

Background: Heparin-treated patients often develop antibodies, but only a subset cause heparin-induced thrombocytopenia.
Results: In a single-molecule assay, a pathogenic monoclonal antibody bound more strongly to cross-linked platelet factor 4 than a non-pathogenic antibody.
Conclusion: Oligomerization of platelet factor 4 may enhance binding of pathogenic antibodies.
Significance: A molecular basis for specificity of pathogenic antibodies in heparin-induced thrombocytopenia is provided.
Keywords: Antibodies, Heparin-binding Protein, Platelets, Single Molecule Biophysics, Thrombosis
Abstract
Heparin-induced thrombocytopenia (HIT) is a thrombotic complication of heparin therapy mediated by antibodies to complexes between platelet factor 4 (PF4) and heparin or cellular glycosaminoglycans. However, only a fraction of patients with anti-PF4-heparin antibodies develop HIT, implying that only a subset of these antibodies is pathogenic. The basis for the pathogenic potential of anti-PF4-heparin antibodies remains unclear. To elucidate the intrinsic PF4-binding properties of HIT-like monoclonal antibody (KKO) versus non-pathogenic antibody (RTO) at the single-molecule level, we utilized optical trap-based force spectroscopy to measure the strength and probability of binding of surface-attached antibodies with oligomeric PF4 to simulate interactions on cells. To mimic the effect of heparin in bringing PF4 complexes into proximity, we chemically cross-linked PF4 tetramers using glutaraldehyde. Analysis of the force histograms revealed that KKO-PF4 interactions had ∼10-fold faster on-rates than RTO-PF4, and apparent equilibrium dissociation constants differed ∼10-fold with similar force-free off-rates (koff = 0.0031 and 0.0029 s−1). Qualitatively similar results were obtained for KKO and RTO interacting with PF4-heparin complexes. In contrast to WT PF4, KKO and RTO showed lower and similar binding probabilities to cross-linked PF4K50E, which forms few if any oligomers. Thus, formation of stable PF4 polymers results in much stronger interactions with the pathogenic antibody without a significant effect on the binding of the non-pathogenic antibody. These results suggest a fundamental difference in the antigen-binding mechanisms between model pathogenic and non-pathogenic anti-PF4 antibodies that might underlie their distinct pathophysiological behaviors.
Introduction
Heparin-induced thrombocytopenia (HIT)2 is an autoimmune thrombotic disorder caused by antibodies (Abs) to complexes between platelet factor 4 (PF4) released from activated platelets and heparin (1) or cellular glycosaminoglycans (GAGs) (2–5). Binding of antibody leads to formation of immune complexes that activate platelets through FcRγIIa receptors in vivo and initiate procoagulant responses by monocytes, endothelial cells, and probably other cell types (6, 7). Remarkably, anti-PF4 Abs develop in many patients exposed to heparin in settings characterized by intense platelet activation and inflammation, such as coronary bypass surgery, yet only ∼1% of patients develop clinical HIT (4, 8–10).
The reason why only a subset of anti-PF4-heparin Abs is associated with HIT is unclear and only partially explained by IgG isotype and titer. We hypothesize that pathogenic potential is largely determined by the affinities of HIT Abs with specific epitopes that are brought into proximity during heparin- or GAG-induced oligomerization of PF4 complexes (11, 12). These interactions may lead to the formation of very large pathogenic immune complexes that promote cellular injury responses.
We have previously generated and characterized two isotype-matched murine anti-human PF4-heparin monoclonal Abs that mimic their human counterparts (13): KKO, which causes HIT in an animal model (7), and RTO, which does not cause HIT in vivo (12). Importantly, ELISA-positive plasma samples from patients suspected of having HIT contain Abs that show heparin-induced binding to PF4, like KKO, and compete with KKO for binding to heparin/PF4 and activate platelets in a heparin- and FcRγIIA-dependent manner, whereas samples that are ELISA-positive but do not activate platelets behave like RTO, show less inhibition of KKO binding to PF4, and do not activate platelets (12, 14, 15). KKO and RTO do not compete for binding to PF4. KKO, unlike RTO, causes PF4 to oligomerize in solution, forming ultralarge complexes (ULCs) that cluster on cell surfaces, which probably activate the cells and predispose to Ab-induced thrombosis (14). This suggests that detailed comparisons between KKO and RTO might help to define the difference between the structure and specificity of pathogenic and non-pathogenic human anti-PF4 antibodies that underlie their dissimilar clinical impact. However, the information obtained from conventional bulk techniques that make use of large ensembles of molecules in equilibrium cannot distinguish subtle functional distinctions of different sets of anti-PF4 antibodies, which, for example, bind to PF4 with the same Bmax in ELISA. The comparison could be substantially enhanced when the studies were performed at the single-molecule level with precise and novel qualitative and quantitative characterization of protein-protein interactions.
We posit that the difference in the behavior of the two types of anti-PF4 Abs is attributable to the capacity of heparin or GAGs to bring the binding site of KKO on individual tetramers in proximity, which substantially enhances its avidity, without affecting the site recognized by RTO (11, 12). Preliminary evidence in support of this explanation for the distinct functionality of KKO and RTO was obtained using nanomechanical measurements of the interactions between surface-bound antibodies and chemically stabilized PF4 tetramers (12). The goal of this study is to further elucidate the molecular basis of the difference between two types of anti-PF4 Abs, one that causes HIT (KKO) and one that does not (RTO) to shed light on the molecular and cellular mechanisms that contribute to pathogenesis.
We used optical trap-based force spectroscopy developed in our laboratory (16, 17) to determine the differences in specificity and the two-dimensional kinetics of pathogenic versus non-pathogenic monoclonal Abs with PF4 at the strictly confirmed single-molecule level. Because these studies measured bimolecular interactions, we were able to detect and measure the strength and probability of binding among individual molecular partners within a complex and distinguish among multiple interacting partners, unlike typical biochemical assays that measure aggregate properties averaged over all interactions occurring simultaneously. We demonstrate directly that the difference in specificity of KKO and RTO is due to polymerization of PF4 that results in ∼10-fold faster kon and stronger interactions with the pathogenic Ab without a significant effect on the binding of the non-pathogenic Ab. These data suggest an amplification reaction in which KKO, unlike RTO, binds preferentially to PF4 tetramers (or higher order complexes), promotes their superoligomerization, and binds with progressively higher avidity as the antigenic complexes grow in size.
EXPERIMENTAL PROCEDURES
Expression and Purification of Human PF4 and Anti-PF4 Monoclonal Antibodies
Wild type (WT) hPF4 and PF4K50E in plasmid pMT/BiP/V5-His (Invitrogen) were expressed using the Drosophila Expression System (Invitrogen), purified, and characterized as described (12). Briefly, the protein was collected in serum-free medium Insect-Xpress (Lonza, Walkersville, MD) and isolated by affinity chromatography using a HiTrap Heparin HP column (GE Healthcare) on an AKTA Prime system (GE Healthcare) at 4 °C and eluted at 1.8 m NaCl (WT PF4) or 1.3 m NaCl (PF4K50E) using a linear gradient. Fractions containing purified PF4 detected by silver staining of 12% polyacrylamide gels (SDS-PAGE) were pooled, concentrated, and buffer-exchanged into 50 mm HEPES, 0.5 m NaCl, pH ∼7.2, using an Amicon Ultra filter (3000 molecular weight cut-off; Millipore). Protein was quantified using a BCA assay (Pierce). KKO and RTO are both mouse IgG2bκ monoclonal anti-human PF4 antibodies (13). The IgG fractions were purified from conditioned PFHM-II media (Invitrogen) using protein A-agarose (Invitrogen) as recommended by the manufacturer. IgG purity was demonstrated by SDS-PAGE on NuPAGE 4–12% BisTris gel (Invitrogen). Fab fragments were generated by digestion with papain using the Pierce® Fab Preparation Kit (Thermo Scientific, Rockford, IL) essentially as recommended by the manufacturer and purified by performing three rounds of removing Fc fragments with protein A-agarose beads and then additional purification with anti-mouse IgG (Fc-specific) (Sigma M4280) and anti-mouse IgG (Fab-specific) Sigma M4155 antibodies bound to CNBr-activated Sepharose 4 Fast Flow beads (Amersham Biosciences) as recommended by the manufacturer.
The Model System to Study Bimolecular Protein-Protein Interactions
To compare the reactivity of IgG antibodies KKO and RTO toward PF4 at the single-molecule level, we applied an optics-based model system to study individual protein-protein interactions (supplemental Fig. S1). This system permits the measurement of discrete rupture forces produced by surface-bound molecular pairs during repeated intermittent contact (16, 17). For these studies, an antigen (WT PF4 or its derivatives) was covalently bound to stationary 5-μm pedestals anchored to the inner surface of a flow chamber. 2-μm latex beads coated covalently with KKO, RTO, or Fab KKO fragment were then flowed into the chamber. One of the latex beads was trapped by a focused laser beam and moved in an oscillatory manner so that the bead was intermittently in contact with a stationary pedestal. The tension produced when an antibody on the latex bead interacted with an antigen on the anchored pedestal was sensed and displayed as a force signal that was proportional to the strength of protein-protein binding (supplemental Figs. S2 and S3). Rupture forces from many interactions were collected and displayed as normalized force distribution histograms for each experimental condition. Details of the optical trap-based single-molecule force spectroscopy and its applications for various interacting molecular pairs can be found elsewhere (12, 16–23).
Measurement of Rupture Forces and Data Processing
Measurements were performed in 100 mm HEPES buffer, pH 7.4, containing 150 mm NaCl, 2 mg/ml BSA, and 0.1% (v/v) Triton X-100. The position of the optical trap and hence a KKO-, RTO-, or Fab KKO-coated latex bead was oscillated in a triangular waveform at 10 Hz with a loading rate of 1600 pN/s. To maximize single-molecule interactions while decreasing the likelihood of multiple interactions during repeated touching events between the protein-coated particles, the surface densities of reacting proteins were deliberately decreased so that the fraction of specific interactions between the two proteins was about 10% of bead-pedestal contacts or less. Because only a small percentage of contact/detachment cycles resulted in effective antigen-antibody binding/unbinding, data from at least 10 bead-pedestal pairs for each experimental condition, representing 6 × 103 to 1.2 × 104 individual measurements, were combined. Optical artifacts observed with or without trapped latex beads produced signals that appeared as forces below 10 pN. Accordingly, rupture forces in this range were not considered when the data were analyzed. Individual forces measured during each contact-detachment cycle were collected into 10-pN-wide bins. The number of events in each bin was plotted against the average force for that bin after normalizing for the total number of interaction cycles. The percentage of events in a particular force range (bin) represents the frequency (probability density) of rupture events at that tension. Details of optical trap design and calibration and the experimental procedure can be found in Ref. 17.
Coating Surfaces with the Proteins
Surfaces coated with the interacting proteins were prepared basically as described previously (17) with modifications. WT PF4 or PF4K50E alone or preincubated with unfractionated heparin (BD PosiFlushTM heparin at a ratio of 50 μg of PF4/1 unit of heparin for 30 min at 37 °C) was bound covalently to spherical silica pedestals 5 μm in diameter anchored to the bottom of a chamber. Pedestals coated with a thin layer of polyacrylamide were activated with 10% glutaraldehyde (4 °C, 10 h), after which the proteins were immobilized for 2 h at 4 °C from 2 mg/ml solution in 50 mm HEPES with 0.5 m NaCl, pH 7.2. After washing, 2 mg/ml bovine serum albumin (BSA) in 0.055 m borate buffer, pH 8.5, was added as a blocker. Before the measurements, the chambers were washed with 20 volumes of 100 mm HEPES, pH 7.4, containing 150 mm NaCl, 2 mg/ml BSA, and 0.1% (v/v) Triton X-100. Antibodies KKO, RTO, or the Fab KKO fragment were bound covalently to carboxylate-modified 2-μm latex beads (Bangs Laboratories, Inc., Fishers, IN) activated in water suspension (0.5% solids) with N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (3 mg/ml, 15 min at room temperature). After washing the beads with 0.055 m borate buffer, pH 8.5, by centrifugation followed by resuspension, the immobilization of antibodies lasted 30 min at room temperature in the same buffer. Solutions of KKO and RTO in the immobilizing mixture with the same freshly activated beads were used in equimolar concentrations (0.13 μm) to keep the same or similar surface densities. Fab KKO was immobilized from a 2-fold higher molar concentration to equalize the surface densities of divalent KKO, RTO, and monovalent Fab KKO by the number of paratopes. BSA was used as a blocker.
Oligomerization and Chemical Cross-linking of PF4
To minimize the potential weaker force signals due to non-covalent PF4-PF4 interactions revealed as a noisy background, the surface-bound PF4 oligomers were covalently cross-linked with 0.5% glutaraldehyde (2 h at 4 °C) followed by blockage with 1 m ethanolamine prior to interaction with an antibody-coated bead. The efficacy of cross-linking was confirmed using electrophoresis of WT PF4 in solution treated with glutaraldehyde at the same conditions as on the surface of a pedestal (supplemental Fig. S4). Briefly, PF4 at 10 μg/ml in PBS was incubated with either glutaraldehyde (final concentration 0.1 or 0.5%) or bis(sulfosuccinimidyl) suberate (final concentration 0.2 mm) for 30 min at room temperature. The reaction was stopped by adding NuPAGE lithium dodecyl sulfate sample buffer and denatured by heating to 70 °C for 10 min according to the manufacturer's instructions. Aliquots (15 μl) were analyzed by SDS-PAGE on a 12% NuPAGE BisTris gel under reducing conditions, and protein was visualized by silver staining. A PageRuler Plus prestained protein ladder (Thermo Fisher Scientific) served as the molecular weight standard. The results confirm that glutaraldehyde formed stable soluble covalently cross-linked oligomers with conserved ability to interact with KKO and RTO.
To rule out a potential destructive effect of glutaraldehyde on the structure of PF4, we took advantage of the PF4K50E mutant that has minimal capacity to form oligomers in the presence of heparin (24). We compared the ability of the PF4K50E mutant to bind KKO before and after treatment with glutaraldehyde using an ELISA format performed essentially as previously described (12). The results presented in supplemental Fig. S5 show that gluteraldehyde increased binding of KKO to PF4. We interpret these results to mean that gluteraldehyde helped to bring the epitopes in PF4K50E into closer proximity and, in doing so, increased antibody avidity. These data establish that glutaraldehyde did not destroy the epitopes seen by KKO.
Analysis of the Kinetics of KKO- and RTO-PF4 Interactions
To extract kinetic parameters of the KKO-PF4 and RTO-PF4 interactions, data analysis was performed using the Bell model, which describes the rupture of non-covalent bonds formed between single molecules subject to mechanical force (25). According to this model, the dependence of the unbinding rate or off-rate (koff) on a tensile force (f), which governs the dissociation transition B → U from the bound state (B) to the unbound (dissociated) state (U), is given by the product, koff = k0exp(σ≠f/kBT), where the prefactor k0 is the force-free bond breakage rate (off-rate at a zero force), σ≠ is the distance between the energy minimum of the bound state and the transition state, and kB and T are the Boltzmann constant and absolute temperature, respectively. Note that σ≠ can be viewed as the minimal bond length at which the bimolecular complex becomes unstable. Also, in the presence of pulling force (f), the probability of rebinding is small, and the kinetic rate for the reverse association process (U → B) or on-rate kon can be safely neglected. Under these conditions, the probability density function of unbinding forces p(f) (i.e. the likelihood of observing the bond breakage at a force f) is given by the equation,
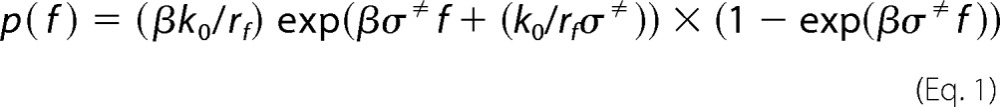 |
where rf is the force-loading rate, and β is the inverse temperature (β = 1/kBT). In the kinetic analysis and modeling of the results of experimental measurements, this equation was used to model the histograms of rupture forces in order to estimate the force-free off-rate k0 and the transition state location σ≠.
The apparent force-free kinetic rate for association (on-rate) k′on was obtained using the experimental value of the binding probability Pb(T) measured at time T (interaction time), expressed in terms of koff and k′on by the following equation (26).
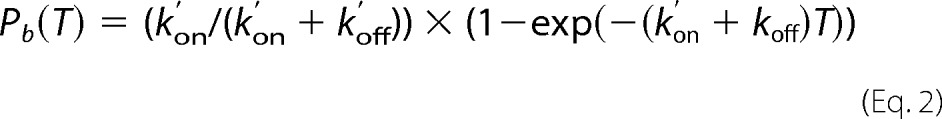 |
Finally, having estimated koff and k′on, we were able to calculate the apparent equilibrium dissociation constant K′d given by Equation 3.
RESULTS
Interactions of KKO and RTO with Uncross-linked Wild Type PF4
To assess directly whether the surface-attached antigen and the antibodies retain the ability to bind each other, surfaces coated with predominantly tetrameric WT PF4 were exposed to either KKO- or RTO-coated beads. Both KKO and RTO were highly reactive toward PF4 and displayed similar rupture force spectra in the range of 10–110 pN with two distinct regimes bordering at about 60 pN (Fig. 1). The lower force regime (10–60 pN) displayed interactions with exponentially decreasing strength, whereas the higher force regime (>60 pN) appeared as a Gaussian-like symmetric peak. Despite the qualitative similarity of the force profiles, the overall cumulative binding probability of KKO-PF4 interactions (13.2%) was significantly higher than that of RTO-PF4 (7.4%, p < 0.05) mainly due to the higher incidence of the weaker forces (Table 1). Although the cumulative probability of the larger forces was small and was similar for KKO-PF4 (0.5%) and RTO-PF4 (0.4%, p > 0.05) interactions, there was a moderate but statistically significant shift in peak position (84 versus 78 pN, p < 0.05), reflecting a difference in binding strength (Fig. 1). When full-length KKO was replaced with its Fab KKO fragment, the rupture force profile with uncross-linked PF4 did not change significantly under the same experimental conditions (supplemental Fig. S6 and Table 1). This result indicates that, despite the bivalent nature of the antibody, the registered binding of KKO to PF4 was monovalent. This is probably due to the inability of the optical trap to measure the strength of bivalent interactions at >130–140 pN and perhaps because of steric limitations originating from surface immobilization.
FIGURE 1.
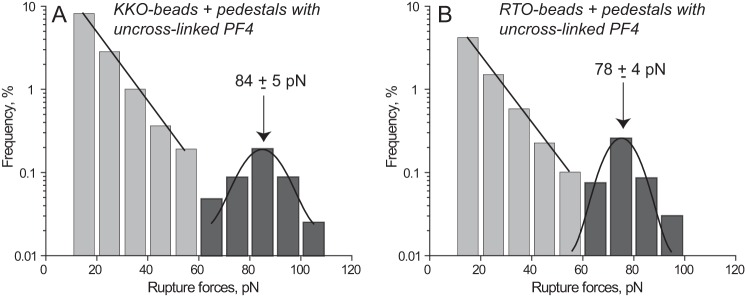
Interactions of KKO (A) and RTO (B) with uncross-linked WT PF4 displayed as normalized rupture force distributions in a semilog plot with two force regimes bordering at 60 pN. The bimodal distribution of rupture forces is fitted with the exponential and Gaussian functions. The total number of contacts (n = 10,429 for A and n = 9,359 for B) is taken to be 100%.
TABLE 1.
Cumulative binding probability for different interacting proteins at various conditions
Values are expressed as means ± S.D.
| Interacting molecular pairs and conditions | Cumulative probability |
Figure(s) | |
|---|---|---|---|
| Forces >10 pN | Forces >60 pNa | ||
| % | % | ||
| Uncross-linked WT PF4b and KKO | 13.2 ± 2.4 | 0.5 ± 0.1 | 1A |
| Uncross-linked WT PF4b and RTO | 7.4 ± 1.4 | 0.4 ± 0.1 | 1B |
| Uncross-linked WT PF4b and Fab KKO | 11.3 ± 2.3 | 0.5 ± 0.1 | S6 |
| (Uncross-linked WT PF4b + PF4A01) and KKO | 10.7 ± 2.4 | 0.8 ± 0.3 | S7A |
| (Uncross-linked WT PF4b + PF4A01) and RTO | 5.2 ± 1.1 | 0.3 ± 0.1 | S7B |
| Cross-linked WT PF4 (0.1 mg/ml) and KKO | 9.2 ± 2.7 | 2.4 ± 0.4 | S9A |
| Cross-linked WT PF4 (0.2 mg/ml) and KKO | 14.6 ± 2.5 | 6.2 ± 0.9 | S7B |
| Cross-linked WT PF4 (1 mg/ml) and KKO | 15.2 ± 3.4 | 9.6 ± 1.8 | 2A and S9C |
| Cross-linked WT PF4 (2 mg/ml) and KKO | 19.0 ± 2.8 | 10.2 ± 1.2 | S9D |
| Cross-linked WT PF4b and RTO | 4.6 ± 1.0 | 1.2 ± 0.5 | 2B |
| Cross-linked WT PF4b and Fab KKO | 9.9 ± 2.2 | 6.5 ± 1.5 | S8 |
| (Cross-linked WT PF4b + free Fab KKO) and KKO | 7.4 ± 1.2 | 1.1 ± 0.3 | 2C |
| Cross-linked WT PF4b and (KKO + free PF4) | 29.2 ± 4.5 | 0.9 ± 0.4 | 2D |
| Uncross-linked PF4 K50Eb and KKO | 17.2 ± 3.6 | 0.3 ± 0.1 | 5A |
| Uncross-linked PF4 K50Eb and RTO | 14.1 ± 2.1 | 0.3 ± 0.1 | 5B |
| Uncross-linked PF4 K50Eb and Fab KKO | 12.7 ± 3.1 | 0.4 ± 0.1 | S10 |
| Cross-linked PF4 K50Eb and KKO | 9.4 ± 2.1 | 3.2 ± 1.1 | 6A |
| Cross-linked PF4 K50Eb and RTO | 4.4 ± 1.1 | 2.0 ± 0.6 | 6A |
| Cross-linked PF4 K50Eb and Fab KKO | 8.3 ± 2.4 | 1.2 ± 0.4 | S11 |
| Uncross-linked WT PF4b-heparin and KKO | 18.9 ± 3.7 | 5.5 ± 1.1 | 7A |
| Uncross-linked WT PF4b-heparin and RTO | 8.0 ± 2.2 | 0.4 ± 0.1 | 7B |
| Cross-linked WT PF4b-heparin and KKO | 15.4 ± 3.3 | 11.8 ± 2.3 | 7C |
| Cross-linked WT PF4b-heparin and RTO | 6.6 ± 2.0 | 3.3 ± 0.8 | 7D |
a Rupture forces of >60 pN have been identified as reflecting specific antigen-antibody interaction between PF4 and KKO or RTO.
b To keep the surface density the same, the concentration of PF4 in the immobilizing mixture was 1 mg/ml, unless otherwise specified.
In our previous studies, specific protein-ligand interactions often appeared as well defined stronger force peaks, whereas the nonspecific background showed up as weaker forces with exponentially decreasing probability (17, 21, 27). Therefore, we assumed that the dominant lower binding strength interactions observed when KKO or RTO was exposed to the mostly tetrameric WT PF4 were from PF4-PF4 bonds, which overwhelmed Ab-PF4 interactions. To assess the importance of the size of the oligomers, WT PF4 was pretreated (100 μm, 37 °C, 30 min, 50 mm Tris-HCl, 0.5 m NaCl, pH 7.2) with a low molecular weight compound, PF4 antagonist PF4A01, previously shown to prevent tetramerization of PF4 from dimers (28). This antagonist reduced the probability of the weaker KKO-PF4 and RTO-PF4 interactions, with a statistically insignificant increase in the higher forces (supplemental Fig. S7). This further supports the concept that the weaker interactions (<60 pN) originate from PF4-PF4 interactions that occur with lower probability among PF4 dimers than tetramers.
Interactions of KKO and RTO with Cross-linked Wild Type PF4
To study antibody-PF4 binding specifically, we prevented rupture of PF4-PF4 bonds by covalently cross-linking the surface-bound PF4 oligomers with glutaraldehyde (see “Experimental Procedures”). The cross-linking of PF4 had a dramatic effect on the force profile both for KKO-PF4 and RTO-PF4 interactions (supplemental Fig. S2 and Figs. 1 and 2). Although the overall probability of KKO binding with uncross-linked versus cross-linked PF4 did not change significantly (13.2 versus 15.2% (p > 0.05), respectively), the fraction of the larger forces >60 pN increased ∼19-fold from 0.5 to 9.6% (p < 0.001), respectively (Table 1). By contrast, the overall probability of RTO binding with uncross-linked versus cross-linked PF4 dropped from 7.4 to 4.6% (p < 0.001), respectively, whereas the share of the larger forces >60 pN increased ∼3-fold from 0.4 to 1.2% (p < 0.001), respectively (Table 1). The force histogram of the KKO-PF4 interactions revealed a redistribution of the lower and larger forces in response to cross-linking of PF4, leading to formation of a new, sharp, well defined peak at 94.5 pN (Fig. 2A). This force profile was quite different from the RTO-PF4 interactions, which displayed a dramatic suppression of both force regimes as a result of PF4 cross-linking with a quite small peak at 82.5 pN (Fig. 2B). Similar to KKO, the Fab KKO fragment exposed to the cross-linked PF4 also formed a strong force peak at 92 pN, but the overall binding probability was substantially lower than seen with full-length KKO (9.9 versus 15.2% (p < 0.05), respectively) (supplemental Fig. S8 and Table 1). Also, interactions of Fab KKO with uncross-linked versus cross-linked PF4 resulted in a striking 13-fold increase in the fraction of forces >60 pN from 0.5 to 6.5% (Table 1). It is noteworthy that all of the observed changes in response to cross-linking of PF4 occurred at identical surface density of the reacting proteins and under the same experimental conditions. These data have two important implications. First, they confirm that the vast majority of signals observed during forced unbinding of the anti-PF4 antibodies and uncross-linked tetrameric PF4 originated from the PF4–PF4 bonds, which was precluded by covalent cross-linking. Second, they show a distinct superiority of KKO over RTO to bind stabilized PF4 oligomers not revealed with uncross-linked PF4. The higher probability of interactions between KKO and PF4 than with Fab KKO (and perhaps RTO) may be explained by the ability of bivalent Ab to bind either of two or more closely located cross-linked tetramers.
FIGURE 2.

The panel of rupture force spectra demonstrating the interactions of KKO (A, C, and D) and RTO (B) with cross-linked WT PF4, including control data (C and D) for nonspecific protein-protein binding displayed as normalized force distributions. A and B have two force regimes, corresponding to nonspecific (<60 pN) and specific (>60 pN) PF4-antibody interactions. C and D are competitive inhibition assays (negative controls to A) with exponentially decreasing rupture forces produced by nonspecifically interacting proteins. The total number of contacts presented in the histograms is n = 11,020 (A), n = 8320 (B), n = 7568 (C), and n = 6876 (D).
Specificity of the KKO-PF4 Interactions
The specificity of rupture forces generated by the surface-bound cross-linked PF4 and KKO was confirmed by competitive inhibition experiments in the presence of free Fab KKO (to block the PF4 epitopes) or free PF4 (to block the KKO paratopes). First, pedestals coated with cross-linked PF4 were preincubated with free Fab KKO (100 μg/ml, 15 min, room temperature). KKO-beads were then inserted into the chamber, and the measurements were performed in the presence of 100 μg/ml Fab KKO. As a result, the overall probability of KKO-PF4 binding decreased from 14.8 to 7.4% (p < 0.01), with the most profound inhibitory effect on the fraction of forces >60 pN, which dropped from 8.1 to 1.1% (p < 0.001) (Table 1). Comparison of the force profiles shown in Fig. 2, A and C (i.e. in the absence and presence of free Fab KKO, respectively), revealed that KKO-PF4 interactions corresponding to the greater force peak were almost completely abolished by excess soluble Fab KKO due to non-equilibrium competitive inhibition. When free WT PF4 (1 mg/ml) was introduced into the reaction chamber to compete with KKO antibodies for surface-attached PF4, it caused a dual effect: a 2-fold increase in the overall binding probability from 13.8 to 29.2% (p < 0.01) combined with almost complete elimination of forces >60 pN (from 9.6 to 0.9%, p < 0.001) to the background level (Table 1). Formation of numerous weak bonds <40 pN (Fig. 2D) probably represents forced rupture of many PF4-PF4 interactions at an interface resulting from adsorption of abundant soluble PF4 on the interacting surfaces. These data confirm that the rupture forces stronger than 60 pN indeed represent specific KKO-PF4 interactions.
Surface Density Dependence of the KKO-PF4 Interactions
To test whether the measured rupture forces represent unbinding of single-molecule KKO-PF4 complexes, we varied the surface density of the pedestal-attached cross-linked PF4 by changing the concentration of PF4 in the binding buffer during immobilization. The results shown in Fig. 3, supplemental Fig. S9, and Table 1 indicate that the probability of specific KKO-PF4 interactions reflected by forces >60 pN increased with surface density of PF4 until it reached a plateau, corresponding to the lack of additional available binding sites (“saturation”) on the surface of KKO-coated beads. This plateau is reached at the concentration of PF4 of 1 mg/ml normally used in our experiments. Importantly, the maximal binding probability of the specific antigen-antibody interactions was only about 10%, meaning that statistically, the likelihood of multiple interactions was negligible. Additional evidence for single-molecule KKO-PF4 interactions came from the finding that bead-pedestal binding strength was unchanged with PF4 surface density in the studied range (Fig. 3 and supplemental Fig. S9), consistent with the interactions of KKO binding to single epitopes (29).
FIGURE 3.
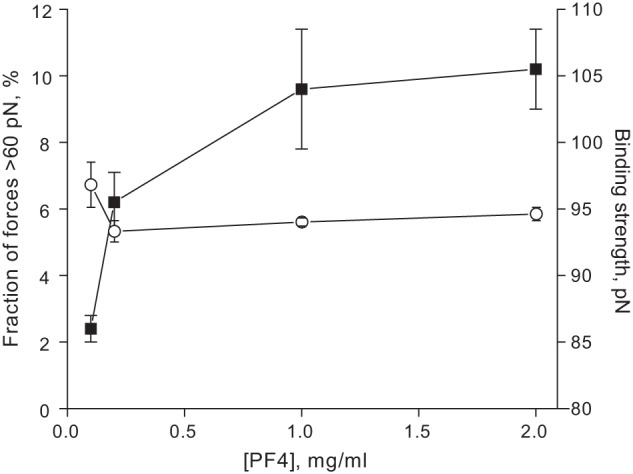
The probability (squares) and the binding strength (circles) of specific KKO-PF4 interactions (recognized as forces >60 pN) plotted against PF4 concentration in the immobilizing mixture (derived from the force histograms shown in supplemental Fig. S9). After immobilization, PF4 was cross-linked with glutaraldehyde. Error bars, S.D.
Kinetics of the KKO-PF4 and RTO-PF4 Interactions
We used Equation 1 for the probability density function of unbinding forces, p(f), to model the experimental histograms of rupture forces for the interactions of KKO and RTO with cross-linked PF4. The results of the numerical fit of the theoretical curve of p(f) to the histograms of rupture forces presented in Fig. 4 show good agreement between the experimental data and theoretical modeling. When performing the fit, we set the values of the force-loading rate to vf = 1600 pN/s (value used in experiments) and the inverse temperature to β = 0.24 pN−1 nm−1 (corresponding to room temperature), in order to estimate the force-free off-rate k0 and the transition state distance σ≠. For the KKO-PF4 interactions, the best fit was obtained with k0 = 3.1 × 10−3 s−1 and σ≠ = 0.55 nm. For the RTO-PF4 interactions, the best fit was obtained with k0 = 2.9 × 10−3 s−1 and σ≠ = 0.6 nm. Using the information about the binding probability Pb(T) as a function of the interaction time T, which took on the value of Pb(T) = 0.090 ± 0.008 for the KKO-PF4 interactions and Pb(T) = 0.0107 ± 0.002 for the RTO-PF4 interactions, we have estimated the apparent on-rate k′on using Equation 2. We obtained k′on = 4.36 ± 0.41 s−1 for formation of the KKO-PF4 complex and k′on = 0.453 ± 0.075 s−1 for formation of the RTO-PF4 complex. Finally, using Equation 3, we estimated the apparent equilibrium dissociation constant K′d. We found that K′d = (0.715 ± 0.065) × 10−3 for KKO-PF4 interactions, and K′d = (6.58 ± 1.09) × 10−3 for RTO-PF4 interactions.
FIGURE 4.
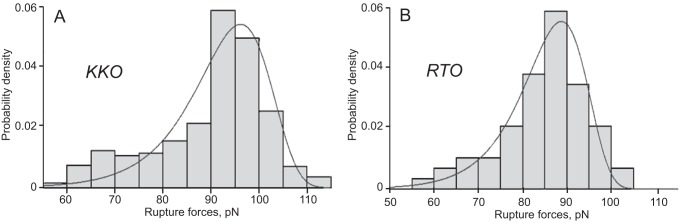
Rupture force distributions of the interactions of KKO (A) and RTO (B) with cross-linked WT PF4 fitted to the Bell function (see “Experimental Procedures”). Signals that appeared as forces below 60 pN were considered nonspecific binding events and excluded from the analysis. The total number of contacts is n = 6401 for A and n = 5590 for B.
Interactions of KKO and RTO with the PF4K50E Mutant
To confirm that the distinct reactivity of KKO and RTO depends on formation of stabilized PF4 oligomers, we studied interactions of these antibodies with the recombinant PF4K50E mutant, previously shown to exist almost exclusively as either monomeric or dimeric structures (24). Both KKO and RTO displayed similar rupture force spectra in the range of 10–110 pN with exponentially decreasing strength and a hardly discernible shoulder at forces of >80 pN (Fig. 5). The overall cumulative binding probability (17.2 and 14.1% (p > 0.05), respectively) as well as the probability of forces of >60 pN (0.3% for both) were indistinguishable for KKO-PF4K50E and RTO-PF4K50E (Table 1). The Fab KKO fragment displayed the same force profile (supplemental Fig. S10) with slightly, but significantly, reduced overall binding probability as compared with full-length KKO (12.7 versus 17.2% (p < 0.05), respectively) (Table 1). This may be due to relatively limited exposure and flexibility of the surface-bound Fab fragments (50 kDa) compared with full-length Abs (150 kDa). The cross-linking of PF4 had a pronounced effect on the force profile both for KKO-PF4K50E and RTO-PF4K50E interactions. The overall probability of KKO and RTO binding with uncross-linked versus cross-linked PF4K50E was significantly reduced (from 17.2 to 9.4% (p < 0.01) for KKO and from 14.1 to 4.4% (p < 0.01) for RTO, respectively; Table 1). In contrast, the fraction of higher forces >60 pN increased after cross-linking of PF4K50E from 0.3 to 3.2% (p < 0.001) for KKO and from 0.3 to 2.0% (p < 0.001) for RTO (Table 1). This relative shift is probably due to elimination of the weaker PF4K50E-PF4K50E interactions <60 pN prevented by chemical cross-linking. There was a moderate but significant difference in the force peak position (86 pN for KKO versus 78 pN for RTO, p < 0.05) (Fig. 6), suggesting that a small fraction of tetrameric PF4K50E still formed. The Fab KKO fragment exposed to the cross-linked PF4K50E formed a relatively small force peak at 85 pN (comprising only 1.2% of the binding probability), although the overall binding probability was similar to that of full-length KKO (8.3 versus 9.4% (p > 0.05), respectively) (Table 1 and supplemental Fig. S11). Thus, in contrast to the WT PF4, KKO and RTO showed lower and relatively similar binding probabilities to glutaraldehyde-treated PF4K50E, which forms few oligomeric complexes, although the binding strength was again slightly but consistently higher for KKO versus RTO. These studies also demonstrate that glutaraldehyde neither destroyed nor created epitopes required for the binding of KKO.
FIGURE 5.
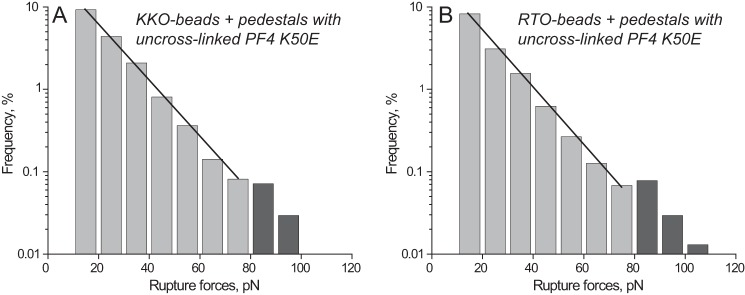
Interactions of KKO (A) and RTO (B) with uncross-linked mutant PF4K50E displayed as normalized rupture force distributions in a semilog plot showing exponential decay of rupture forces with a small shoulder at >80 pN. The total number of contacts is n = 10,193 for A and n = 11,038 for B.
FIGURE 6.
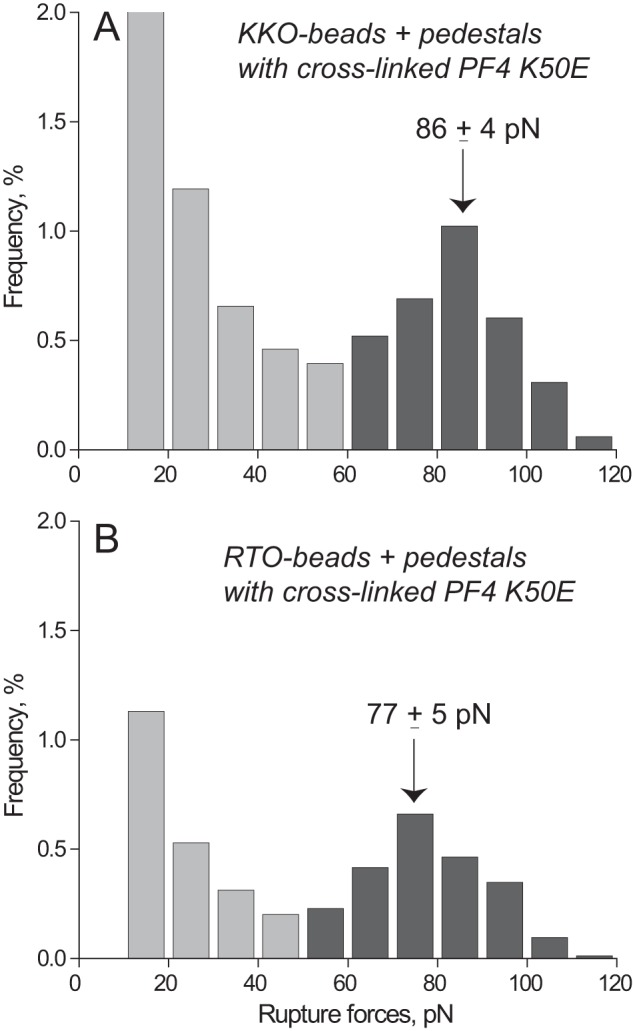
Interactions of KKO (A) and RTO (B) with cross-linked mutant PF4K50E displayed as normalized rupture force distributions with two force regimes bordering at 60 pN. The total number of contacts is n = 9,463 for A and n = 8,320 for B.
Interactions of KKO and RTO with the PF4-Heparin Complex
To see if the differential reactivity of the two Abs could be reproduced in a more complex but physiologically relevant system, we studied interactions of KKO versus RTO with PF4-heparin complexes. The complexes were formed in solution at the optimal PF4/heparin ratio previously determined by ELISA, followed by covalent immobilization on a surface (24). In the presence of heparin, broader and more heterogeneous rupture force profiles were seen, as would be expected based on heparin's biochemical heterogeneity and electrostatic charges. Nevertheless, a relatively small (5.5% cumulative probability) but definitive peak of forces centered at 101 pN was revealed in KKO interactions with PF4-heparin, which was ∼10-fold larger than with RTO (Fig. 7, A and B, and Table 1). To reduce the likelihood of nonspecific non-covalent interactions, we then cross-linked the PF4-heparin complexes chemically with glutaraldehyde, which redistributed the force profile toward stronger interactions while maintaining the significant difference in the reactivity of KKO over RTO (Fig. 7, C and D, and Table 1).
FIGURE 7.

Interactions of KKO (A and C) and RTO (B and D) with WT PF4-heparin complexes before (A and B) and after (C and D) treatment with 0.5% glutaraldehyde. The data are displayed as normalized rupture force distributions with two force regimes. The higher force peak is fitted with a Gaussian function. The total number of contacts (n = 19,421 for A, n = 23,934 for B, n = 23,364 for C, and n = 25,326 for D) is taken to be 100%.
DISCUSSION
An unresolved problem in understanding the pathogenesis of HIT and one that complicates diagnosis is that although antibodies to PF4, a normal host protein released from platelets that binds to heparin-like GAGs, develop so commonly after exposure to heparin, only a subset are associated with clinical HIT. This gap in our knowledge leads to overdiagnosis and overtreatment (30). The present work is aimed at elucidating the functional distinction between two isotype-matched murine anti-human PF4-heparin monoclonal Abs, one of which (KKO) is pathogenic, inducing thrombocytopenia and thrombosis in an animal model (7), and competes with human HIT antibodies for binding to PF4 and the other of which (RTO) is non-pathogenic, neither inducing sequelae in vivo nor showing selective competition with human HIT antibodies (14, 15). Importantly, KKO and RTO, which do not compete with each other, bind to PF4-heparin with a comparable Bmax using ELISA methodology, highlighting our current inability to identify antibodies with the greatest potential to cause human disease.
To extend our previous study on this issue (12), we measured the binding of KKO and RTO to PF4 at the single-molecule level while minimizing effects of avidity and other auxiliary intermolecular interactions. To explore the possibility that KKO and RTO bind PF4 differently at the single-molecule level, we measured their binding interactions more precisely using optical trap-based force spectroscopy, a biophysical methodology that we developed previously and applied successfully to quantify several different protein-ligand interactions (16, 17, 19–21, 23). Basically, in this technique, a microscopic protein-coated latex bead is trapped by a focused laser beam and repeatedly brought into contact with a ligand-coated pedestal. When the protein (in this study KKO or RTO) on the bead binds non-covalently to the ligand (PF4) on the pedestal, the bead is displaced from the moving optical trap center to generate a ramped pulling force that increases linearly until the moment of forced dissociation of the complex. The binding probability and the rupture forces are then measured to characterize the interaction properties of the protein (antibody) and the ligand (antigen) (for details, see Ref. 17).
In a series of initial experiments with tetrameric WT PF4 and either KKO or RTO, we identified a set of lower binding strength interactions of <60 pN probably arising from PF4–PF4 bonds. These were difficult to separate from the interactions that appeared as a relatively small peak at >60 pN, which probably represent antibody-PF4 complexes (Fig. 1). To test our presumption about the nature of the two distinct force regimes, we suppressed the rupture of PF4-PF4 bonds by covalently cross-linking the PF4 tetramers with glutaraldehyde. This eliminated the weaker interactions and produced definite stronger forces of >60 pN that appeared as a prominent peak for KKO and a much less pronounced peak for RTO (Fig. 2, A and B). The specificity of rupture forces >60 pN generated by the surface-bound cross-linked PF4 and KKO was confirmed by competitive inhibition experiments in the presence of free Fab KKO or free PF4 (Fig. 2, C and D). The force histograms revealed that KKO-PF4 interactions occurred with about 8 times higher probability than RTO-PF4 interactions at the same surface densities of the reacting proteins (Table 1), reflecting a much higher reactivity of KKO toward PF4 compared with RTO not evident using bulk equilibrium binding technology, such as ELISAs. The KKO-PF4 interactions were also moderately but significantly stronger, as reflected by the position of the peak at higher rupture force (Fig. 2, A and B). The chemical cross-linking of tetrameric WT PF4 led to formation of stable multimolecular complexes (supplemental Fig. S4). Therefore, the results of force spectroscopy support the hypothesis that further polymerization of PF4 tetramers is crucial to the distinct functionality of KKO and RTO. Indeed, when we used the PF4K50E mutant with impaired ability to oligomerize (24), the reactivity of KKO and RTO was quite similar even after chemical cross-linking (Fig. 6).
The rupture force distributions for KKO-PF4 and RTO-PF4 interactions can be mathematically converted to two-dimensional kinetic parameters, provided the results reflect single-molecule binding and unbinding. Based on several criteria that have been proposed to test whether the observed ruptures were due to single or multiple interactions (16, 31), our data indicate strongly that under the experimental conditions studied, the rupture events were due to single molecules. First, the frequency of the specific Ab-antigen interaction was <10%, implying that the probability of multiple interactions is less than 1% or square probability 0.12 (Fig. 3). Second, if the interactions between beads and pedestals were the result of multiple bonds, then the binding strength would increase at increased surface densities of the interacting proteins. This was not detected for KKO-PF4 (Fig. 3). Third, the rupture of multiple binding sites should proceed as a sequence of multiple steps, whereas the rupture of single molecules should always occur in a single step. Typically, only about 1 of 10 occurred in two steps, manifesting themselves as jagged force signals (supplemental Fig. S3), and only the single-step interactions shown in supplemental Fig. S2 were included in the analysis. Moreover, when PF4 interacted with Fab KKO, unlike full-length KKO, almost no jagged signals were seen, confirming that the jagged signals originate from double/bivalent interactions. It is noteworthy that in our model system, the strength of such multiple Ab-PF4 interactions was beyond the power of the optical trap (the maximal measurable force is ∼130–140 pN) and appeared as episodes of irreversible attachment that were excluded from data analysis.
Based on the Bell theory of forced molecular unbinding (25), we analyzed the rupture force spectra of KKO-PF4 and RTO-PF4 interactions to extract and compare a number of kinetic parameters. One of them is the off-rate extrapolated to spontaneous force-free dissociation (k0), which was ∼3 × 10−3 s−1 for both types of Ab-PF4 complexes, indicating that their strength was similar. For comparison, the reported k0 values are 5.4 × 10−6 s−1 for streptavidin/biotin (32), ∼7 × 10−3 s−1 for avidin/biotin (33), 10−4 s−1 for the A:a knob-hole interaction in fibrin (19), and 1.39–4.3 s−1 for P-selectin/PSGL-1 (34). Thus, the k0 of ∼10−3 s−1 indicates that single KKO-PF4 and RTO-PF4 interactions are relatively stable and very slowly reversible due to low unbinding rate. The main difference between KKO and RTO was revealed in the cumulative binding probability converted to the apparent on-rate k′on, which was much higher for KKO and resulted in a ∼10-fold higher affinity of KKO-PF4 versus RTO-PF4 complexes, reflected by the apparent equilibrium constants K′d. The large difference in on-rates implies that epitope exposure is enhanced by PF4 (super)oligomerization, perhaps as a result of tighter packing of monomers. Another important parameter of a binding interaction is the transition state distance (σ≠), which can be interpreted as the distance of molecular separation at which the bond fails. Of note, the transition state distance for the KKO-PF4 and RTO-PF4 unbinding was ∼0.55–0.6 nm, which is relatively long compared with brittle bonds, such as the A:a knob-hole bonds in fibrin (19) or the platelet integrin αIIbβ3-fibrinogen complexes (35). This parameter may result from the larger size and multimeric nature of the PF4-Ab complexes and reflect their elongation and/or other mechanically induced structural transitions preceding forced dissociation from the binding sites.
To determine if the enhanced binding of KKO to PF4 in the presence of glutaraldehyde was an artifact of chemical cross-linking, we repeated the experiments by forming heparin/PF4 complexes at molar ratios shown previously to form large complexes using size exclusion chromatography and electron microscopy and to optimize the binding of KKO and HIT antibodies (24). The results show the binding of KKO to PF4-heparin and support the potential relevance of the findings to the clinical setting (Fig. 7 and Table 1).
These results support our notion that the biological difference between pathogenic and nonpathogenic anti-PF4 Abs, exemplified by the IgG2bκ mAbs KKO and RTO, might at least in part be explained by differences in epitope specificity that are influenced by the ability of heparin or GAGs to promote superoligomerization of PF4, which increases the avidity of pathogenic Ab by stabilizing ULCs. We hypothesize that the binding avidity of KKO is enhanced when heparin or cellular GAGs promote organization of PF4 into ordered complexes, as observed in this study after chemical cross-linking of the WT PF4. These complexes in turn are further stabilized after binding of KKO (perhaps by Ab-induced conformational rearrangement). Moreover, our data suggest that neither heparin nor GAGs are obligate components of the epitope recognizable by the pathogenic Ab. In other words, we postulate that KKO and heparin or GAGs each promote PF4 oligomerization and act in concert to augment antibody avidity and that additional bridging of PF4 by KKO helps to render the complexes more stable in vitro and in vivo, where they are subject to dilution in flowing blood. In contrast to pathogenic Abs, high titers of non-pathogenic anti-PF4 antibodies may show comparable behavior at equilibrium between PF4 monomers, dimers, or tetramers, but their binding is not enhanced by heparin- or GAG-induced oligomerization, and they bind to sites that do not reinforce oligomer stability.
There are several important limitations to our study. First, the Ab-PF4 interactions observed in our model system are different from physiological conditions in a number of important features, including covalent attachment of both reacting proteins to the surfaces, measurement of only monovalent bimolecular binding events due to limitations of the trap power, and absence of flow. Notwithstanding these differences, we were able to reproduce the physiologically relevant circumstances in which the inherent difference in the reactivity of KKO and RTO toward PF4 becomes obvious (7).
A second important limitation of our study is the use of a monoclonal HIT-like antibody that competes with human HIT antibodies rather than studying polyclonal and likely polyspecific human IgG antibodies directly. Such studies will necessitate methods to segregate the subset of pathogenic antibodies in HIT plasma from the likely far larger mass of RTO-like antibodies that bind PF4 but do not cause disease in vivo. Nevertheless, the results of this study support the concept that heparin and other extended cationic molecules provide a platform that enhances the proximity of PF4 molecules and thereby increases the probability of binding of at least some pathogenic antibodies (36). These studies thereby indicate the importance of epitope specificity and the differential effect of PF4 oligomerization on the binding kinetics and strength of pathogenic and non-pathogenic anti-PF4 antibodies, but they do not exclude other differences in the behavior of human antibodies that might contribute to their propensity to cause disease. In addition to in vivo studies and biochemical experiments in bulk, single-molecule biophysical characterization of PF4-mediated interactions provides a basis to develop novel assays that distinguish pathogenic from nonpathogenic human clinical Abs that in turn should allow more precise identification of who has or is likely to develop HIT and needs therapeutic intervention. Last, functional and structural characteristics of anti-PF4 Abs at the single-molecule level will provide mechanistic insight into the immunology of HIT and may enable the design of HIT-specific Ab-targeted agents to prevent and treat this severe thrombotic disorder.
This work was supported, in whole or in part, by National Institutes of Health Grants HL078726/HL078726-S1 (to B. S. S.), HL099973/HL090774 (to J. W. W.), HL084006 (to D. B. C.), and HL110860 (to B. S. S., L. R., G. M. A., D. B. C., and J. W. W.).

This article contains supplemental Figs. S1–S11.
- HIT
- heparin-induced thrombocytopenia
- Ab
- antibody
- GAG
- glycosaminoglycan
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- pN
- piconewtons
- PF4
- platelet factor 4.
REFERENCES
- 1. Mayo K. H., Ilyina E., Roongta V., Dundas M., Joseph J., Lai C. K., Maione T., Daly T. J. (1995) Heparin binding to platelet factor-4. An NMR and site-directed mutagenesis study. Arginine residues are crucial for binding. Biochem. J. 312, 357–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Greinacher A., Farner B., Kroll H., Kohlmann T., Warkentin T. E., Eichler P. (2005) Clinical features of heparin-induced thrombocytopenia including risk factors for thrombosis. A retrospective analysis of 408 patients. Thromb. Haemost. 94, 132–135 [DOI] [PubMed] [Google Scholar]
- 3. Arepally G. M., Ortel T. L. (2006) Clinical practice. Heparin-induced thrombocytopenia. N. Engl. J. Med. 355, 809–817 [DOI] [PubMed] [Google Scholar]
- 4. Cines D. B., Rauova L., Arepally G., Reilly M. P., McKenzie S. E., Sachais B. S., Poncz M. (2007) Heparin-induced thrombocytopenia. An autoimmune disorder regulated through dynamic autoantigen assembly/disassembly. J. Clin. Apher. 22, 31–36 [DOI] [PubMed] [Google Scholar]
- 5. Warkentin T. E. (2011) HIT paradigms and paradoxes. J. Thromb. Haemost. 9, 105–117 [DOI] [PubMed] [Google Scholar]
- 6. Pouplard C., Iochmann S., Renard B., Herault O., Colombat P., Amiral J., Gruel Y. (2001) Induction of monocyte tissue factor expression by antibodies to heparin-platelet factor 4 complexes developed in heparin-induced thrombocytopenia. Blood 97, 3300–3302 [DOI] [PubMed] [Google Scholar]
- 7. Reilly M. P., Taylor S. M., Hartman N. K., Arepally G. M., Sachais B. S., Cines D. B., Poncz M., McKenzie S. E. (2001) Heparin-induced thrombocytopenia/thrombosis in a transgenic mouse model requires human platelet factor 4 and platelet activation through FcγRIIA. Blood 98, 2442–2447 [DOI] [PubMed] [Google Scholar]
- 8. Amiral J., Bridey F., Wolf M., Boyer-Neumann C., Fressinaud E., Vissac A. M., Peynaud-Debayle E., Dreyfus M., Meyer D. (1995) Antibodies to macromolecular platelet factor 4-heparin complexes in heparin-induced thrombocytopenia. A study of 44 cases. Thromb. Haemost. 73, 21–28 [PubMed] [Google Scholar]
- 9. Warkentin T. E., Sheppard J. A., Horsewood P., Simpson P. J., Moore J. C., Kelton J. G. (2000) Impact of the patient population on the risk for heparin-induced thrombocytopenia. Blood 96, 1703–1708 [PubMed] [Google Scholar]
- 10. Warkentin T. E., Sheppard J. A., Moore J. C., Moore K. M., Sigouin C. S., Kelton J. G. (2005) Laboratory testing for the antibodies that cause heparin-induced thrombocytopenia. How much class do we need? J. Lab. Clin. Med. 146, 341–346 [DOI] [PubMed] [Google Scholar]
- 11. Greinacher A., Gopinadhan M., Günther J. U., Omer-Adam M. A., Strobel U., Warkentin T. E., Papastavrou G., Weitschies W., Helm C. A. (2006) Close approximation of two platelet factor 4 tetramers by charge neutralization forms the antigens recognized by HIT antibodies. Arterioscler. Thromb. Vasc. Biol. 26, 2386–2393 [DOI] [PubMed] [Google Scholar]
- 12. Sachais B. S., Litvinov R. I., Yarovoi S. V., Rauova L., Hinds J. L., Rux A. H., Arepally G. M., Poncz M., Cuker A., Weisel J. W., Cines D. B. (2012) Dynamic antibody-binding properties in the pathogenesis of HIT. Blood 120, 1137–1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Arepally G. M., Kamei S., Park K. S., Kamei K., Li Z. Q., Liu W., Siegel D. L., Kisiel W., Cines D. B., Poncz M. (2000) Characterization of a murine monoclonal antibody that mimics heparin-induced thrombocytopenia antibodies. Blood 95, 1533–1540 [PubMed] [Google Scholar]
- 14. Rauova L., Zhai L., Kowalska M. A., Arepally G. M., Cines D. B., Poncz M. (2006) Role of platelet surface PF4 antigenic complexes in heparin-induced thrombocytopenia pathogenesis. Diagnostic and therapeutic implications. Blood 107, 2346–2353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Cuker A., Rux A. H., Hinds J. L., Dela Cruz M., Yarovoi S. V., Brown I. A., Yang W., Konkle B. A., Arepally G. M., Watson S. P., Cines D. B., Sachais B. S. (2013) Novel diagnostic assays for heparin-induced thrombocytopenia. Blood 121, 3727–3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Litvinov R. I., Shuman H., Bennett J. S., Weisel J. W. (2002) Binding strength and activation state of single fibrinogen-integrin pairs on living cells. Proc. Natl. Acad. Sci. U.S.A. 99, 7426–7431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Litvinov R. I., Bennett J. S., Weisel J. W., Shuman H. (2005) Multi-step fibrinogen binding to the integrin αIIbβ3 detected using force spectroscopy. Biophys. J. 89, 2824–2834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Litvinov R. I., Nagaswami C., Vilaire G., Shuman H., Bennett J. S., Weisel J. W. (2004) Functional and structural correlations of individual αIIbβ3 molecules. Blood 104, 3979–3985 [DOI] [PubMed] [Google Scholar]
- 19. Litvinov R. I., Gorkun O. V., Owen S. F., Shuman H., Weisel J. W. (2005) Polymerization of fibrin. Specificity, strength, and stability of knob-hole interactions studied at the single-molecule level. Blood 106, 2944–2951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Litvinov R. I., Gorkun O. V., Galanakis D. K., Yakovlev S., Medved L., Shuman H., Weisel J. W. (2007) Polymerization of fibrin. Direct observation and quantification of individual B:b knob-hole interactions. Blood 109, 130–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Litvinov R. I., Yakovlev S., Tsurupa G., Gorkun O. V., Medved L., Weisel J. W. (2007) Direct evidence for specific interactions of the fibrinogen αC-domains with the central E region and with each other. Biochemistry 46, 9133–9142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shandler S. J., Korendovych I. V., Moore D. T., Smith-Dupont K. B., Streu C. N., Litvinov R. I., Billings P. C., Gai F., Bennett J. S., DeGrado W. F. (2011) Computational design of a β-peptide that targets transmembrane helices. J. Am. Chem. Soc. 133, 12378–12381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sun J. E., Vranic J., Composto R. J., Streu C., Billings P. C., Bennett J. S., Weisel J. W., Litvinov R. I. (2012) Bimolecular integrin-ligand interactions quantified using peptide-functionalized dextran-coated microparticles. Integr. Biol. (Camb.) 4, 84–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rauova L., Poncz M., McKenzie S. E., Reilly M. P., Arepally G., Weisel J. W., Nagaswami C., Cines D. B., Sachais B. S. (2005) Ultralarge complexes of PF4 and heparin are central to the pathogenesis of heparin-induced thrombocytopenia. Blood 105, 131–138 [DOI] [PubMed] [Google Scholar]
- 25. Bell G. I. (1978) Models for the specific adhesion of cells to cells. Science 200, 618–627 [DOI] [PubMed] [Google Scholar]
- 26. Litvinov R. I., Mekler A., Shuman H., Bennett J. S., Barsegov V., Weisel J. W. (2012) Resolving two-dimensional kinetics of the integrin αIIbβ3-fibrinogen interactions using binding-unbinding correlation spectroscopy. J. Biol. Chem. 287, 35275–35285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gorkun O. V., Litvinov R. I., Veklich Y. I., Weisel J. W. (2006) Interactions mediated by the N-terminus of fibrinogen's Bβ chain. Biochemistry 45, 14843–14852 [DOI] [PubMed] [Google Scholar]
- 28. Sachais B. S., Rux A. H., Cines D. B., Yarovoi S. V., Garner L. I., Watson S. P., Hinds J. L., Rux J. J. (2012) Rational design and characterization of platelet factor 4 antagonists for the study of heparin-induced thrombocytopenia. Blood 119, 5955–5962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhu C., Long M., Chesla S. E., Bongrand P. (2002) Measuring receptor/ligand interaction at the single-bond level. Experimental and interpretative issues. Ann. Biomed. Eng. 30, 305–314 [DOI] [PubMed] [Google Scholar]
- 30. Cuker A., Cines D. B. (2012) How I treat heparin-induced thrombocytopenia. Blood 119, 2209–2218 [DOI] [PubMed] [Google Scholar]
- 31. Litvinov R. I., Vilaire G., Shuman H., Bennett J. S., Weisel J. W. (2003) Quantitative analysis of platelet αvβ3 binding to osteopontin using laser tweezers. J. Biol. Chem. 278, 51285–51290 [DOI] [PubMed] [Google Scholar]
- 32. Chilkoti A., Stayton P. S. (1995) Molecular origins of the slow streptavidin-biotin dissociation kinetics. J. Am. Chem. Soc. 117, 10622–10628 [Google Scholar]
- 33. Evans E. (1999) Looking inside molecular bonds at biological interfaces with dynamic force spectroscopy. Biophys. Chem. 82, 83–97 [DOI] [PubMed] [Google Scholar]
- 34. Rinko L. J., Lawrence M. B., Guilford W. H. (2004) The molecular mechanics of P- and L-selectin lectin domains binding to PSGL-1. Biophys. J. 86, 544–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Litvinov R. I., Barsegov V., Schissler A. J., Fisher A. R., Bennett J. S., Weisel J. W., Shuman H. (2011) Dissociation of bimolecular αIIbβ3-fibrinogen complex under a constant tensile force. Biophys. J. 100, 165–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jaax M. E., Krauel K., Marschall T., Brandt S., Gansler J., Fürll B., Appel B., Fischer S., Block S., Helm C. A., Müller S., Preissner K. T., Greinacher A. (2013) Complex formation with nucleic acids and aptamers alters the antigenic properties of platelet factor 4. Blood 122, 272–281 [DOI] [PMC free article] [PubMed] [Google Scholar]