

Background: β-Glucosidase completes cellulose enzymatic hydrolysis by releasing glucose from cellobiose.
Results: SAXS experiments revealed that Aspergillus niger β-glucosidase has a cellulase-like tadpole molecular shape, uncommon to enzymes that act on the soluble substrates.
Conclusion: We show that AnBgl1 N- and C-terminal domains are linked by a long extended linker.
Significance: Understanding AnBgl1 architecture is useful for comprehension of the enzyme-cell wall interaction and the process of biomass saccharification.
Keywords: Enzyme Mechanisms, Enzyme Structure, Enzymes, Fungi, Glycobiology, Aspergillus niger, SAXS, β-Glucosidase, Glycoside Hydrolase Family 3, Multidomain Proteins
Abstract
Aspergillus niger is known to secrete large amounts of β-glucosidases, which have a variety of biotechnological and industrial applications. Here, we purified an A. niger β-glucosidase (AnBgl1) and conducted its biochemical and biophysical analyses. Purified enzyme with an apparent molecular mass of 116 kDa forms monomers in solution as judged by native gel electrophoresis and has a pI value of 4.55, as found for most of the fungi of β-glucosidases. Surprisingly, the small angle x-ray experiments reveal that AnBgl1 has a tadpole-like structure, with the N-terminal catalytic domain and C-terminal fibronectin III-like domain (FnIII) connected by the long linker peptide (∼100 amino acid residues) in an extended conformation. This molecular organization resembles the one adopted by other cellulases (such as cellobiohydrolases, for example) that frequently contain a catalytic domain linked to the cellulose-binding module that mediates their binding to insoluble and polymeric cellulose. The reasons why AnBgl1, which acts on the small soluble substrates, has a tadpole molecular shape are not entirely clear. However, our enzyme pulldown assays with different polymeric substrates suggest that AnBgl1 has little or no capacity to bind to and to adsorb cellulose, xylan, and starch, but it has high affinity to lignin. Molecular dynamics simulations suggested that clusters of residues located in the C-terminal FnIII domain interact strongly with lignin fragments. The simulations showed that numerous arginine residues scattered throughout the FnIII surface play an important role in the interaction with lignin by means of cation-π stacking with the lignin aromatic rings. These results indicate that the C-terminal FnIII domain could be operational for immobilization of the enzyme on the cell wall and for the prevention of unproductive binding of cellulase to the biomass lignin.
Introduction
The plant cell walls are the largest source of carbohydrates in nature and an important source of renewable material for biofuel generation. Large amounts of the biomass polysaccharides are built of β-linked glucosyl residues; thus, it is not surprising that β-glucosidases play an important role in cell wall conversion into fermentable sugars. The complete hydrolysis of cellulose is generally accomplished by the synergistic action of at least three types of cellulases as follows: endoglucanases, exoglucanases, and β-glucosidases. Each of the enzymes plays a complementary and important role in the biomass depolymerization as follows: endoglucanases (EC 3.2.1.4) randomly cleave the β-1,4-glycosidic linkages of cellulose; cellobiohydrolases (EC 3.2.1.91) attack the reducing and nonreducing ends of a cellulose chain to produce cellobiose (constitutive unit of cellulose), and β-glucosidases (EC 3.2.1.21) hydrolyze cellobiose, releasing two molecules of glucose (1).
In the cellulolytic system, the β-glucosidases are crucial for efficient breaking down of cellulose, performing its complete saccharification by hydrolysis of cellobiose and small cello-oligosaccharides into glucose molecules. In addition, the β-glucosidase activity can synergistically optimize the action of other cellulolytic enzymes, increasing the yield of the final product and decreasing the concentration of cellobiose, which is a strong inhibitor of the cellulolytic complex, especially of cellobiohydrolases, in the reaction mixture (2). Commercially available cellulase preparations are often supplemented with fungal β-d-glucosidases to increase the cellulolytic efficiency on pretreated biomass (3).
Fungal β-d-glucosidases have been isolated from a number of fungal sources: Humicola grisea, Fusarium oxysporum, Gliocladium virens, Neocallimatix frontalis, Botrytis cinerea, Talaromyces emersonii, Trichoderma reesei, Phanerochaete crysosporium, Aspergillus aculeatus, Aspergillus foetidus, Aspergillus roseus, Aspergillus terreus, Aspergillus awamori, and Aspergillus niger, to name a few (4). The most common commercial β-glucosidase preparation (Novozymes SP 188; Novo Nordisk A/S, Bagsvaerd, Denmark) is produced by the cultivation of filamentous fungus A. niger (1).
Although fungal β-glucosidases have been exploited in a variety of biotechnological applications and extensively studied with respect to their production, purification, and kinetic parameters, molecular and structural data about these enzymes continue to be scarce. Based on amino acid sequence homology and structural similarity, fungal β-glucosidases are grouped into two glycoside hydrolase families, GHF1 and GHF3, according to the CAZy database. Both glycoside hydrolase families currently contain over 3000 members. From a structural viewpoint, the enzymes belonging to the GHF1 fold into a (β/α)8-barrel structure that contains their active site and catalyzes substrates following the β-retaining action mechanism that employs Glu as the catalytic nucleophile (5, 6). Although GHF3 has a large number of family members, both structural and enzymatic data are available for only for a handful of enzymes.
Six of the enzymes are from bacteria as follows: β-N-acetylhexosaminidase from Vibrio cholerae (NagZ), PDB2 code 1TR93; exo-1,3–1,4-β-glucanase (ExoP) from Pseudoalteromonas sp. BB1, PDB code 3F93 (7); β-N-acetylglucosaminidase (StNagZ) from Salmonella typhimurium, PDB code 4GVF (8); macrolide β-glycosidase/β-glucosidase (SvDesR) from Streptomyces venezuelae, PDB code 4I3G (9); β-N-acetylglucosaminidase from Bacillus subtilis (YbbD), PDB code 3BMX (10); and a β-glucosidase from Thermotoga neapolitana (TnBgl3B), PDB code 2X40 (11); four are eukaryotic enzymes, exo-β-1,3–1,4-glucanase (ExoI) from Hordeum vulgare subsp. vulgare, PDB code 1EX1 (12); β-glucosidase I (KmBgl1) from Kluyveromyces marxianus NBRC1777, PDB code 3ABZ (13); β-glucosidase 1 (HjBgl1) from T. reesei QM9414, PDB code 3ZYZ4; and β-glucosidase 1 (AaBgl1) from A. aculeatus, PDB code 4IIB (14), and a β-glucosidase (JMB19063) from compost metagenome PDB code 3U4A.5 The domain architectures of these enzymes are very different. NagZ is a one-domain enzyme of the (β/α)8-barrel fold. ExoI consists of two domains as follows: the N-terminal (β/α)8-barrel domain and the C-terminal (α/β)6-sandwich domain. TnBgl3B has a slightly longer C-terminal stretch than ExoI, and this C-terminal extension adopts a fibronectin type III (FnIII)-like fold, which makes TnBgl3B a three-domain enzyme. KmBgl1 has an insertion of a fourth domain, PA14 (residues 392–559) within the (α/β)6-sandwich domain (13). Finally, the AaBgl1 consists of three domains as follows: a catalytic TIM domain, an α/β sandwich domain, and an FnIII domain (14) The (β/α)8-barrel fold domain, (α/β)6-sandwich, and FnIII-like domain are connected by linkers 1 and 2. The members of GHF3 utilize an Asp residue in their nucleophile attack of the substrates. The active site of the GHF3 family is normally found between the (β/α)8-barrel and (α/β)6-sandwich domains, each of which contributes one catalytic carboxylate residue necessary for catalysis (6, 15). Purified AnBgl1 (860 residues) shares 83, 30, 28, and 24% amino acid sequence identities with AaBgl1 (860 residues), TnBgl3B (721 residues), KmBgl1 (845 residues), and ExoI (605 residues), respectively.
Understanding the fundamental structure-function relationships that govern the catalytic activity of β-glucosidases is of crucial importance for elucidating its mechanism of action and for optimization of its potential as an industrial biocatalyst in a variety of biorefinery processes, such as the production of cellulosic bioethanol. The enzymatic hydrolysis of heterogeneous biomass feedstocks requires β-glucosidases with different levels of specificity for di- or oligosaccharides in a biorefinery process, and different glycosidic linkages may require specific β-glucosidases to discriminate between different sugar isomers. Detailed studies of β-glucosidase structure, activity, and specificity are needed to deepen our understanding about the molecular basis of β-glucosidase action.
Here, we applied small angle x-ray scattering (SAXS) techniques to retrieve the molecular form and shape of AnBgl1, which belongs to GHF3 (16), and to determine relative positions of its individual domains and the possible extended conformation of linker 2 that shows a 100-amino acid insertion. We also conducted biochemical analysis of the enzyme aiming to comprehend a functional rationale for such molecular architecture and performed molecular dynamics simulations to investigate the interactions between the FnIII-like domain of AnBgl1 and lignin fragments of plant cell walls.
EXPERIMENTAL PROCEDURES
β-Glucosidase Preparation
A commercial enzyme preparation from A. niger (Novozymes SP 188; Sigma) was used as a source of β-d-glucosidase. For biochemical characterization of the β-glucosidase, solid (NH4)2SO4 was added to the commercial preparation to reach 90% saturation, and the solution was stored overnight at 4 °C under agitation. The precipitate was recovered by centrifugation and washed two times with a 90% (NH4)2SO4 solution. The pellet was dissolved in 50 mm sodium phosphate, pH 7.2, with 1 m (NH4)2SO4 and applied to a hydrophobic interaction column (phenyl-Sepharose 26/10 Fast Flow; GE Healthcare) equilibrated with the same buffer. Proteins were eluted at 3 ml min−1 with a gradient from 1 to 0 m ammonium sulfate using FPLC system (Akta; GE Healthcare). Fractions presenting β-glucosidase activity were joined and concentrated using a centrifuge (Vivaspin 20; Sartorius stedim, Aubagne, France) with molecular mass cutoff of 50 kDa, allowing separation of β-glucosidase and remaining as low molecular weight proteins. The β-glucosidase-enriched sample was applied on a desalting column (HiTrap; GE Healthcare) and eluted with 50 mm sodium acetate buffer, pH 7.2. β-Glucosidase fraction was then injected into an ion exchange column (Mono-Q 5/50GL; GE Healthcare) previously equilibrated with the same buffer. Proteins were eluted at 1 ml min−1 with a gradient from 0 to 500 mm of sodium chloride.
β-Glucosidase Activity
β-Glucosidase activity was determined by measuring the amount of glucose released from the cellobiose used as a substrate. β-Glucosidase assays were performed in triplicate, using 50 μl of cellobiose solution (2% w/v in 50 mm sodium phosphate buffer, pH 5.0) and 50 μl of β-glucosidase preparation, and incubated at 50 °C for 15 min. The reaction was stopped by heating to 95 °C for 10 min. The amount of released glucose was determined by the glucose oxidase/peroxidase method (17).
Native Gel Electrophoresis, SDS-PAGE, and Isoelectric Focusing
The protein content and purity of all chromatographic fractions were verified at each stage by Coomassie Blue-stained SDS-PAGE. Protein was applied on a 10% polyacrylamide gel, and migration of the bands was compared with the molecular mass markers in the range from 14.4 to 212 kDa (electrophoresis LMW calibration kit; GE Healthcare). The protein concentration was determined using the Bradford dye assay (Bio-Rad) (18), with bovine serum albumin as a standard. Purified β-glucosidase, at different concentrations (1, 2.5, 5, and 10 mg/ml), was electrophoresed on an 8–25% (w/v), pH 8.8, gradient polyacrylamide gel at 10 °C using PhastSystem (GE Healthcare), stained with 0.1% PhastGel Blue R-350 solution (GE Healthcare), and destained with 30% methanol and 10% acid acetic solution in distilled water (3:1:6). Isoelectric focusing was performed on the PhastSystem (GE Healthcare) using a precast gel PhastGel isoelectric focusing 3–9. Separations took ∼40 min after which the gel was stained with 0.02% PhastGel Blue R-350 (GE Healthcare) solution containing 30% methanol and 10% acetic acid in distilled water and 0.1% (w/v) CuSO4, fixed with 20% trichloroacetic acid, and destained in a solution containing 30% methanol and 10% acetic acid in distilled water until obtaining a clear background. Isoelectric point was determined using standard pI markers (GE Healthcare).
Deglycosylation of AnBgl1
To deglycosylate the AnBgl1, we applied the anhydrous trifluoromethanesulfonic acid treatment as described by the manufacturer (Sigma) with small modifications. Briefly, 2 mg of protein sample were dried in a SpeedVac at 50 °C and placed on ice for 5 min. The freeze-dry sample was dissolved with 150 μl of trifluoromethanesulfonic acid/anisole (9:1) and left in the freezer for 4 h. The sample was neutralized with 3 volumes of pyridine/methanol/water (3:1:1) in an ice bath for 20 min. One volume of an ammonium bicarbonate (0.5%) solution was added and centrifuged at 13,000 × g for 20 min. The precipitated material was dissolved in water and the protein pattern analyzed by SDS-PAGE.
Protein Digestion
The protein bands were removed from the gel and were submitted to in-gel trypsin (20 ng/μl), chymotrypsin (100 ng/μl), and pepsin A (75 ng/μl) digestion as described previously (19).
Mass Spectrometric Analysis
An aliquot of 4.5 μl of the resulting peptide mixture was analyzed on an ETD-enabled LTQ Orbitrap Velos mass spectrometer (Thermo Fisher Scientific) coupled with LC-MS/MS by an EASY-nLC system (Proxeon Biosystem) through a Proxeon nanoelectrospray ion source as described (20). Peptides were separated by a 2–90% acetonitrile gradient in 0.1% formic acid using an analytical column PicoFrit column (20 cm × inner diameter of 75 μm, 5 μm particle size, New Objective) at a flow rate of 300 nl/min over 27 min for in-gel digestion. The voltage of the nanoelectrospray was set to 2.2 kV, and the source temperature was 275 °C. LTQ Orbitrap Velos was set up in the data-dependent acquisition mode. The full scan MS spectra (m/z 300–1600) were acquired in the Orbitrap analyzer after accumulation to a target value of 1e6. Resolution in the Orbitrap was set to r = 60,000 and the top 20 peptide ions with charge states ≥2 were sequentially isolated to a target value of 5000 and fragmented in the linear ion trap by low energy collision-induced dissociation (normalized collision energy of 35%) or high energy collision dissociation. The signal threshold for triggering an MS/MS event was set to 1000 counts. Dynamic exclusion was enabled with an exclusion size list of 500, exclusion duration of 60 s, and a repeat count of 1. An activation q = 0.25 and activation time of 10 ms were used.
Data Analysis
Raw mass spectrometry files were searched against A. niger database (37,549 sequences, 17,765,295 residues; downloaded in July, 2013 from nonredundant protein database NCBI) using the Peptide Validator search node within Proteome Discoverer (Version 1.3.0.339, Thermo Fisher Scientific) with carbamidomethylation (+57.021 Da) as fixed modification and oxidation of methionine (+15.995 Da) and N-acetylglucosamine (+203.079 Da) as variable modifications; one trypsin missed cleavage and a tolerance of 10 ppm for precursor and 1 Da for fragment ions were also allowed. Only peptides with a minimum of five amino acid residues, which showed significant threshold (p < 0.05) in a Mascot-based score, were considered in the results. MS/MS spectra were manually validated for the b and y ion series.
Multiple Sequence Alignment
Multiple sequence alignment analyses were performed with the following: β-glucosidase 1 from A. niger (AnBgl1); β-glucosidase 1 from A. aculeatus (AaBgl1); thermostable β-glucosidase 1 from Thermoascus aurantiacus (TaBgl1) (21); β-glucosidase from Hypocrea jecorina (HjBgl1) (22); β-glucosidase from T. neapolitana (TnBgl3B) (8); β-glucosidase 1 from K. marxianus (KmBgl1) (10), and exo-β-1,3–1,4-glucanase from H. vulgare subsp. vulgare (ExoI) (9). Clustal X was used for alignment (23), and refinement was carried out with Bioedit sequence tool.
Homology Model of the Isolated Domains of β-Glucosidase
The three-dimensional models of the catalytic domain and FnIII-like fold domain were built by Swiss Model Server (24) using as template the available structure of the T. neapolitana, PDB code 2X40 (8), which has 30% sequence identity with AnBgl1. Use of very recently determined crystallographic structure of A. aculeatus β-glucosidase 1 (14) led to a very similar results (data not shown).
Small Angle X-ray Scattering
SAXS Experiments
The small angle x-ray scattering experiments of the AnBgl1 at 10, 5, 2.5, and 1 mg/ml (in 50 mm sodium acetate buffer, pH 5.0) were carried out at the D02A-SAXS2 beamline of the LNLS using a two-dimensional catalytic core domain detector (MarResearch) and measured at a wavelength of λ = 1.488 Å for the sample detector distance of 1200 mm, resulting in a scattering vector range q of 0.019 to 0.332 Å−1, where q is the magnitude of the q vector defined by q = 4πsinθ/λ(2θ is the scattering angle). To remove any aggregates, the samples were centrifuged for 30 min in an ultracentrifuge, at 23,500 × g, at 4 °C. The scattering curves of the protein solutions and the corresponding buffer were collected in a 1-mm path length cell with mica windows at 10 °C with two successive frames of 300 s each to monitor radiation damage and beam stability. The data were normalized for incident beam intensity and sample absorption and were corrected for detector response. The scattering of the buffer was subtracted from each protein, and SAXS pattern integrations were performed using Fit2D software (25). The protein curves were scaled for protein concentration.
SAXS Data Analysis
Merged SAXS curve was generated using Primus package (26) and using the most diluted and concentrated samples, respectively, for initial and final SAXS curve region (the merging point was at q = 0.10 Å−1). The distance distribution function, p(r), and the maximum diameter, Dmax, were evaluated using Gnom package (27); the radii of gyration, Rg, a global measure of the size and shape of the molecular complex, was approximated using two independent procedures, namely from the Guinier equation, I(q) = |I(0)exp(−Rg2q2/3) (28), and from indirect Fourier transform method also using Gnom. The molecular weight was calculated using a procedure implemented in web tool SAXS MoW (29). The calculation of the molecular weight and, consequently, the protein oligomerization state do not require the measurement of SAXS intensity on an absolute scale and do not involve a comparison with another SAXS curve determined from a known standard protein.
Ab Initio SAXS Model
The dummy atom model (DAM) was calculated from β-glucosidase SAXS curve using the ab initio procedure implemented in DAMMIN package (30). The Crysol package (31) was used to generate the simulated scattering curve from DAM. The evaluation of the Rg and Dmax values was performed with the same package.
Rigid Body Model
The position and orientation of the catalytic lobe, including both (β/α)8-barrel and (α/β)6-sandwich domains relative to the FnIII-like domain, were determined using rigid body modeling against SAXS experimental data. The discrepancy between SAXS curve and homology models were minimized using Bunch (32). Part of linker 2 (amino acids Gly-651–Gly-754) of the rigid body model (RBM), for which no structural information is available, was modeled using dummy atoms. The Crysol package (31) was used to evaluate the RBM parameters and SUPCOMB package (33) to superimpose the AnBgl1 DAM and RBM. Figures for the superposition were generated in the program PyMOL (34).
Protein Specificity on Different Substrates
β-Glucosidase activity was evaluated against 15 different polymeric substrates as follows: debranched arabinan, linear arabinan, sugar beet, galactomannan, β-1,4-mannan, rye arabinoxylan, birchwood xylan, xyloglucan, oat spelled xylan, laminarin, carboxymethylcellulose, β-glucan, lichenan, Avicel PH 101, and Avicel PH102 (Sigma). Enzymatic assay was performed at 50 °C for 60 min, containing 100 μl of reaction solution as follows: 40 μl of 50 mm sodium acetate buffer, pH 5, 10 μl of purified β-glucosidase (10 ng/μl), and 50 μl of substrate solution (100 mg/ml) in the same buffer. Released reducing sugar was quantified using 3,5-dinitrosalicylic acid reagent (35).
Capillary Zone Electrophoresis on Oligosaccharides
β-Glucosidase processivity was evaluated against a synthetic oligosaccharide consisting of five glucose molecules linked by β-1,4-linkages (Megazyme), derivatized with 8-aminopyreno-1,3,6-trisulfonic acid (APTS) by reductive amination, as described previously (36). Capillary electrophoresis was performed on a P/ACE MQD (Beckman Coulter) with laser-induced fluorescence detection. A fused silica capillary (TSP050375, Polymicro Technologies) of internal diameter 50 μm and length 31 cm was used as the separation column for oligosaccharides. The samples were injected by application of 5 p.s.i. pressure for 0.5 s. Electrophoresis conditions were 10 kV, 70–100 μA with the cathode at the inlet, 50 mm sodium phosphate, pH 2.5, as running buffer, and a controlled temperature of 20 °C. The capillary was rinsed with 1 m NaOH followed by running buffer to prevent carryovers. APTS-labeled oligomers were excited at 488 nm, and emission was collected through a 520-nm bandpass filter.
Protein Adsorption
Capacity of β-glucosidase adsorption was evaluated on pure starch, xylan, and Avicel from Sigma, and lignin was obtained from sugarcane bagasse. The assays were conducted with 0.3 mg/ml protein in a 50 mm sodium citrate buffer, pH 5.0, and different polymeric plant-derived compounds (starch, xylan, lignin, and Avicel) at concentrations 0–100 mg/ml. The samples were incubated at room temperature for 1 h under continuous agitation and centrifuged at 13,000 × g for 10 min (37). Centrifugation was repeated twice to ensure the removal of all polymeric compounds. The β-glucosidase activity in the supernatant was measured on cellobiose as described above. Data presented are an average of six replicates.
Molecular Dynamics
We have simulated the FnIII domain and representative small molecular fragments of lignin to investigate how they interact. Small lignin fragments composed of homodimers of guaiacyl (LGG) and syringyl (LSS), the two most abundant units found in the lignin polymers from plants in general (38), were built. The most recurrent linkage between the units, β-O-4′, was adopted for both types of homodimers (39).
Independent molecular dynamics simulations were carried out starting from different initial configurations of the systems as follows: (i) five LGG fragments docked around the FnIII-like domain surface whose binding sites were generated using docking methods with AutoDock Vina software (40) (system 1); (ii) 10 LGG fragments randomly distributed around the FnIII-like domain (system 2); (iii) 10 LGG fragments placed near specific sites of FnIII based on the likelihood of hydrogen bond formation, aromatic ring packing, hydrophobic contacts, and shape complementarity (system 3); (iv) only two lignin dimer fragments randomly placed in solution, far from the FnIII-like domain, so that lignin-lignin interactions were negligible. In this case, we also compared the behavior of LGG (system 4a) and LSS (system 4b) fragments with respect to their interactions with FnIII.
Docking
AutoDock Vina was run setting all LGG covalent bonds as rotatable, except the ones in the aromatic rings, using AutoDockTools (41, 42) to create input files. The exhaustiveness parameter was set to 20. The top five docking poses according to AutoDock scoring were selected to build the LGG-FnIII complexes that compose system 1 (see supplemental material, Molecular Dynamics Simulation).
Molecular Dynamics Simulations
All the systems underwent a basic simulation protocol, described as follows. The simulation boxes were first generated using Packmol (43, 44), before any manipulation of the dimers. The structures were then placed in a cubic box of about 80 Å in each direction and hydrated by 15,000 water molecules. 24 chlorine ions and 20 sodium ions were added to keep the system electrically neutral at 0.15 m salt concentration. The ionization states of ionizable residues (Lys, Arg, His, Asp, and Glu) were determined according to their pKa values at pH 5.0 and the molecular environment (high dielectric constant at the protein surface and low dielectric constant in its interior) using the H++ server (45, 46). As a result, all ionizable residues were considered charged.
The energy of the system was initially minimized by 500 steps of the conjugate gradient method (47, 48), as implemented in NAMD (49) to eliminate bad contacts. After minimization, we performed pre-equilibration runs, following the protocol described elsewhere (50). From the pre-equilibrated systems, we carried out three independent simulations for each system. The simulations were performed using NAMD under periodic boundary conditions. The CHARMM force field was used for the proteins (51, 52) and lignin fragments (53), and the TIP3P model was used for water molecules (54). The temperature and pressure were kept constant at 298 K and 1 bar by means of Langevin dynamics and Nosé-Hoover piston methods (55, 56). The RESPA multiple time step algorithm (57) was used with the shortest time step of 2 fs. All bonds involving a hydrogen atom were kept at fixed bond length using SHAKE (58). A 12-Å cutoff with smooth switching function starting at 10 Å was used for the van der Waals forces, whereas electrostatic forces were treated via the particle mesh Ewald method (59).
The simulation time for system 1 lasted 60 ns. A total of 180 ns of simulation time, divided into three independent 60-ns runs, was performed for each one of the systems 2, 3, 4a, and 4b.
The overall stability and the structural relaxation of the enzymes were monitored by computing the time evolution of the root mean square deviation of the protein Cα atoms along the simulations (data not shown). We observed that, after few nanoseconds, the root mean square deviation of the backbone became stable. For all systems, the first 30 ns of simulation were not considered for calculation of average properties.
RESULTS
Two major protein bands corresponding to two isoforms of the β-glucosidase from A. niger (60), which represents ∼85% of total protein content (1), were clearly identified by SDS-PAGE of the commercial Novozyme 188 preparation. We further purified the β-glucosidase by liquid chromatography using phenyl-Sepharose and Mono Q columns. During the hydrophobic chromatography step, one of the major β-glucosidase peaks was eluted with 0.2 m (NH4)2SO4. Taking into account the high apparent molecular mass of the β-glucosidase (close to 116 kDa), the sample was concentrated using a 50-kDa cutoff membrane to remove smaller proteins. The final β-glucosidase purification step was achieved by ion exchange chromatography using a Mono Q column, with ∼0.3 m of NaCl.
The purity of the enzyme was independently confirmed using three different methods. The molecular mass of the purified β-glucosidase estimated by SDS-PAGE analysis was about 116 kDa (Fig. 1A). A single identical protein band was observed in native gel electrophoresis experiments at all analyzed protein concentrations (10, 5, 2.5, and 1 mg/ml), indicating that the enzyme is in a monomeric state (Fig. 1B). Furthermore, isoelectric focusing showed that the purified enzyme was acidic pI, with the experimentally determined pI value close to 4.55 (Fig. 1C). The calculated mass of AnBgl1, based on its amino acid sequence and without taking into consideration its glycosylation is 93.553 kDa and calculated pI value, is 4.65.
FIGURE 1.
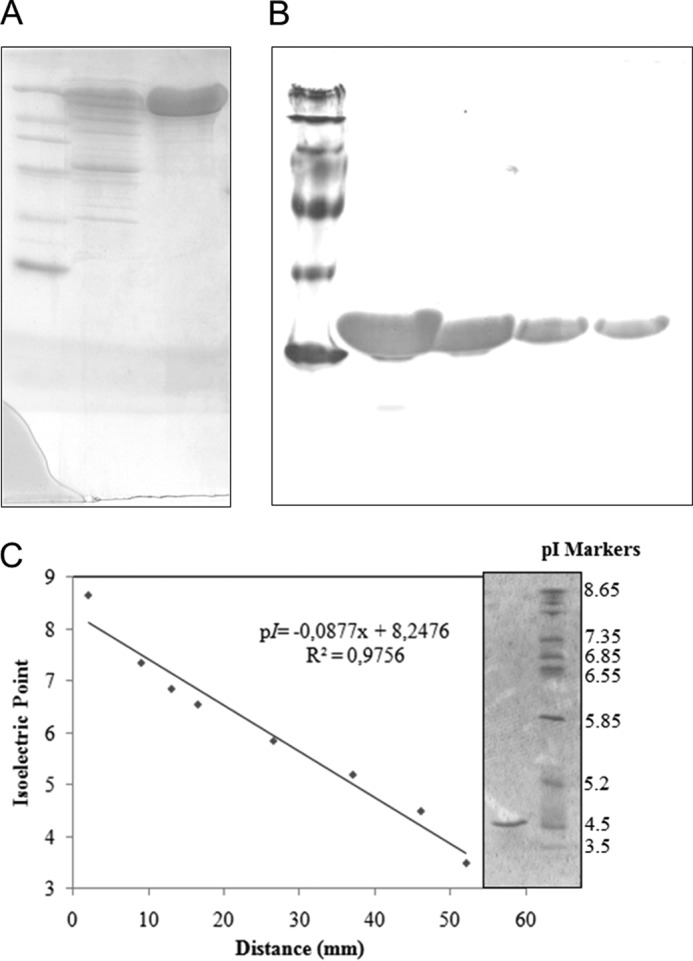
Analysis of purified β-glucosidase from A. niger. A, SDS-PAGE of purified enzyme (1st lane, standard protein markers (14–116 kDa); 2nd lane, commercial preparation Novozymes 188; 3rd lane, purified β-glucosidase). B, native-PAGE of purified enzyme at different concentrations (10, 5, 2.5, and 1 mg/ml). C, isoelectric focusing of purified β-glucosidase (1st lane, standard pI markers; 2nd lane, purified β-glucosidase).
To make sure that the purified enzyme was indeed an A. niger β-glucosidase, its peptide mass fingerprints were identified by mass spectrometry and confirmed by MS/MS analysis. Both nonglycosylated and trifluoromethanesulfonic acid-deglycosylated forms of AnBgl1 were analyzed. The sequence coverage by chymotrypsin, trypsin, and pepsin A digestions were 80, 66, and 45%, respectively. The combined coverage of the amino acid sequence after trypsin, chymotrypsin, and pepsin digestions was 92%, thus clearly confirming identity of the protein sample to A. niger β-glucosidase) (supplemental material, Mass Spectrometry Analysis and supplemental Figs. S1 and S2) (12.
AnBgl1 putative glycosylation sites have been predicted using NetNGlyc 1.0 Server-CBS and identified based on our mass spectrometry analysis (supplemental material, Mass Spectrometry Analysis and supplemental Table S1). Out of 10 putative glycosylation sites identified by the NetNGlyc server (residues Asn-61, Asn-211, Asn-315, Asn-322, Asn-354, Asn-523, Asn-542, Asn-564, Asn-658, and Asn-712), 8 were confirmed (amino acid residues 61, 211, 315, 322, 354, 523, 564, and 712). In addition, three more glycosylation sites were identified for AnBgli1 (at Asn-252, Asn-442, and Asn-690). The later N-glycosylation sites were missed by the NetNGlyc server algorithm. Finally, computationally predicted putative glycosylation at positions Asn-542 and Asn-658 fell into the regions of the amino acid sequence, which were not covered by our mass spectrometry analysis. However, a very recently published crystallographic structure of homologous AaBgl1 revealed that these sites are not glycosylated in the β-glucosidase of A. aculeatus (14). The total number of the confirmed N-glycosylation sites for AnBgl1 (11 sites) is close to the total number of the N-glycan chains (9 sites) in the later structure (supplemental material, Mass Spectrometry Analysis and supplemental Table S1).
Multiple sequence alignment analysis allows comparison of AnBgl1 and other six enzymes, as shown in supplemental material, Multiple Sequence Alignment Analysis. The multiple sequence analysis was performed based on the amino acid sequences of β-glucosidase 1 from the following: A. niger (AnBgl1); β-glucosidase 1 from A. aculeatus (AaBgl1) (14); thermostable β-glucosidase 1 from T. aurantiacus (TaBgl1) (21); β-glucosidase from H. jecorina (HjBgl) (22); β-glucosidase from T. neapolitana (TnBgl3B) (8); β-glucosidase 1 from K. marxianus (KmBgl1) (10); and exo-β-1,3–1,4-glucanase from H. vulgare subsp. vulgare (ExoI) (9). According to Varghese et al. (12), the N-terminal domain of ExoI adopts a canonical (β/α)8-barrel fold. N-terminal domains of TnBgl3B (8), KmBgl1 (10), and AaBlg1 (14) also fold in a (β/α)8-barrel-like structure, in which the first two α-helices corresponding to helix-A and-B of ExoI are absent (supplemental material, Multiple Sequence Alignment Analysis, see gray box). This large deletion is also observed in the sequence alignment of AnBgl1, TaBgl1, and HjBgl. The topology of the N-terminal domain of AnBgl1 can thus be interpreted as a ββ(β/α)6-barrel, in which the second β-strand is antiparallel.
AnBgl1 N-terminal and (α/β)6-sandwich domains contains, respectively, the nucleophile residue Asp-280 and the acid/base residue Glu-509. Both residues are conserved in all five sequences included in the alignment. The other residues that make part of the catalytic site Asp-92, Lys-189, His-190, Met-245, Tyr-248, and Trp-281 are also conserved. Leu-141 residue is conserved in four of the five sequences, whereas ExoI has a mutation to Phe-144 in the respective position, and Ser-436 is absent in HjBgl and ExoI.
Based on the alignment, one can also clearly observe an insertion of ∼100 amino acid residues in the region of AnBgl1 linker 2 (Val-590–Gly-754) as compared with the sequences of available crystal structures, KmBgl1 (10) and TnBgl3B (8). The same 100 amino acid insertion is present in other homologous fungal enzymes exemplified by AaBgl1 (14), TaBgl1 (21), and HjBgl (22).
Homology modeling of the isolated domains of AnBgl1 was performed using the Swiss Model Server (24) The homology model of the AnBgl1 catalytic domain (corresponding to the protein amino acid sequence between Met-1 and Tyr-650) is composed of a ββ(β/α)6-barrel and a (α/β)6-sandwich domain. The enzyme C-terminal part (amino acid residues Asn-755–His-860) of the homology model has an FnIII-like fold. Except for the insertion of the PA14 domain in KmBgl1 and the absence of an FnIII-like fold domain in ExoI, the tertiary structural arrangement and the patterns of the secondary structure of the AnBgl1 homology domains are similar to those of KmBgl1 (10), TnBgl3B (8), and ExoI (9).
SAXS data analysis was performed using the merged SAXS curve (Fig. 2A). The AnBgl1 radius of gyration calculated by the Guinier analysis is 42 Å. The linearity of the Guinier plot indicated that AnBgl1 preparation was monodisperse (Fig. 2A, inset). The pair-distance distribution function p(r) was obtained by the inverse Fourier transform of the AnBgl1 scattering data, using the program Gnom (27). Analysis of the p(r) leads us to conclude that AnBgl1 has an elongated form with a Dmax of 150 Å (Fig. 2B). The value of 41.86 Å for Rg calculated from the distance distribution function is in a good agreement with the estimate derived from the Guinier analysis (Table 1). The molecular mass obtained from experimental SAXS curves using the web tool SAXS MoW (24) was 91 kDa, whereas the theoretical value calculated with basis on the protein amino acid sequence is close to 93.5 kDa. Thus, the discrepancy between the experimental and theoretical molecular weights of the enzyme was only 2.2%. The information leads us to conclude that AnBgl1 is indeed a stable, monodispersive, and monomeric molecular system in solution.
FIGURE 2.
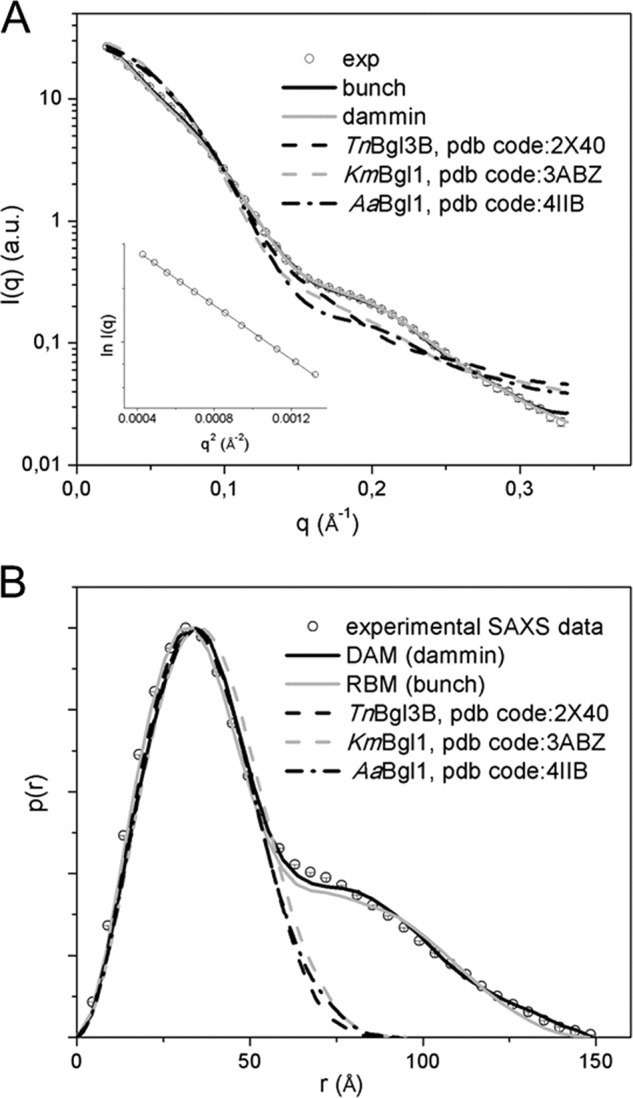
A, small angle x-ray scattering curves, real and reciprocal spaces. Merged experimental SAXS curve (sphere), simulated curves from rigid body model (black line), dummy atom model (gray line), TnBgl3B, PDB code 2X40 (black dashed line), KmBgI1, PDB code 3ABZ (dashed gray line) and AaBgI1, and PDB code 4IIB (black dashed/dotted line) are shown. Inset contains Guinier analysis lnI(q) versus q2. B, distance distribution function p(r), the same symbols were used.
TABLE 1.
Structural parameters derived from small angle x-ray scattering of β-glucosidases
Resolution is 2π/qmax.
| Parameters |
AnBgl |
TnBgl3B PDB code 3ABZ | KmBgl1 PDB code 2X40 | AaBgl1 PDB code 4IIB | ||
|---|---|---|---|---|---|---|
| Exp.a | DAMb | RBMc | ||||
| Dmax (Å) | 150.0 ± 0.50 | 148.9 | 147.4 | 95.60 | 92.65 | 94.56 |
| Rg (Å) | 41.86 ± 0.55 | 41.77 | 40.68 | 27.52 | 26.12 | 26.93 |
| Resolution (Å) | 18.91 | |||||
| Mr, SAXS (kDa)d | 91.00 | |||||
| Χ | 1.73 | 1.30 | 7.54 | 6.67 | 7.69 | |
a This was calculated from experimental data.
b Parameters of the dummy atom models are from the DAMMIN package.
c Parameters were calculated from rigid body model from bunch package.
d This was calculated from SAXS MoW.
Subsequently, AnBgl1 experimental SAXS data were compared with SAXS data obtained from available high resolution structures of the TnBgl3B, from T. neapolitana (8), KmBgl1, from K. marxianus (10), and AaBgl1, from A. aculeatus (14). Structural parameters were obtained from AnBgl1 experimental SAXS curve and from theoretical scattering curves of TnBgl3B, KmBgl1, and AaBgl1 using the program Crysol (31).
The Rg values observed for TnBgl3B, KmBgl1, and AaBgl1 are 26.12, 27.52, and 26.93 Å and the Dmax values are 92.65, 95.60, and 94.56 Å, respectively. These values are much smaller than the correspondent values for AnBgl1. Comparison between simulated SAXS curves obtained for available three-dimensional structures and the AnBgl1 experimental data revealed large discrepancy values for TnBgl3B, KmBgl1, and AaBgl1 of χ = 6.67, 7.54, and 7.69 for, respectively, each of the structures. Furthermore, analysis of p(r) shows that TnBgl3B, KmBgl1, and AaBgl1 exhibit more compact and globular crystal structures than AnBgl1 in solution (Fig. 2; Table 1). As can be observed, TnBgl3B (PDB code 2X40) (Fig. 3A), KmBgl1 (PDB code 3ABZ) (Fig. 3B), and AaBgl1 (PDB code 4IIB) (Fig. 3C) adopt compact structures with atomic contacts at the interface between catalytic and C-terminal domains. These discrepancies were also clarified by computing the AnBgl1 dummy-atom model and ab initio modeling based on our SAXS data.
FIGURE 3.

Three orthogonal view of TnBgl3B (PDB code 2X40) (A), KmBgI1 (PDB code 3ABZ) (B), AaBgI1 (PDB code 4IIB) (C), and superposition of ab initio dummy atom model (gray surface) (D); rigid body model (domains and hinge, respectively, schematic, and dummy atoms). Color patterns are as follows: N-terminal domain (blue); β-sandwich domain (green); PA14 domain (yellow); FnIII-like fold domain (red); first linker (cyan), and second linker (orange); rigid body model hinge 2. The models are rotated from upper models in relation to axis x and y by 90°.
Ab initio modeling as implemented in DAMMIN program (30) was used to obtain information about the molecular shape of AnBgl1 and the relative position of its domains. The retrieved DAM clearly reveals an elongated multidomain structure connected by a long linker in an extended conformation (Fig. 3D). The structural parameters Rg (41.77 Å) and Dmax (148.9 Å) obtained from theoretical scattering curve of DAM are in a close agreement with our experimental SAXS data. Furthermore, DAM showed excellent agreement with the experimental x-ray scattering curve (χ = 1.73; Table 1).
Rigid body modeling was conducted to adjust the catalytic domain and FnIII-like domain homology models within the DAM envelope, with the catalytic domain occupying the larger lobe of the DAM and FnIII-like domain fitting the minor lobe (Fig. 3D). Structural parameters Rg and Dmax obtained from the theoretical scattering curve of the RBM are 40.68 and 147.4 Å, respectively. The linker 2 corresponding to residues Gly-651–Gly-754 (the region absent in the homology-modeled N-terminal catalytic and C-terminal FnIII-like domains) is represented by dummy atoms (Fig. 3D). The tadpole shape is quite common for the enzymes acting upon insoluble polymeric carbohydrates (cellulose and other plant biomass carbohydrates) that frequently possess a large catalytic domain attached to the small cellulose binding module by the flexible and extended linker peptide (61). However, it is quite uncommon for the enzymes active on small soluble substrates.
To verify the possibility of AnBgl1 to be active against a number of insoluble polymeric carbohydrates, we tested its specificity upon different polymeric substrates, aiming to comprehend the functional reasons behind its tadpole-like molecular architecture. However, in contrast with classical exo- and endoglucanases, our results confirmed that purified AnBgl1 has no or very low activity upon tested polymeric compounds, and it has high specificity to cellobiose and small soluble oligosaccharides (Table 2).
TABLE 2.
Substrate specificity of purified AnBgl1
| Substrates | Relative activitya |
|---|---|
| % | |
| Cellobiose | 100 |
| Debranched arabinan | 0.03 |
| Linear arabinan | 0 |
| Sugar beet | 0 |
| Galactomannan | 0 |
| β-1,4-Mannan | 0 |
| Rye arabinoxylan | 0 |
| Xylan birchwood | 0.24 |
| Xyloglucan | 0 |
| Xylan oat spelt | 0.99 |
| Laminarin | 1.02 |
| Carboxymethylcellulose | 0 |
| β-Glucan | 0 |
| Lichenan | 1.01 |
| Avicel PH101 | 1.57 |
| Sigma Cellulose | 1.94 |
| Microcrystalline Cellulose | 1.27 |
a The relative activities are expressed as percentage by normalizing to the cellobiose specific activity (98.7 units/mg).
Enzymatic processivity determined by capillary zone electrophoresis also revealed a catalytic profile characteristic of β-glucosidases. The enzymatic assays using synthetic C5 oligosaccharide as substrate showed that AnBgl1 can release only one glucose molecule per each cycle of hydrolysis. As shown in Fig. 4, the C5 is hydrolyzed to form C4, consecutively until the final product (C1) release. The lower C1 peak area can be explained by the presence of the APTS marker, which obstructs the enzyme access. To gain further insights into putative function of the C-terminal FnIII-like domain, we conducted pulldown assays using major plant biomass fractions (starch, xylan, cellulose, and lignin) as a bite.
FIGURE 4.

AnBgl1 processivity profile analyzed by capillary zone electrophoresis using the synthetic oligosaccharide C5 (five molecules of glucose linked by β-1,4 linkages) as substrate.
Adsorption analyses were performed by pulling down AnBgl1 using four different polymeric substrates and measuring its residual enzymatic activity in the supernatants (Fig. 5). The results showed only weak adsorption (below 20%) of A. niger β-glucosidase on starch, xylan, and Avicel within the range of their concentrations used in our experiments, which might be associated with the enzyme loss during the washing procedures. At the same time, the total β-glucosidase activity in the supernatant decreased significantly with the increase of lignin concentrations applied to the solution. The supernatant relative residual activity of only 17% was observed at the highest lignin concentration used in our experiment, indicating the high capacity of AnBgl1 binding to lignin.
FIGURE 5.

AnBgl1 adsorption assay performed at four different polymeric compounds (pure starch, xylan, and Avicel from Sigma and lignin obtained from sugarcane bagasse) at increasing concentrations (0–100 mg//ml). ●, xylan supernatant; ■, starch supernatant; *, Avicel supernatant; Δ, lignin supernatant.
Molecular dynamics simulations were carried out to assess the atomic level details of the FnIII-like domain interactions with small molecular fragments representative of guaiacyl (LGG) and syringyl (LSS) homodimers of lignin, depicted in Fig. 6.
FIGURE 6.
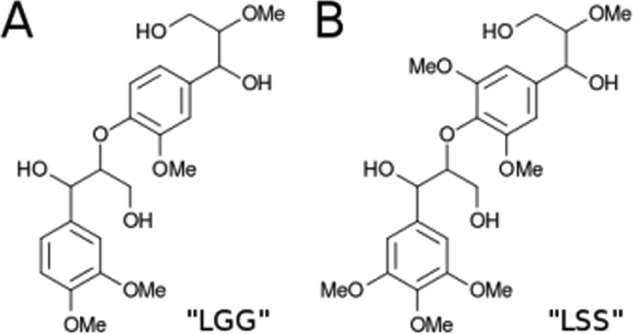
Lignin fragment models used in the molecular dynamics simulations. The molecule in A is a dimer composed of guaiacyl units (LGG), and the molecule in B is a dimer of syringyl units (LSS).
The simulated FnIII domain consisted of a β-sandwich structure, with four strands in each face. As shown in Fig. 7A, it has a small helix and β-strand, followed by a long loop between the E and F strands. The structure does not exhibit any obvious substrate-binding sites, unlike the clefts found in the catalytic core domains or clusters of aromatic residues in the cellulose-binding modules of cellulases. The structures generated with AutoDock Vina (system 1) suggest that a single binding site for lignin fragments may not exist on the surface of FnIII, but instead multiple suitable binding sites are present (supplemental material, Molecular Dynamics Simulation and supplemental Fig. S1). The LGG fragments remained docked in the vicinity of locations S2, S4, and S5 (Fig. 7B) during the entire course of the simulations, but they diffused away from the other initial positions into solution within the first 10 ns of simulation time. Another striking feature of the FnIII-like domain is that most of its residues are well exposed to solvent. The simulations show that 89% of its residues remain hydrated by at least five water molecules. As shown in Fig. 7C, the structure is rich in charged residues but also exhibits exposed hydrophobic amino acid residues.
FIGURE 7.

Tridimensional structure of the FnIII-like domain of AnBgl1 and putative lignin-binding sites. A, β-sandwich-like structure of the FnIII domain. B, five main lignin-binding sites generated by the docking calculations with AutoDock Vina. Simulations of system 1 started from LGG fragments (purple) at indicated positions S1–S5. C, van der Waals surface of FnIII viewed from different angles, showing the exposed residues of different nature (see inner legend).
The interaction energy between FnIII and the LGG or LSS lignin fragments was monitored for the whole set of 12 independent simulations reported here. The results are presented individually for systems 1–4 (supplemental material, Molecular Dynamics Simulation and supplemental Fig. S2). All different approaches revealed that there are multiple regions of favorable interactions between FnIII and the lignin fragments. In the simulations with 10 dimers, especially in the case in which they were randomly distributed (system 2), the statistics of interaction with the FnIII were limited by the frequent interaction among the lignin fragments themselves. This tendency was predicted from the simulations of Petridis and Smith (53), in which the collapse of the lignin polymer was detected at 300 K.
Collecting the average interaction energy between FnIII residues and lignin fragments from the simulations of systems 1–4 on the same graph (Fig. 8A), one clearly sees that some residues emerge as frequent lignin binders and, therefore, are likely binding site candidates. We draw attention to the important role played by arginine residues in the interaction of the lignin fragments with FnIII. The peaks marked X in Fig. 8A, reveal the high occurrence of such interactions. Further inspection shows that dispersive interactions and hydrophobic contacts predominate over hydrogen bonding between FnIII arginine residues and LGG or LSS fragments. Therefore, the aromatic rings of lignin mostly stack parallel to the guanidinium cation plane of the arginines.
FIGURE 8.

Interaction between lignin fragments and the FnIII domain obtained from the simulations. A, interaction energy between LGG and LSS dimers and FnIII residues corresponding to the different approaches adopted to construct the initial disposition of the dimers. Black line, system 1; orange, system 2; red, system 3; blue and green, system 4a/4b. The X marks indicate the arginine residues, which are located in regions of FnIII that emerge recurrently as likely binding sites, independently of the approach adopted for the initial lignin disposition. B–D are snapshots from the simulations representative of the most important lignin-FnIII binding modes. The frequency of cation-π and hydrogen bond interactions along the simulations are indicated.
Considering the frequency of occurrence of each interaction type along the simulations, three important subdomains in FnIII structure emerge. The first subdomain, depicted in Fig. 8B, corresponds to the region of the long loops between C and D and between E and F strands (cf. site S1 in Fig. 7B). This region is attractive to the lignin analogs because it is partially isolated from water, being surrounded by the planar guanidinium and indole groups from arginines (Arg-99 and Arg-100) and tryptophans (Trp-105 and Trp-112), respectively. Clustering of other LGG dimers around this region is also observed in the simulations, mainly because of the π-stacking between the lignin fragments and tryptophans aromatic rings.
Another important site of interaction between FnIII and lignin is depicted in Fig. 8C and corresponds to region S3 that appears in the docking structure (cf. Fig. 7B). In this region, one or two dimers are often found hydrogen bonded to Tyr-64 and Leu-77 and also make hydrophobic contacts with Arg-128. Fig. 8D, in turn, shows an interaction mode frequently observed between LGG or LSS and Arg-44. This binding mode resembles the S5-binding site obtained from the docking experiments. A hydrogen bond between one of the lignin hydroxyl groups and the main chain of Arg-44 is also frequently observed.
DISCUSSION
In this paper, we describe the purification and characterization of native AnBgl1. The experimentally determined apparent molecular mass of the enzyme is 116 kDa, which is close to the molecular mass estimates of almost all purified A. niger β-glucosidases reported so far that are in the range between 90 and 130 kDa (4). The enzyme was found to be monomeric, as are most of the reported glycoside hydrolase family 3 enzymes, including Exo1, that exist in solution as monomers. The experimentally determined pI value is also in accordance with the theoretical value of AnBgl1 pI and with the pI range (3.5–4.5) found in the literature for most of fungal β-glucosidases (62).
Surprisingly, however, the ab initio SAXS modeling, based on our experimental x-ray scattering data, clearly reveals that AnBgl1 has an elongated multidomain structure connected by a long linker (linker 2) in an extended conformation (Fig. 3D), with an RBM that fits very well the experimental x-ray scattering curve (χ = 1.30; Table 2). This kind of architecture is characteristic for canonical cellulases (exo- and endoglucanases) but very different to the molecular form of other structurally characterized β-glucosidases such as TnBgl3B, KmBgl1, and AaBgl1 (Fig. 3, A–C).
The longer linker 2 revealed by SAXS studies is associated to almost 100 amino acid insertion, observed for the fungi enzymes on the multiple alignment sequence. We argue that precisely due to the linker length, AnBgl1 is capable of adopting extended conformation, which is very different from compact TnBgl3B (PDB code 2X40) and KmBgl1 (PDB code 3ABZ) structures. Shorter length of their linker peptides might explain dense packing of the structural domains of TnBgl3B and KmBlg1. C-terminal FnIII-like domain of these structures wraps around the back side of the molecule far from the active site, interacting with the second linker and with N-terminal domain. However, its contact with the β-sandwich domain is more restricted (8).
Linker 2 constitutes the main interface between β-sandwich and C-terminal domains in TnBgl3B molecule. The 25 residues (555–579) on TnBgl3B linker 2 are extended toward the N terminus and provide a considerable number of interactions with this domain. These residues are reasonably conserved in KmBgl1, at positions 678–699. In this way, TnBgl3B residues 568–571 form a short parallel β-sheet with residues 178–180 on the surface of N-terminal domain (supplemental material, Multiple Sequence Alignment Analysis, see black boxes). On the contrary, the correspondent amino acid residues that interact with the N-terminal domain in KmBgl1 and TnBgl3B structures are not conserved in AnBgl1. Two hydrophobic amino acids, Val-570 and Val-571 in TnBgl3B and Val-688 and Ile-689 in KmBgl1, are mutated to residuals Glt-621 and Ala-622 in AnBgl1. Multiple sequence alignment reveals that the same occurs in TaBgl1, AaBlg1, and HjBgl. This is consistent with the notion that their catalytic and FnIII-like domains do not form extensive molecular interfaces. In addition, all three compared fungal β-glucosidases (from T. aurantiacus, H. jecorina, and A. niger) have the same insertion of ∼100 amino acid residues (Thr-656–Asn-755) between the C-terminal catalytic domains and the N-terminal FnIII-like domains, which is not present in the structurally studied enzymes. Based on these observations, we argue that similar two-lobe tadpole-like extended conformations revealed for AnBgl1 might be common for other GHF3 fungal β-glucosidases. Such long and flexible linkers would clearly impede crystallization of the enzymes in their extended conformation and would force the FnIII-like domain to pack against the body of the β-glucosidase in the crystal settings. We advocate that a variety of conformations adopted by the FnIII-like domain in the crystal structures of AaBgl1 (PDB code 4IIB) and HjBgl1 (PDB code 3ZYZ) might be a consequence of the different crystallographic packing.
The extended linker conformation observed in AnBgl1 confers flexibility to the protein and relative mobility of the N-terminal catalytic domain and the C-terminal FnIII domain, which is evident in the SAXS model. The average of different positions of two separate domains results in large volumes for C- and N-terminal lobes of the DAM as compared with their high resolution models. This higher protein flexibility might be relevant to its enzymatic activity.
The extended liker conformation and the multidomain organization revealed for AnBgl1 is similar to many other cellulases, such as cellobiohydrolases and endoglucanases. The cellulose binding domain (CBM) of the cellulases, usually located at the N or C termini of the enzyme, is bound to a catalytic core domain by a linker region enriched with proline, threonine, and serine residues (63). The multidomain cellulases have a tadpole molecular shape, as revealed by earlier SAXS studies (64, 65). The CBM warrants the cellulase displacement along cellulose fibers, optimizing its hydrolytic activity on long and insoluble cellulose. The cellulose binding is generally observed only for cellulases or other enzymes that act upon complex insoluble substrates. CBMs increase cellulase affinities to cellulose, improve the polymeric substrate recognition, and contribute to the enzymes' processivity. However, such molecular organization, which includes extended conformation of two separate domains, is not at all common for the enzymes that act upon soluble substrates, such as β-glucosidases.
β-Glucosidases mainly catalyze by hydrolysis of the β-1,4-glycosidic linkages in oligosaccharides and disaccharides and amino-, alkyl-, or aryl-β-d-glucosides. In bacteria and fungi, β-glucosidases are mainly a component of the cellulase enzyme system and responsible for the breakdown of short chain oligosaccharides and cellobiose into glucose, and the enzyme activity decreases as the glucose chain length increases (66, 67). According to our results, AnBgl1 can be classified as a true cellobiase, once it has a high specificity to cellobiose. Our enzymatic assays confirm that AnBgl1 does not efficiently hydrolyze any of the tested polymeric carbohydrates when compared with cellobiose, despite its multidomain architecture, which resembles that of canonical cellobiohydrolases. Furthermore, our capillary electrophoresis analysis showed that AnBgl1 has a processivity profile characteristic of β-glucosidases, releasing only one glucose molecule for each hydrolysis cycle.
Because AnBgl1 has no (or very little) activity against long and insoluble carbohydrates, we also performed the pulldown assays using major biomass fractions as bites to comprehend the functionality of the AnBgl1 molecular shape. These assays confirmed that AnBgl1 has no adsorption capability to Avicel, xylan, and starch but adsorbs strongly to lignin. Zhu and Sathitsuksanoh (37) performed adsorption experiments with cellulases, β-glucosidase, and BSA utilizing different substrates, such as Avicel, xylan, and regenerated amorphous cellulose. Similarly, they also found no β-glucosidase absorption on any of the tested compounds. Our results are in further agreement with several biomass conversion studies that demonstrated that lignin represents a significant barrier to biomass hydrolysis, because it inhibits enzyme access to polysaccharides due high irreversible protein adsorption, thus decreasing efficiency of the cellulolytic mixtures (37, 68).
The putative functional role of AnBgl1 FnIII is not entirely clear. The C-terminal FnIII domain was also observed in the crystal structures of the thermostable β-glucosidase from T. neapolitana (8) and β-glucosidases from A. aculeatus (14) and T. reesei. The authors (8, 14) argue that the position of this domain excludes its direct participation in the recognition of small soluble substrates, although it may be involved in anchoring the enzyme on large polymeric substrates and in thermostability. The insertion loop was advocated to contribute to the solubility and protection from proteolytic cleavage of AaBgl1 (14). Furthermore, functional studies of hybrid enzymes generated by FnIII domain shuffling confirm its role in the cell wall adhesion (69).
The AnBgl1 FnIII has a considerable solvent-exposed surface, with a large number of charged residues and extensive nonpolar portions. The structure is rich in arginine residues exposed to the solvent, both in this enzyme and in its homolog from T. neopolitana, with eight and nine exposed arginines, respectively. Our molecular dynamics experiments suggested that these positively charged residues may adhere to the noncellulosic lignin polymeric matrix, mainly through cation-π stacking between the guanidinium cation and the aromatic rings from LGG or LSS. This kind of interaction has been identified in the literature to be an important auxiliary factor in protein stability and ligand-protein interactions (70, 71). Moreover, it has been suggested (72) that the molecular structure of arginine in solution is responsible for its capacity to enhance solubility of small organic drugs and proteins. These authors (70–72) have shown that arginine solubilizes fullerene molecules preferentially through hydrophobic and dispersion interactions of the guanidinium group, whereas the polar part of arginine points toward the solvent and creates a hydrophilic shell, working as an amphiphilic molecule. Our simulations suggest that FnIII might bind lignin in a nonspecific manner at the multiple binding sites in which cation-π stacking and hydrogen bonding may simultaneously contribute to stabilize lignin-FnIII adhesion through the arginine residues. In addition, the hydrophobic contacts between the indole groups of tryptophan residues and the lignin aromatic rings are also important stabilizing factors of lignin-FnIII binding.
The fibronectin type III domain is found in a number of proteins, frequently involved in mediation of protein-protein interactions. Often, this domain appears in multiple copies among different families of bacterial glycohydrolases, serving also as a long spacer between catalytic and substrate-binding modules (73).
Kataeva et al. (74) demonstrated the effects of the FnIII module from the Clostridium thermocellum cellobiohydrolase CbhA on cellulose. The authors (74) showed that FnIII might be able to exfoliate and separate cellulose chains, exposing additional sites of cellulose for hydrolysis by catalytic domain. The same authors (74) also demonstrated that stronger effects of FnIII are observed when it is covalently attached to the catalytic domain. It might be that this domain has low binding affinity to cellulose, which explains the weaker effects found when the FnIII was simply mixed with the catalytic during the hydrolysis. Zhou et al. (75) showed that deletion of the FnIII domain between GH9-CBM3c and CBM2 in Thermobifida fusca Cel9A decreased activity on the carboxymethylcellulose and on the crystalline substrate bacterial microcrystalline cellulose.
A novel multidomain GHF10 xylanase (XylK1) from Cellulosimicrobium sp., which was isolated from the digestive tract of the earthworm Eisenia fetida, was recently described (76). The isolated enzyme consists of three putative functional domains (an N-terminal GHF10 domain, an FnIII domain, and C-terminal CBM2), and an FnIII domain was shown to be an important factor for cellulose hydrolysis. The molecular architecture of XylK1 indicates that it is a unique GHF10 enzyme with an FnIII domain, and also the one presenting important activities that differentiate it from the other known GH10 xylanases. At the same time, the presence of FnIII had no measurable effect on thermostability or cellulase activity of chimeras constructed by linking cellulases Clostridium cellulolyticum Cel9G, T. fusca Cel9A, and C. thermocellum Cel9I to T. fusca FnIII (77).
Our molecular dynamics simulations suggest that the AnBgl1 FnIII is important for anchoring the domain to the lignin matrix. Our results revealed that, although there is no specificity in the complexation of the FnIII to lignin, there are important lignin-binding sites. One such relevant region is the longest loops of the domain (loops between C and D and between E and F strands), which easily accommodate one or more lignin dimers by forming a hydrophobic cage surrounded by aromatic residues and arginines. Our results suggest the existence of specific residues whose mutations would likely decrease the affinity of the AnBgl1 FnIII-like domain toward lignin. It would be interesting to conduct site-directed mutagenesis experiments to test this hypothesis.
In summary, we describe here for the first time the extended cellulase-like molecular organization of AnBgl1 and argue that its modular architecture composed of the C-terminal catalytic domain connected to a FnIII-like C-terminal domain by a long and extended linker might serve to direct the enzyme to the lignin portion of the cell wall and to bind to it. Such β-glucosidase anchoring would not only be useful for preventing unproductive binding of exo- and endocellulases to the lignin matrix, but also to facilitate immobilization of the AnBgl1 on the cell wall, a place where cellobiose is produced in the process of biomass depolymerization by the concerted action of exo- and endoglucanases. Because all these enzyme types are simultaneously produced extracellularly by the fungal source, such biochemical synergy in biomass saccharification might be evolutionary advantageous. Clearly, further biochemical studies are needed to verify this hypothesis.
Acknowledgment
We thank the Laboratório Nacional de Luz Síncrotron for beamline time and help with the x-ray data collection.
This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo Grants 08/56255-9, 09/54035-4, and 10/16542-9], Conselho Nacional de Desenvolvimento Científico e Tecnológico Grant 490022/2009-0, Coordenação de Aperfeiçoamento de Pessoal de Nivel Superior and Instituto Nacional de Ciência e Tecnologia do Bioetanol.

This article contains supplemental Figs. S1 and S2 and Table S1.
J. Gorman and L. Shapiro, unpublished data.
M. Sandgren, T. Kaper, N. E. Mikkelsen, H. Hansson, K. Piens, M. Gudmundsson, E. Larenas, B. Kelemen, and S. Karkehabadi, unpublished data.
R. P. McAndrew, J. I. Park, W. Reindl, G. D. Friedland, P. D'haeseleer, T. Northen, K. L. Sale, B. A. Simmons, and P. D. Adams, unpublished data.
- PDB
- Protein Data Bank
- FnIII
- fibronectin III-like domain
- SAXS
- small angle x-ray scattering
- APTS
- 8-aminopyreno-1,3,6-trisulfonic acid
- DAM
- dummy atom model
- RBM
- rigid body model
- LGG
- guaiacyl
- LSS
- syringyl
- CBM
- cellulose binding domain.
REFERENCES
- 1. Chauve M., Mathis H., Huc D., Casanave D., Monot F., Lopes Ferreira N. (2010) Comparative kinetic analysis of two fungal β-glucosidases. Biotechnol. Biofuels 3, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Korotkova O. G., Semenova M. V., Morozova V. V., Zorov I. N., Sokolova L. M., Bubnova T. M., Okunev O. N., Sinitsyn A. P. (2009) Isolation and properties of fungal β-glucosidases. Biochemistry 74, 569–577 [DOI] [PubMed] [Google Scholar]
- 3. Jeoh T., Michener W., Himmel M. E., Decker S. R., Adney W. S. (2008) Implications of cellobiohydrolase glycosylation for use in biomass conversion. Biotechnol. Biofuels 1, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Le Traon-Masson M. P., Pellerin P. (1998) Purification and characterization or two β-d-glucosidases from an Aspergillus niger enzyme preparation: affinity and specificity toward glucosylated compounds of the processing of fruits. Enzyme Microbiol. Technol. 22, 374–382 [Google Scholar]
- 5. Wang Q., Trimbur D., Graham R., Warren R. A., Withers S. G. (1995) Identification of the acid/base catalyst in Agrobacterium faecalis β-glucosidase by kinetic analysis of mutants. Biochemistry 34, 14554–14562 [DOI] [PubMed] [Google Scholar]
- 6. Jeng W. Y., Wang N. C., Lin M. H., Lin C. T., Liaw Y. C., Chang W. J., Liu C. I., Liang P. H., Wang A. H. (2011) Structural and functional analysis of three β-glucosidases from bacterium Clostridium cellulovorans, fungus Trichoderma reesei and termite Neotermes koshunensis. J. Struct. Biol. 173, 46–56 [DOI] [PubMed] [Google Scholar]
- 7. Nakatani Y., Cutfield S. M., Cowieson N. P., Cutfield J. F. (2012) Structure and activity of exo-1,3/1,4-β-glucanase from marine bacterium Pseudoalteromonas sp. BB1 showing a novel C-terminal domain. FEBS J. 279, 464–478 [DOI] [PubMed] [Google Scholar]
- 8. Bacik J.-P., Whitworth G. E., Stubbs K. A., Vocadlo D. J., Mark B. L. (2012) Active site plasticity within the glycoside hydrolase NagZ underlies a dynamic mechanism of substrate distortion. Chem. Biol. 19, 1471–1482 [DOI] [PubMed] [Google Scholar]
- 9. Zmudka M. W., Thoden J. B., Holden H. M. (2013) The structure of DesR from Streptomyces venezuelae, a β-glucosidase involved in macrolide activation. Protein Sci. 22, 883–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Litzinger S., Fischer S., Polzer P., Diederichs K., Welte W., Mayer C. (2010) Structural and kinetic analysis of Bacillus subtilis N-acetylglucosaminidase reveals a unique Asp-His dyad mechanism. J. Biol. Chem. 285, 35675–35684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pozzo T., Pasten J. L., Karlsson E. N., Logan D. T. (2010) Structural and functional analyses of β-glucosidase 3B from Thermotoga neapolitana: a thermostable three-domain representative of glycoside hydrolase 3. J. Mol. Biol. 397, 724–739 [DOI] [PubMed] [Google Scholar]
- 12. Varghese J. N., Hrmova M., Fincher G. B. (1999) Three-dimensional structure of a barley β-d-glucan exohydrolase, a family 3 glycosyl hydrolase. Structure 7, 179–190 [DOI] [PubMed] [Google Scholar]
- 13. Yoshida E., Hidaka M., Fushinobu S., Koyanagi T., Minami H., Tamaki H., Kitaoka M., Katayama T., Kumagai H. (2010) Role of a PA14 domain in determining substrate specificity of a glycoside hydrolase family 3 β-glucosidase from Kluyveromyces marxianus. Biochem. J. 431, 39–49 [DOI] [PubMed] [Google Scholar]
- 14. Suzuki K., Sumitani J., Nam Y.-W., Nishimaki T., Tani S., Wakagi T., Kawaguchi T., Fushinobu S. (2013) Crystal structures of glycoside hydrolase family 3 β-glucosidase 1 from Aspergillus aculeatus. Biochem. J. 452, 211–221 [DOI] [PubMed] [Google Scholar]
- 15. Ketudat Cairns J. R., Esen A. (2010) β-Glucosidases. Cell. Mol. Life Sci. 67, 3389–3405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dan S., Marton I., Dekel M., Bravdo B. A., He S., Withers S. G., Shoseyov O. (2000) Cloning, expression, characterization, and nucleophile identification of family 3, Aspergillus niger β-glucosidase. J. Biol. Chem. 275, 4973–4980 [DOI] [PubMed] [Google Scholar]
- 17. Kunst A., Draeger B., Ziegenhorn J. (1984) Colorimetric methods with glucose oxidase and peroxidase. Methods Enzymatic Analysis, 178–185 [Google Scholar]
- 18. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 19. Vaz Meirelles G., Ferreira Lanza D. C., da Silva J. C., Santana Bernachi J., Paes Leme A. F., Kobarg J. (2010) Characterization of hNek6 interactome reveals an important role for its short N-terminal domain and colocalization with proteins at the centrosome. J. Proteome Res. 9, 6298–6316 [DOI] [PubMed] [Google Scholar]
- 20. Aragão A. Z., Nogueira M. L., Granato D. C., Simabuco F. M., Honorato R. V., Hoffman Z., Yokoo S., Laurindo F. R., Squina F. M., Zeri A. C., Oliveira P. S., Sherman N. E., Paes Leme A. F. (2012) Identification of novel interaction between ADAM17 (a disintegrin and metalloprotease 17) and thioredoxin-1. J. Biol. Chem. 287, 43071–43082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hong J., Tamaki H., Kumagai H. (2007) Cloning and functional expression of thermostable β-glucosidase gene from Thermoascus aurantiacus. Appl. Microbiol. Biotechnol. 73, 1331–1339 [DOI] [PubMed] [Google Scholar]
- 22. Foreman P. K., Brown D., Dankmeyer L., Dean R., Diener S., Dunn-Coleman N. S., Goedegebuur F., Houfek T. D., England G. J., Kelley A. S., Meerman H. J., Mitchell T., Mitchinson C., Olivares H. A., Teunissen P. J., Yao J., Ward M. (2003) Transcriptional regulation of biomass-degrading enzymes in the filamentous fungus Trichoderma reesei. J. Biol. Chem. 278, 31988–31997 [DOI] [PubMed] [Google Scholar]
- 23. Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 [DOI] [PubMed] [Google Scholar]
- 24. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 25. Hammersley A. P. (1997) FIT2D V10.2 Reference Manual V4.0. ESRF Internal Report, ESRF97HA02T. pp. 1–306, European Synchrotron Radiation Facility, Grenoble, France [Google Scholar]
- 26. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H., Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 27. Svergun D. (1992) Determination of the regularization parameter in indirect transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 28. Guinier A., Fournet G. (eds) (1955) Small Angle Scattering of X-rays, pp. 1–268, John Wiley & Sons, Inc., New York [Google Scholar]
- 29. Fischer H., de Oliveira Neto M., Napolitano H. B., Polikarpov I., Craievich A. F. (2010) Determination of the molecular weight of proteins in solution from a single small-angle x-ray scattering measurement on a relative scale. J. Appl. Crystallogr. 43, 101–109 [Google Scholar]
- 30. Svergun D. I. (1999) Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 76, 2879–2886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Svergun D., Barberato C., Koch M. H. (1995) CRYSOL–A program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 28, 768–773 [Google Scholar]
- 32. Petoukhov M. V., Svergun D. I. (2005) Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 89, 1237–1250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Svergun D. I., Petoukhov M. V., Koch M. H. (2001) Determination of domain structure of proteins from x-ray solution scattering. Biophys. J. 80, 2946–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. DeLano W. L. (2002) The PyMOL Molecular Graphics System, Version 1.5, DeLano Scientific, San Carlos, CA [Google Scholar]
- 35. Wood T. M., Bhat K. M. (1988) Methods for measuring cellulase activities. Methods Enzymol. 160, 87–112 [Google Scholar]
- 36. Chen F. T., Evangelista R. A. (1995) Analysis of mono- and oligosaccharide isomers derivatized with 9-aminopyrene-1,4,6-trisulfonate by capillary electrophoresis with laser-induced fluorescence. Anal. Biochem. 230, 273–280 [DOI] [PubMed] [Google Scholar]
- 37. Zhu Z., Sathitsuksanoh N. (2009) Direct quantitative determination of adsorbed cellulase on lignocellulosic biomass with its application to study cellulase desorption for potential recycling. Analyst 134, 2267–2272 [DOI] [PubMed] [Google Scholar]
- 38. Vanholme R., Demedts B., Morreel K., Ralph J., Boerjan W. (2010) Lignin biosynthesis and structure. Plant Physiol. 153, 895–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Sangha A. K., Petridis L., Smith J. C., Ziebell A., Parks J. M. (2012) Molecular simulation as a tool for studying lignan. Environ. Prog. Sustainable Energy 31, 47–54 [Google Scholar]
- 40. Trott O., Olson A. J. (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sanner M. (1999) Python: a programming language for software integration and development. J. Mol. Graph. Model. 17, 57–61 [PubMed] [Google Scholar]
- 42. Morris G. M., Huey R., Lindstrom W., Sanner M. F., Belew R. K., Goodsell D. S., Olson A. J. (2009) AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Martínez J. M., Martínez L. (2003) Packing optimization for automated generation of complex system's initial configurations for molecular dynamics and docking. J. Comput. Chem. 24, 819–825 [DOI] [PubMed] [Google Scholar]
- 44. Martínez L., Andrade R., Birgin E. G., Martínez J. M. (2009) PACKMOL: a package for building initial configurations for molecular dynamics simulations. J. Comput. Chem. 30, 2157–2164 [DOI] [PubMed] [Google Scholar]
- 45. Bashford D., Karplus M. (1990) pKa's of ionizable groups in proteins: atomic detail from a continuum electrostatic model. Biochemistry 29, 10219–10225 [DOI] [PubMed] [Google Scholar]
- 46. Gordon J. C., Myers J. B., Folta T., Shoja V., Heath L. S., Onufriev A. (2005) H++: a server for estimating pKa's and adding missing hydrogens to macromolecules. Nucleic Acids Res. 33, W368–W371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fletcher R., Reeves C. M. (1964) Function minimization by conjugate gradients. Comput. J. 7, 149–154 [Google Scholar]
- 48. Hestenes M., Stiefel E. (1952) Methods of conjugate gradients for solving linear systems. J. Res. Natl. Bureau Stand. 49, 409–436 [Google Scholar]
- 49. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Bleicher L., Prates E. T., Gomes T. C., Silveira R. L., Nascimento A. S., Rojas A. L., Golubev A., Martínez L., Skaf M. S., Polikarpov I. (2011) Molecular basis of the thermostability and thermophilicity of laminarinases: x-ray structure of the hyperthermostable laminarinase from Rhodothermus marinus and molecular dynamics simulations. J. Phys. Chem. B 115, 7940–7949 [DOI] [PubMed] [Google Scholar]
- 51. Mackerell A. D., Jr., Feig M., Brooks C. L., 3rd (2004) Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 25, 1400–1415 [DOI] [PubMed] [Google Scholar]
- 52. MacKerell A. D., Bashford D., Bellott R., Dunbrack L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T., Mattos C., Michnick S., Ngo T., Nguyen D. T., Prodhom B., Reiher W. E., Roux B., Schlenkrich M., Smith J. C., Stote R., Straub J., Watanabe M., Wiórkiewicz-Kuczera J., Yin D., Karplus M. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. 102, 3586–3616 [DOI] [PubMed] [Google Scholar]
- 53. Petridis L., Smith J. C. (2009) A molecular mechanics force field for lignin. J. Comput. Chem. 30, 457–467 [DOI] [PubMed] [Google Scholar]
- 54. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L. (1983) Comparison of simple potential functions for stimulating liquid water. J. Chem. Phys. 79, 926–935 [Google Scholar]
- 55. Schneider T., Stoll E. (1978) Molecular dynamics study of a three-dimensional one-component model for distortive phase transitions. Phys. Rev. 17, 1302–1322 [Google Scholar]
- 56. Martyna G. J. (1994) Remarks on “Constant-temperature molecular dynamics with momentum conservation. Phys. Rev. 50, 3234–3236 [DOI] [PubMed] [Google Scholar]
- 57. Tuckerman M., Berne B. J., Martyna G. J. (1992) Reversible multiple time scale molecular dynamics. J. Chem. Phys. 97, 1990–2001 [Google Scholar]
- 58. Ryckaert J. P., Ciccotti G., Berendsen H. (1977) Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 [Google Scholar]
- 59. Darden T., York D., Pedersen L. (1993) Particle mesh Ewald–an N.Log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 [Google Scholar]
- 60. Himmel M. E., Adney W. S., Fox J. W., Mitchell D. J., Baker J. O. (1993) Isolation and characterization of two forms of β-d-glucosidase from Aspergillus niger. Appl. Biochem. Biotechnol. 39, 213–225 [DOI] [PubMed] [Google Scholar]
- 61. Nimlos M. R., Beckham G. T., Matthews J. F., Bu L., Himmel M. E., Crowley M. F. (2012) Binding preferences, surface attachment, diffusivity, and orientation of a family 1 carbohydrate-binding module on cellulose. J. Biol. Chem. 287, 20603–20612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sorensen A. (2010) A New Highly Efficient β-Glucosidase from the Novel Species Aspergillus Saccharolyticus, Ph.D. thesis Aalborg University, Copenhagen [Google Scholar]
- 63. Lymar E. S., Li B., Renganathan V. (1995) Purification and characterization of a cellulose-binding β-glucosidase from cellulose-degrading cultures of phanerochaete chrysosporium. Appl. Environ. Microbiol. 61, 2976–2980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Receveur V., Czjzek M., Schülein M., Panine P., Henrissat B. (2002) Dimension, shape, and conformational flexibility of a two domain fungal cellulase in solution probed by small angle x-ray scattering. J. Biol. Chem. 277, 40887–40892 [DOI] [PubMed] [Google Scholar]
- 65. Pilz I., Schwarz E., Kilburn D. G., Miller R. C., Jr., Warren R. A., Gilkes N. R. (1990) The tertiary structure of a bacterial cellulase determined by small-angle x-ray-scattering analysis. Biochem. J. 271, 277–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Bhatia Y., Mishra S., Bisaria V. S. (2002) Microbial β-glucosidases: cloning, properties, and applications. Crit. Rev. Biotechnol. 22, 375–407 [DOI] [PubMed] [Google Scholar]
- 67. Kubicek C. P., Messner R., Gruber F., Mach R. L., Kubicek-Pranz E. M. (1993) The Trichoderma cellulase regulatory puzzle: from the interior life of a secretory fungus. Enzyme Microb. Technol. 15, 90–99 [DOI] [PubMed] [Google Scholar]
- 68. Tu M., Chandra R. P., Saddler J. N. (2007) Evaluating the distribution of cellulases and the recycling of free cellulases during the hydrolysis of lignocellulosic substrates. Biotechnol. Prog. 23, 398–406 [DOI] [PubMed] [Google Scholar]
- 69. Marín-Navarro J., Gurgu L., Alamar S., Polaina J. (2011) Structural and functional analysis of hybrid enzymes generated by domain shuffling between Saccharomyces cerevisiae (var. diastaticus) Sta1 glucoamylase and Saccharomycopsis fibuligera Bgl1 β-glucosidase. Appl. Microbiol. Biotechnol. 89, 121–130 [DOI] [PubMed] [Google Scholar]
- 70. Martis R. L., Singh S. K., Gromiha M. M., Santhosh C. (2008) Role of cation-π interactions in single chain “all-α” proteins. J. Theor. Biol. 250, 655–662 [DOI] [PubMed] [Google Scholar]
- 71. Dougherty D. A. (1996) Cation-pi interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp. Science 271, 163–168 [DOI] [PubMed] [Google Scholar]
- 72. Li J., Garg M., Shah D., Rajagopalan R. (2010) Solubilization of aromatic and hydrophobic moieties by arginine in aqueous solutions. J. Chem. Phys. 133, 054902 [DOI] [PubMed] [Google Scholar]
- 73. Campbell I. D., Spitzfaden C. (1994) Building proteins with fibronectin type III modules. Structure 2, 333–337 [DOI] [PubMed] [Google Scholar]
- 74. Kataeva I. A., Seidel R. D., 3rd, Shah A., West L. T., Li X. L., Ljungdahl L. G. (2002) The fibronectin type 3-like repeat from the Clostridium thermocellum cellobiohydrolase CbhA promotes hydrolysis of cellulose by modifying its surface. Appl. Environ. Microbiol. 68, 4292–4300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Zhou W., Irwin D. C., Escovar-Kousen J., Wilson D. B. (2004) Kinetic studies of Thermobifida fusca Cel9A active site mutant enzymes. Biochemistry 43, 9655–9663 [DOI] [PubMed] [Google Scholar]
- 76. Kim do Y., Han M. K., Park D. S., Lee J. S., Oh H. W., Shin D. H., Jeong T. S., Kim S. U., Bae K. S., Son K. H., Park H. Y. (2009) Novel GH10 xylanase, with a fibronectin type 3 domain, from Cellulosimicrobium sp. strain HY-13, a bacterium in the gut of Eisenia fetida. Appl. Environ. Microbiol. 75, 7275–7279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Mingardon F., Bagert J. D., Maisonnier C., Trudeau D. L., Arnold F. H. (2011) Comparison of family 9 cellulases from mesophilic and thermophilic bacteria. Appl. Environ. Microbiol. 77, 1436–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]