

Background: Proteinase-activated receptor-1 (PAR1) is a proteolytically activated G protein-coupled receptor. Neutrophil-derived enzymes might regulate PAR1 signaling.
Results: Neutrophil elastase and proteinase-3 cleave and activate PAR1 signaling that is distinct from thrombin-triggered responses. Neutrophil elastase and proteinase-3 signaling through PAR1 modulates endothelial cell signaling.
Conclusion: Neutrophil enzymes are Gαi-biased agonists for PAR1.
Significance: Biased PAR1-activating compounds may prove of value as therapeutic agents to treat cardiovascular and inflammatory diseases.
Keywords: Calcium Signaling, Endothelial Cell, G Protein-coupled Receptors (GPCR), Intracellular Trafficking, MAP Kinases (MAPKs), Neutrophil, Thrombin, Biased Signaling, Proteinase-activated Receptor
Abstract
Neutrophil proteinases released at sites of inflammation can affect tissue function by either activating or disarming signal transduction mediated by proteinase-activated receptors (PARs). Because PAR1 is expressed at sites where abundant neutrophil infiltration occurs, we hypothesized that neutrophil-derived enzymes might also regulate PAR1 signaling. We report here that both neutrophil elastase and proteinase-3 cleave the human PAR1 N terminus at sites distinct from the thrombin cleavage site. This cleavage results in a disarming of thrombin-activated calcium signaling through PAR1. However, the distinct non-canonical tethered ligands unmasked by neutrophil elastase and proteinase-3, as well as synthetic peptides with sequences derived from these novel exposed tethered ligands, selectively stimulated PAR1-mediated mitogen-activated protein kinase activation. This signaling was blocked by pertussis toxin, implicating a Gαi-triggered signal pathway. We conclude that neutrophil proteinases trigger biased PAR1 signaling and we describe a novel set of tethered ligands that are distinct from the classical tethered ligand revealed by thrombin. We further demonstrate the function of this biased signaling in regulating endothelial cell barrier integrity.
Introduction
Proteinase-activated receptors (PARs)3 are a family of G protein-coupled receptors that are activated by limited proteolysis of the receptor N terminus to unmask a receptor-activating motif that is referred to as the tethered ligand (TL) (1, 2). PAR1, the first of the four PARs to be cloned (3, 4), is activated by thrombin, which triggers the receptor by exposing the TL sequence, S42FLLRN—” in the human receptor (4). Moreover, synthetic peptides based on that thrombin-revealed TL sequence (e.g. SFLLRN-NH2 and the more PAR1-selective peptide, TFLLR-NH2) were found to activate PAR1 in the absence of proteolysis (4, 5). Similarly, the cloning of PARs 2 and 4 showed that the N-terminal sequences revealed by serine proteinase cleavage at a specific arginine target site recognized by trypsin in PAR2 (6–8) or by thrombin in PAR4 (9, 10) can activate these receptors. More recently, it has become apparent that PARs 1 and 2 can be proteolytically activated by N-terminal cleavage at sites distinct from the canonical arginine/serine site targeted by thrombin (PAR1) or trypsin (PAR2). For instance, matrix metalloproteinase-1 (MMP-1) can cleave upstream of the thrombin cleavage site in PAR1 to unmask the novel TL sequence, “PRSFLLR—,“ which can stimulate PAR1-mediated cell invasion and platelet aggregation (11, 12). Similarly, activation of PAR1 by activated protein-C (APC) reveals a novel tethered ligand that is different from the one unmasked by thrombin and that stimulates a distinct cell response (13, 14). Our own work has now shown that neutrophil elastase (NE) can activate PAR2 to generate signaling that differs from the canonical trypsin-triggered response (15). The activation of distinct signaling profiles through the same G protein-coupled receptor (GPCR) in a ligand-dependent manner is termed agonist-biased signaling and has now been described for numerous GPCRs (16–19).
In the setting of acute inflammation, neutrophil influx represents one of the first indices of tissue damage (20). The subsequent degranulation and release of neutrophil proteinases has a dramatic impact on tissue function in part by regulating PAR activity via either activation or disarming. Cathepsin-G can disarm thrombin activation of PAR1 by cleaving downstream of the thrombin cleavage site (21) and proteinase-3 (PR3) can inhibit APC activation of PAR1 through inactivating the co-receptor, endothelial protein C receptor (22).
In this study we have investigated whether neutrophil-derived enzyme processing of PAR1 at non-canonical cleavage sites can, in addition to silencing the Gαq-coupled calcium signaling, activate other distinct signaling pathways. We have also investigated the effect of this non-canonical cleavage on PAR1 trafficking. Finally, we have developed novel biased PAR1 ligands based on the neutrophil enzyme-derived tethered ligands and examined their role in modulating endothelial cell function. Our data indicate that both NE and PR3 can activate MAPK signaling but not calcium signaling through PAR1 and can thus act as biased receptor-activating proteinases. Furthermore, by mapping the neutrophil enzyme cleavage site on PAR1, we have identified two novel ligands for PAR1 that are biased toward Gαi-coupled MAPK signaling downstream of PAR1. In endothelial cells, these novel PAR1 activating peptides trigger increased actin stress fiber formation and NE-derived peptide (NE-TL-AP) can reverse thrombin-stimulated increases in cell monolayer permeability.
MATERIALS AND METHODS
Chemicals and Other Reagents
Thrombin from human plasma (catalogue number 605195; 2800 NIH units/mg) was from EMD Biosciences (San Diego, CA). A concentration of 1 unit/ml was calculated to be 10 nm thrombin. All agonists peptides used in this study (Table 1) were synthesized by the Peptide Synthesis Facility, University of Calgary. Peptide purity was verified by HPLC (>95%), mass spectrometry, and amino acid analysis. The neutrophil enzymes, NE and PR3, purified from human sputum, were obtained from Elastin Products (Owensville, MO) with specific activities of 875 and 3.1 IU/mg, respectively, and verified to be free of trypsin contamination (15). These specific activities were used to calculate the molar concentration of each enzyme (PR3, 1 unit/ml is 300 nm and NE, 10 units/ml is 300 nm).
TABLE 1.
Synthetic peptides studied as PAR1 activating peptides and sequences of synthetic peptides used as substrates for identifying fragments generated by neutrophil enzyme proteolysis
| Peptide name | Peptide sequence |
|---|---|
| PAR1-AP | TFLLR-NH2 |
| NE-TL-AP | RNPNDKYEPF-NH2 |
| PR3-TL-AP | TLDPRSF-NH2 |
| MMP1-TL-AP | PRSFLLRN-NH2 |
| APC-TL-AP | NPNDKYEPF-NH2 |
| PAR129–41 | PESKATNATLDPR |
| PAR135–53 | NATLDPRSFLLRNPNDKYE |
| PAR146–65 | RNPNDKYEPFWEDEEKNESG |
| PAR164–83 | SGLTEYRLVSINKSSPLQKQ |
| PAR182–102 | KQLPAFISEDASGYLTSSWLT |
Mouse monoclonal anti phospho-p42/44 MAPK antibody, rabbit monoclonal anti-p42/44 MAPK antibody, and the secondary HRP-linked anti-mouse IgG and anti-rabbit IgG antibodies were from Cell Signaling Technology (Danvers, MA). Hybond-P PVDF membrane and ECL advanced detection kit were purchased from GE Healthcare Canada. Pertussis toxin (PTX) was from Tocris Bioscience (Ellisville, MO). FuGENE-6 transfection reagent was obtained from Roche Applied Science. All other chemicals were purchased from Sigma unless otherwise specified.
Cell Culture
All cell media and serum were purchased from Invitrogen Corp. KNRK, HEK293, and MCF7 cell lines were routinely grown in DMEM supplemented with 1 mm sodium pyruvate, 10% fetal bovine serum, and 2.5 μg/ml of Plasmocine (InvivoGen, San Diego, CA) on Nunclon δ Surface Cell culture flasks at 37 °C in a 5% CO2 humidified incubator. The cells were routinely subcultured by dissociating with enzyme-free buffer (isotonic phosphate-buffered saline, pH 7.4, containing 1 mm EDTA) and plated in multiwell plates (Nunc) or glass bottom Petri dishes (MatTek) prior to experiments.
Prior to proceeding with measurements of receptor dynamics at the cell surface and monitoring PAR1-triggered MAPK and calcium signaling, it was necessary to optimize the serum starvation conditions for receptor expression and signaling measurements. Substantial trial-and-error development was required to maximize the cell surface expression of transfected PAR1, to minimize the background MAPK signal, which was variable from one set of cultures to another, and to maximize the detection of PAR1-stimulated calcium signaling and MAPK activation. KNRK cells have a low endogenous expression of PAR1 and PAR2 and were therefore used to study PAR1 signaling. Signaling was deemed to be PAR1 dependent if it was detected in PAR1-transfected cells but not in empty vector-transfected cells. KNRK is an immortalized rat kidney epithelial cell line with an activated viral k-ras oncgene that exhibits constitutively activated MAPK signaling. Thus, to analyze the ability of PAR1 to activate MAPK, it was necessary to reduce the background of MAPK activity using forskolin, a stimulator of MAPK phosphatase (23). The background level of phospho-MAPK was significantly reduced by pretreatment of the KNRK-PAR1-eYFP cells with forskolin (1 μm) for 60 min prior to the addition of a PAR1 agonist. We also suspect that endogenously produced Kirsten virus-transformed rat kidney cell (KNRK)-secreted extracellular proteinases can process PAR1 resulting in a loss of cell surface receptor expression and a desensitization of signaling. Thus, inclusion of 5 μm of the cysteine proteinase inhibitor E64 substantially enhanced cell surface receptor expression and responsiveness to PAR1 agonists. Finally, for experiments utilizing thrombin activation, 5 units/ml of heparin was found to enhance thrombin action, most likely through preventing the localization of the serine proteinase inhibitor, proteinase nexin-1, to the cell surface (24) (data not shown). Thus, prior to the signaling and trafficking experiments, cells were routinely placed in serum-free DMEM supplemented with 1 μm forskolin, 5 units/ml of heparin, and/or 5 μm E64 for the specified times as detailed in the relevant sections.
Cloning and Transfection
The plasmid encoding the human PAR1 receptor (wt-hPAR1) cloned in pCDNA3.1 was obtained from Missouri S&T cDNA Resource Center (Rolla, MO). A short N-terminal tetra-cysteine tag (CCPGCC) (25) encoding motif was introduced at Ala29 of hPAR1 and a C-terminal eYFP (enhanced YFP) tag was fused in-frame with the PAR1 sequence by mutating the PAR1 stop codon to tyrosine and insertion of the eYFP fragment with flanking XhoI and XbaI restriction enzyme sites at the C terminus of the hPAR1 coding sequence. To generate the dually tagged mCherry-PAR1-eYFP clone, restriction enzyme sites for BspEI and BamHI were created at Ser34 of hPAR1. Then, the mCherry PCR fragment with flanking BspEI and BamHI restriction enzyme sites was inserted in-frame in the PAR1-eYFP cDNA. The resulting clone encoded an N-terminal mCherry tag with a glycine serine linker for the BamHI and BspEI restriction cleavage sites followed by the sequence for human PAR1 and eYFP. Plasmid DNA mutations were created using the QuikChange Lightning Multi Site-directed Mutagenesis Kit (Agilent Technologies, Mississauga, ON) to generate all mutants including the mCherry-PAR1L44E,L45E-eYFP construct. All constructs were confirmed by direct sequencing (University of Calgary DNA sequencing facility).
KNRK cells were transfected with the human PAR1-eYFP-expressing construct (KNRK-PAR1-eYFP), dually tagged mCherry-eYFP PAR1 construct (KNRK-mCherry-PAR1-eYFP), or empty vector (KNRK-pcDNA3) with FuGENE 6 (Roche) in 24-well multiplates. Transfected cells were subcultured under G418 selection, sorted by flow cytometry, and cell stocks were maintained in liquid nitrogen for future experiments. Extensive passage of the cells resulted in a loss of mCherry-PAR1-eYFP constructs from the cells. Thus, cells used for experiments were replaced from frozen stock every 15 passages. Comparable eYFP or mCherry-PAR1-eYFP constructs were similarly expressed in an HEK293 or MCF-7 cell background for comparison with the receptor expressing KNRK cells.
Calcium Mobilization Assay
Calcium signaling experiments were performed essentially as described before (15). Cells from a T75 flask were detached in enzyme-free cell dissociation buffer, re-suspended in 18 ml of serum-containing growth medium, and 100 μl/well of the cell suspension was plated in black-walled cell culture-treated clear bottom 96-well plates (Corning) and cultured overnight. The next day, adherent cells were washed and switched to serum-free DMEM containing 5 units/ml of heparin and 5 μm E64 (a cysteine proteinase inhibitor) (Sigma) overnight. Culture medium was then replaced with isotonic Hanks' buffered salt solution containing 1.5 mm CaCl2, 1.5 mm MgSO4, 15 mm HEPES, pH 7.0, and Fluo-4-AM no wash calcium indicator dye (Invitrogen). Intracellular fluorescence (excitation 480 nm; emission recorded at 530 nm) was monitored for 2 min after the injection of agonists (enzymes or PAR-activating peptides) into each well, using a Victor X4 plate reader (PerkinElmer Life Sciences). The increase in fluorescence emission monitored at 530 nm was used as an index of increases in intracellular calcium as described in detail previously (15). Where appropriate, responses were normalized to the calcium signal generated by 2 μm A23178, a calcium ionophore.
Detection of MAPK Activation
MAPK activation was monitored by Western blotting essentially as described (15). PAR1 expressing cells were plated in 24-well multiwell plates, cultured overnight, and then washed and placed in serum-free DMEM containing 5 units/ml of heparin for 18 h. The cells were then placed in serum-free DMEM containing 1 μm forskolin and 5 μm E64 for 2 h. Cells were then stimulated with agonists, rapidly rinsed with ice-cold isotonic phosphate-buffered saline, and placed on ice. Total protein was extracted by adding 50 μl of ice-cold lysis buffer (20 mm Tris-HCl, pH 7.5, 100 mm NaCl, 10 mm MgCl2, 1 mm EDTA, 1 mm EGTA, 0.5% Nonidet P-40, 2.5 mm sodium pyrophosphate, 1 mm β-glycerophosphate, 1 mm Na3VO4, 25 mm NaF, 1 μg/ml of leupeptin, 1 μg/ml of aprotinin, 1 mm PMSF, and 1 mm DTT) to each well. The cell lysate was transferred to 1.5-ml microcentrifuge tubes and cleared by centrifugation (15,000 × g for 10 min). The protein samples were heat-denatured at 92 °C for 3 min in denaturing Laemmli buffer and resolved on 8–20% gradient SDS-PAGE gels (Invitrogen TG gel). The resolved proteins were transferred to PVDF membrane, blocked in PBST buffer (PBS with 0.1% (v/v) Tween 20) supplemented with 1% ECL Advance Blocking Agent (GE Healthcare) and 0.02% sodium azide for 1 h at room temperature. p42/44 MAPK phosphorylation was detected with phospho-p42/44 (p-p42/44) specific antibodies diluted 1/5,000 in PBST with 1% ECL Advance Blocking Agent and 0.02% sodium azide overnight at 4 °C. p-p42/44 immunoreactivity was detected using the HRP-conjugated anti-mouse secondary antibody (1/20,000 in PBST, 1% ECL Advance Blocking Agent for 1 h). After washing the membrane with PBST, the peroxidase activity was detected by chemiluminescence reagent ECL Advance (GE Healthcare) and KODAK Image Station 4000MM. Blots were then blocked in PBST with 1% ECL Advance Blocking Agent and 0.02% sodium azide for 1 h at room temperature before incubation with Total-p42/44 (t-p42/44) antibody (1/5,000 in PBST with 1% ECL Advance Blocking Agent and 0.02% sodium azide) overnight at 4 °C. t-p42/44 immunoreactivity was detected using the horseradish peroxidase-conjugated anti-rabbit secondary antibody (1/20,000 in PBST, 1% ECL Advance Blocking Agent for 1 h). After washing the membrane with PBST, the peroxidase activity was detected using a chemiluminescence reagent (ECL-Advance) with Kodak Image Station 4000MM. Band intensities were quantified using the ImageJ quantification software. p-p42/44 MAP kinase levels were normalized for differences in protein loading by expressing the data as a percentage of the corresponding t-p42/44 MAPK signal.
Neutrophil Elastase and Proteinase-3 Cleavage of Synthetic PAR1-derived Peptides
A set of five overlapping synthetic peptides representing the complete N-terminal extracellular sequence of human PAR1 was synthesized (Table 1) and evaluated for hydrolysis by NE and PR3 as described previously (15, 26, 27). Overlapping peptides spanning the entire PAR1 N terminus (100 μm in a total volume of 150 μl) were incubated with NE or PR3 (200 nm) for timed intervals for up to 30 min at 37 °C. Reactions were stopped by adding 150 μl of ice-cold 0.1% trifluoroacetic acid (TFA) in water. Samples were fractionated by reverse-phase high-performance liquid chromatography (5–40% acetonitrile gradient in 0.1% TFA over 30 min at a flow rate of 1 ml/min), and eluted peptides were analyzed by MALDI mass spectrometry to deduce the cleavage products.
PAR1 Intracellular Trafficking Analysis
Dually tagged PAR1 was transiently transfected in HEK cells (HEK-mCherry-PAR1-eYFP or mCherry-PAR1L44E,L45E-eYFP), subcultured into 35-mm glass bottom culture dishes (MatTek), and placed in growth media containing 5 units/ml of heparin and 5 μm E64 for 4 to 16 h, and then stimulated with agonists for specified times. The cells were fixed with 10% buffered formalin and receptor dynamics were monitored with a Olympus FV1000 confocal microscope system on an Olympus IX70 microscope using the fluoview system software. Optimal transfection for visualizing receptor expression was observed in HEK cells and comparable experiments were also performed in KNRK-mCherry-PAR1-eYFP cells.
Endothelial Cell F-actin Structure and Permeability Analysis
Human umbilical vein endothelial cells (HUVECs), basal media (Medium 200), and growth factor mixture (Large Vessel Endothelial supplement) were obtained from Invitrogen. HUVECs were expanded 4-fold at each passage and cells stocks at passage three were then stored in liquid nitrogen for subsequent use in experiments up to passage 8. 150 μl of growth media containing 2 × 104 HUVECs was seeded in the upper chamber of a 0.4-μm 24-well Transwell support (Corning, NY). 750 μl of growth media was added to the lower chamber. After 3 days, culture inserts were transferred to the wells containing 750 μl of fresh growth media and cultured for another 2 days. After 5 days in culture, confluent monolayers were treated with PAR1 agonists from the apical side of the cell layer by adding 50 μl of supplement-free media containing 4-fold concentrated test agonists and 8 mg/ml of FITC-dextran (Sigma, 71.2 kDa). The FITC-dextran flux from the upper insert chamber to the lower well was measured at 10 and 30 min after agonist(s) treatment. The change in permeability was expressed as a percentage increase or decrease in FITC-dextran flux to the lower chamber caused by agonists normalized (% control), relative to the average fluorescence reading obtained from lower wells for untreated monolayers not exposed to any agonists. The F-actin stress fiber formation in HUVEC cells was monitored in cells cultured on collagen-coated glass bottom Petri dishes (MatTek, Ashland, MA) and treated with the specified agonists prior to fixation and labeling with Alexa 647-conjugated phalloidin (Invitrogen).
Statistical Analysis
Statistical analysis of data and curve fitting were done with Prism 5 software (GraphPad Software, San Diego, CA). Statistical significance was assessed using the Student's t test or analysis of variance with Tukey's post-test.
RESULTS
Neutrophil Enzymes Disarm Thrombin-triggered PAR1-dependent Calcium Signaling but Stimulate MAPK Activation
To study PAR1 function and receptor dynamics, we used a KNRK expression system, as we have done previously for evaluating PAR2 signaling (15). KNRK-PAR1-eYFP cells exhibit robust calcium signaling triggered by either thrombin or the PAR1-activating peptide, TFLLR-NH2 (TF) (Fig. 1A), whereas pcDNA-transfected KNRK cells did not shown any detectable calcium signaling (not shown). NE and PR3 did not elicit a calcium signal in the KNRK-PAR1-eYFP cells. However, pretreatment of the cells with either enzyme diminished the cellular response to a subsequent thrombin-triggered activation of calcium signaling (Fig. 1, A–C). The neutrophil proteinases disarmed thrombin-triggered calcium signaling through PAR1 in a concentration-dependent manner (Fig. 1, B and C). At a concentration of 300 nm, NE diminished thrombin-mediated PAR1 calcium signaling by over 80% (Fig. 1B), with a 40% reduction seen in 600 nm PR3-treated cells (Fig. 1C).
FIGURE 1.
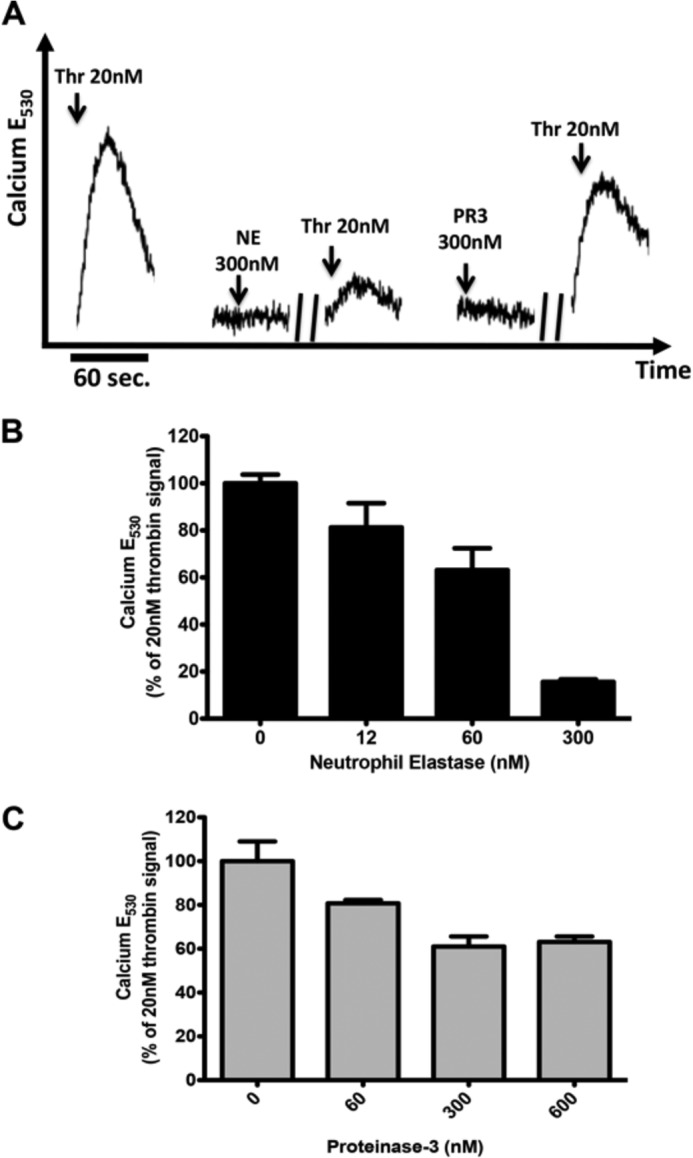
Neutrophil enzymes disarm PAR1 to attenuate thrombin-stimulated calcium signaling. A, representative traces showing calcium signaling in KNRK-PAR1-eYFP cells in response to TFLLR-NH2 (TF), thrombin (Thr), NE, and PR3. // indicates 20 min. B, histogram quantifying thrombin-dependent calcium signaling in KNRK-PAR1-eYFP cells following NE treatment for 20 min. C, histogram quantifying thrombin-dependent calcium signaling in KNRK-PAR1-eYFP cells following PR3 treatment for 20 min. Data are expressed as mean ± S.D. as a percentage of the signal triggered by 20 nm thrombin.
We next monitored the ability of thrombin, TFLLR-NH2, NE, and PR3 to trigger PAR1-dependent MAPK signaling. Both thrombin and TFLLR-NH2 robustly activated the p44/42 MAPK pathway in KNRK-PAR1-eYFP cells but not in empty vector-transfected pcDNA3-KNRK cells (Fig. 2A). Of note, both NE and PR3 also activated p42/44 MAPK signaling in PAR1-eYFP-KNRK cells but not in the KNRK-pcDNA3 cells (Fig. 2, B and C). The level of MAPK activation by neutrophil enzymes at 60 nm was comparable with the levels triggered by 20 nm thrombin or 10 μm TFLLR-NH2. The time course for MAPK activation by NE (Fig. 3C) and PR3 (Fig. 3D) was comparable with that stimulated by either thrombin (Fig. 3A) or TFLLR-NH2 (Fig. 3B), with maximal activation seen 5 min after the addition of agonists and a gradually declining signal, which returned toward baseline levels at 60 min (Fig. 3E). Both neutrophil enzymes also caused similar MAPK activation in PAR1-eYFP-transfected MCF-7 cells (Fig. 2, D and E). Although the MCF-7 cells do not endogenously express PAR1, they do express low levels of PAR2, which may account for the MAPK activation observed in the NE-treated pCDNA3-MCF-7-transfected cells (Fig. 2D).
FIGURE 2.

Neutrophil enzymes activate MAPK signaling in KNRK-PAR1-eYFP cells. A, activation of p42/44 MAP kinase by thrombin (Thr) and TFLLR-NH2 (TF) in KNRK-PAR1-eYFP cells but not in KNRK-pCDNA3 cells. B, concentration-dependent activation of p42/44 MAP kinase by NE in KNRK-PAR1-eYFP cells but not in KNRK-pCDNA3 cells. C, concentration-dependent activation of p42/44 MAP kinase by PR3 in KNRK-PAR1-eYFP cells but not in KNRK-pCDNA3 cells. D, concentration-dependent activation of p42/44 MAP kinase by NE in MCF7-PAR1-eYFP cells. E, concentration-dependent activation of p42/44 MAP kinase by PR3 in MCF7-PAR1-eYFP cells but not in MCF7-pCDNA3 cells. All MAPK signaling data are shown following 10 min of agonist treatment. Blots are representative of data from at least 3 experiments performed in independently cultured cells.
FIGURE 3.
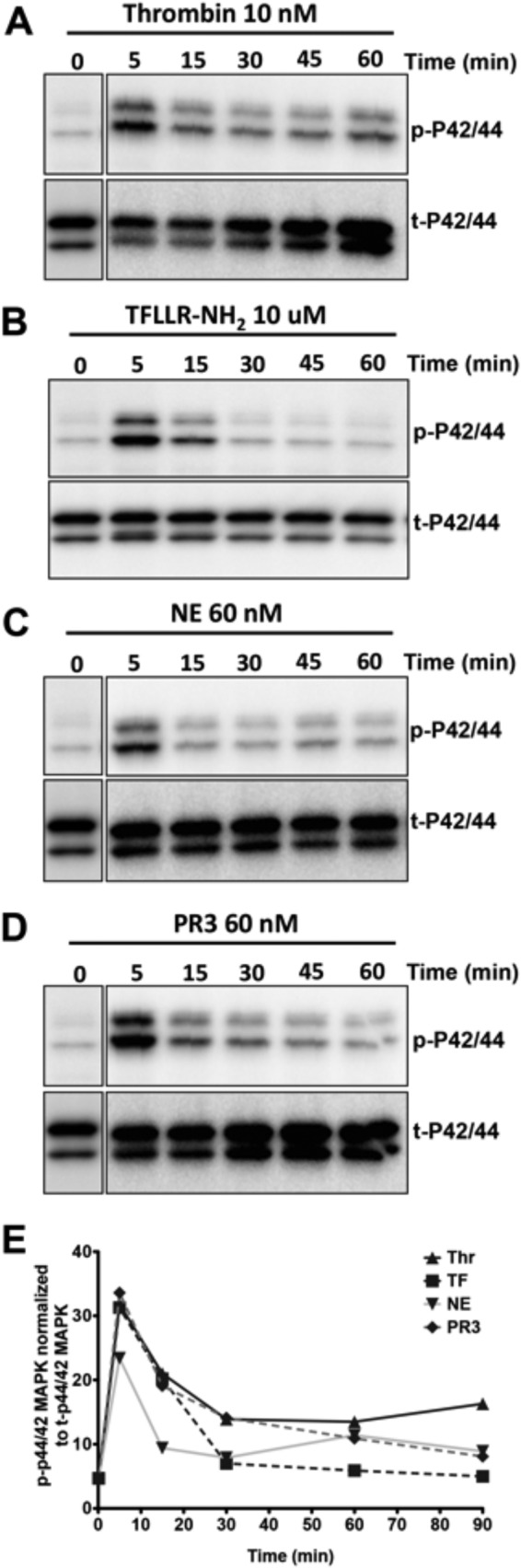
Time course of neutrophil enzyme-activated MAPK signaling in KNRK-PAR1-eYFP cells. Activation of p42/44 MAP kinse by: A, thrombin (Thr); B, TFLLR-NH2 (TF); C, NE; and D, PR3 in KNRK-PAR1-eYFP cells. E, graph showing densitometry analysis of normalized p42/44 MAP kinase activation in KNRK-PAR1-eYFP cells over 90 min. Data are expressed as mean ± S.E., n = 3.
Neutrophil Enzymes Cleave PAR1 but Do Not Cause Receptor Internalization
PAR1 activation by thrombin or TFLLR-NH2 leads to rapid internalization of the receptor and targeting to intracellular vesicles. To verify simultaneously that the neutrophil enzymes were cleaving the PAR1 N terminus and were affecting the trafficking of the receptor following such cleavage, we generated a dually labeled PAR1 construct tagged with mCherry (red pseudocolor) at the N terminus and eYFP (green pseudocolor) at the C terminus (Fig. 4A). Thus, the intact dually tagged receptor shows a co-localization of mCherry and eYFP (yellow pseudocolor) when visualized by confocal microscopy (Fig. 4B). The cleavage and release of the N-terminal mCherry tag due to proteolysis would result in a C-terminal eYFP-tagged receptor (green pseudocolor).
FIGURE 4.
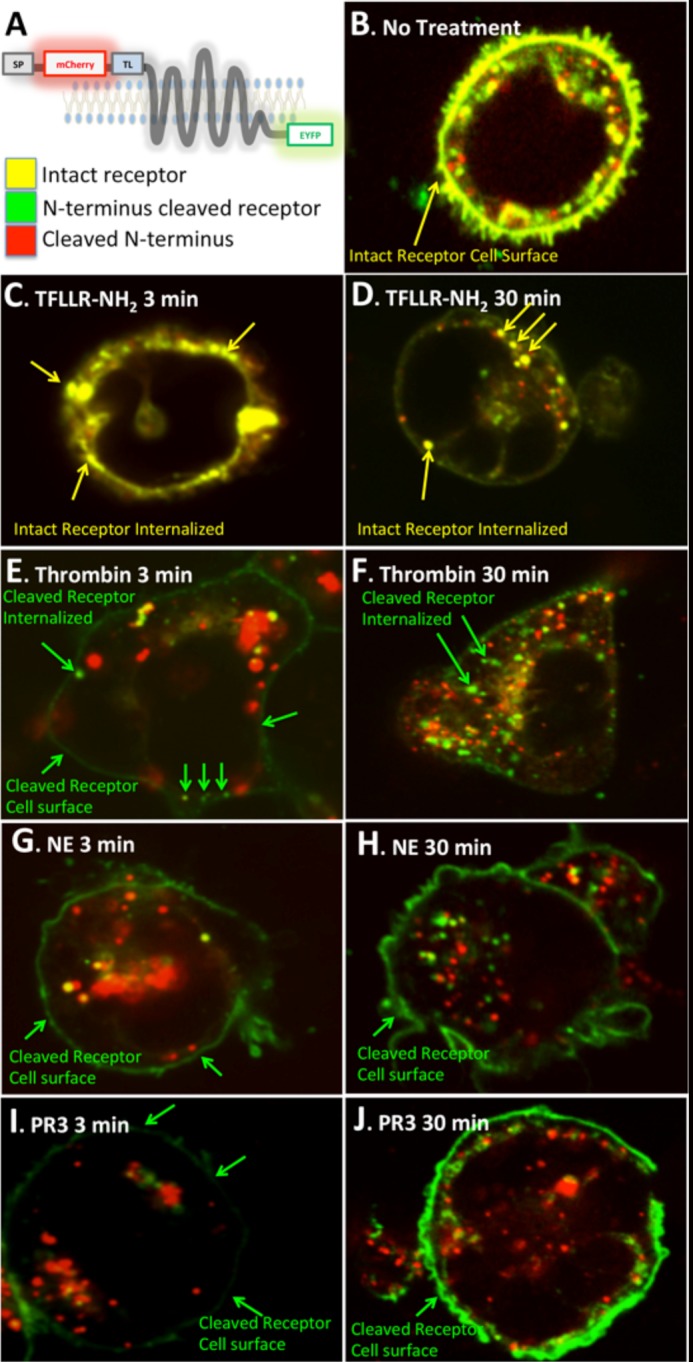
Confocal imaging of PAR1 dynamics following activation with TFLLR-NH2, thrombin, NE, and PR3. A, a schematic depicting the structure of the mCherry-hPAR1-eYFP construct and the expected pseudo-color image when the receptor is intact or proteolytically cleaved. SP is signal peptide; TL is thrombin-revealed tethered ligand. B, untreated HEK-mCherry-PAR1-eYFP cell showing intact hPAR1 on the cell surface. C, HEK-mCherry-PAR1-eYFP cells treated with TFLLR-NH2 for 3 min showing the intact receptor clustering on the cell membrane. D, HEK-mCherry-PAR1-eYFP cells treated with TFLLR-NH2 for 30 min showing the intact receptor internalized. E, HEK-mCherry-PAR1-eYFP cells treated with thrombin for 3 min showing the N terminus-cleaved receptor on the cell surface. F, HEK-mCherry-PAR1-eYFP cells treated with thrombin for 30 min showing the N terminus-cleaved receptor internalized. G, HEK-mCherry-PAR1-eYFP cells treated with NE for 3 min showing the N terminus-cleaved receptor on the cell surface. H, HEK-mCherry-PAR1-eYFP cells treated with NE for 30 min showing the N terminus-cleaved receptor on the cell surface. I, HEK-mCherry-PAR1-eYFP cells treated with PR3 for 3 min showing the N terminus-cleaved receptor on the cell surface. J, HEK-mCherry-PAR1-eYFP cells treated with PR3 for 30 min showing the N terminus-cleaved receptor on the cell surface. Internalized red structures are presumed to represent the proteolytically released-internalized mCherry-tagged receptor N-terminal fragment.
Treatment of HEK-mCherry-PAR1-eYFP cells with TFLLR-NH2, which activates the receptor without proteolytic cleavage, triggered a rapid clustering at the cell membrane within 3 min (Fig. 4C) and a subsequent internalization of intact receptor (yellow pseudocolor) (Fig. 4D). Activation with thrombin resulted in the cleavage of the N-terminal tag as early as 3 min after agonist addition (Fig. 4E) and cleaved (green pseudocolor) internalized receptors were seen at 30 min (Fig. 4F). Similarly, with NE and PR3 treatments, PAR1 N-terminal cleavage was evident at 3 min after addition of the proteinases (Fig. 4, G and I). However, the cleaved receptors were retained on the cell surface even after 30 min (Fig. 4, H and J). Similar results were observed in KNRK-mCherry-PAR1-eYFP cells (not shown). Activation with thrombin, NE, and PR3 also resulted in the detection of an internalized mCherry signal that likely represents the N-terminal receptor fragment (Fig. 4, E, G, and I) (28).
Thus, over a 5–30-min time frame where both neutrophil elastase and proteinase-3 stimulated MAPK activation in the cells, there was no receptor internalization of PAR1, in contrast with thrombin activation, which triggered substantial PAR1 internalization. We conclude that although both thrombin-cleaved and neutrophil elastase/proteinase-3-cleaved PAR1 can release an N-terminal sequence in parallel with MAPK activation, the receptor internalization dynamics accompanying the cellular response are different for thrombin and the neutrophil enzymes.
Identification of Neutrophil Elastase (NE-TL) and Proteinase-3 (PR3-TL)-tethered Ligand Sequences
Because we established that both NE and PR3 could cleave and release an N-terminal motif from human PAR1, we sought to identify the cleavage sites through HPLC and mass spectral peptide mapping. We monitored the cleavage of a set of 5 overlapping peptides (PAR129–41, PAR135–53, PAR146–65, PAR164–83, and PAR182–102) representing the entire N-terminal extracellular domain of PAR1 (Table 1). Predominant NE and PR3 cleavage targets were identified in the peptides corresponding to PAR135–53 and PAR129–41, respectively. None of the other peptides representing the PAR1 extracellular N-terminal were hydrolyzed under the cleavage conditions described under “Materials and Methods” (data not shown). Isolation by HPLC and mass spectral identification of the hydrolysis products revealed the major NE cleavage site of PAR135–53 to be at Leu45/Arg46 (Fig. 5A). PR3 cleavage of PAR129–41 occurred at Ala36/Thr37 (Fig. 5B) with these proteolysis products being generated rapidly and detectable within 5 min of adding the substrate peptides to the enzymes (data not shown).
FIGURE 5.

Mapping neutrophil proteinase cleavage of PAR1 N terminus. Five overlapping synthetic peptides covering the entire first extracellular domain of human PAR1 (PAR129–41, PESKATNATLDPR; PAR135–53, NATLDPRSFLLRNPNDKYE; PAR146–65, RNPNDKYEPFWEDEEKNESG; PAR164–83, SGLTEYRLVSINKSSPLQKQ; and PAR182–102, KQLPAFISEDASGYLTSSWLT) (Table 1) were synthesized and subject to proteolysis by NE and PR3. A, HPLC trace showing peaks corresponding to the major cleavage products for PAR135–53 subject to NE proteolysis. Peptide identities corresponding to each peak were obtained by mass spectrometry. B, HPLC trace showing peaks corresponding to the major cleavage products for PAR129–41 subject to PR3 proteolysis. Peptide identities corresponding to each peak were obtained by mass spectrometry. C, schematic depicting the PAR1 N-terminal region showing the major identified cleavage sites for NE and PR3 as well as known cleavage sites for MMP-1, thrombin, and APC. No significant cleavage of PAR135–53 was seen with PR3 and NE failed to cleave PAR129–41. Neither proteinase cleaved PAR146–65, PAR164–83, nor PAR182–102.
Thus, PAR1 cleavage by NE and PR3 would generate the “non-canonical tethered ligands” R46NPNDKY53E— (NE-TL) and T37LDP41RSFLLRN— (PR3-TL), respectively (Table 1). In addition, cleavage of the PAR135–53 peptide by neutrophil elastase would result in the removal of the thrombin cleavage-activation site and thus disarm the receptor to thrombin activation, whereas the minor cleavage of the N-terminal PAR1 sequence by PR3 that was also detected (Phe43/Leu44 and Leu44/Leu45, not shown) would result in a partial disarming of thrombin activation of PAR1. The cleavage of PAR1 by NE and PR3 is thus localized principally to an area of the PAR1 N terminus that is now known to be a target for a number of proteinases including MMP-1 (12) and APC (13, 14) (Fig. 5C).
To confirm Leu45/Arg46 as the neutrophil elastase cleavage site, we also performed site-directed mutagenesis on the mCherry-PAR1-eYFP clone to generate mCherry-PAR1L44E,L45E-eYFP, where the leucine residue in the P1 and P2 positions of the putative elastase cleavage site was mutated to glutamic acid, a change that is predicted to be unfavorable for NE cleavage at that site (MEROPS database). Confocal microscopy revealed that mCherry-PAR1L44E,L45E-eYFP is expressed on the cell surface (Fig. 6A) but can no longer be cleaved by NE to release the N-terminal mCherry tag (Fig. 6, C and D). Of note, thrombin cleavage and removal of the mCherry tag in mCherry-PAR1L44E,L45E-eYFP is still observed, even after a short exposure to the enzyme (Fig. 6B).
FIGURE 6.
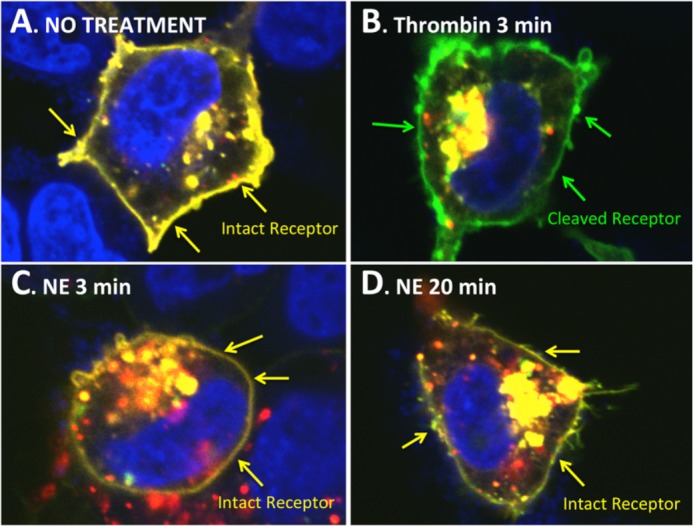
Confocal imaging of PAR1 cleavage by thrombin and NE in mCherry-PAR1L44E,L45E-eYFP transfected cells. A, untreated HEK-mCherry-PAR1L44E,L45E-eYFP cell showing intact receptor on the cell surface (yellow pseudocolour from overlay of N-terminal mCherry (red) and C-terminal eYFP (green) tags. B, thrombin-treated (3 min) HEK-mCherry-PAR1L44E,L45E-eYFP cell showing removal of the N-terminal mCherry tag (image showing green pseudocolor from C-terminal eYFP tag). C, 3-min NE-treated HEK-mCherry-PAR1L44E,L45E-eYFP cell showing intact receptor on the cell surface (yellow pseudocolour from overlay of N-terminal mCherry (red) and C-terminal eYFP (green) tags. D, 20-min NE-treated HEK-mCherry-PAR1L44E,L45E-eYFP cell showing intact receptor on the cell surface (yellow pseudocolour from overlay of N-terminal mCherry (red) and C-terminal eYFP (green) tags.
The Neutrophil Enzyme-revealed TL-derived Peptides, NE-TL-AP and PR3-TL-AP Are Biased Agonists of PAR1
The peptide cleavage data predicted the exposure of two distinct tethered ligand sequences by neutrophil elastase (RNPNDKYE … ) and proteinase-3 (TLDPRSFL … ). We thus synthesized receptor-activating peptides representing these non-canonical PAR1-tethered ligands (NE-TL-AP, RNPNDKYEPF-NH2 for a NE-tethered ligand-derived activating peptide, and PR3-TL-AP, TLDPRSF-NH2 for a PR3-tethered ligand-derived activating peptide) (Table 1). We evaluated the ability of these novel agonist peptides to trigger the activation of PAR1 calcium and MAPK signaling. Neither NE-TL-AP nor PR3-TL-AP caused calcium-signaling responses in KNRK-PAR1-eYFP cells. This result is in contrast to the response seen with the MMP-1 revealed tethered ligand-derived peptide (MMP1-TL-AP) PRSFLLRN-NH2 (50–200 μm) (12), which stimulated calcium signaling (not shown). Both NE-TL-AP and PR3-TL-AP stimulated MAPK signaling in KNRK-PAR1-eYFP cells (Fig. 7, A and B) but not in KNRK-pcDNA3 cells. NE-TL-AP and PR3-TL-AP are thus novel MAPK-activating biased ligands for PAR1. The activation of MAPK by both of these peptides was also observed in PAR1-eYFP-transfected MCF7 cells (data not shown) as well as in HEK293 cells that endogenously express PAR1 (Fig. 7, C and D). In HEK cells, the PAR1 antagonist SCH79797 blocked MAPK activation by NE-TL-AP but did not block PR3-TL-AP activation of MAPK at higher concentrations (Fig. 7D).
FIGURE 7.
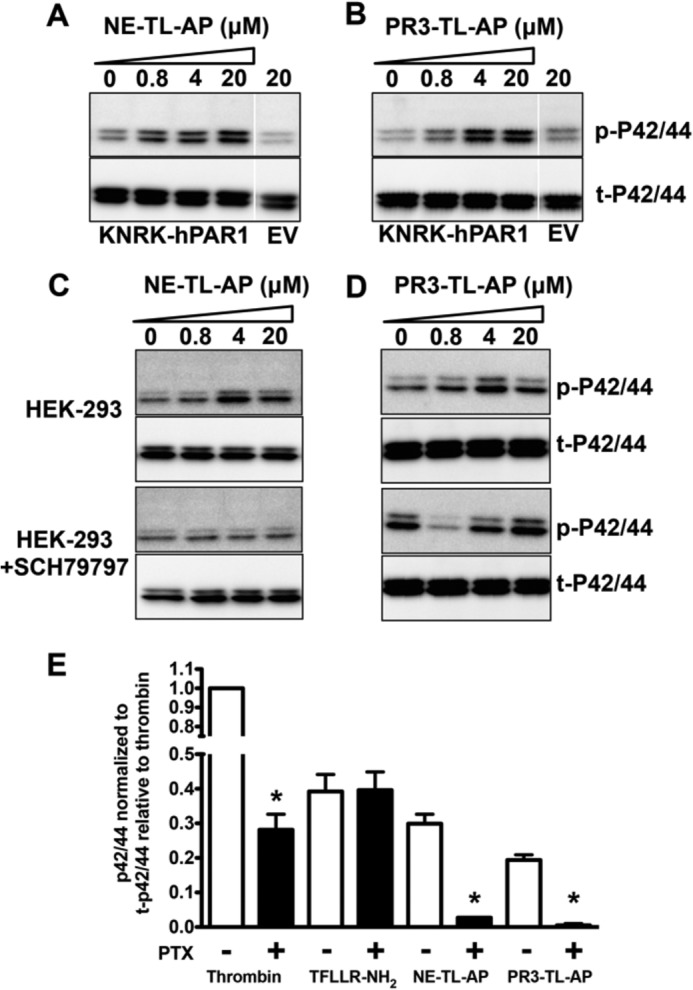
Peptides derived from neutrophil enzyme-revealed tethered ligands activate PAR1-dependent MAP kinase signaling in a Gαi-dependent manner. A, neutrophil elastase-tethered ligand (NE-TL-AP, RNPNDKYEPF-NH2), and B, proteinase-3-tethered ligand (PR3-TL-AP, TLDPRSF-NH2) peptides activate KNRK-PAR1-eYFP cell MAPK in a concentration-dependent manner. MAPK activation is not seen in empty vector (EV)-transfected KNRK-pCDNA3 cells treated with 20 μm NE-TL-AP or PR3-TL-AP. C, MAPK activation by NE-TL-AP, and D, PR3-TL-AP in endogenous PAR1-expressing HEK-293 cells, but not in HEK-293 cells treated with the PAR1 antagonist SCH79797 (2 μm). E, histogram quantifying the PTX inhibition of MAPK signaling caused by thrombin, NE-TL-AP, and PR3-TL-AP in KNRK-PAR1-eYFP cells. Data are expressed as mean ± S.E. * indicates significant difference (p < 0.05, n = 3) from non-PTX-treated cell MAPK signal.
We also examined the possibility that NE-TL-AP and PR3-TL-AP could antagonize PAR1 activation by TFLLR-NH2 and thrombin. However, the calcium-signaling response triggered by TFLLR-NH2 and thrombin were identical in cells that had been pre-treated or not with NE-TL-AP (100 μm) and PR3-TL-AP (100 μm) (data not shown), suggesting that the binding determinants for these new TL-derived peptides are different from those for the conventional tethered ligand sequence.
The lack of calcium signaling activation by the neutrophil enzymes (NE or PR3) and their corresponding tethered ligand-derived peptides (NE-TL-AP or PR3-TL-AP) indicated that these agonists do not drive PAR1 coupling to Gαq. However, PAR1 signaling by thrombin is known to involve coupling to other G proteins as well. Thus, we hypothesized that the biased PAR1 signaling caused by the neutrophil enzymes might be due to a selective interaction with Gαi. To evaluate this possibility, we tested the ability of pertussis toxin, known to block Gαi signaling, to inhibit MAPK signaling by NE, PR3, NE-TL-AP, and PR3-TL-AP. Treatment of cells with pertussis toxin partially blocked MAPK activation by thrombin, did not inhibit TFLLR-NH2-triggered MAPK activation and completely abrogated MAPK activation by NE, PR3, NE-TL-AP, and PR3-TL-AP (Fig. 7E).
NE-TL-AP and PR3-TL-AP Induce Actin Stress Fiber Formation in Endothelial Cells and Modulate Endothelial Barrier Permeability
PAR1 activation on endothelial cells is known to modulate endothelial cell stress fiber formation and barrier permeability. We therefore investigated the ability of the biased PAR1 agonist peptides NE-TL-AP and PR3-TL-AP to modulate these parameters in HUVECs. Treatment of HUVEC cultures with NE-TL-AP (Fig. 8C) or PR3-TL-AP (Fig. 8D) induced a rapid rearrangement and appearance of actin stress fibers, as did thrombin (Fig. 8B). In endothelial cells cultured on transwell permeable supports, we detect an increase in transcellular flux of FITC-dextran when cells are treated with thrombin (Fig. 8, E–H). The increased endothelial monolayer permeability was also seen in cells treated with PR3-TL-AP (Fig. 7F), but not with NE-TL-AP (Fig. 8E). Significantly, NE-TL-AP treatment was able to diminish the thrombin-stimulated increase in endothelial barrier permeability (Fig. 8E) as was a peptide corresponding to the tethered ligand derived from APC activation of PAR1 (APC-TL-AP) (Table 1) (Fig. 8G). In contrast, the peptide derived from the MMP-1-revealed TL sequence (MMP-TL-AP) (Table 1) increased FITC-dextran flux through endothelial cell monolayers (Fig. 8H) and like PR3-TL-AP (Fig. 8F) enhanced the endothelial cell barrier disruption by thrombin.
FIGURE 8.

Induction of F-actin stress fiber formation and modulation of permeability in HUVECs by NE-TL-AP and PR3-TL-AP. Confocal microscopy image of phalloidin-labeled F-actin (red) and DAPI-labeled nucleus (blue) in HUVECs treated with no agonist (A), thrombin (5 nm) (B), NE-TL-AP (20 μm) (C), and PR3-TL-AP (4 μm) (D). Integrity of the endothelial barrier was monitored by tracking the flux of FITC-dextran across HUVEC monolayers cultured on transwell supports following treatment with thrombin (5 nm) in combination with NE-TL-AP (20 μm) (E), PR3-TL-AP (4 μm) (F), APC-TL-AP (100 μm) (G), and MMP-TL-AP (20 μm) (H) as indicated. Permeability (% control) was normalized to the fluorescent reading obtained in the lower chamber for cells that had not been treated with agonists (expressed as 100%). Data are expressed as mean ± S.E. * indicates significant difference (p < 0.05) compared with agonist-untreated cells and # indicates significant difference (p < 0.05) compared with thrombin-treated cells (n = 6 for E and F; n = 3 for G and H).
DISCUSSION
We report here the ability of neutrophil-derived proteinases, NE and PR3 to trigger signaling through PAR1 in a manner that is distinct from thrombin activation of this receptor. NE and PR3 unmask two different non-canonical TL sequences, NE-TL and PR3-TL, that lead to the activation of Gαi-coupled p42/44 MAPK signaling downstream of PAR1 activation, but not Gαq-coupled calcium signaling. Thus, in an inflammatory setting, enzymes released by infiltrating neutrophils would modulate PAR1 signaling through silencing PAR1 calcium signaling and activating MAPK pathways. Furthermore, synthetic peptides derived from the predicted NE-TL and PR3-TL sequences (designated NE-TL-AP and PR3-TL-AP) were also able to act as biased agonists for PAR1 to stimulate MAPK but not calcium signaling. In endothelial cells both NE-TL-AP and PR3-TL-AP induced formation of actin stress fibers. NE-TL-AP did not induce an increase in endothelial cell barrier permeability, whereas PR3-AP-TL treatment did. Significantly, NE-TL-AP suppressed the thrombin-stimulated increase in endothelial barrier permeability mimicking the action of the APC-generated non-canonical PAR1-activating peptide. In contrast, PR3-TL-AP treatment potentiated the thrombin-dependent transendothelial flux of FITC-dextran. The mechanisms that result in these distinct actions of the PAR1-activating peptides and their relationship to biased PAR1 signaling by the neutrophil proteinase remains to be fully elucidated.
Based on our current work and previous studies it is now evident that PAR1 activation by proteolytic cleavage is a process with much greater complexity than first envisioned for the action of thrombin, which selectively unmasks a unique activating sequence in PAR1 (R41/S42FLLRNPN—) (4). Previous studies have identified the ability of MMP-1 to process the PAR1 N terminus to reveal the TL D39/P40RSFLLRN— (11, 12), whereas APC reveals yet another TL (R46/N47PNDKYE—) (13, 14). In cardiac cells MMP-13 is reported to cleave PAR1 to reveal the TL S42/F43LLRNPND— (29). Our data described here identify two more unique PAR1 activating TL sequences produced by NE (L45/R46NPNDKYE—) and PR3 (A36/T37LDPRSFLLR—) cleavage. The NE cleavage site was further verified by site-directed mutagenesis studies. Importantly, each of these sequences appear to be capable of driving a distinct subset of PAR1 signals presumably by inducing unique conformational changes in the receptor. Thus thrombin is known to trigger PAR1 coupling to Gαq, Gαi, Gα12/13, and β-arrestin (2, 30–32). MMP-1 and MMP-13 appear to trigger Gαq-dependent MAPK and RhoA activation (12, 29). APC activation of PAR1 is believed to be a G-protein independent process that is dependent on β-arrestin recruitment (33). In this study we have found that NE and PR3 trigger MAPK in a Gαi-dependent manner. The concept of agonist-biased signaling through GPCRs is now widely recognized and accepted to be relevant to our understanding of GPCR function and therapeutic targeting (16, 17). In the case of proteolytically activated receptors such as PAR1, activation by multiple proteinases, and the triggering of distinct signaling pathway subsets presents a challenge as well as a novel targeting strategy for drug development.
PAR1 activation by stromal cell-derived MMP-1 promotes invasion and growth of breast cancer cells (11) as well as invasion and metastasis of melanoma (34). The increased invasion and growth of cancer cells by MMP-1 activation of PAR1 occurs through up-regulation of pro-angiogenic genes (35), and activation of the Akt survival pathway in cancer cells (36). It is interesting to note that thrombin and MMP-1 up-regulate distinct but complementary sets of pro-angiogenic genes through activating PAR1 (35). The MMP-1 PAR1 signaling axis is also shown to have an important role in regulating platelet and endothelial barrier function. Platelet MMP-1 could promote aggregation through cleaving and activating platelet PAR1 in a manner that favors triggering of Rho-GTP pathways (12). PAR1 activation by MMP-1 is also implicated in the adverse outcomes in sepsis. MMP-1 activation of PAR1 causes endothelial cell contraction in a Rho-dependent manner, leading to an increase in vascular permeabiltity in wild-type but not in PAR1 knock-out mice (37).
In contrast with MMP-1, APC triggers Rac-1 signaling through PAR1, resulting in a tightening of the endothelial cell barrier (13, 14, 38, 39). APC activation of PAR1 requires the expression of endothelial cell protein C receptor as a co-receptor that presents protein C to the thrombin-thrombomodulin complex and enhances APC generation (38). Based on the inability of APC to induce endothelial barrier protection in PTX-treated endothelial cells it has been proposed that APC-PAR1 signaling is Gαi dependent (40). A subsequent study has, however, pointed out that PTX treatment on its own induces endothelial barrier dysfunction and showed: 1) that ERK activation due to APC stimulation of PAR1 could not be blocked by PTX treatment, and 2) that APC signaling through PAR1 was dependent on β-arrestin and disheveled-2 (33). Significantly, the APC-mediated activation of PAR1 is also dependent on caveolin-1, suggesting that compartmentalization of PAR1 in the caveolar microdomain is necessary for cytoprotective APC-PAR1 signaling (41, 42).
The ability of the new elastase/proteinase-3-unmasked tethered ligand sequences to signal has been verified by monitoring the PAR1-targeted activities of synthetic peptides derived from the revealed sequences. What cannot be determined is if the novel enzyme-exposed TL sequences tethered to the intact receptor activate the same set of downstream signaling, as do the synthetic peptides. In the case of MMP-1 activation of PAR1 on platelets, both the enzyme and the tethered ligand-derived peptide appear to signal in an identical manner (12). However, we have previously found significant differences in signaling by a PAR2-tethered ligand sequence functioning either as a part of the intact receptor or as a synthetic peptide (43, 44). Further work will be required to clarify this issue.
Thus, it now appears that for both PARs 1 and 2, the original tethered ligand mechanism, suggesting that a selective serine proteinase cleavage unmasks a single receptor-activating sequence, must now be modified to take into account the biased signaling pathways that can be triggered by cleavage at multiple different ”tethered ligand-generating“ sites, each with distinct N-terminal sequences. Whether unique cleavages of the N-terminal sequences of PARs 3 and 4 apart from those sites hydrolyzed by thrombin and trypsin (PAR4) can confer biased signaling remains to be determined.
The impact of this kind of biased PAR signaling by neutrophil or other proteinases on the overall inflammatory response is not easy to predict. Thus, a target like the human platelet, which expresses PAR1 but not PAR2, would be subject only to PAR1-biased signaling caused by neutrophil elastase, which can in principle activate both PARs 1 and 2. This neutrophil-activated platelet PAR1 response, which remains to be characterized, would presumably differ from the effects triggered by either thrombin or activated protein C. In contrast, both PARs 1 and 2, which are present on vascular endothelial cells and intestinal epithelial cells, can be targeted either by coagulation proteinases or neutrophil enzymes, which could in principle trigger distinct biased signals in different pathological settings. Based on our current and previous data (15) we predict that very different biased PAR1-PAR2 combined cell responses will be generated, depending on which proteinases are released at the sites of inflammation.
In contrast with NE signaling via PAR2 that involves Gα12/13 (15), biased PAR1 signaling stimulated by this enzyme is mediated via Gαi. Yet, when activated by neutrophil elastase, PAR1, like PAR2 (15), remains at the cell surface (Fig. 4H), but signals via a distinct G-protein interaction to trigger MAPK. This observation is also in keeping with the lack of internalization observed following PAR1 activation by APC (45). Very possibly the different modes of biased MAPK activation by PARs 1 and 2 involve additional as yet unidentified receptor interacting proteins that regulate the different signaling and cellular responses. This issue merits further study. Furthermore, it will be of importance to define the domains in the two PARs responsible for their differential interactions with Gαi (PAR1) and Gα12/13 (PAR2). These motifs may prove of value for the design of PAR-selective signal-biased receptor modulating compounds.
The biased signaling by PAR1 via distinct neutrophil-unmasked tethered ligands also raises questions about the impact of previously developed PAR1 antagonists (see Ref. 46 for a discussion). These antagonists were likely discovered by calcium signaling-based screening paradigms that depend on blocking the activation of PAR1 calcium signaling when stimulated either by thrombin (47), or a PAR1-activating peptide representing the thrombin-exposed tethered ligand sequence (48, 49). Because the PAR1 antagonists developed to date are presumed to block the docking of the thrombin-revealed tethered ligand, it is an open question as to whether those compounds will also block the biased actions of the distinct tethered ligands triggered by various proteinases. It can be noted that in HEK cells the PAR1 antagonist SCH79797 can inhibit the MAPK activation triggered by NE, PR3 (data not shown), and NE-TL-AP but not by PR3-TL-AP (Fig. 7, C and D). Similarly, the MMP-1-TL-AP-induced activation of platelets can be blocked by the PAR1 antagonist RWJ-56110 (12).
In summary, we describe novel signal-biased PAR1-activating peptide sequences that are unmasked by the actions of neutrophil elastase and proteinase-3. Although these proteinases partially disarm PAR1 for thrombin-dependent calcium signaling, they themselves selectively activate MAPK. These newly discovered PAR1-tethered ligand sequences that cause biased receptor signaling may thus play a unique role in the setting of inflammation. Indeed, the NE-derived TL peptide acts on endothelial cells to counter the thrombin-stimulated increase in barrier permeability. Biased PAR1-activating peptides may therefore prove of value as novel therapeutic agents to treat cardiovascular and inflammatory diseases.
Acknowledgments
The microscopy work was supported by the Snyder Institute Live Cell Imaging Facility funded by an equipment and infrastructure grant from the Canadian Foundation Innovation and the Alberta Science and Research Authority.
This work was supported in part by grants from the Canadian Institutes of Health Research (CIHR) and the Heart and Stroke Foundation of Alberta, Northwest Territories, and Nunavut (to M. D. H.).
- PAR
- proteinase-activated receptor
- TL
- tethered ligand
- APC
- activated protein-C
- MMP-1
- matrix metalloproteinase-1
- NE
- neutrophil elastase
- GPCR
- G protein-coupled receptor
- PR3
- proteinase-3
- PTX
- pertussis toxin
- KNRK
- Kirsten virus-transformed rat kidney cell
- HUVEC
- human umbilical vein endothelial cell
- eYFP
- enhanced yellow fluorescent protein.
REFERENCES
- 1. Ramachandran R., Hollenberg M. D. (2008) Proteinases and signalling. Pathophysiological and therapeutic implications via PARs and more. Br. J. Pharmacol. 153, S263–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Adams M. N., Ramachandran R., Yau M. K., Suen J. Y., Fairlie D. P., Hollenberg M. D., Hooper J. D. (2011) Structure, function and pathophysiology of protease activated receptors. Pharmacol. Ther. 130, 248–282 [DOI] [PubMed] [Google Scholar]
- 3. Rasmussen U. B., Vouret-Craviari V., Jallat S., Schlesinger Y., Pagès G., Pavirani A., Lecocq J. P., Pouysségur J., Van Obberghen-Schilling E. (1991) cDNA cloning and expression of a hamster α-thrombin receptor coupled to Ca2+ mobilization. FEBS Lett. 288, 123–128 [DOI] [PubMed] [Google Scholar]
- 4. Vu T. K., Hung D. T., Wheaton V. I., Coughlin S. R. (1991) Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 64, 1057–1068 [DOI] [PubMed] [Google Scholar]
- 5. Vu T. K., Wheaton V. I., Hung D. T., Charo I., Coughlin S. R. (1991) Domains specifying thrombin-receptor interaction. Nature 353, 674–677 [DOI] [PubMed] [Google Scholar]
- 6. Nystedt S., Emilsson K., Larsson A. K., Strömbeck B., Sundelin J. (1995) Molecular cloning and functional expression of the gene encoding the human proteinase-activated receptor 2. Eur. J. Biochem. 232, 84–89 [DOI] [PubMed] [Google Scholar]
- 7. Nystedt S., Emilsson K., Wahlestedt C., Sundelin J. (1994) Molecular cloning of a potential proteinase activated receptor. Proc. Natl. Acad. Sci. U.S.A. 91, 9208–9212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nystedt S., Larsson A. K., Aberg H., Sundelin J. (1995) The mouse proteinase-activated receptor-2 cDNA and gene. Molecular cloning and functional expression. J. Biol. Chem. 270, 5950–5955 [DOI] [PubMed] [Google Scholar]
- 9. Kahn M. L., Zheng Y. W., Huang W., Bigornia V., Zeng D., Moff S., Farese R. V., Jr., Tam C., Coughlin S. R. (1998) A dual thrombin receptor system for platelet activation. Nature 394, 690–694 [DOI] [PubMed] [Google Scholar]
- 10. Xu W. F., Andersen H., Whitmore T. E., Presnell S. R., Yee D. P., Ching A., Gilbert T., Davie E. W., Foster D. C. (1998) Cloning and characterization of human protease-activated receptor 4. Proc. Natl. Acad. Sci. U.S.A. 95, 6642–6646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boire A., Covic L., Agarwal A., Jacques S., Sherifi S., Kuliopulos A. (2005) PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell 120, 303–313 [DOI] [PubMed] [Google Scholar]
- 12. Trivedi V., Boire A., Tchernychev B., Kaneider N. C., Leger A. J., O'Callaghan K., Covic L., Kuliopulos A. (2009) Platelet matrix metalloprotease-1 mediates thrombogenesis by activating PAR1 at a cryptic ligand site. Cell 137, 332–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schuepbach R. A., Madon J., Ender M., Galli P., Riewald M. (2012) Protease-activated receptor-1 cleaved at R46 mediates cytoprotective effects. J. Thromb. Haemost. 10, 1675–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mosnier L. O., Sinha R. K., Burnier L., Bouwens E. A., Griffin J. H. (2012) Biased agonism of protease-activated receptor 1 by activated protein C caused by noncanonical cleavage at Arg46. Blood 120, 5237–5246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ramachandran R., Mihara K., Chung H., Renaux B., Lau C. S., Muruve D. A., DeFea K. A., Bouvier M., Hollenberg M. D. (2011) Neutrophil elastase acts as a biased agonist for proteinase-activated receptor-2 (PAR2). J. Biol. Chem. 286, 24638–24648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kenakin T. (2013) New concepts in pharmacological efficacy at 7TM receptors. IUPHAR review 2. Br. J. Pharmacol. 168, 554–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kenakin T., Christopoulos A. (2013) Signalling bias in new drug discovery. Detection, quantification and therapeutic impact. Nat. Rev. Drug. Discov. 12, 205–216 [DOI] [PubMed] [Google Scholar]
- 18. Kenakin T. (2011) Functional selectivity and biased receptor signaling. J. Pharmacol. Exp. Ther. 336, 296–302 [DOI] [PubMed] [Google Scholar]
- 19. Rajagopal S., Rajagopal K., Lefkowitz R. J. (2010) Teaching old receptors new tricks. Biasing seven-transmembrane receptors. Nat. Rev. Drug. Discov. 9, 373–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pham C. T. (2006) Neutrophil serine proteases. Specific regulators of inflammation. Nat. Rev. Immunol. 6, 541–550 [DOI] [PubMed] [Google Scholar]
- 21. Molino M., Blanchard N., Belmonte E., Tarver A. P., Abrams C., Hoxie J. A., Cerletti C., Brass L. F. (1995) Proteolysis of the human platelet and endothelial cell thrombin receptor by neutrophil-derived cathepsin G. J. Biol. Chem. 270, 11168–11175 [DOI] [PubMed] [Google Scholar]
- 22. Villegas-Mendez A., Montes R., Ambrose L. R., Warrens A. N., Laffan M., Lane D. A. (2007) Proteolysis of the endothelial cell protein C receptor by neutrophil proteinase 3. J. Thromb. Haemost. 5, 980–988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Burgun C., Esteve L., Humblot N., Aunis D., Zwiller J. (2000) Cyclic AMP-elevating agents induce the expression of MAP kinase phosphatase-1 in PC12 cells. FEBS Lett. 484, 189–193 [DOI] [PubMed] [Google Scholar]
- 24. Leroy-Viard K., Jandrot-Perrus M., Tobelem G., Guillin M. C. (1989) Covalent binding of human thrombin to a human endothelial cell-associated protein. Exp. Cell Res. 181, 1–10 [DOI] [PubMed] [Google Scholar]
- 25. Martin B. R., Giepmans B. N., Adams S. R., Tsien R. Y. (2005) Mammalian cell-based optimization of the biarsenical-binding tetracysteine motif for improved fluorescence and affinity. Nat. Biotechnol 23, 1308–1314 [DOI] [PubMed] [Google Scholar]
- 26. Knecht W., Cottrell G. S., Amadesi S., Mohlin J., Skåregärde A., Gedda K., Peterson A., Chapman K., Hollenberg M. D., Vergnolle N., Bunnett N. W. (2007) Trypsin IV or mesotrypsin and p23 cleave protease-activated receptors 1 and 2 to induce inflammation and hyperalgesia. J. Biol. Chem. 282, 26089–26100 [DOI] [PubMed] [Google Scholar]
- 27. Oikonomopoulou K., Hansen K. K., Saifeddine M., Tea I., Blaber M., Blaber S. I., Scarisbrick I., Andrade-Gordon P., Cottrell G. S., Bunnett N. W., Diamandis E. P., Hollenberg M. D. (2006) Proteinase-activated receptors, targets for kallikrein signaling. J. Biol. Chem. 281, 32095–32112 [DOI] [PubMed] [Google Scholar]
- 28. Zania P., Gourni D., Aplin A. C., Nicosia R. F., Flordellis C. S., Maragoudakis M. E., Tsopanoglou N. E. (2009) Parstatin, the cleaved peptide on proteinase-activated receptor 1 activation, is a potent inhibitor of angiogenesis. J. Pharmacol. Exp. Ther. 328, 378–389 [DOI] [PubMed] [Google Scholar]
- 29. Jaffré F., Friedman A. E., Hu Z., Mackman N., Blaxall B. C. (2012) β-Adrenergic receptor stimulation transactivates protease-activated receptor 1 via matrix metalloproteinase 13 in cardiac cells. Circulation 125, 2993–3003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Verrall S., Ishii M., Chen M., Wang L., Tram T., Coughlin S. R. (1997) The thrombin receptor second cytoplasmic loop confers coupling to like Gq-G proteins in chimeric receptors. Additional evidence for a common transmembrane signaling and G protein coupling mechanism in G protein-coupled receptors. J. Biol. Chem. 272, 6898–6902 [DOI] [PubMed] [Google Scholar]
- 31. McLaughlin J. N., Shen L., Holinstat M., Brooks J. D., Dibenedetto E., Hamm H. E. (2005) Functional selectivity of G protein signaling by agonist peptides and thrombin for the protease-activated receptor-1. J. Biol. Chem. 280, 25048–25059 [DOI] [PubMed] [Google Scholar]
- 32. Ayoub M. A., Trinquet E., Pfleger K. D., Pin J. P. (2010) Differential association modes of the thrombin receptor PAR1 with Gαi1, Gα12, and β-arrestin 1. FASEB J. 24, 3522–3535 [DOI] [PubMed] [Google Scholar]
- 33. Soh U. J., Trejo J. (2011) Activated protein C promotes protease-activated receptor-1 cytoprotective signaling through β-arrestin and dishevelled-2 scaffolds. Proc. Natl. Acad. Sci. U.S.A. 108, E1372–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Blackburn J. S., Liu I., Coon C. I., Brinckerhoff C. E. (2009) A matrix metalloproteinase-1/protease activated receptor-1 signaling axis promotes melanoma invasion and metastasis. Oncogene 28, 4237–4248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Blackburn J. S., Brinckerhoff C. E. (2008) Matrix metalloproteinase-1 and thrombin differentially activate gene expression in endothelial cells via PAR-1 and promote angiogenesis. Am. J. Pathol. 173, 1736–1746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yang E., Boire A., Agarwal A., Nguyen N., O'Callaghan K., Tu P., Kuliopulos A., Covic L. (2009) Blockade of PAR1 signaling with cell-penetrating pepducins inhibits Akt survival pathways in breast cancer cells and suppresses tumor survival and metastasis. Cancer Res. 69, 6223–6231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tressel S. L., Kaneider N. C., Kasuda S., Foley C., Koukos G., Austin K., Agarwal A., Covic L., Opal S. M., Kuliopulos A. (2011) A matrix metalloprotease-PAR1 system regulates vascular integrity, systemic inflammation and death in sepsis. EMBO Mol. Med. 3, 370–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Riewald M., Petrovan R. J., Donner A., Mueller B. M., Ruf W. (2002) Activation of endothelial cell protease activated receptor 1 by the protein C pathway. Science 296, 1880–1882 [DOI] [PubMed] [Google Scholar]
- 39. Schuepbach R. A., Feistritzer C., Fernández J. A., Griffin J. H., Riewald M. (2009) Protection of vascular barrier integrity by activated protein C in murine models depends on protease-activated receptor-1. Thromb. Haemost. 101, 724–733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bae J. S., Yang L., Manithody C., Rezaie A. R. (2007) The ligand occupancy of endothelial protein C receptor switches the protease-activated receptor 1-dependent signaling specificity of thrombin from a permeability-enhancing to a barrier-protective response in endothelial cells. Blood 110, 3909–3916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Russo A., Soh U. J., Paing M. M., Arora P., Trejo J. (2009) Caveolae are required for protease-selective signaling by protease-activated receptor-1. Proc. Natl. Acad. Sci. U.S.A. 106, 6393–6397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bae J. S., Yang L., Rezaie A. R. (2008) Lipid raft localization regulates the cleavage specificity of protease activated receptor 1 in endothelial cells. J. Thromb. Haemost. 6, 954–961 [DOI] [PubMed] [Google Scholar]
- 43. Al-Ani B., Wijesuriya S. J., Hollenberg M. D. (2002) Proteinase-activated receptor 2. Differential activation of the receptor by tethered ligand and soluble peptide analogs. J. Pharmacol. Exp. Ther. 302, 1046–1054 [DOI] [PubMed] [Google Scholar]
- 44. Ramachandran R., Mihara K., Mathur M., Rochdi M. D., Bouvier M., Defea K., Hollenberg M. D. (2009) Agonist-biased signaling via proteinase activated receptor-2. Differential activation of calcium and mitogen-activated protein kinase pathways. Mol. Pharmacol. 76, 791–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schuepbach R. A., Feistritzer C., Brass L. F., Riewald M. (2008) Activated protein C-cleaved protease activated receptor-1 is retained on the endothelial cell surface even in the presence of thrombin. Blood 111, 2667–2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ramachandran R., Noorbakhsh F., Defea K., Hollenberg M. D. (2012) Targeting proteinase-activated receptors. Therapeutic potential and challenges. Nat. Rev. Drug. Discov. 11, 69–86 [DOI] [PubMed] [Google Scholar]
- 47. Andrade-Gordon P., Maryanoff B. E., Derian C. K., Zhang H. C., Addo M. F., Darrow A. L., Eckardt A. J., Hoekstra W. J., McComsey D. F., Oksenberg D., Reynolds E. E., Santulli R. J., Scarborough R. M., Smith C. E., White K. B. (1999) Design, synthesis, and biological characterization of a peptide-mimetic antagonist for a tethered-ligand receptor. Proc. Natl. Acad. Sci. U.S.A. 96, 12257–12262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chackalamannil S., Xia Y., Greenlee W. J., Clasby M., Doller D., Tsai H., Asberom T., Czarniecki M., Ahn H. S., Boykow G., Foster C., Agans-Fantuzzi J., Bryant M., Lau J., Chintala M. (2005) Discovery of potent orally active thrombin receptor (protease activated receptor 1) antagonists as novel antithrombotic agents. J. Med. Chem. 48, 5884–5887 [DOI] [PubMed] [Google Scholar]
- 49. Chackalamannil S., Wang Y., Greenlee W. J., Hu Z., Xia Y., Ahn H. S., Boykow G., Hsieh Y., Palamanda J., Agans-Fantuzzi J., Kurowski S., Graziano M., Chintala M. (2008) Discovery of a novel, orally active himbacine-based thrombin receptor antagonist (SCH 530348) with potent antiplatelet activity. J. Med. Chem. 51, 3061–3064 [DOI] [PubMed] [Google Scholar]