficient at inducing IP3R up-regulation, the results from Thaps argue that a general cytoplasmic Ca2+ rise is sufficient to induce up-regulation of the IP3R in VSM.
FIGURE 4.
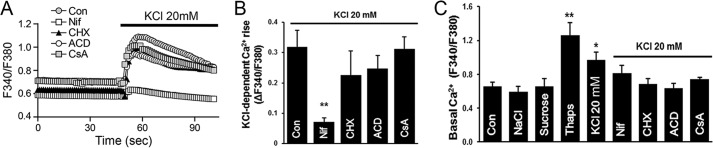
Ca2+ dynamics in response to acute KCl depolarization of A7r5 cells. Cells were loaded with Fura 2-AM and bathed in Ca2+-containing HBS. Imaging was performed as described under “Experimental Procedures.” A, acute membrane depolarization by K20 results in a global rise in intracellular Ca2+, which is inhibited by Nif (10 μm) but not by ACD (10 μm), CHX (10 μg/liter), or CsA (10 μm). Traces represent average fluorescence intensity of 15–20 individual cells. B, quantitation of the K20-induced Ca2+ rise as maximal Fura-2 ratio from base line (ΔF340/F380, n = 4–6; **, p < 0.01). C, basal intracellular Ca2+ levels were significantly increased in A7r5 cells after 24 h exposure to K20 or 1 μm Thaps. The osmolarity controls NaCl and sucrose did not alter resting Ca2+ levels in A7r5 cells. The addition of Nif (10 μm), CHX (10 μg/liter), ACD (10 μm), or CsA (10 μm) before depolarization by K20 prevents the elevation of basal Ca2+ level (n = 3–12; *, p < 0.05; **, p < 0.01). Error bars, S.E.
Calcium Influx Triggers IP3R Expression through Calcineurin-NFAT Signaling
The establishment of the A7r5 cell model allowed us to identify the signal transduction cascade that mediates depolarization-induced IP3R up-regulation. Studies using quantitative RT-PCR revealed a 2.2 ± 0.4-, 2.1 ± 0.3-, and 1.5 ± 0.3-fold increase of α1C, IP3R1, and the type 3 IP3R (IP3R3) transcripts, respectively, in A7r5 cells exposed to K20 for 24 h (Fig. 3F). Similar results were observed in semiquantitative PCR experiments (supplemental Fig. 1), whereas the type 2 IP3R (IP3R2) transcript was not detected.
Block of protein translation with 10 μg/liter cycloheximide (CHX) prevented K20-induced up-regulation of IP3R at 24 h (Fig. 3, D and E). Subsequently, nonspecific block of transcription with 10 μm actinomycin D (ACD) also prevented K20-induced IP3R enrichment (Fig. 3, C and D). Given that the Ca2+-calmodulin dependent phosphatase, calcineurin, promotes IP3R abundance in neurons (49), we treated A7r5 cells next with 10 μm cyclosporine A (CsA), a specific calcineurin blocker. CsA also abrogated the increased expression of IP3R caused by K20 (Fig. 3E).
Calcineurin regulates gene expression by dephosphorylating the transcription factor NFAT, which translocates to the nucleus to promote transcription (50). To directly support a role for NFAT in IP3R up-regulation, we imaged NFATc1 localization in control or K20-depolarized A7r5 cells. Control A7r5 cells transfected with GFP-NFATc1 exhibit a cytosolic distribution (Fig. 3G). The addition of K20 induced NFATc1 translocation to the nucleus within 15–20 min. Depolarization-induced NFATc1 translocation was prevented by block of LTCC by Nif or by inhibition of calcineurin activity with CsA (Fig. 3G). However, NFATc1 translocation to the nucleus persisted in K20-exposed A7r5 cells pretreated with either CHX or ACD, suggesting that these agents prevent depolarization-induced up-regulation of IP3R by acting at a site downstream of NFATc1 activation. Notably, we verified that CHX, ACD, and CsA per se do not blunt IP3R up-regulation by directly inhibiting 20K-induced Ca2+ influx through LTCCs, whereas Nif blocked the response as expected (Fig. 4, A and B). Consistent with our earlier finding that exposure of A7r5 cells to Thaps induces IP3R expression, Thaps treatment also resulted in NFATc1 translocation to the nucleus (Fig. 3G). These data support the concept that Ca2+ influx through LTCCs activates calcineurin, resulting in translocation of NFATc1 to the nucleus to trigger IP3R1 transcription.
Given the important role of cytoplasmic Ca2+ in mediating IP3R up-regulation, we were interested in determining the effect of the different treatments on basal Ca2+ levels in A7r5 cells. Compared with untreated cells, exposure of A7r5 cells for 24 h to the osmolarity controls (NaCl and sucrose) did not alter basal Ca2+ levels (Fig. 4C). In contrast, 20K and Thaps significantly increased basal Ca2+ levels at 24 h (Fig. 4C), findings that correlate well with IP3R up-regulation under these conditions (Fig. 3E). Interestingly, the observed increase in basal Ca2+ following 20K treatment was inhibited when cells were pretreated with Nif, CHX, ACD, or CsA (Fig. 4C). These results argue that the increased basal Ca2+ levels observed after prolonged KCl treatment are not due to Ca2+ influx through LTCCs but rather depend critically on IP3R up-regulation.
IP3R Up-regulation Sensitizes IP3-dependent Ca2+ Release
To test the consequence of IP3R up-regulation on IP3-dependent Ca2+ release in response to agonist stimulation, we performed a PE concentration-response curve, which revealed significantly larger Ca2+ transients at PE concentration of ≥5 μm in K20-treated cells compared with control (Con) (Fig. 5A). This enhanced Ca2+ transient would be predicted to promote VSM contraction. However, the enhanced Ca2+ release in response to PE could be due to the remodeling of other components of the signal transduction cascade leading to Ca2+ release during the 24-h incubation period and as such may not directly reflect IP3R function.
FIGURE 5.

IP3R up-regulation sensitizes IP3-dependent Ca2+ release and VSM contraction. A, PE concentration-response curve for Ca2+ release in control (Con) A7r5 cells or similar cells depolarized by K20 for 24 h to up-regulate IP3R. Cells were loaded with Fura2-AM and bathed in Ca2+-containing HBS, and images were acquired using an epifluorescence microscope. At low PE concentrations, the two groups of cells had similar fluorescence amplitudes. At PE concentrations above 5 μm, K20-pretreated cells showed higher fluorescence amplitude than control. Values are the mean of fluorescence intensity of 15–20 individual cells for each group given as the relative change in Fura2 fluorescence ratio (ΔF340/F380) (n = 3–5, mean ± S.E.; *, p < 0.05; **, p < 0.01). B–E, kinetics of IP3-dependent Ca2+ release evoked by UV photorelease of ciIP3 in A7r5 cells bathed in Ca2+-containing HBS and loaded with ciIP3 and Ca-Green-1. ciIP3 was uncaged by repeated exposure of cells to UV illumination (100 ms every 1.5 s), and the fluorescence of Ca-Green was recorded in real time. B, in a subset of cells, ciIP3 uncaging caused an initial global sustained Ca2+ transient followed by Ca2+ oscillations in both control and 20K-pretreated cells. Cells in both groups also responded to ciIP3 uncaging by a monotonic Ca2+ rise without Ca2+ oscillations. C, the maximal IP3-dependent Ca2+ release was increased in K20-pretreated cells exhibiting enriched IP3R (n = 8; *, p < 0.05). D, the percentage of oscillating cells (n = 8–10; ***, p < 0.0001), oscillation frequency/min (n = 5–8; ***, p < 0.0001), and duration of oscillations (n = 5–7; *, p = 0.05) were enhanced in K20-pretreated cells. E, the time to threshold Ca2+ signal was shortened in K20-pretreated cells compared with control (n = 8; **, p < 0.001). F, contractile responses of A7r5 cells to PE (20 μm) in Ca2+-containing HBS were evaluated by imaging of contractile fiber formation. PE results in a transient contractile response in control cells, whereas the contractile response in 20K-pretreated cells is more pronounced and sustained. G, statistical analysis of the maximal contractile response to PE of control and K20-pretreated A7r5 cells (n = 4; **, p < 0.05). Error bars, S.E.
To directly assess IP3R function under conditions where the receptor is up-regulated, we followed the kinetics of IP3-dependent Ca2+ release in A7r5 cells loaded with caged IP3 (ciIP3) and exposed to UV illumination to rapidly release IP3, thus mimicking the rapid responses of the receptor/PLC signaling system. Cells were also loaded with the Ca2+-sensitive dye Ca-Green-1 to visualize the ensuing Ca2+ dynamics (Fig. 5B). We uncaged ciIP3 repeatedly for 100 ms every 1.5 s, which resulted in an initial global sustained Ca2+ transient in both control and K20-treated cells (Fig. 5B). In a subset of control and K20-treated cells, this was followed by Ca2+ oscillations presumably triggered by the oscillatory IP3 uncaging (Fig. 5B). All aspects of IP3R function exhibited dramatic sensitization in cells with depolarization-induced enriched IP3R. As expected, maximal Ca2+ release measured in both cells that oscillate and those that respond with a monotonic Ca2+ rise was significantly increased (Fig. 5C). In addition, A7r5 cells exposed to K20 for 24 h to up-regulate IP3Rs were significantly more likely to oscillate compared with control cells; their oscillation frequency was enhanced dramatically as well as the duration of the oscillations (Fig. 5D).
The increased amplitude of Ca2+ release in response to IP3 uncaging is consistent with an increased number of IP3Rs (Fig. 5C). However, the pronounced oscillatory behavior of the K20-treated cells argues that IP3R sensitivity is also enhanced in cells with up-regulated IP3R expression. This predicts that K20-treated cells would release Ca2+ at lower IP3 threshold concentrations. To test whether this is the case, we evaluated the time to threshold, which was defined as the time required to produce a Ca2+ release signal significantly above the resting base line (empirically determined to F/F0 ≥ 1.04). The time to threshold is significantly shorter in K20-treated versus control cells, consistent with the conclusion that IP3R sensitivity is enhanced in A7r5 cells showing overabundant IP3R.
Therefore, up-regulation of the IP3R is functionally significant and sensitizes Ca2+ release. However, the mechanisms regulating this increased sensitivity require additional investigation. For example, IP3R up-regulation could be sufficient to mediate the increased sensitivity of IP3-dependent Ca2+ release. Alternatively, the increased mass of the IP3R could be coupled to post-translational modifications or protein-protein interactions that mediate the enhanced sensitivity of the receptor. Nonetheless, because Ca2+ release in our experiments was induced using caged IP3, the observed enhanced sensitivity of IP3-dependent Ca2+ release cannot be due to modulation of the signal transduction cascade upstream of IP3 production.
Consistent with our findings, an increased IP3 binding capacity has been reported in the aorta of SHR (51). In addition, in cerebral arteries, the IP3R was shown to activate Ca2+ influx through TRPC3, leading to depolarization and muscle contraction (40). This mechanism, however, is unlikely to contribute to PE-induced Ca2+ influx in A7r5 cells that do not express any TRPC3 channels, based on our Western blot analysis (supplemental Fig. 2) and as reported by others (52, 53).
Up-regulation of IP3R Enhances VSM Contractility
VSM contraction establishes vascular tone and peripheral resistance. Because IP3R up-regulation is associated with enhanced Ca2+ signaling in A7r5 cells, we wondered whether this finding correlates with increased contraction. Thus, we examined the contractile responsiveness of A7r5 cells to PE. A7r5 cells were of the elongated form, and nearly 100% of them contracted upon the addition of PE (20 μm). The extent of contraction was quantified using an imaging approach outlined under “Experimental Procedures,” which exploited the appearance of contractile fibers following PE addition. PE results in a transient contractile response in control cells (Fig. 5F), whereas A7r5 cells with up-regulated IP3R (KCl 20 mm) show a significantly more pronounced contractile response that is sustained for the duration of the recording (Fig. 5F). Consistently, the mean value of maximal contraction was increased 2 ± 0.02-fold in K20-treated cells when compared with control (Fig. 5G).
Collectively, our results show that increased expression of the IP3R in arterial VSM is consistently associated with hypertension. Depolarization-induced IP3R up-regulation in A7r5 VSM cells is associated with a higher resting Ca2+ level, enhanced response to agonist stimulation, and more robust and sustained contraction. Hence, IP3R up-regulation observed in VSM cells during hypertension is likely to be an important contributor to enhanced vascular reactivity. As such, the IP3R represents an attractive potential drug target for the treatment of hypertension.
An increased myogenic response and enhanced reactivity to vasoconstrictor agonists are characteristics of hypertension and contribute significantly to increased blood pressure (54). Multiple factors may act in concert to contribute to the enhanced vascular tone, including an overabundance of Ca2+-conducting channels, such as α1C and TRPC3, alterations in the Ca2+ sensitivity of the contractile apparatus in VSM (55), and structural remodeling of arteries (54). With the recognition that the VSM is the mediator of the myogenic response and that a rise in cytoplasmic Ca2+ is essential for smooth muscle contraction, it is significant, as shown in this study, that the IP3R up-regulates in VSM during hypertension, and this event is associated with enhanced IP3R-mediated Ca2+ signaling and VSM contraction. Enrichment of IP3R can be recapitulated in cultured VSM cells by depolarization-induced opening of LTCC and Ca2+-dependent activation of the calcineurin-NFAT pathway.
Acknowledgments
We are grateful to Aleksandra Pesic, Miodrag Pesic, and Shirley Haun for performing the Western blots in Fig. 2C.
This work was supported by a grant from the Qatar National Research Fund, National Priority Research Program Grant NPRP 08-392-3-087. The laboratory of K. M. is also supported by the Biomedical Research Program at Weill Cornell Medical College in Qatar, a program funded by Qatar Foundation.

This article contains supplemental Figs. 1 and 2.
- VSM
- vascular smooth muscle
- IP3
- inositol 1,4,5-trisphosphate
- IP3R
- IP3 receptor
- NFAT
- nuclear factor of activated T cells
- AHT
- angiotensin II-induced hypertension
- SAL
- saline
- ciIP3
- caged IP3
- ciIP3
- d-2,3-O-isopropylidene-6-O-(2-nitro-4,5-dimethoxy) benzyl-myo-inositol 1,4,5-triphosphate-hexakis(propionoxylmethyl) ester
- K20
- K40, and K60, 20, 40, and 60 mm KCl, respectively
- PLC
- phospholipase C
- LTCC
- L-type Ca2+ channel
- SHR
- spontaneously hypertensive rat(s)
- Ang II
- angiotensin II
- SBP
- systolic blood pressure
- MA
- mesenteric arteries
- PSS
- physiological salt solution
- PE
- phenylephrine
- HBS
- HEPES-buffered saline
- 2-APB
- 2-aminoethoxydiphenyl borate
- vWF
- von Willebrand factor
- SOCE
- store-operated Ca2+ entry
- PMCA
- plasma membrane Ca2+ pump
- Nif
- nifedipine
- Thaps
- thapsigargin
- CHX
- cycloheximide
- ACD
- actinomycin D
- CsA
- cyclosporine A.
REFERENCES
- 1. Chockalingam A. (2008) World Hypertension Day and global awareness. Can. J. Cardiol. 24, 441–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Oparil S., Zaman M. A., Calhoun D. A. (2003) Pathogenesis of hypertension. Ann. Intern. Med. 139, 761–776 [DOI] [PubMed] [Google Scholar]
- 3. Rizzoni D., Agabiti-Rosei E. (2012) Structural abnormalities of small resistance arteries in essential hypertension. Intern. Emerg. Med. 7, 205–212 [DOI] [PubMed] [Google Scholar]
- 4. Lemarié C. A., Tharaux P. L., Lehoux S. (2010) Extracellular matrix alterations in hypertensive vascular remodeling. J. Mol. Cell Cardiol. 48, 433–439 [DOI] [PubMed] [Google Scholar]
- 5. House S. J., Potier M., Bisaillon J., Singer H. A., Trebak M. (2008) The non-excitable smooth muscle. Calcium signaling and phenotypic switching during vascular disease. Pflugers Arch. 456, 769–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bosnjak Z. J. (1993) Ion channels in vascular smooth muscle. Physiology and pharmacology. Anesthesiology 79, 1392–1401 [DOI] [PubMed] [Google Scholar]
- 7. Hill M. A., Zou H., Potocnik S. J., Meininger G. A., Davis M. J. (2001) Arteriolar smooth muscle mechanotransduction. Ca2+ signaling pathways underlying myogenic reactivity. J. Appl. Physiol. 91, 973–983 [DOI] [PubMed] [Google Scholar]
- 8. Hirst G. D., Edwards F. R. (1989) Sympathetic neuroeffector transmission in arteries and arterioles. Physiol. Rev. 69, 546–604 [DOI] [PubMed] [Google Scholar]
- 9. Folkow B. (1982) Physiological aspects of primary hypertension. Physiol. Rev. 62, 347–504 [DOI] [PubMed] [Google Scholar]
- 10. Cox R. H., Rusch N. J. (2002) New expression profiles of voltage-gated ion channels in arteries exposed to high blood pressure. Microcirculation 9, 243–257 [DOI] [PubMed] [Google Scholar]
- 11. Ziegler M. G., Mills P., Dimsdale J. E. (1991) Hypertensives' pressor response to norepinephrine. Analysis by infusion rate and plasma levels. Am. J. Hypertens. 4, 586–591 [PubMed] [Google Scholar]
- 12. Pesic A., Madden J. A., Pesic M., Rusch N. J. (2004) High blood pressure upregulates arterial L-type Ca2+ channels. Is membrane depolarization the signal? Circ. Res. 94, e97–e104 [DOI] [PubMed] [Google Scholar]
- 13. Pratt P. F., Bonnet S., Ludwig L. M., Bonnet P., Rusch N. J. (2002) Upregulation of L-type Ca2+ channels in mesenteric and skeletal arteries of SHR. Hypertension 40, 214–219 [DOI] [PubMed] [Google Scholar]
- 14. Nelson M. T., Patlak J. B., Worley J. F., Standen N. B. (1990) Calcium channels, potassium channels, and voltage dependence of arterial smooth muscle tone. Am. J. Physiol. 259, C3–C18 [DOI] [PubMed] [Google Scholar]
- 15. Moosmang S., Schulla V., Welling A., Feil R., Feil S., Wegener J. W., Hofmann F., Klugbauer N. (2003) Dominant role of smooth muscle L-type calcium channel Cav1.2 for blood pressure regulation. EMBO J. 22, 6027–6034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Somlyo A. V., Bond M., Somlyo A. P., Scarpa A. (1985) Inositol trisphosphate-induced calcium release and contraction in vascular smooth muscle. Proc. Natl. Acad. Sci. U.S.A. 82, 5231–5235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hashimoto T., Hirata M., Itoh T., Kanmura Y., Kuriyama H. (1986) Inositol 1,4,5-trisphosphate activates pharmacomechanical coupling in smooth muscle of the rabbit mesenteric artery. J. Physiol. 370, 605–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Smith J. B. (1986) Angiotensin-receptor signaling in cultured vascular smooth muscle cells. Am. J. Physiol. 250, F759–F769 [DOI] [PubMed] [Google Scholar]
- 19. Perez J. F., Sanderson M. J. (2005) The contraction of smooth muscle cells of intrapulmonary arterioles is determined by the frequency of Ca2+ oscillations induced by 5-HT and KCl. J. Gen. Phys. 125, 555–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Miriel V. A., Mauban J. R., Blaustein M. P., Wier W. G. (1999) Local and cellular Ca2+ transients in smooth muscle of pressurized rat resistance arteries during myogenic and agonist stimulation. J. Physiol. 518, 815–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Matsumoto H., Baron C. B., Coburn R. F. (1995) Smooth muscle stretch-activated phospholipase C activity. Am. J. Physiol. 268, C458–C465 [DOI] [PubMed] [Google Scholar]
- 22. Narayanan J., Imig M., Roman R. J., Harder D. R. (1994) Pressurization of isolated renal arteries increases inositol trisphosphate and diacylglycerol. Am. J. Physiol. 266, H1840–H1845 [DOI] [PubMed] [Google Scholar]
- 23. Kulik T. J., Bialecki R. A., Colucci W. S., Rothman A., Glennon E. T., Underwood R. H. (1991) Stretch increases inositol trisphosphate and inositol tetrakisphosphate in cultured pulmonary vascular smooth muscle cells. Biochem. Biophys. Res. Commun. 180, 982–987 [DOI] [PubMed] [Google Scholar]
- 24. Davis M. J., Hill M. A. (1999) Signaling mechanisms underlying the vascular myogenic response. Physiol. Rev. 79, 387–423 [DOI] [PubMed] [Google Scholar]
- 25. Inscho E. W., Cook A. K., Mui V., Imig J. D. (1998) Calcium mobilization contributes to pressure-mediated afferent arteriolar vasoconstriction. Hypertension 31, 421–428 [DOI] [PubMed] [Google Scholar]
- 26. Osol G., Laher I., Kelley M. (1993) Myogenic tone is coupled to phospholipase C and G protein activation in small cerebral arteries. Am. J. Physiol. 265, H415–H420 [DOI] [PubMed] [Google Scholar]
- 27. Watanabe J., Karibe A., Horiguchi S., Keitoku M., Satoh S., Takishima T., Shirato K. (1993) Modification of myogenic intrinsic tone and [Ca2+]i of rat isolated arterioles by ryanodine and cyclopiazonic acid. Circ. Res. 73, 465–472 [DOI] [PubMed] [Google Scholar]
- 28. Kharade S. V., Sonkusare S. K., Srivastava A. K., Thakali K. M., Fletcher T. W., Rhee S. W., Rusch N. J. (2013) The β3 subunit contributes to vascular calcium channel upregulation and hypertension in angiotensin II-infused C57BL/6 mice. Hypertension 61, 137–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Monkawa T., Hayashi M., Miyawaki A., Sugiyama T., Yamamoto-Hino M., Hasegawa M., Furuichi T., Mikoshiba K., Saruta T. (1998) Localization of inositol 1,4,5-trisphosphate receptors in the rat kidney. Kidney Int. 53, 296–301 [DOI] [PubMed] [Google Scholar]
- 30. Grayson T. H., Haddock R. E., Murray T. P., Wojcikiewicz R. J., Hill C. E. (2004) Inositol 1,4,5-trisphosphate receptor subtypes are differentially distributed between smooth muscle and endothelial layers of rat arteries. Cell Calcium 36, 447–458 [DOI] [PubMed] [Google Scholar]
- 31. Wang Y., Chen J., Wang Y., Taylor C. W., Hirata Y., Hagiwara H., Mikoshiba K., Toyo-oka T., Omata M., Sakaki Y. (2001) Crucial role of type 1, but not type 3, inositol 1,4,5-trisphosphate (IP3) receptors in IP3-induced Ca2+ release, capacitative Ca2+ entry, and proliferation of A7r5 vascular smooth muscle cells. Circ. Res. 88, 202–209 [DOI] [PubMed] [Google Scholar]
- 32. Lee C. H., Poburko D., Sahota P., Sandhu J., Ruehlmann D. O., van Breemen C. (2001) The mechanism of phenylephrine-mediated [Ca2+]i oscillations underlying tonic contraction in the rabbit inferior vena cava. J. Physiol. 534, 641–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maruyama T., Kanaji T., Nakade S., Kanno T., Mikoshiba K. (1997) 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J. Biochem. 122, 498–505 [DOI] [PubMed] [Google Scholar]
- 34. Freichel M., Suh S. H., Pfeifer A., Schweig U., Trost C., Weissgerber P., Biel M., Philipp S., Freise D., Droogmans G., Hofmann F., Flockerzi V., Nilius B. (2001) Lack of an endothelial store-operated Ca2+ current impairs agonist-dependent vasorelaxation in TRP4−/− mice. Nat. Cell Biol. 3, 121–127 [DOI] [PubMed] [Google Scholar]
- 35. Bergdahl A., Gomez M. F., Dreja K., Xu S. Z., Adner M., Beech D. J., Broman J., Hellstrand P., Swärd K. (2003) Cholesterol depletion impairs vascular reactivity to endothelin-1 by reducing store-operated Ca2+ entry dependent on TRPC1. Circ. Res. 93, 839–847 [DOI] [PubMed] [Google Scholar]
- 36. Xu S. Z., Beech D. J. (2001) TrpC1 is a membrane-spanning subunit of store-operated Ca2+ channels in native vascular smooth muscle cells. Circ. Res. 88, 84–87 [DOI] [PubMed] [Google Scholar]
- 37. Takata Y., Hutchinson J. S. (1983) Exaggerated hypotensive responses to calcium antagonists in spontaneously hypertensive rats. Clin. Exp. Hypertens. A 5, 827–847 [DOI] [PubMed] [Google Scholar]
- 38. Bergdahl A., Gomez M. F., Wihlborg A. K., Erlinge D., Eyjolfson A., Xu S. Z., Beech D. J., Dreja K., Hellstrand P. (2005) Plasticity of TRPC expression in arterial smooth muscle. Correlation with store-operated Ca2+ entry. Am. J. Physiol. Cell Physiol. 288, C872–C880 [DOI] [PubMed] [Google Scholar]
- 39. DeHaven W. I., Smyth J. T., Boyles R. R., Bird G. S., Putney J. W., Jr. (2008) Complex actions of 2-aminoethyldiphenyl borate on store-operated calcium entry. J. Biol. Chem. 283, 19265–19273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xi Q., Adebiyi A., Zhao G., Chapman K. E., Waters C. M., Hassid A., Jaggar J. H. (2008) IP3 constricts cerebral arteries via IP3 receptor-mediated TRPC3 channel activation and independently of sarcoplasmic reticulum Ca2+ release. Circ. Res. 102, 1118–1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cahalan M. D. (2009) STIMulating store-operated Ca2+ entry. Nat. Cell Biol. 11, 669–677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stekiel W. J., Contney S. J., Lombard J. H. (1986) Small vessel membrane potential, sympathetic input, and electrogenic pump rate in SHR. Am. J. Physiol. 250, C547–C556 [DOI] [PubMed] [Google Scholar]
- 43. Stekiel W. J., Contney S. J., Lombard J. H. (1991) Sympathetic neural control of vascular muscle in reduced renal mass hypertension. Hypertension 17, 1185–1191 [DOI] [PubMed] [Google Scholar]
- 44. Rusch N. J., Stekiel W. J. (1991) Ionic channels of vascular smooth muscle in hypertension. in Cellular and Molecular Mechanisms of Hypertension (Cox R. H., ed) pp. 1–7, Plenum Press, New York: [DOI] [PubMed] [Google Scholar]
- 45. Stekiel W. J., Contney S. J., Rusch N. J. (1993) Altered β-receptor control of in situ membrane potential in hypertensive rats. Hypertension 21, 1005–1009 [DOI] [PubMed] [Google Scholar]
- 46. Potier M., Gonzalez J. C., Motiani R. K., Abdullaev I. F., Bisaillon J. M., Singer H. A., Trebak M. (2009) Evidence for STIM1- and Orai1-dependent store-operated calcium influx through ICRAC in vascular smooth muscle cells. Role in proliferation and migration. FASEB J. 23, 2425–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Thastrup O., Dawson A. P., Scharff O., Foder B., Cullen P. J., Drøbak B. K., Bjerrum P. J., Christensen S. B., Hanley M. R. (1989) Thapsigargin, a novel molecular probe for studying intracellular calcium release and storage. Agents Actions 27, 17–23 [DOI] [PubMed] [Google Scholar]
- 48. Parekh A. B., Putney J. W., Jr. (2005) Store-operated calcium channels. Physiol. Rev. 85, 757–810 [DOI] [PubMed] [Google Scholar]
- 49. Genazzani A. A., Carafoli E., Guerini D. (1999) Calcineurin controls inositol 1,4,5-trisphosphate type 1 receptor expression in neurons. Proc. Natl. Acad. Sci. U.S.A. 96, 5797–5801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rao A., Luo C., Hogan P. G. (1997) Transcription factors of the NFAT family. Regulation and function. Annu. Rev. Immunol. 15, 707–747 [DOI] [PubMed] [Google Scholar]
- 51. Bernier S., Guillemette G. (1993) Increased inositol 1,4,5-trisphosphate binding capacity in vascular smooth muscle of spontaneously hypertensive rats. Am. J. Hypertens. 6, 217–225 [PubMed] [Google Scholar]
- 52. Jung S., Strotmann R., Schultz G., Plant T. D. (2002) TRPC6 is a candidate channel involved in receptor-stimulated cation currents in A7r5 smooth muscle cells. Am. J. Physiol. Cell Physiol. 282, C347–C359 [DOI] [PubMed] [Google Scholar]
- 53. Soboloff J., Spassova M., Xu W., He L. P., Cuesta N., Gill D. L. (2005) Role of endogenous TRPC6 channels in Ca2+ signal generation in A7r5 smooth muscle cells. J. Biol. Chem. 280, 39786–39794 [DOI] [PubMed] [Google Scholar]
- 54. Baek E. B., Kim S. J. (2011) Mechanisms of myogenic response. Ca2+-dependent and -independent signaling. J. Smooth Muscle Res. 47, 55–65 [DOI] [PubMed] [Google Scholar]
- 55. Schubert R., Lidington D., Bolz S. S. (2008) The emerging role of Ca2+ sensitivity regulation in promoting myogenic vasoconstriction. Cardiovasc. Res. 77, 8–18 [DOI] [PubMed] [Google Scholar]