

Background: Family 6 glycoside hydrolases represent an important, diverse enzyme class in cellulolytic organisms.
Results: We solved structures of two Thermobifida fusca Cel6B (TfuCel6B) cellobiohydrolase mutants and examined ligand dynamics and product release with simulation.
Conclusion: These results suggest mechanisms for product release in TfuCel6B.
Significance: This study further elucidates the mechanism of a unique cellobiohydrolase with an extended, enclosed active site tunnel.
Keywords: Cellulase, Enzyme Structure, Glycoside Hydrolases, Molecular Dynamics, X-ray Crystallography, Thermobifida fusca, Cellobiohydrolase
Abstract
Cellobiohydrolases (CBHs) are typically major components of natural enzyme cocktails for biomass degradation. Their active sites are enclosed in a tunnel, enabling processive hydrolysis of cellulose chains. Glycoside hydrolase Family 6 (GH6) CBHs act from nonreducing ends by an inverting mechanism and are present in many cellulolytic fungi and bacteria. The bacterial Thermobifida fusca Cel6B (TfuCel6B) exhibits a longer and more enclosed active site tunnel than its fungal counterparts. Here, we determine the structures of two TfuCel6B mutants co-crystallized with cellobiose, D274A (catalytic acid), and the double mutant D226A/S232A, which targets the putative catalytic base and a conserved serine that binds the nucleophilic water. The ligand binding and the structure of the active site are retained when compared with the wild type structure, supporting the hypothesis that these residues are directly involved in catalysis. One structure exhibits crystallographic waters that enable construction of a model of the α-anomer product after hydrolysis. Interestingly, the product sites of TfuCel6B are completely enclosed by an “exit loop” not present in fungal GH6 CBHs and by an extended “bottom loop”. From the structures, we hypothesize that either of the loops enclosing the product subsites in the TfuCel6B active site tunnel must open substantially for product release. With simulation, we demonstrate that both loops can readily open to allow product release with equal probability in solution or when the enzyme is engaged on cellulose. Overall, this study reveals new structural details of GH6 CBHs likely important for functional differences among enzymes from this important family.
Introduction
Plants comprise the majority of terrestrial, organic carbon on earth, and organisms across all kingdoms of life have developed myriad strategies to depolymerize plant cell wall polymers for food (1–3). Enzyme cocktails from fungi and bacteria, the primary degraders of plant biomass in nature, have been widely studied for biomass degradation potential, often with the aim to harness these enzymes in support of renewable fuels portfolios (2, 4, 5). The primary organism studied for enzymatic breakdown of biomass to date is the ascomycete fungus, Hypocrea jecorina (Trichoderma reesei) (6, 7). Driven mainly by studies of H. jecorina and similar organisms, a model of cellulose deconstruction by cellobiohydrolases (CBHs),6 endoglucanases, and β-glucosidases emerged (8). Namely, endoglucanases act on amorphous regions of cellulose to create points for attachment and detachment of processive CBHs (9), which hydrolyze successive units of the cellulose chain via a multistep mechanism involving processive motion, hydrolysis, and product expulsion (8, 10–13). β-Glucosidases subsequently hydrolyze soluble cellobiose to glucose to reduce CBH product inhibition (14, 15).
The aerobic filamentous bacterium Thermobifida fusca has received significant attention as a biomass degrader in heated organic materials (16). T. fusca secretes at least eight polysaccharide-degrading enzymes (17), including two glycoside hydrolase Family 6 enzymes (GH6): the endoglucanase TfuCel6A and the CBH TfuCel6B. GH6 enzymes are important components of many fungal and bacterial enzyme cocktails for biomass degradation (16, 18–21). Additionally, there is substantial structural and sequence diversity in GH6 enzymes, further suggesting a broad range of activity, function, and stability. Fig. 1 shows a surface representation of known structures of GH6 CBHs (22–25) and endoglucanases (26, 27). The T. fusca GH6 enzymes represent the extremes of known structural diversity in terms of active site enclosure. TfuCel6A has the shortest and most open cleft, whereas TfuCel6B surprisingly exhibits the longest and most enclosed tunnel.
FIGURE 1.
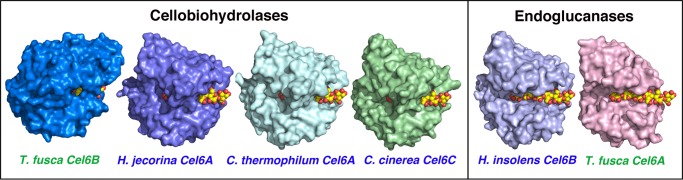
Surface model comparison of GH Family 6 enzymes with a cellohexaose ligand spanning the active site tunnel. The enzymes with blue labels are from fungi (H. jecorina, Chaetomium thermophilum, Coprinopsis cinerea, and Humicola insolens) (22, 24, 25, 27), and enzymes labeled with green are from bacteria (T. fusca) (23, 26). The first four listed GH6 cellulases are cellobiohydrolases, and the last two are endoglucanases.
In addition to substantial structural diversity of GH6 enzymes, the catalytic mechanism remains a question of interest as the identity of a well defined catalytic base remains elusive (28). The GH6 catalytic acid has been identified as a conserved aspartic acid (Asp-221 in H. jecorina Cel6A (HjeCel6A) and Asp-274 in TfuCel6B) (29, 30). Several potential titratable residues nearby have been proposed to be the catalytic base (29–32), but none of these residues have been found in crystal structures within the necessary distance to act directly as the base. Instead, the predominant mechanism for GH6 hydrolysis proposed to date from several structural studies involves a water wire or Grotthuss mechanism. Several structures reveal a nucleophilic water molecule near the anomeric carbon stabilized by a serine residue on the “active center” loop (residues 175–183 in HjeCel6A and 226–234 in TfuCel6B) (23, 30). This water molecule is hydrogen-bonded to a second water molecule, which in turn hydrogen-bonds to a conserved aspartate (Asp-175 in HjeCel6A and Asp-226 in TfuCel6B). The mechanism for GH6-inverting hydrolysis has thus been proposed to proceed via a water wire mechanism wherein the nucleophilic water attacks the anomeric carbon and transfers a proton to a second water molecule, which then transfers a proton to the putative catalytic base (30). The data to support this mechanism primarily come from structural studies, but enzymes wherein the hypothesized catalytic base has been mutated to alanine retain partial activity (29), warranting further study.
In this study, we extend our previous studies on TfuCel6B (23, 29). TfuCel6B is an interesting GH6 model system because of its extended and more enclosed tunnel, and also because Wilson and co-workers (33, 34) have conducted considerable experimental work on its catalytic mechanism, substrate specificity, and synergy with other T. fusca enzymes, thus providing significant mutational data for mechanistic interpretations. TfuCel6B is a bimodular enzyme with an N terminus Family 2 carbohydrate-binding module attached to the catalytic domain via a Pro/Ser-rich linker. In a previous study, Asp-274 was shown to be the catalytic acid (29). As with HjeCel6A, no direct catalytic base was identified in TfuCel6B. Mutation of the conserved Asp-226 to alanine resulted in lower activity on insoluble substrates, yet the D226A mutant enzyme exhibits slightly higher carboxymethyl cellulose (CMC) activity than the wild type enzyme. Furthermore, the double mutant D226A/S232A was completely inactive but showed rescued activity on CMC by a low concentration of sodium azide (29).
Recently, we published ligand-free and ligand-bound structures of the wild type TfuCel6B catalytic domain, which are the first reported GH6 CBH structures from a bacterium (23), shown in Fig. 2. The TfuCel6B catalytic domain has ∼100 more residues than its fungal counterparts, and the majority of extra residues are located to insertions at six discrete sections of the sequence (referred to as sections I to VI, and labeled in Fig. 2). In one of the structures obtained by co-crystallization with cellobiose, we could model an intact cellohexaose molecule in the active site tunnel in chain A from the +4 to the −2 glucopyranose-binding sites (with a cellotetraose spanning +2 to −2 in chain B). The substrate-binding path is significantly longer and more enclosed than in fungal GH6 CBHs due to these insertions of which one is an extra loop at the exit of the tunnel. In the ligand-free enzyme structure, this “exit loop” (section I) is disordered (residues 185–197), but in the ligand-bound enzyme structure, it completely encloses the product subsites, suggesting that the exit loop likely opens for product release.
FIGURE 2.

Overall structure of TfuCel6B. Sectional insertions compared with fungal GH6 CBHs are labeled I to VI. The exit loop (I) and bottom loop (V) are colored red and magenta, respectively. The catalytic center loop is colored cyan, and the loop pair of sections II and VI are colored green and light green. Sections III and IV (blue and light blue) are not further discussed in this study. Left, bottom view of the enzyme. Right, side view of the enzyme.
In this study, we employ x-ray crystallography to examine two catalytically deficient mutants (D274A and D226A/S232A) of TfuCel6B (29–32). Based on the new structures presented here and the previously published wild type structures (23), we conduct various molecular dynamics (MD) simulations to examine questions of enzyme and ligand dynamics relative to HjeCel6A, cellobiose product release after hydrolysis, and the complexation of the TfuCel6B catalytic domain on the surface of a cellulose Iβ crystal.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
We expressed TfuCel6B variants D226A/S232A and D274A in Escherichia coli and prepared them as described previously (29). To remove the carbohydrate-binding module, the full-length enzymes were subjected to papain digestion, and the resulting catalytic domains were purified using ion exchange chromatography on a Source 30Q column as described previously (23) followed by size-exclusion chromatography on a Superdex 200 column.
Crystallization and Data Collection
We co-crystallized the catalytic domains of TfuCel6B variants (6 mg/ml) with 10 mm cellobiose using the hanging drop method as described previously (23). The gradient seeding method was necessary for obtaining crystals and improving quality of the crystals. Both 20% ethylene glycol and 10% glycerol were added to the mother liquid as cryoprotectants. The crystals were flash-frozen in liquid nitrogen prior to the collection of x-ray diffraction data.
Two diffraction data sets were collected, one for each of the TfuCel6B mutants (D226A/S232A and D274A), from single crystals on beamlines ID14-4 and ID23-1, respectively, at European Synchrotron Radiation Facility (ESRF; Grenoble, France). The datasets were reduced and scaled using iMosflm (35) and Scala in CCP4 program suite (36).
Model Building and Refinement
The structures were solved by molecular replacement using Molrep (auto-MR function) in the CCP4 program suite (37, 38) with the structure of wild type TfuCel6B as a search model. For the model refinement, REFMAC5 (39) was used and the models were manually adjusted using Coot (40) by inspection of 2mFo − DFc and mFo − DFc σA-weighted maps. Solvent molecules were automatically added using the ARP/wARP water-adding function within Refmac5 (41) and manually checked by inspection of electron density in 2mFo − DFc map during refinement. The ligands were introduced by fitting cellohexaose, cellotetraose, or glucose to the electron density in different structure models at a late stage, when the refinement of the protein part was nearly finished.
Structural Preparation
The MD simulations were conducted with wild type TfuCel6B. Three ligand-bound structures and the ligand-free wild type structure were studied. For the cellodextrin-bound enzymes, we consider two different binding scenarios: a cellobiose bound at the −2, −1 subsites and a cellotetraose bound from the +1 to +4 subsites, as shown in the crystal structure, and a single cellohexaose bound in the tunnel from the −2 to +4 subsites. Additionally, a previously equilibrated cellulose Iβ microfibril (42, 43) was used to construct a TfuCel6B-cellulose complex (Fig. 3). In our previous studies, we have examined catalytic engagement of H. jecorina Cel7A and HjeCel6A on the crystalline cellulose Iβ microfibril by manually docking the enzymes on the cellulose [100] surface (44). In this work, we superimposed TfuCel6B on the equilibrated structure of HjeCel6A-cellulose complex to generate the initial structure of TfuCel6B-cellulose complex. The microfibril was composed of four layers, which consist of three, four, five, and six cellulose chains from the top layer to the bottom layer, respectively. Each cellulose chain has 16 glucose residues. The enzyme was placed on the hydrophobic surface at the nonreducing end. The cellodextrin chain from the crystal was bonded to the ligand at the +4 position manually, and the threaded chain was an “edge” chain, which are easier to decrystallize relative to “middle” chains (13). The positions of the cellobiose and the cellotetraose bound at the tunnel were the same as in the TfuCel6B-cellodextrin system.
FIGURE 3.
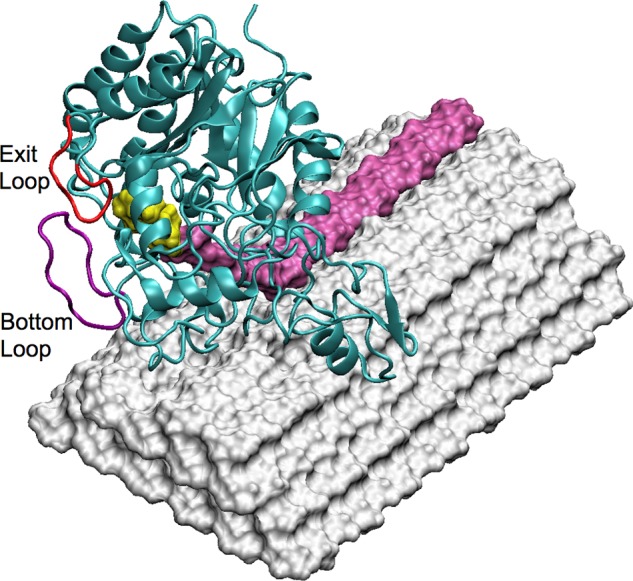
TfuCel6B catalytically engaged on the crystalline cellulose surface. The enzyme was placed on the hydrophobic surface at the nonreducing end. An edge chain was pulled up from the cellulose surface and bound in the catalytic tunnel. The product cellobiose is shown in yellow, and the bottom and exit loops are labeled.
MD Simulations
All the systems were solvated in an orthogonal box of TIP3P water molecules (45, 46) using CHARMM (47). The CHARMM22 force field parameters (47, 48) with the CMAP correction (49–51) were used for the enzyme, and the C35 carbohydrate force field parameters (52, 53) were used for the substrates. Ions were added into the systems to maintain charge neutrality. The final system size was ∼56,000 atoms for TfuCel6B-cellodextrin complex and 81,000 atoms for TfuCel6B-cellulose complex. In the TfuCel6B-cellulose complex, all atoms except hydrogen atoms of the six cellulose chains at the bottom of the microfibril were harmonically restrained with a weak force constant of 2 kcal/(mol × Å2). The four systems were equilibrated in the NPT ensemble at 300 K with a Nosé-Hoover thermostat (54, 55) and 1 atm for 100 ps with a step size of 1 fs. SHAKE was used to fix the covalent bonds to hydrogen atoms in all MD simulations (56). The nonbonded interactions were zeroed at 10 Å with a switching function. The electrostatic interactions were calculated using the particle mesh Ewald method (57) with a sixth order b-spline interpolation, a Gaussian distribution width of 0.34 Å, and a mesh size of 96 × 96 × 96 for ligand-free enzyme structure and TfuCel6B-cellodextrin complexes and a mesh size of 120 × 90 × 90 for TfuCel6B-cellulose complex. Following the equilibration, the production MD simulations were conducted in the NVT ensemble for 260 ns at 300 K with a step size of 2 fs using NAMD (58).
Steered Molecular Dynamics (SMD) Simulations
The protocols for conducting SMD simulations on the TfuCel6B-cellodextrin complex and the TfuCel6B-cellulose complex follow our previous work on product inhibition (15, 59). CHAMBER (61) was used to convert the protein structure file, coordinate file, and force field files in CHARMM format to Amber format. Each system was simulated in the NVT ensemble for 20 ns at 300 K with a step size of 2 fs using the PMEMD engine in Amber (62). Subsequently, a set of 100 starting structures was selected from the 20-ns simulation of the complex system as the initial structures of 100 SMD simulations, which were conducted using the Jarzynski implementation in Amber (62). During the SMD simulations, an external force was applied to the cellobiose ligand, and the distance between the two dummy atoms shown in Fig. 4, d (also referred to as the reaction coordinate), was increased with a speed of 1 Å/ns in 14 ns. We examined different pulling speeds, and 1 Å/ns was found to yield converged results similar to our previous work (15, 59). The force constant is 5000 kcal/(mol × Å2). The general work needed to drive the system from the cellobiose bound state to the unbound state is calculated as a function of the distance d for each SMD simulation. The Jarzynski equality ΔG = −kT ln〈exp(−W/kT)〉 was used to calculate the binding free energy ΔG (63–65) where W is the work associated with the distance d in each SMD simulation. The exponential average of the 100 work profiles results in the determination of the free energy (i.e. the potential of mean force along the reaction coordinate, d). The SMD measurement uncertainty was estimated with bootstrapping (66).
FIGURE 4.
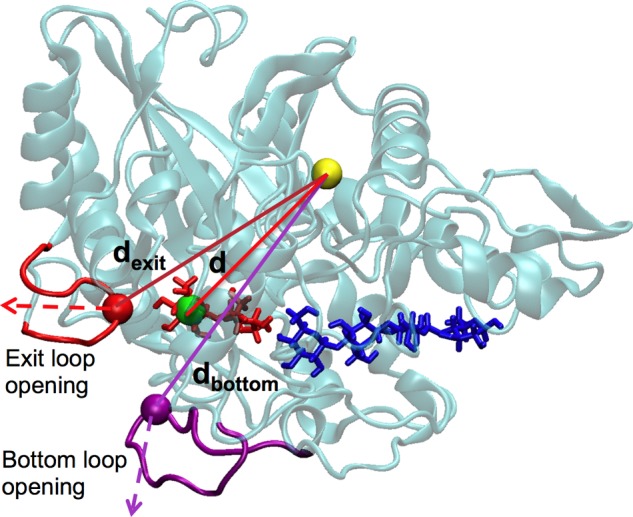
SMD simulation setup for TfuCel6B. Two dummy atoms, representing the endpoints of the pulling distance d, are shown as a green sphere (representing the center-of-mass of glucose at the −2 site in TfuCel6B), as well as a yellow sphere (representing the center-of-mass of 79 Cα atoms from TfuCel6B residues 318:324, 333:348, 358:363, 398:418, 420:426, 478:488, and 543:553). These residues were chosen to represent the position of the enzyme because they form stable secondary structures and have small RMSF values (see data below). The green and yellow dummy atoms align between the exit loop opening pathway and the bottom loop opening pathway to avoid any bias. Two additional dummy atoms, representing the positions of the tip of the exit loop and the bottom loop, are shown as a red sphere (representing the exit loop), as well as a purple sphere (representing the bottom loop). The distances dexit and dbottom are used to determine the amplitude of loop opening.
As shown in Fig. 4, two dummy atoms were chosen to represent the positions of the exit loop and the bottom loop (section V) (23). The distance between the red/purple dummy atom, representing the position of the exit/bottom loop, and the yellow dummy atom, representing the protein position, was measured from the 200-ns unbiased MD simulation of the TfuCel6B-cellodextrin complex. During the 100 SMD simulations, we measured the increased distance as a function of time, i.e. Δd(t) = d(t) − d(0), where d(t) is the distance of the two dummy atoms at time t and d(0) is the initial distance before the SMD simulation starts. The largest value of Δd(t) was chosen for each SMD trajectory to characterize the extent of loop opening.
RESULTS
Overall TfuCel6B Structural Results
The catalytic modules of two catalytically deficient mutants of TfuCel6B, D274A and D226A/S232A (the latter is dubbed the “double mutant”), were crystallized in the presence of cellobiose in the space group P21. Both the crystal packing (one molecule/asymmetric unit) and the unit cell dimensions were different from that of wild type TfuCel6B structure (two molecules/asymmetric unit) (23). The Cel6B D274A and D226A/S232A structures (Protein Data Bank (PDB) codes 4AVO and 4AVN, respectively) were solved and refined to 2.0 and 1.8 Å resolution and final Rwork/Rfree of 15.5/19.1% and 15.4/19.8%, respectively. The statistics of the x-ray diffraction data and structure refinement are summarized in Table 1. Overall, the two structures are similar to one another and to the wild type structure. The exit loop (residues 185–197) and the active center loop (226–234), which were open in the wild type ligand-free structure, are closed in both mutant structures as in the ligand structure of the wild type enzyme. Salient differences between the published structures (23) and the present structures are discussed below.
TABLE 1.
Crystallographic data for TfuCel6B catalytic mutants co-crystallized with cellobiose
| D274A (PDB code 4AVO) | D226A/S232A (PDB code 4AVN) | |
|---|---|---|
| Data quality | ||
| Beamlinea | ID23–1 | ID14–4 |
| Space group | P21 | P21 |
| Cell dimensions | ||
| a, b, c (Å) | 42.7 92.2 49.6 | 42.5 92.1 49.8 |
| β (°) | 112.0 | 111.9 |
| Resolutionb | 20.0–1.8 (1.9–1.8) | 30.0–2.0 (2.1–2.0) |
| Rmergeb,c | 0.102 (0.296) | 0.102 (0.335) |
| Mean I/σ (I)b | 10.7 (4.9) | 15.3 (5.7) |
| Completeness (%)b | 99.9 (99.9) | 99.4 (99.9) |
| Multiplicityb | 4.1 (4.1) | 7.1 (7.2) |
| Refinement | ||
| Rcryst | 0.155 | 0.154 |
| Rfree | 0.191 | 0.198 |
| No. of unique reflections | 32986 | 23941 |
| No. of protein residues | 420 | 420 |
| No. of water molecules | 304 | 275 |
| No. of carbohydrates | 6 | 5 |
| RMSDd | ||
| Bond length (Å) | 0.007 | 0.009 |
| Bond angles (°) | 1.116 | 1.08 |
| Average B-factor (Å2)e | ||
| Proteine | 11.1 | 13.4 |
| Ligande | 11.8 | 23.7 |
| Watere | 21.3 | 20.7 |
| Ramachandran outliers(%)f | 0.25 | 0.48 |
a Beamlines at European Synchrotron Radiation Facility (ESRF), Grenoble, France.
b Values in parentheses are those for the highest resolution shell.
c Rmerge = ΣhklΣi|I − 〈I〉|ΣhklΣi|I|.
d RMSD, root mean square deviation. From Engh and Huber (78).
e Calculated using MOLEMAN2 (79).
f Calculated using a strict-boundary Ramachandran definition given by Kleywegt and Jones (60).
Both of the TfuCel6B variant structures have carbohydrate ligands bound in the active site tunnel. The glycan-binding subsites are numbered from the nonreducing end to the reducing end of the glycan chain, −2, −1, +1, +2, +3, +4, with the glycosidic bond to be cleaved between sites −1 and +1. The product subsites are at −1 and −2 because they harbor the cellobiose moiety that is cleaved off from the nonreducing end of a cellulose chain during processive action. In both of the structures, there is distinct electron density for one glucose unit in subsite −2 and contiguous density for four glucose units from +1 to +4 subsites. The electron densities of the glycans were successively weaker from the nonreducing to the reducing end, which correlates with increasing temperature factors of the glucopyranosyl atoms. The D274A structure also displays partially occupied density for a glucose residue at site −1 (Fig. 5, top), but the D226A/S232A structure does not (Fig. 5, bottom). Consecutive glucose residues could be connected with β-1,4-glycosidic bonds in all cases without violation of bond distance and angle restraints. Therefore, a cellohexaose molecule was modeled into TfuCel6B D274A, spanning subsites −2 to +4, whereas the D226A/S232A structure contains a glucose residue in subsite −2 and cellotetraose in subsites +1 to +4 (Fig. 5).
FIGURE 5.
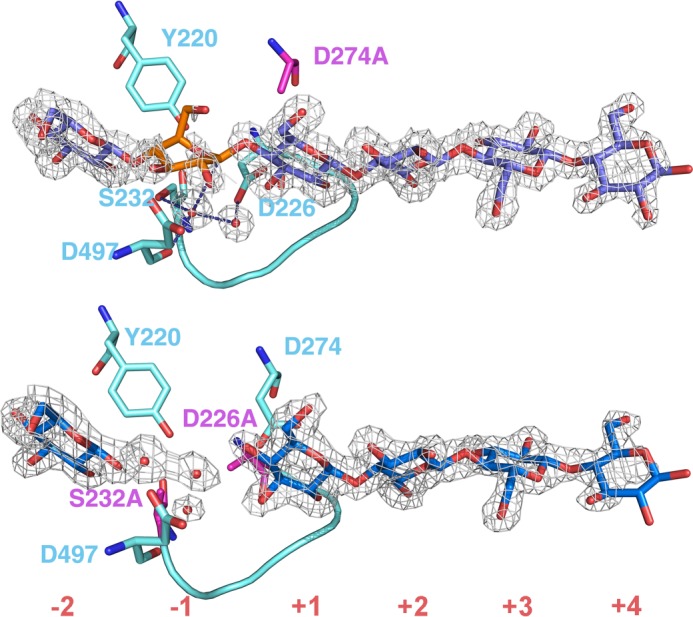
Electron density maps of bound glucopyranose residues in the active site tunnel of the TfuCel6B mutants. Top, D274A, the catalytic acid mutant. Bottom, D226A/S232A double mutant. Subsites of the substrate-binding tunnel are numbered from nonreducing end to reducing end as −2, −1, +1, +2, +3, +4. The 2Fo − Fc electron density maps are contoured at 0.46 electrons/Å3 (1σ). Cyan-colored residues around the active site and the active center loops are shown with a stick and a graphic representation, respectively. Water molecules are shown as red spheres. Residues in magenta were mutated to alanine in each mutant. The distorted glucopyranose residue at the −1 subsite of D274A mutant (top) is highlighted in orange.
The catalytic acid mutant D274A structure in complex with glycans is similar to the wild type enzyme. The −1 glucopyranosyl residue refines in the same 2SO skew boat conformation (Fig. 5, top). The density for a glucopyranosyl residue at this position is weak, which makes the 2SO conformation less certain. However, there is no support for any other sugar conformation, and the 2SO skew boat conformation was previously observed in wild type TfuCel6B and GH6 enzymes (23, 32, 67, 68). Two water molecules are retained between the anomeric carbon and the proposed catalytic base, Asp-226, which adopts the “active” conformation.
The substrate binds similarly in the D226A/D232A complex. The catalytic acid, Asp-274, adopts the “inactive” conformation, not pointing toward 4OH of the +1 glucose unit, i.e. the position for the glycosidic oxygen to be protonated. It is noteworthy that the inactive conformation is adopted despite the lack of a hydrogen bond to the proposed catalytic base, as observed in GH6 structures where the latter residue has not been mutated (38). We observe electron density for a water molecule bound to the backbone carbonyl of Asp-497, corresponding to the position of the nucleophilic water, but not for the second water molecule in the proposed mechanism, suggesting that the D226A and D232A mutations have disrupted the putative Grotthuss proton transfer chain for nucleophilic attack on the anomeric carbon.
Closer examination of the electron density in subsite −1 of the D226A/D232A glycan complex reveals that water molecules bind at the expected sites for hydroxyl groups of a reducing end glucose residue. Using these water molecules, an α-cellobiose molecule placed in sites −2 and −1 could be refined to provide a model of how the product binds after hydrolysis of the glycosidic bond. After refinement, the α-glucose residue at site −1 adopts a 2,5B boat conformation (Fig. 6, green), consistent with the proposed conformational itinerary for the inverting mechanism of GH6 enzymes, from a 2SO skew via an oxocarbenium ion-like transition state to a 2,5B boat conformation (30, 69, 70). This result indicates that the −1 ring pucker should undergo a conformational change from 2So to 2,5B conformations through the transition state. Comparison of the glycan ring distortion at site −1 suggests that the glucose ring is held in place via the 4-O-glycosidic bond, the C2 and C3 hydroxyl groups, and through the steric restraints imposed on the C5-C6-O6 arm by Tyr-220. The anomeric carbon migrates ∼1.2 Å from the β-bond with the glycosidic oxygen, over the transition state, to an α-linkage with the nucleophilic water molecule.
FIGURE 6.
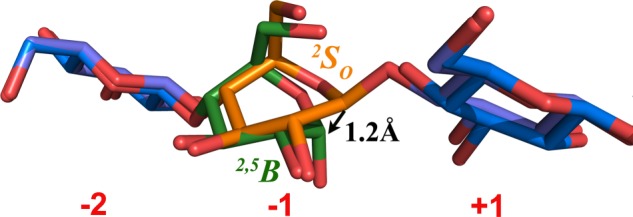
Water molecules present in the double mutant structure enable construction of an α-cellobiose model in the product of TfuCel6B in the 2,5B conformation. The orange glucopyranose residue is from the TfuCel6B D274A structure in the 2So conformation. The green glucose residue was built in the TfuCel6B D226AA/S232A structure as a product-binding model in the 2,5B boat conformation. The distance between two anomeric carbons in these structures is 1.2 Å, which suggests the itinerary of the anomeric carbon during nucleophilic attack by the water molecule and concomitant glycosidic bond cleavage.
Computational Investigation of Enzyme and Ligand Dynamics
From the structural data, two loops in particular are noteworthy for their putative action during product expulsion and the differences that these loops may impart specifically to TfuCel6B ligand and enzyme dynamics. Namely, the exit loop (residues 185–197) is disordered and presumably flexible in the ligand-free structure, but in all of the holo structures, it closes over the product subsites. Moreover, the bottom loop (residues 501–510) forms the other section of the enzyme along with the exit loop that closes over the cellobiose product. Based on the structural results for the exit loop and on the proximity of the bottom loop to the product subsites, we hypothesize that either of these loops must open during product expulsion.
To examine this hypothesis, we first conducted long unbiased MD simulations of TfuCel6B to examine the flexibility of the ligand, the enzyme, and the loops surrounding the tunnel exit and active site. Three MD simulations were conducted in solution: a ligand-free MD simulation, an MD simulation with a cellotetraose spanning +4 to +1 with a cellobiose in −1 and −2 to mimic the post-hydrolysis system, and an MD simulation with a cellohexaose spanning the active site tunnel from +4 to −2 to model the catalytically active state. Moreover, catalytic engagement of TfuCel6B on a solid cellulose crystal may influence the conformation and flexibility of the tunnel-forming loops. Thus, we examined whether substrate complexation on the cellulose surface can block either of the loops opening by running an MD simulation (Fig. 3) to model the post-hydrolysis system with a cellobiose in the −1 and −2 subsites and a cellotetraose spanning the +4 to +1 subsites, which is further bonded to the cellulose chain being decrystallized from the crystal surface.
The root mean square fluctuation (RMSF) values of each residue in the ligand-free enzyme and the two TfuCel6B-cellodextrin complexes are shown in Fig. 7A. The exit loop exhibits an RMSF value of ∼2 Å larger than that of the bottom loop for the simulations of TfuCel6B in solution. However, when the enzyme is catalytically engaged on the cellulose surface, the RMSF value of the bottom loop increases significantly by ∼3 Å. The bottom loop does not interact with the cellulose surface, but the subsequent portion of the section VI loop (residues 520–530) binds to the cellulose surface, which in turn dramatically modifies the dynamics of the bottom loop. As shown in the residue cross-correlation maps (supplemental Fig. S1), the fluctuations of the bottom loop are not correlated with the motions of other regions in TfuCel6B after catalytic engagement, during which the interaction energy between the bottom loop and other regions in TfuCel6B decreased by ∼30 kcal/mol (supplemental Table S1), suggesting the loss of a significant number of protein-protein contacts. As a result, the bottom loop becomes more flexible after TfuCel6B is engaged on the cellulose surface. Our results suggest that catalytic engagement of TfuCel6B on a cellulose surface may influence the flexibility of the tunnel-forming loops in TfuCel6B differently from that in solution, and thus product expulsion was examined in both cases.
FIGURE 7.
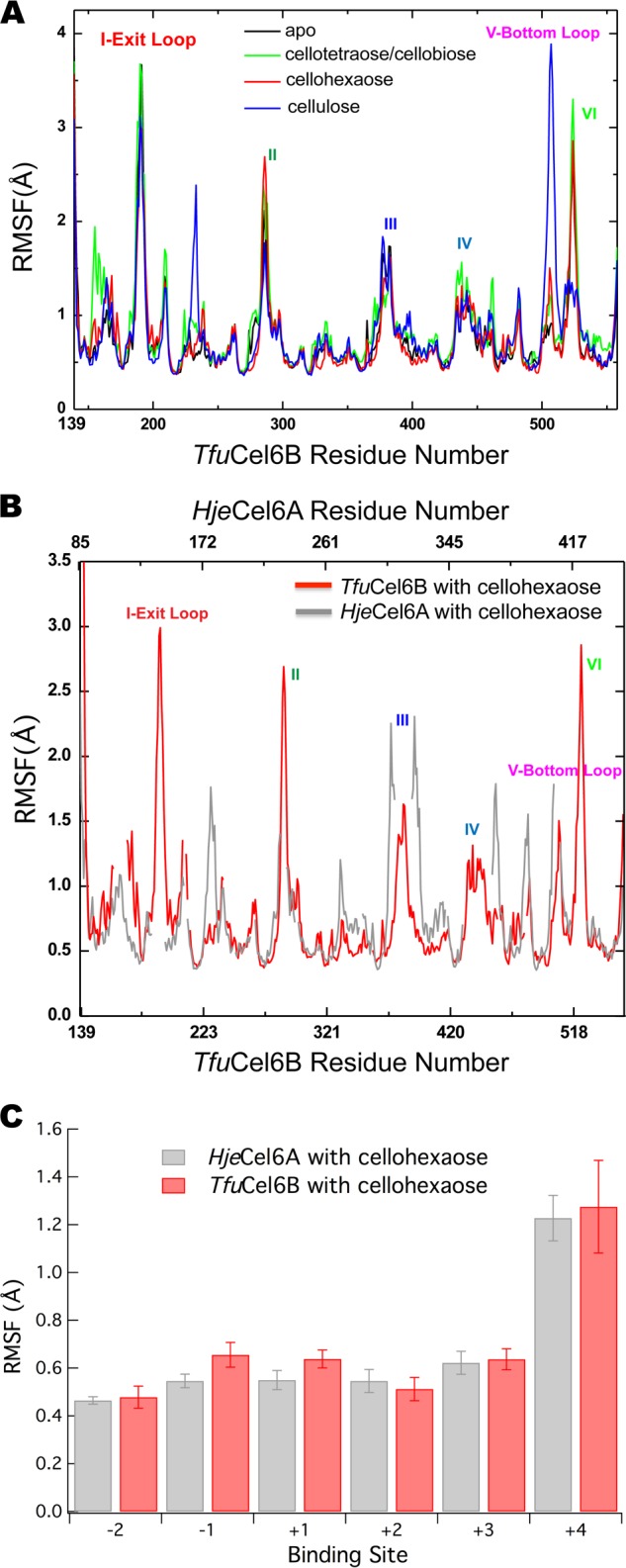
Enzyme and ligand dynamics in TfuCel6B compared with HjeCel6A. A, RMSF values of the four TfuCel6B complexation states. B, RMSF values of the cellohexaose-bound TfuCel6B and HjeCel6A. C, RMSF values of the ligand as a function of binding site for TfuCel6B and HjeCel6A with a cellohexaose ligand bound to the enzymes in solution. Data for HjeCel6A are from Ref. 71.
Additionally, we compared the overall enzyme fluctuations of TfuCel6B with HjeCel6A to understand how the more enclosed active site tunnel may affect enzyme and ligand dynamics. As mentioned above, the exit and bottom loops are absent in HjeCel6A, and overall the TfuCel6B fluctuations are on average higher (Fig. 7B). Additionally, we compared the fluctuations of the ligands between HjeCel6A and TfuCel6B in the cellohexaose-bound structures (Fig. 7B). The HjeCel6A data are taken from previous work (71). As shown in Fig. 7B, the ligand RMSF is surprisingly similar along the entire active site tunnel for both enzymes despite the fact that TfuCel6B exhibits a significantly more enclosed tunnel. As with HjeCel6A, the +4 subsite is the most flexible, likely due to greater solvent exposure along the active site tunnel.
Product Expulsion Pathways
To investigate whether the bottom loop or exit loop open after hydrolysis in TfuCel6B, we conducted 100 SMD simulations to slowly pull the product from the tunnel, both with TfuCel6B in solution and with TfuCel6B complexed on cellulose. The product expulsion pathways from the 100 SMD simulations in both scenarios were analyzed to identify the most probable pathway that the cellobiose follows by measuring the positions of the exit/bottom loops relative to the protein during the SMD simulations. The results are summarized in Fig. 8. The data suggest that the exit loop can open up to ∼14 Å relative to its initial position, whereas the bottom loop can open up to ∼16 Å. The exit loop opens largely, i.e. beyond 10 Å, in 19 simulations, whereas the bottom loop opens beyond 10 Å in 20 simulations. Interestingly, there is no overlap between these two subsets of the 100 trajectories, i.e. under no circumstances do both loops open more than 10 Å simultaneously. Most of the product expulsion pathways involve both moderate opening of the exit and bottom loops, suggesting that both the exit and the bottom loop open with equal probability for product expulsion from the closed catalytic tunnel of TfuCel6B. Moreover, there is no clear difference in the propensity for either loop to open more than another in solution relative to when TfuCel6B is complexed on cellulose.
FIGURE 8.

SMD results for loop opening in TfuCel6B. A, a two-dimensional matrix representation of loop opening in the 100 SMD simulations of TfuCel6B (red, in solution; blue, complexed on cellulose). Each square represents the extent of loop opening during an individual SMD simulation defined by the largest distance increment between two loops and a chosen position in protein, relative to the initial crystal structure. The equally distributed squares along the diagonal indicate no preference between the opening either of the two loops. B, two extreme pathways involve a large opening of either the exit loop or the bottom loop. 19 and 20 SMD trajectories are superimposed for the exit loop opening pathway (cellobiose is shown in mauve sticks) and the bottom loop opening pathway (cellobiose is shown in blue sticks), respectively. The initial and final conformations of the exit loop are shown in yellow and red, respectively, whereas the initial and final conformations of the bottom loop are shown in cyan and purple, respectively.
Fig. 9 illustrates the potential of mean force of cellobiose expulsion from the catalytic domain of TfuCel6B. The computed reversible thermodynamic work required to remove cellobiose from the product sites of the enzyme is the negative cellobiose binding free energy. Thus, the overall binding free energy of cellobiose calculated using all 100 SMD trajectories from TfuCel6B-cellodextrin complex and TfuCel6B-cellulose complex is −21.2 ± 1.1 and −19.8 ± 0.5 kcal/mol, respectively, which is consistent with the binding free energy of cellobiose to the catalytic domain of HjeCel6A, −22.4 ± 0.3 kcal/mol, as characterized in our previous study (15). Specifically, for the TfuCel6B-cellodextrin complex, the calculated binding free energy is −25.9 ± 1.1 kcal/mol by using 19 trajectories involving only the large opening of the exit loop and −25.4 ± 0.2 kcal/mol by using 20 trajectories involving only the large opening of the bottom loop, respectively (supplemental Fig. S2). The quantified free energy of product expulsion via the two “extreme” pathways supports the observation in Fig. 8 that no preference exists in opening of the exit loop and the bottom loop.
FIGURE 9.
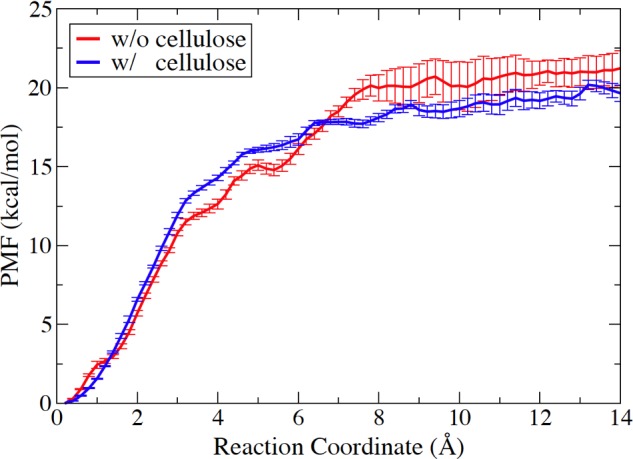
Potential of mean force (PMF) profiles of cellobiose expulsion from the catalytic domain of TfuCel6B with or without binding to the crystalline cellulose.
DISCUSSION
In this study, we have characterized two structures of TfuCel6B catalytic mutants. These structures are noteworthy for understanding GH6 hydrolysis mechanisms as mutation of the catalytic acid (D274A) or mutation of the putative catalytic base along with the residue that stabilizes the nucleophilic water (D226A/S232A) does not disrupt the structure of the active site considerably. This result in turn supports the hypothesized role of Asp-226 and Ser-232 in the catalytic step (30). The results for the double mutant were additionally surprising in that the active center loop remains closed over the cellodextrin despite removal of stabilizing hydrogen bonds, which is hypothesized to be a key interaction during CBH action. TfuCel6B was also chosen as a model GH6 enzyme because it has been extensively studied and its activity profile upon mutations has been well characterized (29, 33, 72), and the structures solved here also suggest a possible explanation for the observed catalytic profiles. A mutant enzyme where only the putative catalytic base is mutated, TfuCel6B D226A, exhibits higher activity on CMC than the wild type enzyme (29). Analysis of the TfuCel6B structures reveals that the active site of the mutant D226A could harbor a 3-hydroxy-substituted carboxymethyl group on the glucopyranose residue in the +1 subsite. Fig. 10 shows a model for how such a carboxymethyl substitution could be in a position to form a hydrogen bond directly with the nucleophilic water in the active site of a D226A mutant. The Grotthuss mechanism may thus be retained despite the lack of an aspartic acid side chain. In the D226A/S232A double mutant, the proton network would be disrupted due to the lack of Ser-232, which explains the loss of activity on CMC (29). For this mutant, activity could partially be regained by adding sodium azide (29). The structure of the double mutant shows that in the absence of the hydroxyl group, there is space to accommodate an azide molecule. A nucleophilic water molecule functionally similar to that of the wild type enzyme may thus be present with the help of a carboxymethyl group on the 3-hydroxyl group in the subsite +1 and the azide molecule.
FIGURE 10.
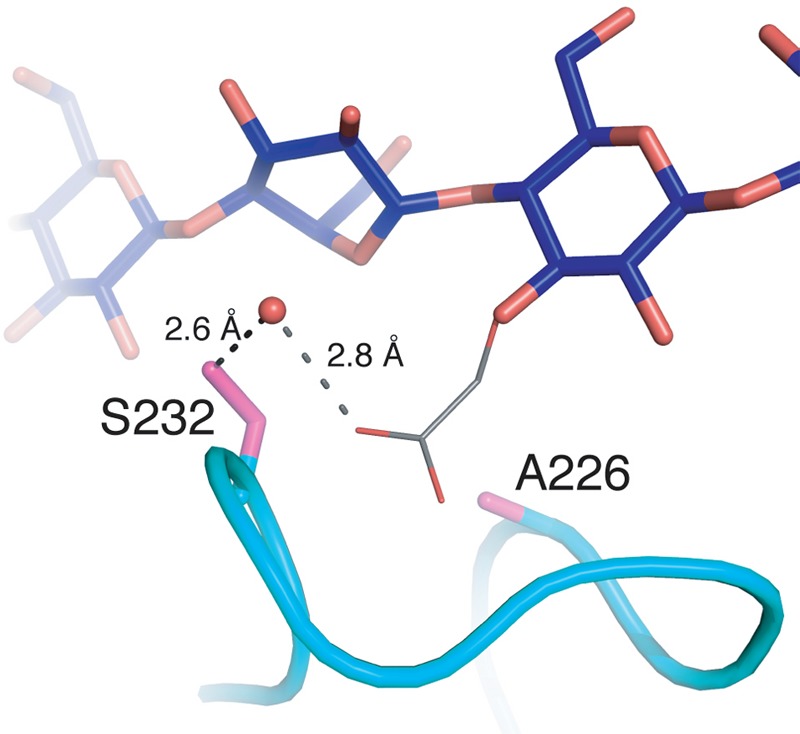
Hypothetical model for CMC activity by the TfuCel6B D226A mutant. A carboxymethyl group on the 3-hydroxyl group of the glucopyranose in subsite +1 can be accommodated at the catalytic center and can be suitably positioned to activate the water molecule for nucleophilic attack of the anomeric carbon in the −1 subsite.
Additionally, the exit loop and bottom loop are not found in fungal GH6 enzymes, and it is of interest to understand the evolutionary pressure to utilize a completely enclosed product region; e.g. the presence of a loop corresponding to the bottom loop has previously been linked to a more cellobiohydrolase-like behavior of the GH6 enzyme cbhA from Cellulomonas fimi (73). As illustrated in Fig. 1, the product regions are much more closed in TfuCel6B than in fungal counterparts. From the results shown in Fig. 7, it is surprising given the extent of ligand contacts in TfuCel6B relative to HjeCel6A that the ligand fluctuations are not lower (i.e. the enzyme stabilizes the ligand more) in TfuCel6B, which has been correlated in chitinases (74) and GH7 cellulases (75) to higher degrees of processivity. However, it is perhaps noteworthy that the −3 and −4 subsites of TfuCel6B are blocked by the presence of the exit loop. We hypothesize that a potential functional reason for blocking the −3 and −4 subsites in a GH6 enzyme could be to minimize the ability of the enzyme to perform endo-like initiation or endoglucanase-like cuts in cellulose, but rather force it to act more in a CBH-only mode. This result may explain why there is surprisingly no difference in ligand fluctuations, and yet TfuCel6B may still be more processive than HjeCel6A. Testing this hypothesis will require careful measurements of endo-initiation and processivity on cellulose substrates (76, 77), which to date has proven challenging for nonreducing end-specific CBHs.
Moreover, we conducted an extensive sequence alignment of GH6 cellulases. By comparing the exit and bottom loops together, as shown in supplemental Fig. S3, we find 7 basic groups of bacterial GH6s in terms of sequence diversity of these two loops. Neither of the loops is present in fungal GH6 cellulases. The first two groups comprise the majority of the available sequences (Fig. 11). Namely, we find that the long exit loop such as that in TfuCel6B of 10–11 extra residues is found only in bacteria from the order Actinomycetales, and mainly clustered in Streptosporangineae, Pseudonocardineae and Micromonosporaceae. A second group present only in Streptomycineae contains an ∼6-residue insertion with either a conserved asparagine or a conserved threonine residue, but no structures exist from this second class of GH6 enzymes to date. Conversely in most cases, the bottom loop in bacterial cellulases examined in the sequence alignment is ∼4 residues longer than in HjeCel6A with only a few entries that are significantly longer.
FIGURE 11.
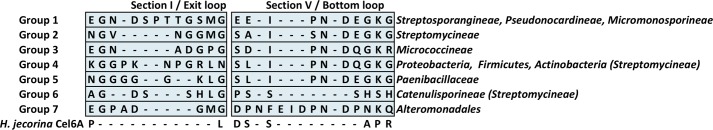
Representative sequence profiles. One representative of each of the seven identified groups is shown. To the right of each representative, the major occurrence within bacteria is given. Additional minor occurrences are written within parentheses. For comparison, the sequence profiles of the corresponding loops in HjeCel6A are also shown.
The subsequent section VI is a surface loop located at the entrance to the active site tunnel that pairs with the section II loop on the opposite side of the tunnel (23, 30). The structure formed by sections II and VI and with the bottom loop are a large part of the active site tunnel roof and not present at all in bacterial endoglucanases. In our simulations of the TfuCel6B-cellulose complex, section VI interacts with the cellulose surface. This section is much more conserved than the exit and bottom loops as shown in supplemental Fig. S4. More than half of the available sequences share a PXYXGNXRN pattern. Of the conserved residues, only the tyrosine and the arginine side chains could potentially interact with the cellulose surface. The conserved arginine (Arg-524) adopts several different side chain conformations among the different structure models of TfuCel6B. One of these is a direct interaction with the open active site loop in the ligand-free structure (23, 30), an interaction that is not detected in any of the structure models with a closed active site loop. The active site loop of a GH6 CBH needs to open and close for processive action on the cellulose chain. Arg-524 may thus be a link between an interaction of the enzyme with the cellulose surface via section VI and the processive catalytic activity, which will be examined in a future study.
Overall, this study sheds new light on the structure of the active site and role of long loops near the product site of a unique class of GH6 CBHs. However, many questions remain to be answered generally for GH6 action regarding the mechanisms of hydrolysis and processivity as well as regarding the role of long loops that block the product sites in processive action, which will be addressed in future structural and computational studies. Understanding the functional diversity in this important class of GH enzymes will be essential for the development of structure-activity relationships and to understand why certain organisms evolved seemingly such specialized features on a common enzyme fold and reaction mechanism.
Acknowledgments
Computer time for this research was provided by the Texas Advanced Computing Center Stampede cluster under National Science Foundation Extreme Science and Engineering Discovery Environment (XSEDE) Grant number MCB090159 and the National Renewable Energy Laboratory (NREL) Computational Sciences Center supported by the Department of Energy Efficiency and Renewable Energy (EERE) under Contract DE-AC36-08GO28308.
This work was partly supported by the Faculty for Natural Resources and Agriculture at the Swedish University of Agricultural Sciences through the research program MicroDrivE.

This article contains supplemental Table S1 and Figs. S1–S4.
The atomic coordinates and structure factors (codes 4AVO and 4AVN) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- CBH
- cellobiohydrolase
- GH6
- glycoside hydrolase Family 6 enzymes
- CMC
- carboxymethyl cellulose
- RMSF
- root mean square fluctuation
- MD
- molecular dynamics
- SMD
- steered molecular dynamics.
REFERENCES
- 1. Floudas D., Binder M., Riley R., Barry K., Blanchette R. A., Henrissat B., Martínez A. T., Otillar R., Spatafora J. W., Yadav J. S., Aerts A., Benoit I., Boyd A., Carlson A., Copeland A., Coutinho P. M., de Vries R. P., Ferreira P., Findley K., Foster B., Gaskell J., Glotzer D., Górecki P., Heitman J., Hesse C., Hori C., Igarashi K., Jurgens J. A., Kallen N., Kersten P., Kohler A., Kües U., Kumar T. K., Kuo A., LaButti K., Larrondo L. F., Lindquist E., Ling A., Lombard V., Lucas S., Lundell T., Martin R., McLaughlin D. J., Morgenstern I., Morin E., Murat C., Nagy L. G., Nolan M., Ohm R. A., Patyshakuliyeva A., Rokas A., Ruiz-Dueñas F. J., Sabat G., Salamov A., Samejima M., Schmutz J., Slot J. C., St John F., Stenlid J., Sun H., Sun S., Syed K., Tsang A., Wiebenga A., Young D., Pisabarro A., Eastwood D. C., Martin F., Cullen D., Grigoriev I. V., Hibbett D. S. (2012) The paleozoic origin of enzymatic lignin decomposition reconstructed from 31 fungal genomes. Science 336, 1715–1719 [DOI] [PubMed] [Google Scholar]
- 2. Lynd L. R., Weimer P. J., van Zyl W. H., Pretorius I. S. (2002) Microbial cellulose utilization: fundamentals and biotechnology. Microbiol. Mol. Biol. Rev. 66, 506–577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kirk T. K., Farrell R. L. (1987) Enzymatic “combustion”: the microbial degradation of lignin. Annu. Rev. Microbiol. 41, 465–505 [DOI] [PubMed] [Google Scholar]
- 4. Himmel M. E., Ding S. Y., Johnson D. K., Adney W. S., Nimlos M. R., Brady J. W., Foust T. D. (2007) Biomass recalcitrance: engineering plants and enzymes for biofuels production. Science 315, 804–807 [DOI] [PubMed] [Google Scholar]
- 5. Percival Zhang Y. H., Himmel M. E., Mielenz J. R. (2006) Outlook for cellulase improvement: Screening and selection strategies. Biotechnol. Adv. 24, 452–481 [DOI] [PubMed] [Google Scholar]
- 6. Reese E. T. (1976) History of the cellulase program at the U. S. Army Natick Development Center. Biotechnol. Bioeng. Symp. 6, 9–20 [PubMed] [Google Scholar]
- 7. Foreman P. K., Brown D., Dankmeyer L., Dean R., Diener S., Dunn-Coleman N. S., Goedegebuur F., Houfek T. D., England G. J., Kelley A. S., Meerman H. J., Mitchell T., Mitchinson C., Olivares H. A., Teunissen P. J., Yao J., Ward M. (2003) Transcriptional regulation of biomass-degrading enzymes in the filamentous fungus Trichoderma reesei. J. Biol. Chem. 278, 31988–31997 [DOI] [PubMed] [Google Scholar]
- 8. Teeri T. T., Koivula A., Linder M., Wohlfahrt G., Divne C., Jones T. A. (1998) Trichoderma reesei cellobiohydrolases: why so efficient on crystalline cellulose? Biochem. Soc. Trans. 26, 173–178 [DOI] [PubMed] [Google Scholar]
- 9. Jalak J., Kurašin M., Teugjas H., Väljamäe P. (2012) Endo-exo synergism in cellulose hydrolysis revisited. J. Biol. Chem. 287, 28802–28815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chundawat S. P. S., Beckham G. T., Himmel M. E., Dale B. E. (2011) Deconstruction of lignocellulosic biomass to fuels and chemicals. Annu. Rev. Chem. Biomol. Eng. 2, 121–145 [DOI] [PubMed] [Google Scholar]
- 11. Bansal P., Hall M., Realff M. J., Lee J. H., Bommarius A. S. (2009) Modeling cellulase kinetics on lignocellulosic substrates. Biotechnol. Adv. 27, 833–848 [DOI] [PubMed] [Google Scholar]
- 12. Beckham G. T., Bomble Y. J., Bayer E. A., Himmel M. E., Crowley M. F. (2011) Applications of computational science for understanding enzymatic deconstruction of cellulose. Curr. Opin. Biotechnol. 22, 231–238 [DOI] [PubMed] [Google Scholar]
- 13. Beckham G. T., Matthews J. F., Peters B., Bomble Y. J., Himmel M. E., Crowley M. F. (2011) Molecular-level origins of biomass recalcitrance: Decrystallization free energies for four common cellulose polymorphs. J. Phys. Chem. B 115, 4118–4127 [DOI] [PubMed] [Google Scholar]
- 14. Gruno M., Väljamäe P., Pettersson G., Johansson G. (2004) Inhibition of the Trichoderma reesei cellulases by cellobiose is strongly dependent on the nature of the substrate. Biotechnol. Bioeng. 86, 503–511 [DOI] [PubMed] [Google Scholar]
- 15. Bu L., Nimlos M. R., Shirts M. R., Ståhlberg J., Himmel M. E., Crowley M. F., Beckham G. T. (2012) Product binding varies dramatically between processive and nonprocessive cellulase enzymes. J. Biol. Chem. 287, 24807–24813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wilson D. B. (2004) Studies of Thermobifida fusca plant cell wall degrading enzymes. Chem. Rec. 4, 72–82 [DOI] [PubMed] [Google Scholar]
- 17. Irwin D. C., Spezio M., Walker L. P., Wilson D. B. (1993) Activity studies of eight purified cellulases: Specificity, synergism, and binding domain effects. Biotechnol. Bioeng. 42, 1002–1013 [DOI] [PubMed] [Google Scholar]
- 18. Herpoël-Gimbert I., Margeot A., Dolla A., Jan G., Mollé D., Lignon S., Mathis H., Sigoillot J. C., Monot F., Asther M. (2008) Comparative secretome analyses of two Trichoderma reesei RUT-C30 and CL847 hypersecretory strains. Biotechnol. Biofuels 1, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tian C., Beeson W. T., Iavarone A. T., Sun J., Marletta M. A., Cate J. H. D., Glass N. L. (2009) Systems analysis of plant cell wall degradation by the model filamentous fungus Neurospora crassa. Proc. Natl. Acad. Sci. U.S.A. 106, 22157–22162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Couturier M., Navarro D., Olivé C., Chevret D., Haon M., Favel A., Lesage-Meessen L., Henrissat B., Coutinho P. M., Berrin J. G. (2012) Post-genomic analyses of fungal lignocellulosic biomass degradation reveal the unexpected potential of the plant pathogen Ustilago maydis. BMC Genomics 13, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ribeiro D. A., Cota J., Alvarez T. M., Brüchli F., Bragato J., Pereira B. M. P., Pauletti B. A., Jackson G., Pimenta M. T. B., Murakami M. T., Camassola M., Ruller R., Dillon A. J., Pradella J. G., Paes Leme A. F., Squina F. M. (2012) The Penicillium echinulatum secretome on sugar cane bagasse. PLoS One 7, e50571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rouvinen J., Bergfors T., Teeri T., Knowles J. K. C., Jones T. A. (1990) 3-Dimensional structure of cellobiohydrolase II from Trichoderma reesei. Science 249, 380–386 [DOI] [PubMed] [Google Scholar]
- 23. Sandgren M., Wu M., Karkehabadi S., Mitchinson C., Kelemen B. R., Larenas E. A., Ståhlberg J., Hansson H. (2013) The structure of a bacterial cellobiohydrolase: the catalytic core of the Thermobifida fusca family GH6 cellobiohydrolase Cel6B. J. Mol. Biol. 425, 622–635 [DOI] [PubMed] [Google Scholar]
- 24. Varrot A., Hastrup S., Schülein M., Davies G. (1999) Crystal structure of the catalytic core domain of the family 6 cellobiohydrolase II, Cel6A, from Humicola insolens, at 1.92 Ä resolution. Biochem. J. 337, 297–304 [PMC free article] [PubMed] [Google Scholar]
- 25. Tamura M., Miyazaki T., Tanaka Y., Yoshida M., Nishikawa A., Tonozuka T. (2012) Comparison of the structural changes in two cellobiohydrolases, CcCel6A and CcCel6C, from Coprinopsis cinerea – a tweezer-like motion in the structure of CcCel6C. FEBS J. 279, 1871–1882 [DOI] [PubMed] [Google Scholar]
- 26. Spezio M., Wilson D. B., Karplus P. A. (1993) Crystal structure of the catalytic domain of a thermophilic endocellulase. Biochemistry 32, 9906–9916 [DOI] [PubMed] [Google Scholar]
- 27. Davies G. J., Brzozowski A. M., Dauter M., Varrot A., Schülein M. (2000) Structure and function of Humicola insolens family 6 cellulases: structure of the endoglucanase, Cel6B, at 1.6 angstrom resolution. Biochem. J. 348, 201–207 [PMC free article] [PubMed] [Google Scholar]
- 28. Vuong T. V., Wilson D. B. (2010) Glycoside hydrolases: catalytic base/nucleophile diversity. Biotechnol. Bioeng. 107, 195–205 [DOI] [PubMed] [Google Scholar]
- 29. Vuong T. V., Wilson D. B. (2009) The absence of an identifiable single catalytic base residue in Thermobifida fusca exocellulase Cel6B. FEBS J. 276, 3837–3845 [DOI] [PubMed] [Google Scholar]
- 30. Koivula A., Ruohonen L., Wohlfahrt G., Reinikainen T., Teeri T. T., Piens K., Claeyssens M., Weber M., Vasella A., Becker D., Sinnott M. L., Zou J. Y., Kleywegt G. J., Szardenings M., Ståhlberg J., Jones T. A. (2002) The active site of cellobiohydrolase Cel6A from Trichoderma reesei: the roles of aspartic acids D221 and D175. J. Am. Chem. Soc. 124, 10015–10024 [DOI] [PubMed] [Google Scholar]
- 31. Damude H. G., Withers S. G., Kilburn D. G., Miller R. C., Jr., Warren R. A. J. (1995) Site-directed mutation of the putative catalytic residues of endoglucanase cena from Cellulomonas fumi. Biochemistry 34, 2220–2224 [DOI] [PubMed] [Google Scholar]
- 32. Zou J.y., Kleywegt G. J., Ståhlberg J., Driguez H., Nerinckx W., Claeyssens M., Koivula A., Teeri T. T., Jones T. A. (1999) Crystallographic evidence for substrate ring distortion and protein conformational changes during catalysis in cellobiohydrolase Ce16A from Trichoderma reesei. Structure. 7, 1035–1045 [DOI] [PubMed] [Google Scholar]
- 33. Vuong T. V., Wilson D. B. (2009) Processivity, synergism, and substrate specificity of Thermobifida fusca Cel6B. Appl. Environ. Microbiol. 75, 6655–6661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jung H., Wilson D. B., Walker L. P. (2003) Binding and reversibility of Thermobifida fusca Cel5A, Cel6B, and Cel48A and their respective catalytic domains to bacterial microcrystalline cellulose. Biotechnol. Bioeng. 84, 151–159 [DOI] [PubMed] [Google Scholar]
- 35. Battye T. G. G., Kontogiannis L., Johnson O., Powell H. R., Leslie A. G. W. (2011) iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 67, 271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 [DOI] [PubMed] [Google Scholar]
- 37. Potterton E., Briggs P., Turkenburg M., Dodson E. (2003) A graphical user interface to the CCP4 program suite. Acta Crystallogr. D Biol. Crystallogr. 59, 1131–1137 [DOI] [PubMed] [Google Scholar]
- 38. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 39. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 40. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 41. Evrard G. X., Langer G. G., Perrakis A., Lamzin V. S. (2007) Assessment of automatic ligand building in ARP/wARP. Acta Crystallogr. D Biol. Crystallogr. 63, 108–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Payne C. M., Himmel M. E., Crowley M. F., Beckham G. T. (2011) Decrystallization of oligosaccharides from the cellulose Iβ surface with molecular simulation. J. Phys. Chem. Lett. 2, 1546–1550 [Google Scholar]
- 43. Beckham G. T., Dai Z., Matthews J. F., Momany M., Payne C. M., Adney W. S., Baker S. E., Himmel M. E. (2012) Harnessing glycosylation to improve cellulase activity. Curr. Opin. Biotechnol. 23, 338–345 [DOI] [PubMed] [Google Scholar]
- 44. Payne C. M., Resch M. G., Chen L., Crowley M. F., Himmel M. E., Taylor L. E., 2nd, Sandgren M., Ståhlberg J., Stals I., Tan Z., Beckham G. T. (2013) Glycosylated linkers in multimodular lignocellulose-degrading enzymes dynamically bind to cellulose. Proc. Natl. Acad. Sci. U.S.A. 110, 14646–14651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Durell S. R., Brooks B. R., Bennaim A. (1994) Solvent-induced forces between 2 hydrophilic groups. J. Phys. Chem. 98, 2198–2202 [Google Scholar]
- 46. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 [Google Scholar]
- 47. Brooks B. R., Brooks C. L., 3rd, Mackerell A. D., Jr., Nilsson L., Petrella R. J., Roux B., Won Y., Archontis G., Bartels C., Boresch S., Caflisch A., Caves L., Cui Q., Dinner A. R., Feig M., Fischer S., Gao J., Hodoscek M., Im W., Kuczera K., Lazaridis T., Ma J., Ovchinnikov V., Paci E., Pastor R. W., Post C. B., Pu J. Z., Schaefer M., Tidor B., Venable R. M., Woodcock H. L., Wu X., Yang W., York D. M., Karplus M. (2009) CHARMM: the biomolecular simulation program. J. Comput. Chem. 30, 1545–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. MacKerell A. D., Bashford D., Bellott M., Dunbrack R. L., Evanseck J. D., Field M. J., Fischer S., Gao J., Guo H., Ha S., Joseph-McCarthy D., Kuchnir L., Kuczera K., Lau F. T. K., Mattos C., Michnick S., Ngo T., Nguyen D. T., Prodhom B., Reiher W. E., III, Roux B., Schlenkrich M., Smith J. C., Stote R., Straub J., Watanabe M., Wiórkiewicz-Kuczera J., Yin D., Karplus M. (1998) All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B 102, 3586–3616 [DOI] [PubMed] [Google Scholar]
- 49. Feig M., MacKerell A. D., Brooks C. L. (2003) Force field influence on the observation of π-helical protein structures in molecular dynamics simulations. J. Phys. Chem. B 107, 2831–2836 [Google Scholar]
- 50. Mackerell A. D., Jr., Feig M., Brooks C. L., 3rd (2004) Extending the treatment of backbone energetics in protein force fields: Limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem. 25, 1400–1415 [DOI] [PubMed] [Google Scholar]
- 51. MacKerell A. D., Jr., Feig M., Brooks C. L., 3rd (2004) Improved treatment of the protein backbone in empirical force fields. J. Am. Chem. Soc. 126, 698–699 [DOI] [PubMed] [Google Scholar]
- 52. Guvench O., Greene S. N., Kamath G., Brady J. W., Venable R. M., Pastor R. W., Mackerell A. D. (2008) Additive empirical force field for hexopyranose monosaccharides. J. Comput. Chem. 29, 2543–2564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Guvench O., Hatcher E. R., Venable R. M., Pastor R. W., Mackerell A. D. (2009) CHARMM additive all-atom force field for glycosidic linkages between hexopyranoses. J. Chem. Theory Comput. 5, 2353–2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hoover W. G. (1985) Canonical dynamics – equilibrium phase-space distributions. Phys. Rev. A 31, 1695–1697 [DOI] [PubMed] [Google Scholar]
- 55. Nose S. (1984) A unified formulation of the constant temperature molecular-dynamics methods. J. Chem. Phys. 81, 511–519 [Google Scholar]
- 56. Ryckaert J. P., Ciccotti G., Berendsen H. J. C. (1977) Numerical-integration of Cartesian equations of motion of a system with constraints – Molecular-dynamics of N-alkanes. J. Comput. Phys. 23, 327–341 [Google Scholar]
- 57. Essmann U., Perera L., Berkowitz M. L., Darden T., Lee H., Pedersen L. G. (1995) A smooth particle mesh Ewald method. J. Chem. Phys. 103, 8577–8593 [Google Scholar]
- 58. Phillips J. C., Braun R., Wang W., Gumbart J., Tajkhorshid E., Villa E., Chipot C., Skeel R. D., Kalé L., Schulten K. (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem. 26, 1781–1802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Bu L., Beckham G. T., Shirts M. R., Nimlos M. R., Adney W. S., Himmel M. E., Crowley M. F. (2011) Probing carbohydrate product expulsion from a processive cellulase with multiple absolute binding free energy methods. J. Biol. Chem. 286, 18161–18169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kleywegt G. J., Jones T. A. (1996) Phi/psi-chology: Ramachandran revisited. Structure 4, 1395–1400 [DOI] [PubMed] [Google Scholar]
- 61. Crowley M. F., Williamson M. J., Walker R. C. (2009) CHAMBER: Comprehensive support for CHARMM force fields within the AMBER software. Int. J. Quantum. Chem. 109, 3767–3772 [Google Scholar]
- 62. Case D. A., Cheatham T. E., 3rd, Darden T., Gohlke H., Luo R., Merz K. M., Jr., Onufriev A., Simmerling C., Wang B., Woods R. J. (2005) The Amber biomolecular simulation programs. J. Comput. Chem. 26, 1668–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Hummer G. (2001) Fast-growth thermodynamic integration: Error and efficiency analysis. J. Chem. Phys. 114, 7330–7337 [Google Scholar]
- 64. Hummer G., Szabo A. (2001) Free energy reconstruction from nonequilibrium single-molecule pulling experiments. Proc. Natl. Acad. Sci. U.S.A. 98, 3658–3661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Jarzynski C. (1997) Nonequilibrium equality for free energy differences. Phys. Rev. Lett. 78, 2690–2693 [Google Scholar]
- 66. Efron B., Tibshirani R. (1986) Bootstrap methods for standard errors, confidence intervals, and other measures of statistical accuracy. Statistical Science 1, 54–75 [Google Scholar]
- 67. Larsson A. M., Bergfors T., Dultz E., Irwin D. C., Roos A., Driguez H., Wilson D. B., Jones T. A. (2005) Crystal structure of Thermobifida fusca endoglucanase Cel6A in complex with substrate and inhibitor: The role of tyrosine Y73 in substrate ring distortion. Biochemistry 44, 12915–12922 [DOI] [PubMed] [Google Scholar]
- 68. Varrot A., Frandsen T. P., Driguez H., Davies G. J. (2002) Structure of the Humicola insolens cellobiohydrolase Cel6A D416A mutant in complex with a non-hydrolysable substrate analogue, methyl cellobiosyl-4-thio-β-cellobioside, at 1.9 angstrom. Acta Crystallogr. D Biol. Crystallogr. 58, 2201–2204 [DOI] [PubMed] [Google Scholar]
- 69. Varrot A., Macdonald J., Stick R. V., Pell G., Gilbert H. J., Davies G. J. (2003) Distortion of a cellobio-derived isofagomine highlights the potential conformational itinerary of inverting β-glucosidases. Chem. Commun. 946–947 [DOI] [PubMed] [Google Scholar]
- 70. Vocadlo D. J., Davies G. J. (2008) Mechanistic insights into glycosidase chemistry. Curr. Opin. Chem. Biol. 12, 539–555 [DOI] [PubMed] [Google Scholar]
- 71. Payne C. M., Bomble Y. J., Taylor C. B., McCabe C., Himmel M. E., Crowley M. F., Beckham G. T. (2011) Multiple functions of aromatic-carbohydrate interactions in a processive cellulase examined with molecular simulation. J. Biol. Chem. 286, 41028–41035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhang S., Irwin D. C., Wilson D. B. (2000) Site-directed mutation of noncatalytic residues of Thermobifida fusca exocellulase Cel6B. Eur. J. Biochem. 267, 3101–3115 [DOI] [PubMed] [Google Scholar]
- 73. Meinke A., Damude H. G., Tomme P., Kwan E., Kilburn D. G., Miller R. C., Jr., Warren R. A., Gilkes N. R. (1995) Enhancement of the endo-β-1,4 glucanase activity of an exocellobiohydrolase by deletion of a surface loop. J. Biol. Chem. 270, 4383–4386 [DOI] [PubMed] [Google Scholar]
- 74. Payne C. M., Baban J., Horn S. J., Backe P. H., Arvai A. S., Dalhus B., Bjørås M., Eijsink V. G. H., Sørlie M., Beckham G. T., Vaaje-Kolstad G. (2012) Hallmarks of processivity in glycoside hydrolases from crystallographic and computational studies of the Serratia marcescens chitinases. J. Biol. Chem. 287, 36322–36330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Momeni M. H., Payne C. M., Hansson H., Mikkelsen N. E., Svedberg J., Engström Å., Sandgren M., Beckham G. T., Ståhlberg J. (2013) Structural, biochemical, and computational characterization of the glycoside hydrolase family 7 cellobiohydrolase of the tree-killing fungus Heterobasidion irregulare. J. Biol. Chem. 288, 5861–5872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kurasin M., Väljamäe P. (2011) Processivity of cellobiohydrolases is limited by the substrate. J. Biol. Chem. 286, 169–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Ståhlberg J., Johansson G., Pettersson G. (1993) Trichoderma reesei has no true exo-cellulase – all intact and truncated cellulases produce new reducing end groups on cellulose. Biochim. Biophys. Acta 1157, 107–113 [DOI] [PubMed] [Google Scholar]
- 78. Engh R. A., Huber R. (1991) Accurate bond and angle parameters for x-ray protein-structure refinement. Acta Crystallogr. Sect. A 47, 392–400 [Google Scholar]
- 79. Kleywegt G. J. (1997) Validation of protein models from C-α coordinates alone. J. Mol. Biol. 273, 371–376 [DOI] [PubMed] [Google Scholar]