

Background: The role of Slfn genes in antineoplastic responses is yet to be defined.
Results: Murine Slfn genes are up-regulated in an IFN-dependent manner in melanoma, and RCC cells and members of this family differentially suppress proliferation and anchorage-independent malignant cell growth.
Conclusion: Slfns play important roles in controlling malignant melanoma and RCC tumorigenesis.
Significance: Modulation of Slfn expression may provide a novel approach for the treatment of malignancies.
Keywords: Antiviral Agents, Cell Death, Innate Immunity, Interferon, Signal Transduction, Signaling, Schlafen
Abstract
There is emerging evidence that the IFN-inducible family of Slfn genes and proteins play important roles in cell cycle progression and control of cellular proliferation, but the precise functional roles of different Slfn members in the regulation of tumorigenesis remain unclear. In the present study, we undertook a systematic analysis on the expression and functional relevance of different mouse Slfn genes in malignant melanoma and renal cell carcinoma cells. Our studies demonstrate that several mouse Slfn genes are up-regulated in response to IFN treatment of mouse melanoma and renal cell carcinoma cells, including Slfn1, Slfn2, Slfn4, Slfn5, and Slfn8. Our data show that Slfn2 and Slfn3 play essential roles in the control of mouse malignant melanoma cell proliferation and/or anchorage-independent growth, suggesting key and non-overlapping roles for these genes in the control of malignant melanoma tumorigenesis. In renal cell carcinoma cells, in addition to Slfn2 and Slfn3, Slfn5 also exhibits important antineoplastic effects. Altogether, our findings indicate important functions for distinct mouse Slfn genes in the control of tumorigenesis and provide evidence for differential involvement of distinct members of this gene family in controlling tumorigenesis. They also raise the potential of future therapeutic approaches involving modulation of expression of members of this family of genes in malignant melanoma and renal cell carcinoma.
Introduction
The family of Schlafen (Slfn) genes was first described in mouse cells, in studies that demonstrated up-regulation of members of this family during thymocyte maturation and T cell development (1). At that time, it was shown that Slfn expression in thymoma cells and fibroblasts suppresses cell proliferation (1). It is now well established that members of the Slfn family are widely expressed in mammals (2). There are currently many known Slfn genes in mice and humans (2). Although the precise function of the different Slfn genes and proteins remains to be established, there is emerging evidence that several members of the family are involved in development, immune response, and cell proliferation (2).
In mouse cells, Slfns are classified in three major groups based on their molecular masses. Slfn1 and -2 belong to group I or short Slfns; Slfn3 and -4 belong to group II or intermediate Slfns; and Slfn5, -8, -9, and -14 to group III or long Slfns (2, 3). All Slfns share a specific slfn box domain, localized adjacent to an AAA domain (3, 4). Groups II and III Slfns share an additional Slfn-specific domain defined by the sequence Ser-Trp-Ala-Asp-Leu, called the SWADL domain (3, 4). Finally, members of group III exhibit a C-terminal extension containing motif, homologous to the superfamily I of DNA/RNA helicases (5–7). Additionally, the C-terminal extensions also harbor a nuclear localization signal (RKRRR) further supporting the notion of nuclear function for the group of long Slfns (7).
The mouse members of the Slfn family that have been most extensively studied are Slfn1, Slfn2, and Slfn3 (4, 8–15). Slfn2 protein appears to play a role in maintaining quiescence in cells of the immune response, as mice harboring a Slfn2 mutation (elektra) are more susceptible to both bacterial and viral infections, which is a consequence of T cells undergoing apoptosis via the intrinsic pathway (10). More evidence that Slfn2 is involved in the immune response is the observation that this member of the Slfn family is up-regulated by LPS, in an NF-kB- and AP-1-dependent manner (6). There is also recent evidence that Slfn2 is an IFN-inducible gene and suppresses cyclin D1 expression, suggesting that it plays an important role in the control of cell cycle progression (9). Additionally, Slfn2 has been shown to be induced by RANKL, a member of the TNF family of cytokines, and is involved in osteoclastogenesis through increasing c-Jun activation and NFATc1 expression (8). Regulation of cell cycle via control of cyclin D1 expression has been also described in the case of Slfn1 (4). Slfn3 has been mostly implicated in differentiation as it is involved in the control of differentiation of the intestinal epithelial cells as well as in T cell differentiation (13–15), whereas it has been postulated that Slfn3 may provide a new marker for T cell activation (15). The only evidence for regulatory effects of Slfn3 on cell growth stems from prior work, in which ectopic expression of this Slfn member in human colon cancer cells inhibited cellular growth (16). The function of Slfn4 is not well known at this time, as there is only one report showing its involvement in macrophage activation and differentiation (17). Similarly, the functions of the remaining members of the Slfn family are largely unknown.
As there are no consistent homology patterns between mouse and human Slfn genes, it has been difficult to define the evolution of this gene family and its precise role in immune surveillance against cancer. In previous studies, we demonstrated that among human SLFNs, SLFN5 is selectively up-regulated in response to IFN treatment in malignant melanoma cells (18). In the present study, we undertook a systematic analysis on the IFN-inducible expression and biological relevance of different mouse Slfn genes in mouse malignant melanoma and renal cell carcinoma (RCC)2 cells. Our results demonstrate that Slfns 1, 2, 4, 5, and 8 are induced by IFNα in malignant melanoma as well as in RCC cells. Knockdown of group I and II Slfns (Slfn2, Slfn3) results in enhanced cell proliferation and anchorage-independent malignant melanoma cell growth, suggesting important roles for these Slfn subgroups in the regulation tumorigenesis and the control of neoplastic cell growth. However, in renal cell carcinoma, a member of group III Slfns, Slfn5, also appears to regulate cell proliferation and anchorage-independent growth suggesting differential involvement and requirements for distinct Slfn proteins in controlling malignant cell proliferation in different cell types.
MATERIALS AND METHODS
Cell Lines, Reagents, and Antibodies
Mouse melanoma cells (B16-F1) were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum and antibiotics. The melanocytic cell line (L10BIOBR) was cultured in Ham's F-10 medium supplemented with 50 ng/ml phorbol 12-myristate 13-acetate with 7% horse serum and antibiotics. The Cloudman S91 melanoma cells (clone M3) were cultured in high-glucose Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 2 mm l-glutamine, and antibiotics. The renal cell carcinoma cell line (RENCA) was maintained in RPMI 1640 supplemented with 10% fetal bovine serum, antibiotics, sodium pyruvate, nonessential amino acids, and l-glutamine. The murine kidney carcinoma cell line (CLS-CD3575) was grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 2 mm l-glutamine, and antibiotics. A polyclonal antibody against Slfn2 has been described previously (9). Antibodies to GAPDH were obtained from Millipore.
Cell Proliferation Assays
Cell counting was performed using a Scepter Handheld Automatic Cell Counter (version 2.0, EMD Millipore). Assays using the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-tetra-zolium bromide method were performed after treating cells with IFNα for 5 to 7 days, as in our previous studies (9, 18).
RNAi-mediated Knockdown of Target Genes
Transient knockdown of Slfn2, Slfn3, and Slfn5 was performed using a pool of three target-specific siRNAs for each Slfn as well as non-targeting control pool siRNA. Validated, inventoried siRNA pools were purchased from Santa Cruz Biotechnology and used according to the manufacturer's instructions. The siRNA used were as follows: Slfn2, sc-40925; Slfn3, sc-40926; Slfn5, sc-153590; and control siRNA, sc-44230. siRNA was introduced to the cells using Lipofectamine RNAiMAX according to the manufacturer's instructions (Invitrogen).
RNA Isolation and Real Time PCR
Cells were treated with 1 × 103 IU/ml IFNα, as indicated. Isolation of RNA and conversion into cDNA was performed using the respective kits from Qiagen, according to the manufacturer's instructions. Validated, inventoried probes and primers for real time PCR and Taqman PCR master mix were purchased from Applied Biosystems and used according to the manufacturer's instructions. The probes and primers used were as follows: GAPDH, Mm99999915_g1; Slfn1, Mm00488306_m1; Slfn2, Mm00488307_m1; Slfn3, Mm00488309_g1; Slfn4, Mm01298330_m1; Slfn5, Mm00806095_m1; Slfn8, Mm00824405_m1. GAPDH was used as an internal control.
Soft Agar Assays
Anchorage-independent cell growth was analyzed in soft agar assays as described previously (9, 18, 19). Equal number of cells were plated in soft agar and allowed to form colonies for 14 days, and colony formation was assessed.
Cell Lysis and Immunoblotting
Cell lysis in phosphorylation lysis buffer and immunoblotting using an ECL kit for protein detection were performed as described previously (20, 21).
Statistical Analysis
Results were expressed as means ± S.E., with the number of experiments indicated in each figure. Statistical significance was assessed using paired t test analysis.
RESULTS
To determine the inducible patterns of expression of Slfns in response to type I IFN treatment of murine melanoma cells, we assessed the mRNA expression profile of different Slfns in B16-F1 cells treated with IFNα for different times. Slfn1, Slfn2, Slfn4, Slfn5, and Slfn8 were all strongly IFN-inducible, whereas there was no significant induction of Slfn3 (Fig. 1, A–F). The peak of induction for all inducible Slfn genes was seen at 6 h of treatment (Fig. 1). Notably, in the case of Slfn2, a biphasic pattern of induction was seen with a second peak noticeable at 24 h (Fig. 1A). The intensity of induction was similar for Slfn2, Slfn4, Slfn8, and Slfn1 (Fig. 1, A, C, E, and F), whereas the induction of Slfn5 was the highest (Fig. 1D). When the induction of different mouse Slfns in another murine melanoma cell line, clone M3, was studied, we found very high levels of expression of Slfn5 (Fig. 2A). Slfn1, 2, and 4 transcripts were undetectable at base line, but Slfn2 and 4 increased after IFN treatment (Fig. 2B). Similarly, when the immortalized murine melanocyte cell line L10BIOBR was studied, we found IFN-dependent up-regulation of Slfn2, 3, 4, and 8 (Fig. 2C). Slfn1 and 5 transcripts were undetectable at base line in these cells, but the expression of both of them increased and was detectable after IFN treatment (Fig. 2D).
FIGURE 1.

Type I IFN-dependent expression of mouse Slfn genes in malignant melanoma cells. B16-F1 cells were treated with IFNα for the indicated times. Expression of Slfn2 (A), Slfn3 (B), Slfn4 (C), Slfn5 (D), Slfn8 (E), and Slfn1 (F) was measured by quantitative real-time RT-PCR using specific primers and was normalized to GAPDH. Data are expressed as fold increase compared with untreated samples and represent means ± S.E. of the indicated number of experiments: Slfn2 (n = 4), Slfn3 (n = 4), Slfn4 (n = 4), Slfn5 (n = 4), Slfn8 (n = 4), and Slfn1 (n = 2).
FIGURE 2.

Type I IFN-dependent expression of mouse Slfn genes in malignant melanoma and immortalized murine melanocyte cells. Clone M3 and L10BIOBR cells were treated with IFNα for the indicated time. Expression of Slfn3, Slfn5, and Slfn8 (A); Slfn1, Slfn2, and Slfn4 (B) in clone M3; and the expression of Slfn2, Slfn3, Slfn4, and Slfn8 (C); and Slfn1 and Slfn5 (D) in L10BIOBR were measured by quantitative real time RT-PCR using specific primers and were normalized to GAPDH. Data are expressed as fold increase compared with untreated (UT) samples and represent means ± S.E. of the indicated number of experiments: Slfn3, Slfn5, Slfn8, Slfn1, and Slfn4 (n = 4) and Slfn2 (n = 3). Und, undetected.
To determine the potential regulatory effects of different Slfns on proliferation and survival of malignant melanoma cells, we used specific siRNAs to knock down the expression of selected Slfn genes (supplemental Fig. S1) and assess responses in their presence or absence. Using siRNAs against specific sequences in distinct Slfns, selective targeting and specificity was achievable to a large extent (supplemental Fig. S1), allowing evaluation of distinct Slfns in the generation of growth inhibitory responses in B16-F1 malignant melanoma cells. As shown in Fig. 3A, mouse melanoma cells exhibited higher proliferation rates when Slfn2 and Slfn3 were individually knocked down, whereas mouse Slfn5 knockdown did not significantly affect malignant melanoma cell proliferation (Fig. 3A). Similar results were obtained in studies in which cell viability/cell proliferation was assessed using MTT assays. Knockdown of Slfn2 (Fig. 3B) or Slfn3 (Fig. 3C) melanoma cells resulted in enhanced cellular proliferation. Also, cells in which either Slfn2 or Slfn3 were knocked down were less sensitive to IFN-induced anti-proliferative responses (Fig. 3, B and C). However, selective knockdown of mouse Slfn5 (Fig. 3D) did not augment malignant melanoma cell growth and did not reverse the inhibitory effects of mouse IFNα (Fig. 3D).
FIGURE 3.

Effect of knockdown of different mouse Slfns on cell proliferation of malignant melanoma cells. A, B16-F1 cells were transfected with control siRNAs or siRNAs specific for Slfn2, Slfn3, or Slfn5 as indicated. 48 h post-transfection, an equal number of cells was plated and allowed to proliferate for 72 h, at which time point the cells were counted. Data are expressed as % control siRNA samples and represent means ± S.E. of four experiments. *, p < 0.05 versus control siRNA cells. B–D, B16-F1 cells transfected with siRNAs against the indicated Slfn genes were treated with indicated doses of IFNα, and proliferation was measured by MTT assays. Data are expressed as % untreated (UT) control (CTL) siRNA samples and represent means ± S.E. of the several experiments. *, p < 0.05, Slfn2 (n = 5), Slfn3 (n = 5), and Slfn5 (n = 3). Und, undetected.
To assess the potential regulatory effects of different mouse Slfn genes on anchorage-independent malignant melanoma cell growth, different Slfn genes were knocked down using specific siRNAs, and colony formation in soft agar was determined (9, 20). In cells in which either Slfn2 or Slfn3 were knocked down, there was marked increase in colony numbers, reflecting anchorage-independent cell growth (Fig. 4, A–D), whereas Slfn5 knockdown did not have significant effects (Fig. 4, C and D). Similarly, knockdown of Slfn3 increased colony formation from clone M3 cells, whereas as in the case of B16-F1 cells, Slfn5 knockdown had no significant effects on colony formation (Fig. 4E). Thus, members of the mouse Slfn family appear to play regulatory roles on anchorage-independent malignant melanoma cell growth, underscoring their relevance in malignant melanoma tumorigenesis.
FIGURE 4.

Effects of knockdown of different Slfn genes on anchorage-independent malignant melanoma cell growth. A, B16-F1 cells were transfected with control siRNA or Slfn2 siRNA. Equal number of cells were plated in soft agar and allowed to form colonies for 14 days. Representative images of the soft agar wells are shown. B, quantitation of colonies from four independent experiments, including the one shown in A. Data are expressed as % control siRNA transfected cells and represent means ± S.E. of four experiments. *, p < 0.05. C, B16-F1 cells were transfected with control siRNA or Slfn3 or Slfn5 siRNAs. An equal number of cells was plated in soft agar and allowed to form colonies for 14 days. Representative images of the soft agar wells are shown. D, quantitation of colonies from four independent experiments, including the one shown in C. Data are expressed as % control siRNA transfected cells and represent means ± S.E. of four experiments. *, p < 0.05. E, clone M3 cells were transfected with control (CTL) siRNA or Slfn3 or Slfn5 siRNAs. An equal number of cells was plated in soft agar, and colonies from four independent experiments were quantified after 14 days. Data are expressed as % control siRNA-transfected cells and represent means ± S.E. of four experiments. *, p < 0.05.
Beyond malignant melanoma, one of the most IFN-responsive malignant tumors is renal cell carcinoma (22). To examine the patterns of expression of different mouse Slfns in response to type I IFN treatment, RENCA cells were treated for different times, and Slfn mRNA expression was assessed by real-time RT-PCR. As shown in Fig. 5, all different mouse Slfn genes examined (Slfn 2, 3, 4, 5, 8, 1) were induced at variable levels in mouse RENCA renal cell carcinoma cells (Fig. 5). The highest levels of IFN-induced gene expression were seen in the case of Slfn5 (Fig. 5D), whereas Slfn3 was the least inducible member of the family in these cells (Fig. 5B). Different degrees of IFN-dependent up-regulation of different Slfns were also seen in the mouse CD3575 kidney cell carcinoma cell line (Fig. 6). To examine whether different Slfns play regulatory roles on cell proliferation of RCC cells, we knocked down Slfn2, Slfn3, and Slfn5 using specific siRNAs (supplemental Fig. S2). As in the case of melanoma cells, knockdown of either Slfn2 or Slfn3 resulted in enhanced RCC cell proliferation (Fig. 7A). However, in contrast to what was observed in melanoma cells, knockdown of Slfn5 increased the proliferation of RENCA cells (Fig. 7B). In addition, when the regulatory effects of distinct mouse Slfns on anchorage-independent growth of RCC cells were assessed, we found that knockdown of Slfn2, Slfn3, or Slfn5 resulted in substantial enhancement of malignant cell colony formation in RENCA cells (Fig. 8, A and B). Similarly, knockdown of Slfn3 and Slfn5 resulted in significantly enhanced colony formation in CD3575 cells (Fig. 8C). We also performed studies in which we examined the effects of overexpression of Slfn2 (Fig. 9, A and B) on anchorage-independent CD3575 malignant cell growth. Slfn2 overexpression suppressed malignant cell colony formation (Fig. 9, C and D), further establishing a role for Slfns on anchorage-independent malignant cell growth.
FIGURE 5.

Type I IFN-dependent expression of mouse Slfn genes in renal cell carcinoma. A–E, RENCA cells were treated with IFNα for the indicated times. mRNA expression for Slfn2 (A), Slfn3 (B), Slfn4 (C), Slfn5 (D), Slfn8 (E), and Slfn1 (F) was measured by quantitative real-time RT-PCR using specific primers and was normalized to GAPDH. Data are expressed as fold increase over untreated samples. Data represent means ± S.E. of several experiments for each Slfn gene as follows: Slfn2 (n = 4), Slfn3 (n = 4), Slfn4 (n = 4), Slfn5 (n = 3), Slfn8 (n = 4), and Slfn1 (n = 3).
FIGURE 6.
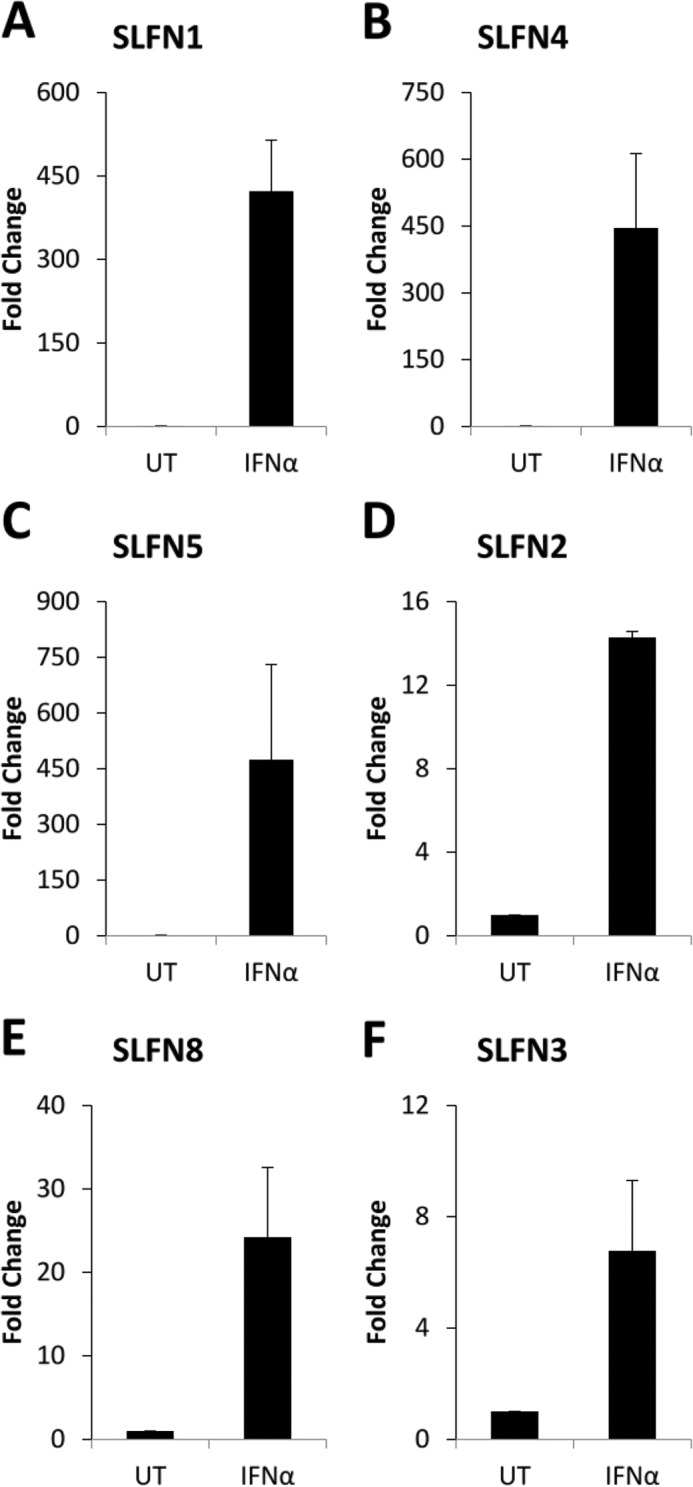
Type I IFN-dependent expression of mouse Slfn genes in kidney carcinoma cells. CD3575 cells were treated with mouse IFNα for the indicated time. Expression of Slfn1 (A), Slfn4 (B), Slfn5 (C), Slfn2 (D), Slfn8 (E), and Slfn3 (F) in CD3575 was measured by quantitative real-time PCR using specific primers and was normalized to GAPDH. Data are expressed as fold increase compared with untreated (UT) samples and represent means ± S.E. of three independent experiments.
FIGURE 7.
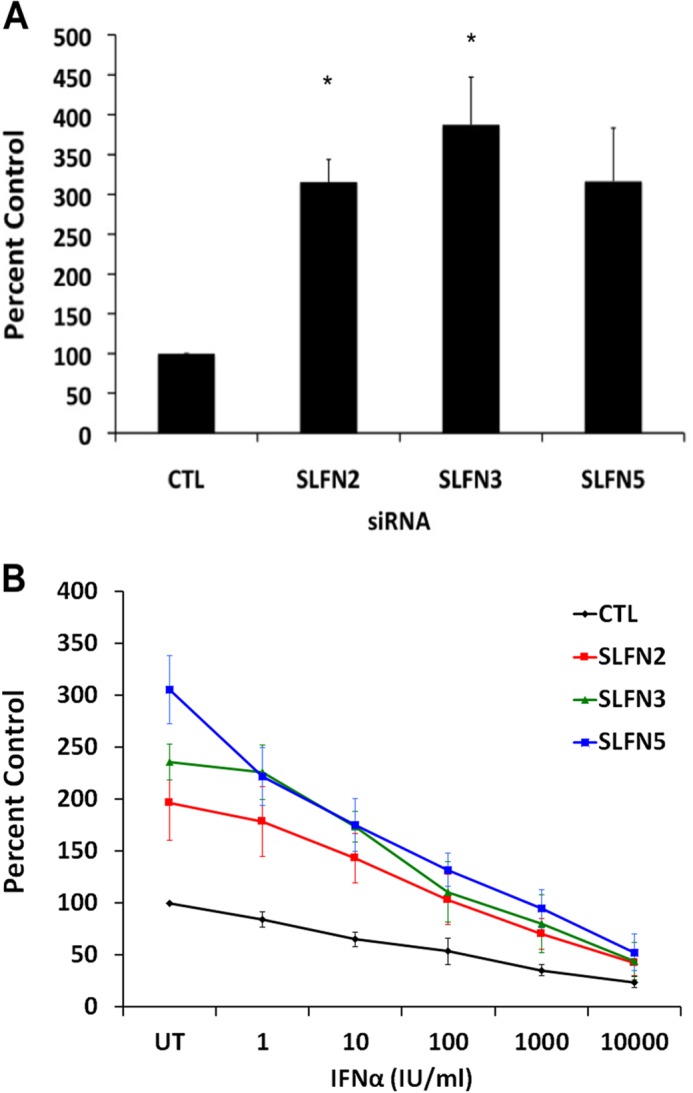
Effects of knockdown of different mouse Slfns on cell proliferation of mouse renal cell carcinoma cells. A, RENCA cells were transfected with control siRNAs or siRNAs specific for Slfn2, Slfn3, or Slfn5 as indicated. 48 h post-transfection, an equal number of cells was plated and allowed to proliferate for 72 h, at which time the cells were counted. Data are expressed as % of control (CTL) siRNA-transfected samples and represent means ± S.E. of three experiments. *, p < 0.05 versus control siRNA cells. B, transfected RENCA cells were treated with indicated doses of IFNα, and viability was assessed by MTT assays. Data are expressed as % untreated control (CTL) siRNA samples and represent means ± S.E. of five experiments. *, p < 0.05 control versus Slfn2, control versus Slfn5, **, p < 0.05 control versus Slfn2, control versus Slfn3, control versus Slfn5.
FIGURE 8.

Effects of knockdown of Slfn2, Slfn3, and Slfn5 on anchorage-independent RCC cell growth. A, RENCA cells were transfected with control siRNA or Slfn2, Slfn3, or Slfn5 siRNAs. Equal number of cells were plated in soft agar and allowed to form colonies for 14 days. Representative images of the soft agar wells are shown. B, quantitation of colonies from four independent experiments, including the one shown in A. Data are expressed as % control siRNA-transfected cells and represent means ± S.E. of four experiments. *, p < 0.05. C, CD3575 cells were transfected with control siRNA or Slfn3 or Slfn5 siRNAs. Equal number of cells were plated in soft agar and colonies were quantified after 14 days. Data are expressed as % control (CTL) siRNA-transfected cells and represent means ± S.E. of four experiments.
FIGURE 9.

Effects of overexpression of mouse Slfn2 on cell proliferation and anchorage-independent growth in kidney carcinoma cells. A and B, CD3575 cells were transiently transfected with either empty vector (E.V.) or Slfn2 cDNA. Levels of Slfn2 were quantified at 48 h post-transfection by quantitative RT-PCR (A) or examined by immunoblotting (B). The quantitative RT-PCR data are expressed as fold increase in the Slfn2-transfected samples over empty vector transfected samples normalized to GAPDH and represent means ± S.E. from two independent experiments performed in triplicate. C, CD3575 cells were transiently transfected with either empty vector (E.V.) or Slfn2 cDNA. An equal number of cells was plated in soft agar and allowed to form colonies for 8 days. Representative images of the soft agar wells are shown. D, quantitation of colonies from three independent experiments, including the one shown in A. Data are expressed as % control empty vector transfected cells and represent means ± S.E. of three experiments performed in triplicate. *, p < 0.05.
DISCUSSION
The Slfn family includes a large and diverse group of genes and proteins, with substantial variability and functional diversity among species. There are several murine Slfns and human Slfns, whereas there is only one Slfn in pigs (2). Virtually all mouse Slfns do not have actual human homologues and vice versa (2), raising the question of whether a functional analogy between mouse and human Slfns exists and/or whether the mouse and human SLFN systems play distinct and species-specific roles. Several mouse Slfn genes, including Slfn1 (4), Slfn 2 (9), and Slfn3 (13, 14, 16) have been shown to play key regulatory roles in control of cell proliferation and/or differentiation. Mouse Slfn2 has been implicated in immune cell quiescence (10), whereas Slfn4 has been implicated in the control of myelopoiesis (17).
Although there continues to be emerging evidence that different mouse Slfn genes play important regulatory functions during normal development, as regulators of the immune, hematopoietic, and other systems, very little is known on their potential involvement in tumorigenesis and whether abnormalities in their expression contribute to the pathophysiology of various malignancies. This may be of particular interest, as several Slfn genes appear to mediate growth inhibitory responses, and, therefore, deregulation of Slfn gene expression or other abnormalities in their function may contribute to the malignant phenotype. As our previous studies have demonstrated that Slfn genes are IFN-inducible (9, 18), we have examined the patterns of induction of different mouse Slfn genes and their functions, in malignant melanoma and renal cell carcinoma cells, diseases that exhibit relatively high sensitivity to IFN treatment as compared with several other malignancies (22–24).
Our studies demonstrate that there is IFN-dependent induction of expression of several murine Slfn genes in malignant melanoma and renal carcinoma cells, including Slfn1, Slfn2, Slfn4, Slfn5, and Slfn8. Importantly, selective knockdown of group I and II Slfns (Slfn2, Slfn3) results in enhanced cell proliferation and anchorage-independent cell growth, raising the possibility that group I Slfns are regulators of growth of malignant melanoma cells. However, in renal cell carcinoma cells, Slfns from all three groups appear to regulate cell proliferation and anchorage-independent malignant cell growth, including Slfn5. Notably, our previous studies have shown that in the human system, only SLFN5 is up-regulated by type I IFNs in malignant melanoma cells and that its inducible expression correlates with IFN responses (18). Human SLFN5 contains an RNA/DNA helicase motif, as well as a nuclear localization signal, which is not seen in group I Slfns but only in group III Slfns in the mouse system (3, 25). Although mouse Slfn5 is not a functional homologue of human SLFN5, the fact that murine Slfn5 does not appear to play a key role in the control of proliferation of mouse malignant melanoma cells, further underscores the functional diversity and differences of function between murine versus human SLFN isoforms. It is of particular interest that selective knockdown of either Slfn2 or Slfn3 was sufficient to promote malignant melanoma cell proliferation, indicating that these mouse Slfns exhibit essential but distinct non-overlapping functions. Moreover, in the case of RCC cells, in addition to Slfn2 and Slfn3, there was no compensatory mechanism for the suppressive effects of mouse Slfn5, indicating an essential role for this member of the family of proteins in the suppression of RCC tumorigenesis.
The precise mechanisms accounting for such selective requirements for distinct Slfns in the control of tumorigenesis remain to be defined in future studies. It is possible that some of their common activities reflect effects on elements of cell cycle progression in malignant melanoma or renal cell carcinoma cells. It is also possible that, beyond their suppressive properties on anchorage-independent malignant cell growth, additional, yet unknown, functions for various murine Slfns may exist, which define their unique biological specificities. Beyond delineating such putative functions in future studies, it would be important to expand this work in other types of tumors, beyond malignant melanoma and RCC. For instance, it would be interesting to examine the role of Slfn family in the pathophysiology of various other type I IFN-sensitive solid tumors, such as carcinoids, as well as hematological malignancies such as chronic myeloproliferative neoplasms and cutaneous T cell lymphoma (26–28). Nevertheless, independently of the precise mechanisms accounting for tissue specificity for different Slfns, identification, or design of pharmaceutical agents that may promote expression of distinct Slfns may ultimately provide a novel therapeutic approach for the treatment of such malignancies.
This work was supported, in whole or in part, by National Institutes of Health Grants CA161796, CA155566, and CA77816. This work was also supported by a Merit Review grant from the Department of Veterans Affairs.

This article contains supplemental Figs. S1 and S2.
- RCC
- renal cell carcinoma
- RENCA
- renal cell carcinoma cell line.
REFERENCES
- 1. Schwarz D. A., Katayama C. D., Hedrick S. M. (1998) Schlafen, a new family of growth regulatory genes that affect thymocyte development. Immunity 9, 657–668 [DOI] [PubMed] [Google Scholar]
- 2. Bustos O., Naik S., Ayers G., Casola C., Perez-Lamigueiro M. A., Chippindale P. T., Pritham E. J., de la Casa-Esperón E. (2009) Evolution of the Schlafen genes, a gene family associated with embryonic lethality, meiotic drive, immune processes and orthopoxvirus virulence. Gene 447, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Geserick P., Kaiser F., Klemm U., Kaufmann S. H., Zerrahn J. (2004) Modulation of T cell development and activation by novel members of the Schlafen (slfn) gene family harbouring an RNA helicase-like motif. Int. Immunol. 16, 1535–1548 [DOI] [PubMed] [Google Scholar]
- 4. Brady G., Boggan L., Bowie A., O'Neill L. A. (2005) Schlafen-1 causes a cell cycle arrest by inhibiting induction of cyclin D1. J. Biol. Chem. 280, 30723–30734 [DOI] [PubMed] [Google Scholar]
- 5. Bell T. A., de la Casa-Esperón E., Doherty H. E., Ideraabdullah F., Kim K., Wang Y., Lange L. A., Wilhemsen K., Lange E. M., Sapienza C., de Villena F. P. (2006) The paternal gene of the DDK syndrome maps to the Schlafen gene cluster on mouse chromosome 11. Genetics 172, 411–423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sohn W. J., Kim D., Lee K. W., Kim M. S., Kwon S., Lee Y., Kim D. S., Kwon H. J. (2007) Novel transcriptional regulation of the schlafen-2 gene in macrophages in response to TLR-triggered stimulation. Mol. Immunol. 44, 3273–3282 [DOI] [PubMed] [Google Scholar]
- 7. Neumann B., Zhao L., Murphy K., Gonda T. J. (2008) Subcellular localization of the Schlafen protein family. Biochem. Biophys. Res. Commun. 370, 62–66 [DOI] [PubMed] [Google Scholar]
- 8. Lee N. K., Choi H. K., Yoo H. J., Shin J., Lee S. Y. (2008) RANKL-induced schlafen2 is a positive regulator of osteoclastogenesis. Cell Signal. 20, 2302–2308 [DOI] [PubMed] [Google Scholar]
- 9. Katsoulidis E., Carayol N., Woodard J., Konieczna I., Majchrzak-Kita B., Jordan A., Sassano A., Eklund E. A., Fish E. N., Platanias L. C. (2009) Role of Schlafen 2 (SLFN2) in the generation of interferon α-induced growth inhibitory responses. J. Biol. Chem. 284, 25051–25064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Berger M., Krebs P., Crozat K., Li X., Croker B. A., Siggs O. M., Popkin D., Du X., Lawson B. R., Theofilopoulos A. N., Xia Y., Khovananth K., Moresco E. M., Satoh T., Takeuchi O., Akira S., Beutler B. (2010) An Slfn2 mutation causes lymphoid and myeloid immunodeficiency due to loss of immune cell quiescence. Nat. Immunol. 11, 335–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Horton M. R., Powell J. D. (2010) Quieting T cells with Slfn2. Nat. Immunol. 11, 281–282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ahmadi S., Veinotte L. L. (2011) Effect of Schlafen 2 on natural killer and T cell development from common T/natural killer progenitors. Pak. J. Biol. Sci. 14, 1002–1010 [DOI] [PubMed] [Google Scholar]
- 13. Patel V. B., Yu Y., Das J. K., Patel B. B., Majumdar A. P. (2009) Schlafen-3: a novel regulator of intestinal differentiation. Biochem. Biophys. Res. Commun. 388, 752–756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yuan L., Yu Y., Sanders M. A., Majumdar A. P., Basson M. D. (2010) Schlafen 3 induction by cyclic strain regulates intestinal epithelial differentiation. Am. J. Physiol. Gastrointest. Liver Physiol. 298, G994-G1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Condamine T., Le Luduec J. B., Chiffoleau E., Bériou G., Louvet C., Heslan M., Tilly G., Cuturi M. C. (2010) Characterization of Schlafen-3 expression in effector and regulatory T cells. J. Leukoc. Biol. 87, 451–456 [DOI] [PubMed] [Google Scholar]
- 16. Patel B. B., Yu Y., Du J., Rishi A. K., Sarkar F. H., Tarca A. L., Wali A., Majumdar A. P. (2009) Schlafen 3, a novel gene, regulates colonic mucosal growth during aging. Am. J. Physiol. Gastrointest. Liver Physiol. 296, G955–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van Zuylen W. J., Garceau V., Idris A., Schroder K., Irvine K. M., Lattin J. E., Ovchinnikov D. A., Perkins A. C., Cook A. D., Hamilton J. A., Hertzog P. J., Stacey K. J., Kellie S., Hume D. A., Sweet M. J. (2011) Macrophage activation and differentiation signals regulate schlafen-4 gene expression: evidence for Schlafen-4 as a modulator of myelopoiesis. PloS One 6, e15723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Katsoulidis E., Mavrommatis E., Woodard J., Shields M. A., Sassano A., Carayol N., Sawicki K. T., Munshi H. G., Platanias L. C. (2010) Role of interferon α (IFNα)-inducible Schlafen-5 in regulation of anchorage-independent growth and invasion of malignant melanoma cells. J. Biol. Chem. 285, 40333–40341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hamburger A. W., Salmon S. E. (1977) Primary bioassay of human tumor stem cells. Science 197, 461–463 [DOI] [PubMed] [Google Scholar]
- 20. Uddin S., Majchrzak B., Woodson J., Arunkumar P., Alsayed Y., Pine R., Young P. R., Fish E. N., Platanias L. C. (1999) Activation of the p38 mitogen-activated protein kinase by type I interferons. J. Biol. Chem. 274, 30127–30131 [DOI] [PubMed] [Google Scholar]
- 21. Verma A., Deb D. K., Sassano A., Uddin S., Varga J., Wickrema A., Platanias L. C. (2002) Activation of the p38 mitogen-activated protein kinase mediates the suppressive effects of type I interferons and transforming growth factor-β on normal hematopoiesis. J. Biol. Chem. 277, 7726–7735 [DOI] [PubMed] [Google Scholar]
- 22. Bukowski RM. (2010) Systemic therapy for metastatic renal cell carcinoma in treatment naïve patients: a risk-based approach. Expert Opin. Pharmacother. 11, 2351–2362 [DOI] [PubMed] [Google Scholar]
- 23. Ascierto P. A., Gogas H. J., Grob J. J., Algarra S. M., Mohr P., Hansson J., Hauschild A. (2013) Adjuvant interferon alfa in malignant melanoma: An interdisciplinary and multinational expert review. Crit. Rev. Oncol. Hematol. 85, 149–161 [DOI] [PubMed] [Google Scholar]
- 24. Moschos S., Varanasi S., Kirkwood J. M. (2005) Interferons in the treatment of solid tumors. Cancer Treat. Res. 126, 207–241 [DOI] [PubMed] [Google Scholar]
- 25. Mavrommatis E, Fish EN, Platanias LC. (2013) The schlafen family of proteins and their regulation by interferons. J. Interferon Cytokine Res. 33, 206–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Parmar S., Platanias L. C. (2003) Interferons: mechanisms of action and clinical applications. Curr. Opin. Oncol. 15, 431–439 [DOI] [PubMed] [Google Scholar]
- 27. Bracarda S., Eggermont A. M., Samuelsson J. (2010) Redefining the role of interferon in the treatment of malignant diseases. Eur. J. Cancer 46, 284–297 [DOI] [PubMed] [Google Scholar]
- 28. Tefferi A., Vainchenker W. (2011) Myeloproliferative neoplasms: molecular pathophysiology, essential clinical understanding, and treatment strategies. J. Clin. Oncol. 29, 573–582 [DOI] [PubMed] [Google Scholar]