

Background: Proteasomal degradation of GADD34 dictates cell sensitivity to ER stress.
Results: Phosphorylation at tyrosine 262 controls GADD34 protein turnover and cell fate following ER stress.
Conclusion: Tyrosine 262 phosphorylation controls the cellular levels of GADD34, a key component of the stress-activated eIF2α phosphatase.
Significance: Stress-induced GADD34 gene transcription and subsequent regulated GADD34 protein turnover control the feedback loop that ensures cell survival following ER stress.
Keywords: Apoptosis, Phosphotyrosine, Protein Turnover, Stress, Tyrosine Protein Phosphatase (Tyrosine Phosphatase), GADD34
Abstract
In mammalian cells, metabolic and environmental stress increases the phosphorylation of the eukaryotic translational initiation factor, eIF2α, and attenuates global protein synthesis. Subsequent transcriptional activation of GADD34 assembles an eIF2α phosphatase that feeds back to restore mRNA translation. Active proteasomal degradation of GADD34 protein then reestablishes the sensitivity of cells to subsequent bouts of stress. Mass spectrometry established GADD34 phosphorylation on multiple serines, threonines, and tyrosines. Phosphorylation at tyrosine 262 enhanced the rate of the GADD34 protein turnover. Substrate-trapping studies identified TC-PTP (PTPN2) as a potential GADD34 phosphatase, recognizing phosphotyrosine 262. Reduced GADD34 protein levels in TC-PTP-null MEFs following ER stress emphasized the importance of TC-PTP in determining the cellular levels of GADD34 protein. The susceptibility of TC-PTP-null MEFs to ER stress-induced apoptosis was significantly ameliorated by ectopic expression of GADD34. The data suggested that GADD34 phosphorylation on tyrosine 262 modulates endoplasmic reticulum stress signaling and cell fate.
Introduction
Mammalian cells possess complex mechanisms for detecting transient changes in their environment and respond to unfavorable conditions or stress by activating signaling pathways that ensure cell survival. Errors in the integrated stress response (ISR)2 have been implicated in many chronic human diseases (1–3). Stresses such as nutrient deprivation, viral infection, iron deficiency, radiation, hypoxia, and other insults activate one or more of four mammalian protein kinases, GCN2, PKR, HRI, and PERK, which phosphorylate serine 51 on the eukaryotic translational initiation factor, eIF2α. Phosphorylated eIF2α binds to eIF2B to inhibit its guanine nucleotide exchange function. This in turn prevents the recruitment of the initiator methionyl-tRNA and the ribosomal subunits to assemble the translational initiation complex and thereby inhibits general protein translation. This allows cells to focus their attention on expressing stress proteins and overcome or survive the stress. Among the stress-induced proteins that are translated in the presence of eIF2α phosphorylation are two transcription factors, ATF4 (4) and CHOP (5), which collaborate to induce the expression of GADD34, the protein product of a Growth Arrest and DNA Damage-inducible gene (6). GADD34 recruits the α-subunit of protein phosphatase-1 (PP1α) and assembles an active eIF2α phosphatase (7), which functions in a feedback loop to restore protein synthesis (6, 8). While ISR, and particularly eIF2α phosphorylation (9), allows cells to survive mild or transient bouts of stress, ISR also initiates programmed cell death in response to chronic stress eliminating damaged or dysfunctional cells.
GADD34 and its upstream regulators, ATF4 and CHOP, are transiently expressed in mammalian cells (9), and both their mRNAs and proteins are rapidly turned over following ER stress. Modeling their expression subsequent to ER stress hinted that the turnover of the GADD34 protein may be a particularly important determinant of cell fate. Our prior work established that GADD34 was polyubiquitinated and degraded by the 26S proteasome with a t1/2 < 1 h (10). Later studies (11) established that human GADD34 was equally distributed between ER and cytosol and identified an N-terminal amphipathic helix that mediated its association with ER membranes. Mutations that precluded ER membrane localization increased the stability of the GADD34 protein. Fluorescence recovery after photobleaching studies suggested rapid shuttling of GADD34 on and off the ER membranes. However, the mechanisms that control trafficking of GADD34, its subsequent polyubiquitination and proteasomal degradation are still not fully understood.
Mammalian GADD34 proteins are characterized by multiple PEST repeats in the central region flanked by the N-terminal ER membrane localization domain and the C-terminal PP1α-binding domain. While PEST sequences have been implicated in directing proteasomal degradation of many proteins (12), systematic deletion of the PEST repeats had no effect on GADD34 protein turnover (10). On other hand, metabolic labeling showed that cellular GADD34 was highly phosphorylated with majority of the covalent modification being in the PEST domains (10). Prior studies used phosphopeptide enrichment followed by electron transfer dissociation tandem mass spectrometry to analyze the phosphoproteome in HEK293T cells and identified a phosphopeptide containing phosphotyrosine 262 and phosphoserine 264, in human GADD34, also known as PPP1R15A (13). However, the role of covalent modification in GADD34 function or regulation has not been investigated.
In current studies, vanadate increased eIF2α (serine 51) phosphorylation in GADD34-expressing cells. Mass spectrometry of vanadate- or pervanadate-treated HEK1293T cells ectopically expressing human GADD34 identified numerous phosphorylated serines, threonines, and tyrosines. Site-directed mutagenesis established tyrosine 262 as the major phosphorylated tyrosine in human GADD34. Substitution of tyrosine 262 with a phenylalanine reduced the rate of turnover of GADD34 in cells. Utilizing substrate-trapping mutants of protein tyrosine phosphatases (PTPases) implicated in ER stress (14–17), we identified TC-PTP, as a candidate ER-associated GADD34 (phosphotyrosine 262) phosphatase. Consistent with this, levels of stress-induced GADD34 protein were significantly reduced in TC-PTP-null MEFs compared with WT MEFs, differences that were abolished by inhibition of the proteasome. Our results highlighted that the increased ER stress-induced apoptosis in TC-PTP-null MEFs could be ameliorated by the ectopic expression of GADD34. These studies suggested that ER-association and subsequent tyrosine phosphorylation determine the cellular abundance of the GADD34 protein and cell fate following ER stress.
MATERIALS AND METHODS
Cell Culture, Transfection, Reagents, and Antibodies
Human embryonic kidney epithelial (HEK) 293T, Human cervical cancer epithelial (HeLa) cells, COS-7 African Green Monkey kidney cells and TC-PTP WT (+/+) and KO (−/−) Mouse Embryonic Fibroblasts (MEFs) were maintained in the Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (Invitrogen) unless otherwise stated. Transfections were undertaken using Lipofectamine 2000 (Invitrogen) according to manufacturer's instructions. Transfections on TC-PTP MEFs were carried out by Neon® transfection system (Invitrogen). Thapsigargin, tunicamycin, calyculin A, cycloheximide, Hoechst 33258 were obtained from Sigma, MG132 and okadaic acid from Tocris Bioscience and Na3VO4 from Santa Cruz Biotechnology. The following commercial antibodies were used: anti-GADD34 (Serotec); anti-eIF2α and anti-PTP1B (Santa Cruz Biotechnology); anti-phospho-eIF2α-S51 (Invitrogen); anti-FLAG (Sigma); anti-tubulin (Sigma); anti-CHOP, anti-BiP, anti-cleaved PARP, anti-cleaved-caspase3, and anti-phosphotyrosine PY100 (Cell Signaling), anti-phosphotyrosine pY20 (Millipore); anti-HA (Zymed Laboratories Inc.); anti-TC-PTP (R&D Systems). The anti-TRAPα antibody was provided by Christopher Nicchitta of Duke University. Immunoblot analyses were undertaken as previously described (11).
Site-directed Mutagenesis
The substitutions, Y262A, Y262E, and Y262F, and other point mutations were introduced into GADD34-FLAG using QuikChange® Site-directed Mutagenesis Kit (Stratagene) according to the manufacturer's instructions.
Immunoprecipitation
Plasmids expressing FLAG-tagged GADD34 proteins were transfected in HEK293T cells. Cells were harvested and lysed in 50 mm Tris-HCl pH 7.4 containing 100 mm NaCl, 10% (v/v) glycerol, 1% (v/v) Triton X-100, 1 mm EDTA, and supplemented with protease and phosphatase inhibitors. Lysates were subjected to centrifugation at 16,000 × g for 10 min to remove cell debris. The supernatants were incubated with FLAG-M2 beads for 3 to 5 h at 4 °C. The beads were washed with lysis buffer 3 to 5 times and following the addition of equal volume of 2× SDS-PAGE buffer, heated at 95 °C for 10 min prior to SDS-PAGE and Western immunoblotting.
Substrate-trapping Experiments
HEK293T cells were transfected with plasmids expressing either FLAG-TC-PTP(C/S) or GST-PTP1B(C/S) and WT GADD34-GFP or GADD34(Y262F)-GFP. Cells were lysed and the lysates incubated with either anti-FLAG antibodies and FLAG-M2 beads or GST-Sepharose. The bound proteins were resolved by SDS-PAGE and detected by immunoblotting. In competition experiments, lysates were supplemented with 10 mm Na3VO4 prior to sedimenting the PTPase complexes as described above.
Immunocytochemistry
Cells were grown on coverslips in 6-well or 12-well plates, transfected with plasmids expressing GADD34-GFP proteins. After 24 h, cells were fixed with 4% (v/v) formaldehyde. For immunostaining, the cells, permeablized using 0.2% (v/v) Triton X-100, were incubated with goat serum, followed by the primary antibody and the fluorescent dye-conjugated secondary antibody. Coverslips were rinsed with PBS (phosphate-buffered saline) and stained with Hoechst 33258. The coverslips mounted on glass slides CRYSTAL/MOUNTTM (Biomeda) were viewed using Confocal Scanning Microscope LSM710 (Zeiss) and the images processed by the ZEN 2009 software (Zeiss).
Analysis of Protein Turnover
HEK293T or HeLa cells expressing GADD34 proteins were treated with cycloheximide (30 μg/ml). Cells were harvested at 1 to 2 h intervals, lysed in 2× SDS sample buffer, and subjected to SDS-PAGE and Western immunoblotting.
Real-time Quantitative Polymerase Chain Reaction
Total mRNA was extracted from cells using RNA easy mini kit (Qiagen). The complementary cDNA were synthesized using iScript (Bio-Rad) and qPCR performed using SsoFast kit (Bio-Rad) on iQ5 thermocycler (Bio-Rad). The following primers were used in the PCR reactions: murine GADD34: 5′-gagattcctctaaaagctcgg-3′ and 5′-cagggacctcgacgggcagc-3′ (9); murine CHOP: 5′-gcgacagagccagaataaca-3′ and 5′-gatgcacttccttctggaaca-3′; murine ATF4: 5′-atgatggcttggccagtg-3′ and 5′-ccattttctccaacatccaatc-3′; murine β-actin: 5′-ctaaggccaaccgtgaaaag-3′ and 5′-accagaggcatacagggaca-3′; Unspliced murine XBP1: 5′-tgacgaggttccagaggtg-3′ and 5′-tgcagaggtgcacatagtctg-3′; Spliced murine XBP1: 5′-ctgagtccgaatcaggtgcag-3′ and 5′-gtccatgggaagatgttc-3′. The data were analyzed using iQ5 software (Bio-Rad).
DNA Fragmentation Assay
Mouse embryonic fibroblasts (MEFs) treated with thapsigargin (1 μm) for 24 h, were processed using Suicide Track DNA ladder isolation Kit (Calbiochem). The isolated DNA was subjected to electrophoresis on 1.5% agarose gel. Gel images obtained by Bio-Rad Gel Dock system were analyzed using Quantity One Software.
Cell Death and Viability Assays
Programmed cell death or apoptosis was analyzed by ApoAlertTM Annexin V-FITC Apoptosis Kit (Clontech). Cells were fixed and then stained with propidium iodide (PI) and Annexin V-FITC. The cells were viewed using LSM 710 Zeiss confocal microscope. Cell viability was assessed using the CellTiter-Glo Luminescent Cell Viability assay (Promega), according to manufacturer's instructions.
Phosphopeptide Mapping by LC-MS/MS
HEK 293T cells expressing WT FLAG-GADD34 were treated with either 1 mm sodium orthovanadate or 0.5 mm sodium pervanadate for 30 min. Cells were lysed in 20 mm HEPES (pH 7.4), 137 mm NaCl, 1.5 mm MgCl2, 1 mm EGTA, 10% (v/v) glycerol, 1% (v/v) Triton X-100, protease inhibitors (Roche), and 0.2 mm sodium orthovanadate. The lysates were incubated with FLAG-M2 agarose beads for 2 h at 4 °C, the beads were sedimented by centrifugation, washed with the above buffer and proteins eluted in SDS sample buffer. Following SDS-PAGE, the GADD34 band was excised from Coomassie Blue-stained gels and destained before incubating with trypsin or Glu-C protease. The peptides were separated by Prominence HPLC (Shimadzu) followed by mass spectrometry on LTQ-FT Ultra mass spectrometer (Thermo Fisher) as described (18). Mass Spectrometry analyses were undertaken in the Proteomic Core Facility of the Biological Research Centre in Nanyang Technology University.
Statistical Analysis
Statistical analysis was undertaken using the Student's t test or ANOVA by GraghPad Prism software (GraphPad Inc).
RESULTS
Vanadate Stimulates eIF2α Phosphorylation in Cells Expressing GADD34
Endogenous GADD34 protein levels were elevated in cells following the pharmacological inhibition of the proteasome (10). HeLa cells, treated with vehicle (DMSO), displayed modest phosphorylation of the eukaryotic initiation factor, eIF2α. Induction of GADD34 by exposure to the proteasomal inhibitor MG132 (1 μm for 8 h) resulted in the reduction of basal eIF2α phosphorylation (Fig. 1A), reflecting GADD34's ability to assemble an eIF2α phosphatase. Treatment with the protein serine/threonine phosphatase inhibitors, 100 nm calyculin A (CA) or 1 μm okadaic acid (data not shown) for 30 min, further increased eIF2α phosphorylation, consistent with the inhibition of the GADD34-bound PP1 (8). To our surprise, exposure of MG132-treated HeLa Cells to 1 mm Na3VO4, a PTPase inhibitor, also elevated eIF2α phosphorylation to levels similar to CA (Fig. 1A). This suggested a role for tyrosine phosphorylation in regulating either the eIF2α kinase or phosphatase.
FIGURE 1.
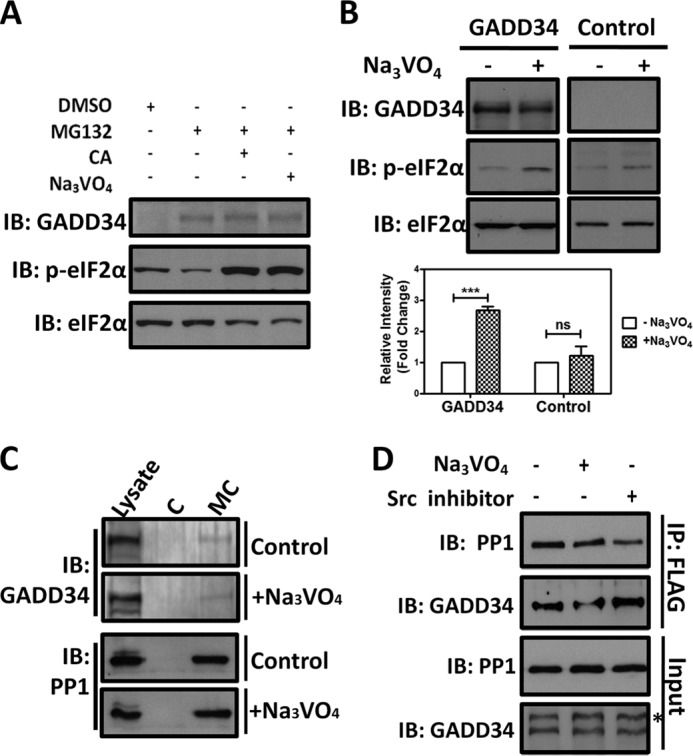
Vanadate enhances basal eIF2α phosphorylation in HeLa cells. Panel A, Hela cells were treated with DMSO or MG132 (1 μm) for 8 h to induce expression of endogenous GADD34. The MG132-treated cells were incubated with medium containing 100 nm calyculin A (CA) or 1 mm Na3VO4 for 30 min. Cell lysates were subjected to SDS-PAGE and GADD34 expression and eIF2α phosphorylation (labeled p-eIF2α) analyzed by immunoblotting. Immunoblotting for total eIF2α established the equivalent loading. Panel B, control HEK293T cells or cells expressing GADD34-HA were treated with vehicle or 1 mm Na3VO4 for 30 min. Cell lysates were subjected to SDS-PAGE and GADD34, total eIF2α and phospho-eIF2α (p-eIF2α) analyzed by immunoblotting. The quantitation of eIF2α phosphorylation in control HeLa cells and HeLa cells ectopically expressing human GADD34 in the presence (+Na3VO4, hatched bars) and absence (−Na3VO4, open bars) of vanadate is summarized from multiple independent experiments as a bar graph with standard errors. The impact of vanadate on the relative intensity of eIF2α phosphorylation following vanadate treatment was statistically analyzed using Student's t test and showed a significance of p = 0.003 in the GADD34-expressing cells (marked with ***). By contrast, the effect of vanadate in control HeLa cells was not significant (ns). Panel C, lysates of control HEK293T cells expressing GADD34-HA and cells treated with 1 mm Na3VO4 for 30 min were incubated with control agarose (C) or Microcystin-agarose (MC). The bound proteins were eluted in SDS sample buffer and subjected to SDS-PAGE and immunoblotted for GADD34 and PP1. Panel D, HEK293T cells expressing GADD34-FLAG were treated with either 1 mm Na3VO4 for 30 min or 10 μm PP1 (labeled Src inhibitor) for 8 h. Cells lysates were subjected to immunoprecipitation (IP) using FLAG-M2 beads, the bound proteins eluted with SDS sample buffer and subjected to SDS-PAGE and immunoblotting (IB). * indicates a nonspecific immunoreactive band.
Ectopic expression of C-terminal hemagglutinin (HA)-fused human GADD34 (GADD34-HA), which, like the endogenous GADD34, was degraded by the 26S proteasome (10), reduced basal eIF2α phosphorylation in HeLa cells (Fig. 1B). Treatment of GADD34-HA-expressing cells with 1 mm Na3VO4 for 30 min increased basal eIF2α phosphorylation above that seen in untreated cells (Fig. 1B). Using microcystin (MC), a protein serine/threonine phosphatase inhibitor, immobilized on agarose, we noted equivalent pulldown of PP1 catalytic subunit and GADD34 from lysates of the control and vanadate-treated cells (Fig. 1C). Neither PP1 nor GADD34 bound to control (C) agarose. Moreover, equivalent amounts of GADD34-FLAG and the associated PP1 catalytic subunit were coimmunoprecipitated from lysates of HEK293T cells, either untreated or treated with 1 mm Na3VO4 or 10 μm PP1, a Src tyrosine kinase inhibitor (Fig. 1D), suggesting that the increased basal eIF2α phosphorylation seen in vanadate-treated cells did not result from the disruption or disassembly of the GADD34/PP1 complex.
Tyrosine 262 Is the Major Phosphorylated Tyrosine in Human GADD34
Metabolic labeling of GADD34-overexpressing HeLa cells with [32P]orthophosphate established that human GADD34 was highly phosphorylated with the majority of phosphate residing in a region encompassed by amino acids 250 to 450 (10). Global phosphoproteome analyses in HEK293T showed the phosphorylation of endogenous GADD34 on tyrosine 262 and serine 264 (13). Multiple Mass Spectrometry experiments analyzed either GADD34-FLAG or FLAG-GADD34 expressed in HEK293T cells and identified 32 phosphoserines, 21 phosphothreonines, and 3 phosphotyrosines (supplemental Fig. S1). However, peptides representing the region between residues 240 and 290 as well as 341 and 371 were not detected. No peptides containing phosphotyrosine 262 and phosphoserine 264 (13), were identified.
Polyclonal and monoclonal antibodies generated against synthetic peptides encompassing phosphotyrosine 262 in human GADD34 recognized the antigen phosphopeptide with >20,000-fold selectivity over the corresponding unphosphorylated peptide but failed to show any immunoreactivity with the full-length human GADD34 protein. To establish the GADD34 phosphorylation on tyrosine 262, human FLAG-GADD34 and the corresponding mutant protein, Y262F, substituting tyrosine 262 with a non-phosphorylatable amino acid, phenylalanine (F), were expressed in HEK293T cells. FLAG-GADD34, immunoprecipitated from control and vanadate-treated cells, was subjected to immunoblotting with an anti-phosphotyrosine antibody. Cells treated with orthovanadate showed an ∼1.4-fold increase in tyrosine phosphorylation of WT FLAG-GADD34. The ratio of phosphotyrosine (P-Tyr) immunoreactivity to that for the FLAG epitope suggested that FLAG-GADD34 (Y262F) was associated with ∼80% reduction in tyrosine phosphorylation compared with WT FLAG-GADD34 (Fig. 2A), highlighting that tyrosine 262 was the major phosphorylated tyrosine in human GADD34.
FIGURE 2.
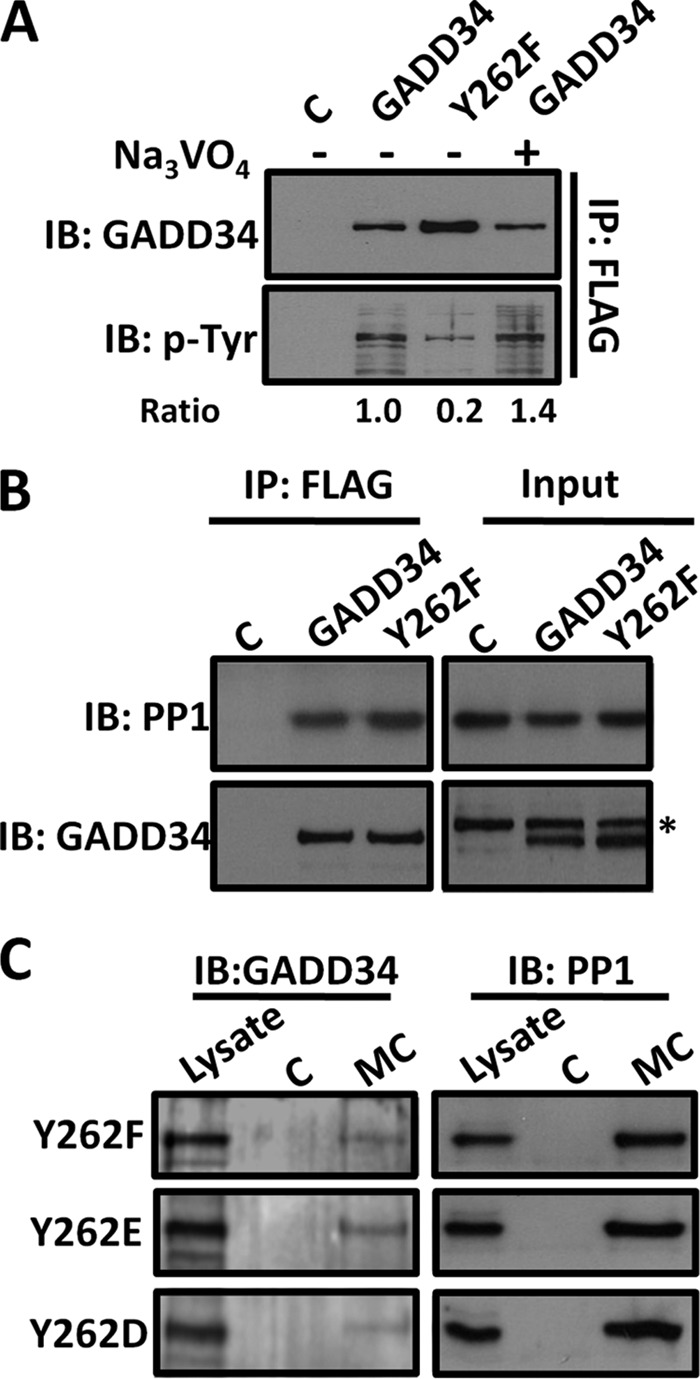
Tyrosine 262 is the major phosphorylated tyrosine in Human GADD34. Panel A, cells expressing control vector (C), WT GADD34-FLAG (GADD34), or the mutant GADD34 (Y262F)-FLAG (Y262F) plasmids were subjected to immunoprecipitation using FLAG-M2 beads. The bound proteins were eluted as described in the methods and expression of GADD34 and its phosphotyrosine content analyzed by immunoblotting. Representative ratios of phosphotyrosine (p-Tyr) content relative to the total GADD34 protein are shown below each lane. Panel B, lysates of HEK293 cells expressing vector control (C), WT GADD34-FLAG (GADD34), or the mutant protein (Y262F) were subjected to immunoprecipitation (IP) using FLAG-M2 beads. * indicates a nonspecific band. Panel C, in separate experiments, lysates of HEK293T cells expressing the mutant GADD34-FLAG proteins, Y262F, Y262E, and Y262D, were incubated with either control agarose (C) or Microcystin-agarose (MC) as described in “Materials and Methods.” The bound proteins were eluted in sample buffer, separated by SDS-PAGE, and immunoblotted for GADD34 and PP1.
Immunoprecipitation of WT FLAG-GADD34 and mutant FLAG-GADD34 (Y262F) established that both proteins bound PP1 (Fig. 2B). Pulldowns using microcystin-coupled agarose (MC) also confirmed the equivalent association of PP1 with mutant GADD34 proteins containing either Y262F or the phosphomimetic substitutions, Y262E or Y262D (Fig. 2C). Together with the data shown in Fig. 1D, this demonstrated that the covalent modification at tyrosine 262 did not impair PP1 binding to GADD34.
Tyrosine 262 Phosphorylation Enhances GADD34 Protein Turnover
GADD34 is a short-lived protein (10, 11). To investigate the potential contribution of tyrosine 262 phosphorylation in GADD34 protein turnover, the decay rate for GADD34 was analyzed following cycloheximide (CHX) treatment of HEK293T expressing either WT GADD34-FLAG of mutant GADD34(Y262F)-FLAG. Unlike the fusion of N-terminal epitopes, the C-terminal FLAG epitope did not alter the rate of GADD34 protein turnover (10). However, compared with WT GADD34-FLAG with t1/2 < 1 h, GADD34(Y262F)-FLAG displayed a significantly increased t1/2 > 2 h (Fig. 3, A and B). Vanadate treatment of HEK293T cells expressing WT GADD34-FLAG had not modest effect on the rate of GADD34 degradation (Fig. 3C) that was not significant in multiple experiments (Fig. 3D). Finally, analysis of the GADD34 mutants e.g. Y118F or S264A, the latter shown to be phosphorylated in cells (13), had no effect on the rate of protein turnover of GADD34-FLAG (Fig. 3E). These data highlighted the unique contribution of tyrosine 262 phosphorylation in enhancing GADD34 protein turnover.
FIGURE 3.
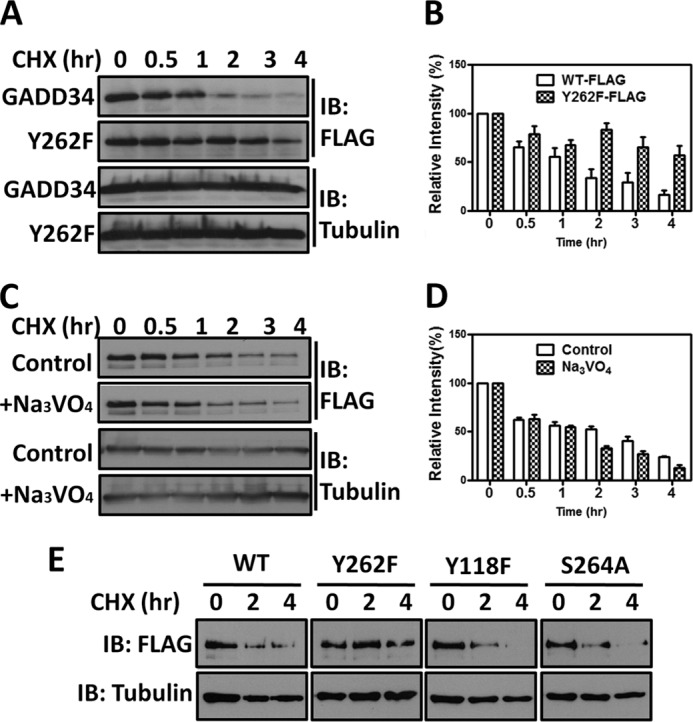
Substitution of tyrosine 262 with phenylalanine stabilizes human GADD34. Panel A, HEK293T cells expressing WT GADD34-FLAG (labeled WT-FLAG) or the mutant GADD34(Y262F)-FLAG (labeled Y262F-FLAG) were treated with 30 μg/ml cycloheximide (CHX). The cells were lysed at various time points in SDS sample buffer, and the samples subjected to SDS-PAGE and immunoblotting (IB) with anti-FLAG antibody. Immunoblotting with anti-tubulin antibody established equivalent protein loading in all lanes. Panel B, multiple independent immunoblotting experiments as described in panel A were quantified for either GADD34 or Y262F levels by scanning and the combined results are shown as a bar graph with standard errors. Panel C, HEK293T cells expressing WT GADD34-FLAG were treated with 30 μg/ml CHX in the presence and absence of 1 mm Na3VO4. Immunoblotting with anti-tubulin established equivalent protein loading. Panel D, quantitation of GADD34 protein in control and Na3VO4-treated cells in multiple independent experiments was undertaken and is shown as a bar graph with standard errors. Panel E, HEK293T cells expressing WT GADD34-FLAG or the mutants Y262F, Y118F, and S264A were exposed to 30 μg/ml CHX and at selected times, lysed in SDS sample buffer. Separated by SDS-PAGE and analyzed by immunoblotting.
Tyrosine 262 Phosphorylation Is Enhanced by GADD34 Association with ER Membranes
An N-terminal amphipathic helix-mediated GADD34 association with ER membranes and the substitution, V25R, redistributed the mutant GADD34 protein to the cytosol, slowing its degradation by the proteasome (12). Analysis of COS7 cells expressing WT GADD34-GFP or GADD34 (Y262F)-GFP established that both proteins were localized at ER, immunostained with anti-TRAPα (Fig. 4A). Thus, subcellular distribution could not account for the increased stability of GADD34 (Y262F)-GFP protein.
FIGURE 4.
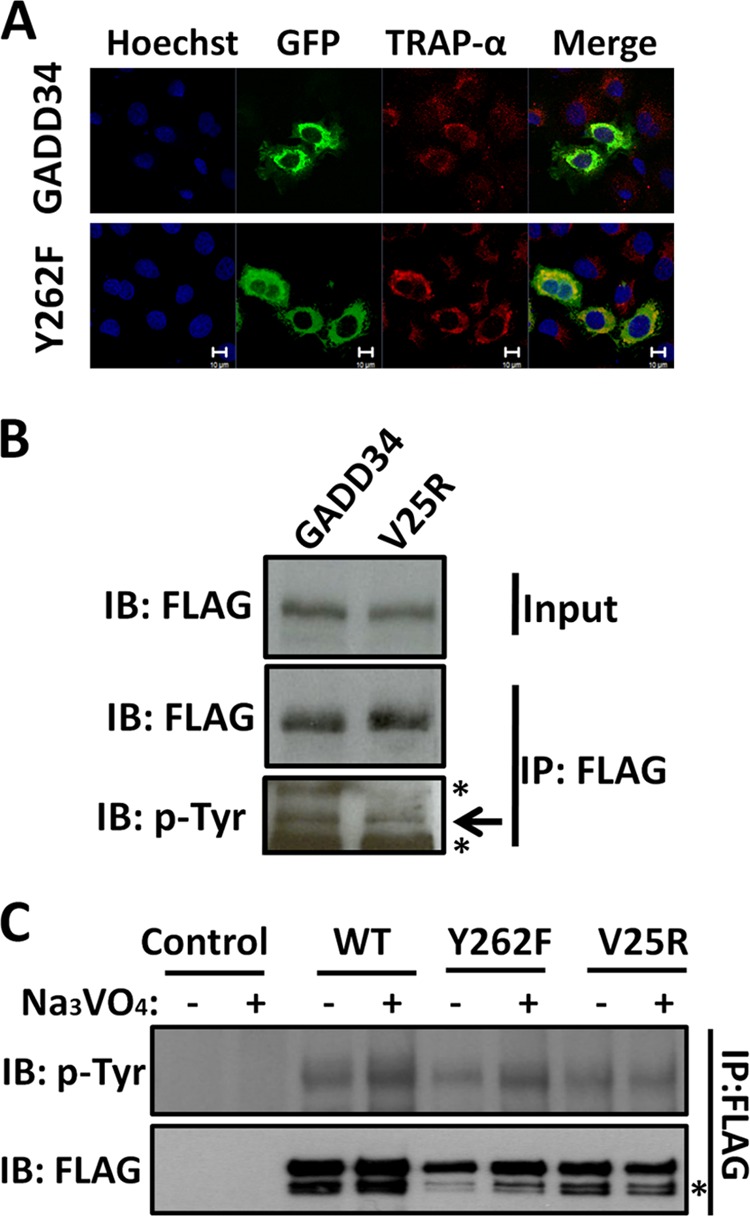
Cellular localization dictates tyrosine phosphorylation of GADD34. Panel A, COS7 cells expressing either GADD34-GFP (labeled GADD34) or GADD34(Y262F)-GFP (labeled Y262F) were fixed with formaldehyde and stained with anti-TRAPα antibody (shown in red), followed by Hoechst dye (shown in blue). The cells were also analyzed for GFP fluorescence as described in “Materials and Methods.” Magnification is shown by the bar representing 10 μm. Panel B, lysates of HEK293T cells expressing either WT GADD34-FLAG (GADD34) or mutant GADD34(V25R)-FLAG plasmids were subjected to immunoprecipitation (IP) using FLAG-M2 beads. The bound proteins were eluted in SDS sample buffer, and along with the lysates (Input) separated by SDS-PAGE and analyzed by immunoblotting (IB) with anti-FLAG (FLAG) and anti-phosphotyrosine (p-Tyr) antibodies. * indicates a nonspecific band. Panel C, control HEK293T cells and cells expressing WT GADD34-FLAG (labeled WT) or the mutants, Y262F and V25R, were treated with 1 mm Na3VO4 for 30 min. The cells were lysed, and the lysates subjected to immunoprecipitation (IP) with FLAG-M2 beads. The bound proteins were eluted with SDS sample buffer, subjected to SDS-PAGE and analyzed by immunoblotting with anti-FLAG (FLAG) and anti-phosphotyrosine (p-Tyr) antibodies. * indicates a nonspecific band.
Comparison of WT GADD34-GFP and GADD34(V25R)-GFP, immunoprecipitated from HEK293T cells, suggested the reduced tyrosine phosphorylation of the largely cytosolic GADD34(V25R)-GFP (Fig. 4B). Vanadate increased the tyrosine phosphorylation of WT GADD34-FLAG and to a lesser degree, GADD34(Y262F)-FLAG. However, no change in tyrosine phosphorylation of GADD34(V25R)-FLAG was observed following vanadate treatment (Fig. 4C). These data suggested that ER association promotes the vanadate-mediated tyrosine phosphorylation of GADD34.
TC-PTP Is a Candidate GADD34 (Tyrosine 262) Phosphatase
TC-PTP (T cell protein tyrosine phosphatase) and PTP-1B (protein tyrosine phosphatase-1B) are structurally related enzymes that are broadly expressed in mammalian tissues. Both PTPases associate with ER and have been implicated in modulating ER stress (14, 16, 17). WT GADD34-GFP but not the mutant GADD34(Y262F)-GFP bound the substrate-trapping mutant, FLAG-TC-PTP(C/S) and was co-immunoprecipitated from HEK293T lysates. Moreover, the binding of WT GADD34-GFP to FLAG-TC-PTP(C/S) was reduced in the presence of vanadate, a competitive active site inhibitor (Fig. 5A). This identified TC-PTP as a potential or candidate GADD34(Y262) phosphatase. By contrast, the substrate-trapping mutant, GST-PTP1B(C/S), did not bind WT GADD34-GFP or GADD34(Y262F)-GFP (Fig. 5B) although some nonspecific binding of GADD34-GFP proteins was observed to glutathione-agarose. This suggested that PTP-1B did not recognize the tyrosine 262 phosphorylated GADD34.
FIGURE 5.
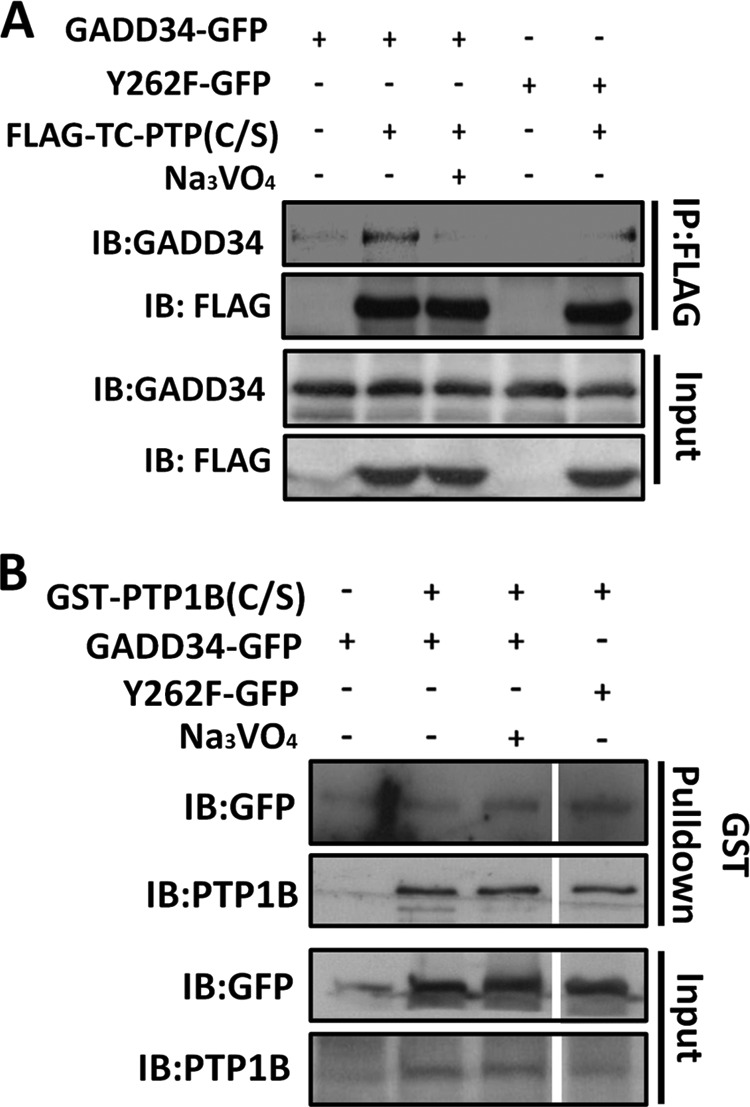
TC-PTP recognizes GADD34 phosphorylated on tyrosine 262. Panel A, lysates of HEK293T cells expressing WT GADD34-GFP or mutant GADD34(Y262F)-GFP with or without the catalytically inactive FLAG-TC-PTP(C/S) containing a serine substitution in place of the catalytic cysteine were incubated with FLAG-M2 beads (IP) with or without 10 mm Na3VO4. The bound proteins were eluted with sample buffer and with lysates (Input) subjected to SDS-PAGE and immunoblotting (IB) with anti-GADD34 and anti-FLAG. Panel B, lysates of HEK293T cells expressing WT GADD34-GFP or the mutant GADD34(Y262F)-GFP either alone or with GST-PTP1B(C/S) were incubated with GST-agarose beads in the presence or absence of 1 mm Na3VO4. The bound proteins were eluted in sample buffer and with lysates (input) subjected to SDS-PAGE prior to immunoblotting with anti-GFP and anti-PTP1B antibodies.
Reduced GADD34 Induction by Stress in TC-PTP-null Cells
GADD34 protein was undetected in WT (+/+) and TC-PTP(−/−) MEFs in the presence of vehicle (DMSO). Exposure to thapsigargin (TG), arsenite (As), and the eIF2α phosphatase inhibitor, Sal003 (19) induced GADD34 in these cells within 24 h. Much lower levels of GADD34 protein were induced in TC-PTP(−/−) compared with WT (+/+) MEFs (Fig. 6A). In numerous independent experiments, thapsigargin (TG) induced ∼70% less GADD34 in TC-PTP-null MEFs compared with WT MEFs. By contrast, the levels of GADD34 protein induced by the proteasome inhibitor, MG132, were comparable in the two cells (Fig. 6, B and C). These data hinted at a potential role for TC-PTP in modulating proteasomal degradation of GADD34.
FIGURE 6.
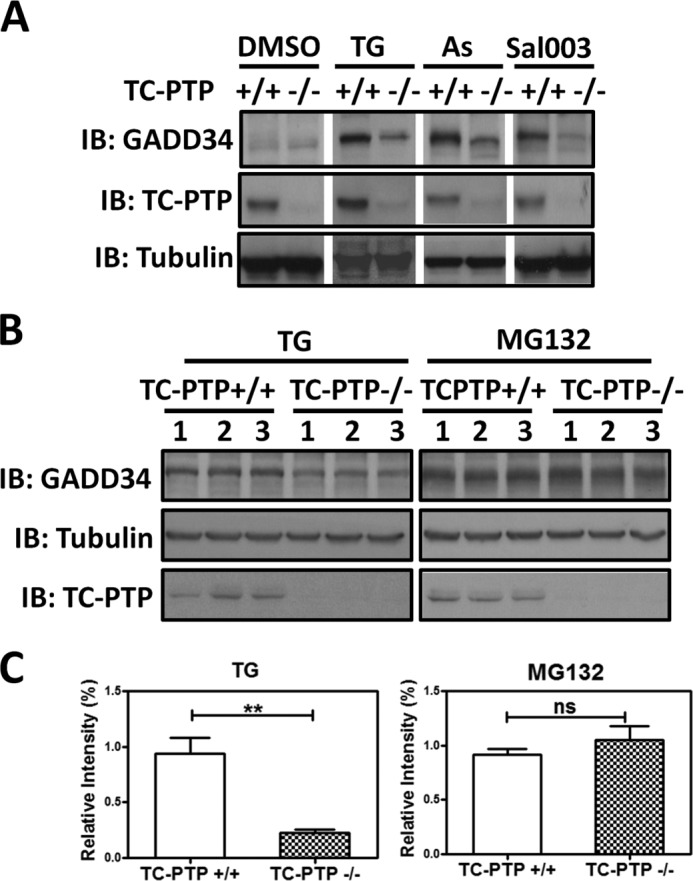
Stress-induced expression of GADD34 in TC-PTP(+/+) and TC-PTP(−/−) MEFs. Panel A, WT TC-PTP(+/+) and mutant TC-PTP(−/−) MEFs were exposed to vehicle (DMSO), 1 μm thapsigaragin (TG), 50 μm arsenite (As), or 75 μm Sal003, a Salubrinal analog, for 24 h. Immunoblotting with anti-tubulin established equivalent protein loading. Panel B, TC-PTP(+/+) and TC-PTP(−/−) MEFs were exposed to 1 μm thapsigargin (TG) or 1 μm MG132 for 24 h. Cell lysates were separated by SDS-PAGE and immunoblotted for GADD34, TC-PTP, and tubulin as described above. Three independent replicates are shown. Panel C, quantification of GADD34 protein levels normalized to tubulin, in three independent experiments in TC-PTP(+/+) and TC-PTP(−/−) MEFs treated with thapsigargin (TG) and MG132 is shown with standard errors. ** indicates p = 0.0043 while ns, represents not significant.
Despite the reduced levels of stress-induced GADD34 protein seen in the TC-PTP(−/−) MEFs at 24 h (Fig. 6A), the early phases of ER stress (0 to 6 h following thapsigargin (TG) treatment), exemplified by eIF2α phosphorylation and expression of ATF4, CHOP, and GADD34, were not significantly different between WT(+/+) and TC-PTP(−/−) MEFs (Fig. 7A). The ER stress-induced changes in mRNA levels for GADD34, ATF4, CHOP, and Xbp1 analyzed in the two cells using RT-qPCR were also not significantly different (Fig. 7B). Finally, no differences in Xbp1 splicing were noted in the two cells (Fig. 7C). Thus, the initiation of ER stress signaling by PERK and IRE1 was not impaired in the TC-PTP-null cells.
FIGURE 7.
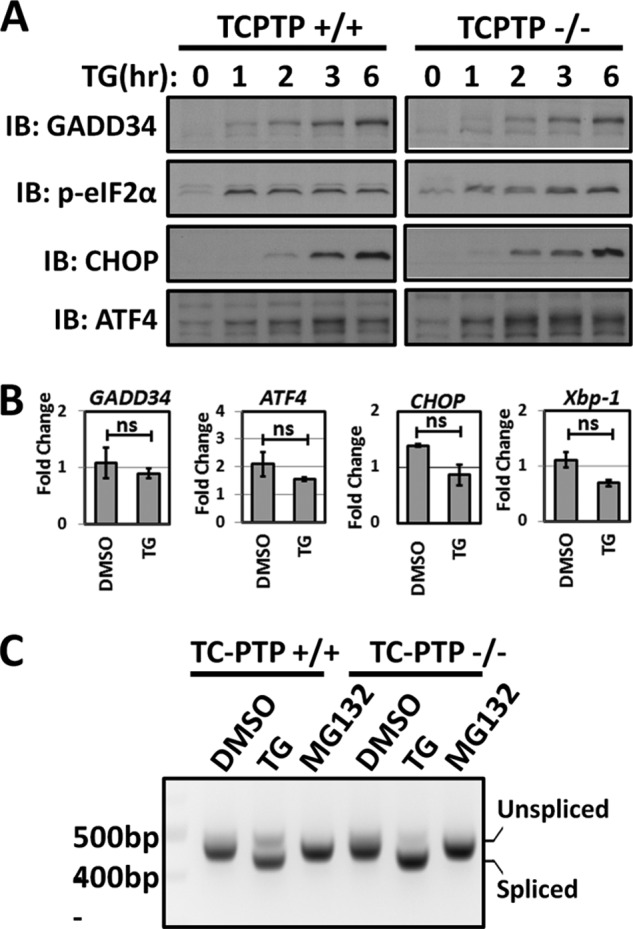
ER stress signaling in TC-PTP(+/+) and TC-PTP(−/−) MEFs. Panel A, TC-PTP(+/+) and TC-PTP(−/−) MEFs were exposed to 1 μm thapsigargin (TG) for various periods (hr). Expression of GADD34, p-eIF2α, CHOP, and ATF4 proteins was analyzed by immunoblotting. Panel B, RT-qPCR of mRNAs extracted from vehicle- (DMSO) and thapsigargin (TG)-treated TC-PTP(+/+) and TC-PTP(−/−) MEFs was used to analyze the ER Stress-induced transcription of GADD34, ATF4, CHOP, and Xbp1. Data were normalized against the β-actin mRNA and were shown as the fold change or ratio of mRNAs from TC-PTP(−/−) or KO MEFs versus WT TC-PTP(+/+) MEFs with standard errors. The data were also statistically analyzed using Student's t test with ns representing not significant. Panel C, Xbp1 mRNAs, spliced and unspliced, were amplified from cDNA of TC-PTP(+/+) and TC-PTP(−/−) MEFs treated with either DMSO, 1 μm thapsigargin (TG), or 1 μm MG132 for 24 h.
Loss of TC-PTP Sensitizes Cells to ER Stress-induced Cell Death
Thapsigargin (TG) and tunicamycin (TN) induced greater rounding and detachment from the culture dish with TC-PTP(−/−) than WT(+/+) MEFs. MG132, which resulted equivalent induction of GADD34 protein in both cells, had no effect on their cell number or morphology (Fig. 8A). However, TC-PTP(−/−) MEFs displayed significantly reduced viability on exposure to thapsigargin (0.5 μm or 1 μm) and tunicamycin (5 μg/ml or 10 μg/ml) compared with WT(+/+) MEFs at 12 and 24 h (Fig. 8B).
FIGURE 8.
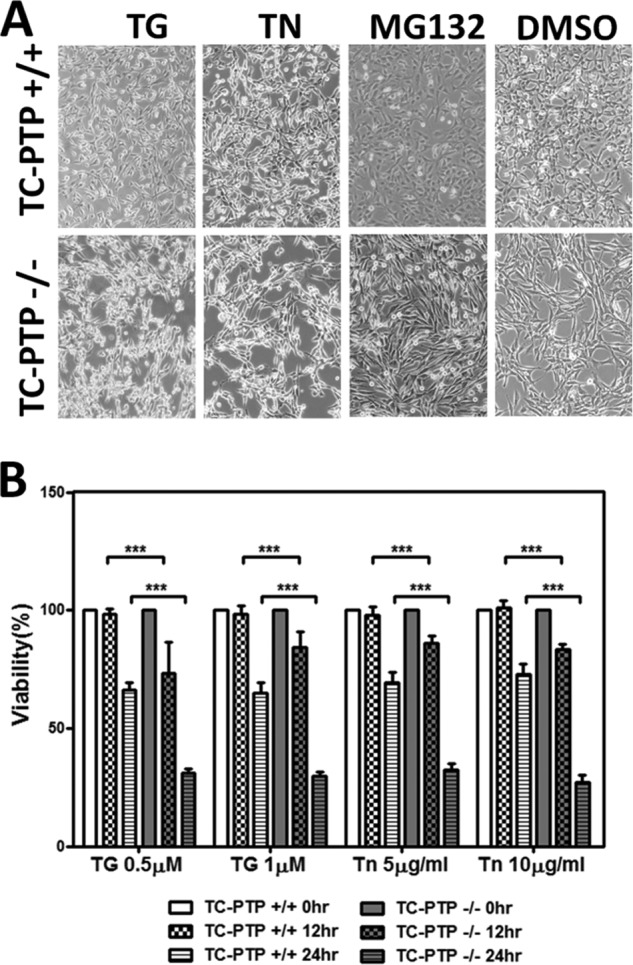
TC-PTP(−/−) MEFs are sensitized to ER stress-induced cell death. Panel A, phase contrast images of TC-PTP(+/+) and TC-PTP(−/−) MEFs treated with 1 μm thapsigargin (TG), 10 μg/ml tunicamycin (TN), 1 μm MG132, and the vehicle (DMSO) for 24 h are shown. Panel B, quantitation of three independent experiments analyzing the viability of TC-PTP(+/+) and TC-PTP(−/−) MEFs treated with 0.5 or 1.0 μm thapsigargin (TG) or 5 or 10 μg/ml tunicamycin (TN) for either 12 or 24 h is shown with standard errors with p value, *** means p < 0.0001.
Thapsigargin-treated TC-PTP(−/−) cells showed increased Annexin V and propidium iodide (PI) staining (Fig. 9A), DNA fragmentation (Fig. 9B), cleaved PARP (Fig. 9C) and cleaved or activated caspase-3 (Fig. 9D) compared with WT(+/+) MEFs. These data suggested the increased activation of apoptosis in TC-PTP(−/−) MEFs by ER stress. Ectopic expression of WT GADD34-GFP in TC-PTP(−/−) MEFs significantly attenuated the programmed cell death induced by thapsigargin (TG) and tunicamycin (TN) (Fig. 9E).
FIGURE 9.
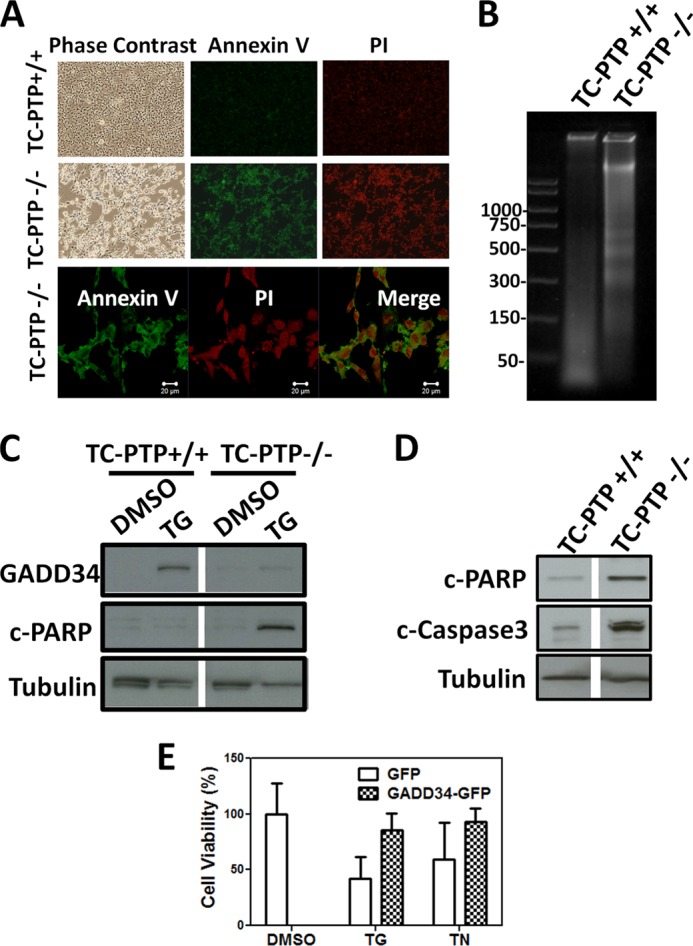
ER stress-induced apoptosis in TC-PTP(−/−) MEFs. Panel A, TC-PTP(+/+) and TC-PTP(−/−) MEFs treated with 1 μm thapsigargin for 24 h are shown in phase contrast as well as after staining with Annexin V-FITC and propidium iodide (PI). Enlarged images of Annexin V and PI-stained TC-PTP(−/−) cells with the merged images are shown below. Magnification is shown by the white bar representing 20 μm. Panel B, DNA was extracted from TC-PTP(+/+) and TC-PTP(−/−) MEFs treated with 1 μm thapsigargin for 24 h, subjected to electrophoresis on agarose gels. Panel C, lysates of TC-PTP(+/+) and TC-PTP(−/−) MEFs treated with vehicle (DMSO) or 1 μm thapsigargin (TG) for 24 h were subjected to SDS-PAGE and immunoblotting for GADD34, cleaved PARP (c-PARP) and tubulin. Panel D, lysates of TC-PTP(+/+) and TC-PTP(−/−) MEFs treated with vehicle (DMSO) or 1 μm thapsigargin (TG) for 24 h were subjected to SDS-PAGE and immunoblotting for cleaved PARP (c-PARP), cleaved or activated caspase-3 (c-caspase-3) and tubulin. Panel E, quantitation of viability of TC-PTP(−/−) MEFs expressing GFP or GADD34-GFP following treatment with vehicle (DMSO) or 1 μm thapsigargin (TG) or 10 μg/ml tunicamycin (TN) is shown with error bars. One representative experiment from four independent replicates is shown.
DISCUSSION
Accumulation of misfolded proteins activates the ER sensors, PERK, ATF6, and IRE1. ATF6 by increasing chaperone expression and IRE1 by enhancing ERAD (ER-associated protein degradation) together precondition cells to better tolerate subsequent bouts of stress (20). By contrast, PERK-mediated eIF2α phosphorylation transiently attenuates general protein synthesis while enhancing expression of the transcription factors, ATF4 (4) and CHOP (21). Emerging evidence points the cooperation of ATF4 and CHOP in activating numerous genes including GADD34 (4, 21), which assembles an eIF2α phosphatase that restores mRNA translation. GADD34 overexpression is also proapoptotic (22), possibly triggering oxidative stress (23, 24). Overexpression of its upstream activators, ATF4 and CHOP, suggested that GADD34-dependent and -independent mechanisms result in increased protein synthesis and oxidative stress to promote apoptosis (Fig. 10)(24). Thus, cells possess robust mechanisms for eliminating ATF4, CHOP and GADD34 mRNAs and proteins following the resolution of ER stress (10).
FIGURE 10.

Protein tyrosine phosphatases in the control of eIF2α phosphorylation and cell fate. ER stress has been extensively documented to activate PERK, which phosphorylates eIF2α to inhibit mRNA translation and general protein synthesis. However, translation of the mRNA encoding the transcription factor, ATF4, is enhanced following eIF2α phosphorylation. ATF4 then promotes the transcription of the gene, encoding CHOP, and ATF4 and CHOP collaborate to promote the expression of GADD34 as well as many autophagy genes (35). Finally, GADD34 assembles an eIF2α phosphatase restores general protein synthesis. ATF4 and CHOP also collaborate to turn on numerous genes that may promote both GADD34-dependent and GADD34-independent protein synthesis (23, 24) and the ensuing oxidative stress plays a key role in triggering programmed cell death (24). CHOP, possibly in conjunction with ATF4, enhances the transcription of numerous proapoptotic genes (36). Thus, the current evidence suggests that both the phosphorylation and dephosphorylation of eIF2α can induce apoptosis (36). In this context, the rapid turnover of mRNAs and proteins encoding ATF4, CHOP, and GADD34, is likely to be a critical factor in determining cell fate (9). While prior work suggested that PTP1B antagonized PERK activity, the current work highlighted the ability of TC-PTP to control GADD34 protein stability, potentially enhancing eIF2α dephosphorylation. Thus, the coordinate regulation of these PTPases by ER stress may fine-tune eIF2α phosphorylation and downstream signaling that determines the cell's decision to survive or die.
Focusing on GADD34, we previously showed that its short half-life (less than 1 h) resulted from polyubiquitination and degradation by the 26S proteasome (10). Moreover, its dynamic trafficking from cytosol to ER accelerated GADD34 protein turnover (11).
Reversible protein phosphorylation also plays a role in modulating protein turnover (25). In this context, our prior work showed that GADD34 is highly phosphorylated in cells, with the region between by amino acids 180 to 460 representing the major site of covalent modifications (10). Current work utilized mass spectrometry of GADD34 ectopically expressed in HEK293T cells to identify 32 phosphoserines, 21 phosphothreonines, and 3 phosphotyrosines (supplemental Fig. S1). While the identified peptides accounted for 80% of the primary sequence of GADD34, no peptides representing amino acids 240 to 289 were detected despite the digestion of GADD34 with different proteases and the phosphorylation of GADD34 at tyrosine 262 (or serine 264) previously shown using electron transfer dissociation (ETD) mass spectrometry (13) could not be confirmed. Efforts to generate phosphospecific antibodies against peptides encompassing Y262 also failed to resolve this issue.
The observation that vanadate, a PTPase inhibitor, enhanced basal eIF2α phosphorylation similar to okadaic acid, an inhibitor of protein serine/threonine phosphatases (Fig. 1) prompted us to investigate the potential role of GADD34 phosphorylation on tyrosines in modulating eIF2α phosphatase activity. Site-directed mutagenesis established that Y262 accounted for 80% of the phosphotyrosine present in GADD34. Neither vanadate treatment nor the Y262F substitution compromised GADD34's ability to recruit PP1α but pulse-chase studies showed that the turnover of the mutant protein, Y262F, was significantly reduced compared with WT GADD34. Eliminating an adjacent phosphorylation in S264A had no effect on turnover of this mutant GADD34 protein. Lack of significant change in the rate of GADD34 turnover in the vanadate-treated cells hinted at significant constitutive phosphorylation of Y262 in unstressed cells.
The N-terminal substitution, V25R, prevented ER localization of this mutant GADD34 and reduced its rate of protein turnover (11). GADD34(Y262F)-GFP, like WT GADD34-GFP, was however targeted to ER membranes. The cytoplasmic protein, GADD34(V25R), showed much reduced tyrosine phosphorylation compared WT GADD34-GFP, suggesting that association with the ER membranes may enhance tyrosine phosphorylation of GADD34. To identify potential ER-bound GADD34 regulators, we analyzed two PTPases, PTP1B and TC-PTP, previously shown to modulate ER stress (16, 17). Comparison of substrate-trapping mutants of TC-PTP(C/S) and PTP-1B(C/S) established that TC-PTP(C/S) but not PTP1B(C/S) bound WT GADD34. This binding was reduced by the Y262F substitution and by the addition of vanadate, a catalytic site-directed PTPase inhibitor (26). Consistent with TC-PTP's role in regulating Y262 phosphorylation and in turn, GADD34 protein turnover, the levels of GADD34 protein induced by stressors in TC-PTP-null MEFs were significantly reduced compared with either WT or PTP1B-null MEFs. The reduced GADD34 levels likely reflected its enhanced protein turnover in the TC-PTP-null cells as proteasome inhibition with MG132 induced similar levesl of GADD34 in both WT and TC-PTP-null MEFs.
The TC-PTP-null MEFs were more sensitive than WT MEFs to ER stress-induced apoptosis, characterized by DNA fragmentation, Annexin V labeling, caspase-3 cleavage, and PARP cleavage. However, the early steps in ER stress signaling were indistinguishable in the WT and TC-PTP-null MEFs. Regardless, the ectopic expression of GADD34 protected the TC-PTP(−/−) cells from ER stress-induced cell death, suggesting a GADD34-mediated mechanism for cell survival (Fig. 9). In contrast, earlier studies used shRNA-mediated knockdown of TC-PTP to show that the loss of TC-PTP expression protected MIN6 cells from ER stress-induced apoptosis (16). This may simply highlight that TC-PTP regulates substrates other than GADD34 to control ER stress in different cell types.
The current studies did not identify the GADD34 (Y262) kinase but cell treatments with a broad specificity Src family kinase inhibitor PP1, reduced GADD34 tyrosine phosphorylation (data not shown) pointing to member(s) of this family as potential GADD34 kinases. Interestingly, the four tyrosines, Y262, Y391, Y434, and Y512, phosphorylated in human GADD34, reside within similar primary sequences with the consensus, Wv/iYr/qPGE, perhaps suggesting a single or common tyrosine kinase. Thus it is noteworthy that prior studies reported GADD34 phosphorylation in B-lymphocytes by Lyn, an ER-localized Src family tyrosine kinase (27). However, these studies did not identify the modified tyrosines and did not investigate the role of tyrosine phosphorylation in Lyn-mediated inhibition of GADD34 function.
The ability of ER stress to modulate tyrosine phosphorylation of ER stress signaling proteins and more specifically, alter the levels and/or activity protein tyrosine phosphatases, has only recently been apparent. For example, prolonged ER stress increased the expression of PTP1B in pancreas (16), muscle (28), and liver (29) while reducing TC-PTP levels in the same cells (16). Moreover, PTP1B and TC-PTP appeared to have opposing effects on ER stress (16). The antagonism between PTP1B and TC-PTP has been highlighted in the regulation of Src. Specifically, PTP1B activates Src, reversing the inhibitory phosphorylation on tyrosine 527 (30, 31). By contrast, TC-PTP inhibits Src by dephosphorylating phosphotyrosine 416, an activating modification (32). The ER stressors, tunicamycin (31) and thapsigargin (33), both activate Src tyrosine kinase to phosphorylate the transcription factor, TFII-I and enhance transcription of the ER luminal chaperone, Bip (33). Thus, Src may be one focal point for the antagonistic actions of PTP1B and TC-PTP to regulate ER stress (16).
Recent studies showed that ER stress also regulates PTPase activity. Specifically, tunicamycin and thapsigargin promoted the transient sulfydration of the catalytic cysteine, conserved in all PTPases, to inhibit PTP1B (34). This activated PERK, facilitating its autophosphorylation on phosphotyrosine 615, and increased eIF2α phosphorylation (34). Paradoxically, in the PTP1B-null mice, ER stress suppresssed eIF2α phosphorylation in liver (29) and muscle (28) but increased this modification in adipose tissue (14). This suggests significant tissue-specific differences, potentially involving other PTP1B substrates, in controlling eIF2α phosphorylation. Current studies demonstrated, for the first time, that a key component of the stress-activated eIF2α phosphatase, GADD34, was also subject to tyrosine phosphorylation and identified TC-PTP as a candidate GADD34 phosphatase. Our studies suggested that TC-PTP by reversing the phosphorylation of Y262 increased cellular GADD34 protein levels. While further studies are needed to establish the utilization of this regulatory mechanism in mammalian tissues, our data provided an example of potential cooperation between PTP1B, which inhibits PERK activity, and TC-PTP, which stabilizes the GADD34 protein, to control eIF2α phosphorylation (Fig. 10) and fine-tune downstream signaling that determines cell fate following ER stress.
Acknowledgments
We thank Dr. Michel L. Tremblay (McGill University) for providing TC-PTP(+/+) and TC-PTP(−/−) MEFs and the substrate-trapping mutant TC-PTP(C/S) expression plasmid. We also thank Dr. Nicholas Tonks (Cold Spring Harbor Laboratory) for providing PTP1B(+/+) and PTP1B(−/−) MEFs and the substrate-trapping mutant PTP1B(C/S) expression plasmid. We thank Dr. Zhang Zhong-Yin (Indiana University, School of Medicine) for small molecule TC-PTP and PTP1B inhibitors also examined during these studies.
This work was supported by funds from the Duke-NUS Graduate Medical School, Singapore and an IRG grant (to S. S.) from the National Medical Research Council, Singapore.

This article contains supplemental Fig. S1.
- ISR
- integrated stress response
- PTP
- protein tyrosine phosphatase
- TG
- thapsigargin
- TN
- tunicamycin
- eIF
- eukaryotic translational initiation factor
- PP
- protein phosphatase
- PI
- propidium iodide
- CA
- calyculin A
- MC
- microcystin
- CHX
- cycloheximide.
REFERENCES
- 1. Prahlad V., Morimoto R. I. (2009) Integrating the stress response: lessons for neurodegenerative diseases from C. elegans. Trends Cell Biol. 19, 52–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Moenner M., Pluquet O., Bouchecareilh M., Chevet E. (2007) Integrated endoplasmic reticulum stress responses in cancer. Cancer Res. 67, 10631–10634 [DOI] [PubMed] [Google Scholar]
- 3. Baird T. D., Wek R. C. (2012) Eukaryotic initiation factor 2 phosphorylation and translational control in metabolism. Adv. Nutr. 3, 307–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ma Y., Hendershot L. M. (2003) Delineation of a negative feedback regulatory loop that controls protein translation during endoplasmic reticulum stress. J. Biol. Chem. 278, 34864–34873 [DOI] [PubMed] [Google Scholar]
- 5. Harding H. P., Novoa I., Zhang Y., Zeng H., Wek R., Schapira M., Ron D. (2000) Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell 6, 1099–1108 [DOI] [PubMed] [Google Scholar]
- 6. Novoa I., Zhang Y., Zeng H., Jungreis R., Harding H. P., Ron D. (2003) Stress-induced gene expression requires programmed recovery from translational repression. EMBO J. 22, 1180–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Connor J. H., Weiser D. C., Li S., Hallenbeck J. M., Shenolikar S. (2001) Growth arrest and DNA damage-inducible protein GADD34 assembles a novel signaling complex containing protein phosphatase 1 and inhibitor 1. Mol. Cell Biol. 21, 6841–6850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Brush M. H., Weiser D. C., Shenolikar S. (2003) Growth arrest and DNA damage-inducible protein GADD34 targets protein phosphatase 1α to the endoplasmic reticulum and promotes dephosphorylation of the α-subunit of eukaryotic translation initiation factor 2. Mol. Cell Biol. 23, 1292–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rutkowski D. T., Arnold S. M., Miller C. N., Wu J., Li J., Gunnison K. M., Mori K., Sadighi Akha A. A., Raden D., Kaufman R. J. (2006) Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 4, e374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brush M. H., Shenolikar S. (2008) Control of cellular GADD34 levels by the 26S proteasome. Mol. Cell Biol. 28, 6989–7000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhou W., Brush M. H., Choy M. S., Shenolikar S. (2011) Association with Endoplasmic Reticulum Promotes Proteasomal Degradation of GADD34 Protein. J. Biol. Chem. 286, 21687–21696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Rechsteiner M. (1990) PEST sequences are signals for rapid intracellular proteolysis. Semin. Cell Biol. 1, 433–440 [PubMed] [Google Scholar]
- 13. Molina H., Horn D. M., Tang N., Mathivanan S., Pandey A. (2007) Global proteomic profiling of phosphopeptides using electron transfer dissociation tandem mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 104, 2199–2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bettaieb A., Matsuo K., Matsuo I., Wang S., Melhem R., Koromilas A. E., Haj F. G. (2012) Protein tyrosine phosphatase 1B deficiency potentiates PERK/eIF2α signaling in brown adipocytes. PLoS ONE 7, e34412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Krishnan N., Fu C., Pappin D. J., Tonks N. K. (2011) H2S-Induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci. Signal 4, ra86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bettaieb A., Liu S., Xi Y., Nagata N., Matsuo K., Matsuo I., Chahed S., Bakke J., Keilhack H., Tiganis T., Haj F. G. (2011) Differential regulation of endoplasmic reticulum stress by protein tyrosine phosphatase 1B and T cell protein tyrosine phosphatase. J. Biol. Chem. 286, 9225–9235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gu F., Nguyên D. T., Stuible M., Dubé N., Tremblay M. L., Chevet E. (2004) Protein-tyrosine phosphatase 1B potentiates IRE1 signaling during endoplasmic reticulum stress. J. Biol. Chem. 279, 49689–49693 [DOI] [PubMed] [Google Scholar]
- 18. Zhang H., Guo T., Li X., Datta A., Park J. E., Yang J., Lim S. K., Tam J. P., Sze S. K. (2010) Simultaneous characterization of glyco- and phosphoproteomes of mouse brain membrane proteome with electrostatic repulsion hydrophilic interaction chromatography. Mol. Cell Proteomics 9, 635–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Robert F., Kapp L. D., Khan S. N., Acker M. G., Kolitz S., Kazemi S., Kaufman R. J., Merrick W. C., Koromilas A. E., Lorsch J. R., Pelletier J. (2006) Initiation of protein synthesis by hepatitis C virus is refractory to reduced eIF2.GTP. Met-tRNA(i) (Met) ternary complex availability. Mol. Biol. Cell 17, 4632–4644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ron D., Walter P. (2007) Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 8, 519–529 [DOI] [PubMed] [Google Scholar]
- 21. Marciniak S. J., Yun C. Y., Oyadomari S., Novoa I., Zhang Y., Jungreis R., Nagata K., Harding H. P., Ron D. (2004) CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 18, 3066–3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hollander M. C., Poola-Kella S., Fornace A. J., Jr. (2003) Gadd34 functional domains involved in growth suppression and apoptosis. Oncogene 22, 3827–3832 [DOI] [PubMed] [Google Scholar]
- 23. Lu P. D., Jousse C., Marciniak S. J., Zhang Y., Novoa I., Scheuner D., Kaufman R. J., Ron D., Harding H. P. (2004) Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2. EMBO J. 23, 169–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Han J., Back S. H., Hur J., Lin Y. H., Gildersleeve R., Shan J., Yuan C. L., Krokowski D., Wang S., Hatzoglou M., Kilberg M. S., Sartor M. A., Kaufman R. J. (2013) ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol. 15, 481–490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Karin M., Ben-Neriah Y. (2000) Phosphorylation meets ubiquitination: the control of NF-κB activity. Annu. Rev. Immunol 18, 621–663 [DOI] [PubMed] [Google Scholar]
- 26. Gordon J. A. (1991) Use of vanadate as protein-phosphotyrosine phosphatase inhibitor. Methods Enzymol. 201, 477–482 [DOI] [PubMed] [Google Scholar]
- 27. Grishin A. V., Azhipa O., Semenov I., Corey S. J. (2001) Interaction between growth arrest-DNA damage protein 34 and Src kinase Lyn negatively regulates genotoxic apoptosis. Proc. Natl. Acad. Sci. U.S.A. 98, 10172–10177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Panzhinskiy E., Hua Y., Culver B., Ren J., Nair S. (2013) Endoplasmic reticulum stress upregulates protein tyrosine phosphatase 1B and impairs glucose uptake in cultured myotubes. Diabetologia 56, 598–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Delibegovic M., Zimmer D., Kauffman C., Rak K., Hong E. G., Cho Y. R., Kim J. K., Kahn B. B., Neel B. G., Bence K. K. (2009) Liver-specific deletion of protein-tyrosine phosphatase 1B (PTP1B) improves metabolic syndrome and attenuates diet-induced endoplasmic reticulum stress. Diabetes 58, 590–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhu S., Bjorge J. D., Fujita D. J. (2007) PTP1B contributes to the oncogenic properties of colon cancer cells through Src activation. Cancer Res. 67, 10129–10137 [DOI] [PubMed] [Google Scholar]
- 31. Bjorge J. D., Pang A., Fujita D. J. (2000) Identification of protein-tyrosine phosphatase 1B as the major tyrosine phosphatase activity capable of dephosphorylating and activating c-Src in several human breast cancer cell lines. J. Biol. Chem. 275, 41439–41446 [DOI] [PubMed] [Google Scholar]
- 32. Wiede F., Shields B. J., Chew S. H., Kyparissoudis K., van Vliet C., Galic S., Tremblay M. L., Russell S. M., Godfrey D. I., Tiganis T. (2011) T cell protein tyrosine phosphatase attenuates T cell signaling to maintain tolerance in mice. J. Clin. Invest. 121, 4758–4774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hong M., Lin M. Y., Huang J. M., Baumeister P., Hakre S., Roy A. L., Lee A. S. (2005) Transcriptional regulation of the Grp78 promoter by endoplasmic reticulum stress: role of TFII-I and its tyrosine phosphorylation. J. Biol. Chem. 280, 16821–16828 [DOI] [PubMed] [Google Scholar]
- 34. Su Q., Wang S., Gao H. Q., Kazemi S., Harding H. P., Ron D., Koromilas A. E. (2008) Modulation of the eukaryotic initiation factor 2α-subunit kinase PERK by tyrosine phosphorylation. J. Biol. Chem. 283, 469–475 [DOI] [PubMed] [Google Scholar]
- 35. B'Chir W., Maurin A. C., Carraro V., Averous J., Jousse C., Muranishi Y., Parry L., Stepien G., Fafournoux P., Bruhat A. (2013) The eIF2α/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 41, 7683–7699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Malhi H., Kaufman R. J. (2011) Endoplasmic reticulum stress in liver disease. J. Hepatol. 54, 795–809 [DOI] [PMC free article] [PubMed] [Google Scholar]