

Background: A specific surface of the TPP1 protein, called the TEL patch, mediates telomerase recruitment in human cells.
Results: TEL patch deficiency caused cancer cell death, which was exacerbated by inhibition of telomerase enzymatic activity.
Conclusion: TEL patch mutations are detrimental to cancer cell viability.
Significance: This work encourages the development of new combinational cancer therapies that involve simultaneous inhibition of telomerase recruitment and telomerase activity.
Keywords: Apoptosis, Cancer, Cell Biology, Telomerase, Telomeres, TPP1
Abstract
Continued proliferation of human cells requires maintenance of telomere length, usually accomplished by telomerase. Telomerase is recruited to chromosome ends by interaction with a patch of amino acids (the TEL patch, for TPP1 glutamate (E) and leucine (L)-rich patch) on the surface of telomere protein TPP1. In previous studies, interruption of this interaction by mutation prevented telomere extension in HeLa cells, but the cell culture continued to grow. We now show that the telomerase inhibitor BIBR1532 acts together with TEL patch mutations to inhibit the growth of HeLa cell lines and that apoptosis is a prominent mechanism of death of these cells. Survivor cells take over the population beginning around 40 days in culture. These cells no longer express the TEL patch mutant TPP1, apparently because of silencing of the expression cassette, a survival mechanism that would not be available to cancer cells. These results provide hope that inhibiting the binding of telomerase to the TEL patch of TPP1, perhaps together with a modest inhibition of the telomerase enzyme, could comprise an effective anticancer therapy for the ∼90% of human tumors that are telomerase-positive.
Introduction
Telomeres are unique protein-DNA complexes that cap the ends of eukaryotic chromosomes to protect them from gross resection and deleterious chromosomal end-to-end fusion (1). Telomeric DNA is prone to erosion during every round of DNA replication because of the inability of replicative DNA polymerases to copy the extreme ends of their genomic DNA templates (2). In the absence of a resetting mechanism, telomeres shrink until they reach a point at which cells stop dividing and enter a non-proliferative state known as replicative senescence. Telomerase, a unique ribonucleoprotein enzyme, is responsible for synthesizing telomeric DNA to reset telomere length during every cell cycle in stem cells and ∼90% of cancers (3). Recent studies (4–9) have revealed gain-of-function mutations in the promoter of the gene for telomerase reverse transcriptase (10, 11), the catalytic protein subunit of telomerase, as a driver of certain cancers, further highlighting the importance of telomerase in cancer and validating this enzyme as a target for anticancer therapeutics.
Telomerase is assembled from its template-containing RNA, telomerase reverse transcriptase, and accessory protein subunits and resides in subnuclear structures known as Cajal bodies (12, 13) before it is recruited to chromosome ends for their elongation. Chromosome ends are normally coated with the six-protein (TRF1, TRF2, Rap1, TIN2, POT1, and TPP1) shelterin complex, which performs the end-capping function at mammalian telomeres (1). The TPP1 protein in shelterin includes an N-terminal oligonucleotide/oligosaccharide-binding domain, which is responsible for binding telomerase and recruiting it to chromosome ends (14–16). Moreover, a surface on the oligonucleotide/oligosaccharide-binding domain of TPP1, called the TEL patch (for TPP1 glutamate (E) and leucine (L)-rich patch), was shown to mediate telomerase binding (17, 18), enzyme processivity (18), and recruitment (18, 19). Consistent with these data, the TEL patch is also required for telomere elongation in cultured cancer cells (18, 19).
Given that the TEL patch is indispensable for telomerase function, which is essential for the proliferation of telomerase-positive cancer cells, mutation of the TEL patch would be expected to impede cancer cell growth. However, cell lines with TEL patch mutations continue to divide over many (> 80) population doublings (18). To further investigate the importance of the TEL patch in cancer cell proliferation, we conducted cell proliferation assays, telomere length measurements, and cell death analysis of TEL patch mutant cell lines over several months in culture and found that the TEL patch of TPP1 is indeed critical for the survival of cancer cells. The appearance of long-term (> 40 days in culture) survivors in the TEL patch mutant cell lines coincides with a complete loss of TEL patch mutant expression. Furthermore, inhibition of telomerase activity with a well characterized telomerase inhibitor, BIBR1532, accelerates the shortening of telomeres and the suppression of cell proliferation of TEL patch mutant cell lines. These studies further highlight the critical importance of the shelterin protein TPP1 in telomerase function and suggest new anticancer therapeutic interventions that involve inhibition of telomerase recruitment to telomeres.
EXPERIMENTAL PROCEDURES
Cell Culture
All stable cell lines used in this study were established and described in a previous study (18). In brief, gene cassettes containing bidirectional Tet-inducible promoters driving both shTPP1 and shRNA-resistant TPP1 (starting from Met-87) genes were integrated as single copies into the genome of the HeLa-EM2–11ht cells (Tet Systems Holdings GmbH & Co. KG) using Flp recombinase. All cell lines were cultured in growth medium containing DMEM supplemented with 10% FBS, 2 mm glutamax (Life Technologies), 1 mm sodium pyruvate, 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C with 5% CO2. Cells were grown in 10-cm dishes and passaged every 3–4 days. At every passage, cells were counted, and 0.3 million cells were seeded/plate in fresh growth medium. For “+BIBR” cultures, BIBR1532 (Santa Cruz Biotechnology, catalog no. 203843) dissolved in dimethyl sulfoxide (0.1% final concentration in culture) was added to 20 μm final BIBR1532 concentration. Dimethyl sulfoxide was adjusted to the same concentration (in the absence of BIBR5) to “−BIBR” control cultures. Doxycycline was added to a final concentration of 200 ng/ml for induction of pTet-driven genes. To generate a positive control for apoptosis, parental HeLa cells were treated with anisomycin (Sigma) at 10 μg/ml and incubated 5.5–6 h before harvesting.
Telomere Length Analysis
Cells were washed with PBS, which removed floating cells, and adherent cells were then dislodged using trypsin and harvested. Genomic DNA was isolated from harvested cells using GenEluteTM mammalian genomic DNA miniprep kits (Sigma), and 1.5 μg of isolated DNA was digested by HinfI and RsaI restriction enzymes for 2 h at 37 °C. The DNA digests were run on a 0.8% agarose gel in 1× Tris borate-EDTA buffer at 70 V for 13–16 h. A 5′-32P-labeled λ DNA/HindIII digest ladder was run as a marker on the same gel. After electrophoresis, the DNA in the gel was denatured and transferred to a nylon membrane as described previously (18). For DNA hybridization, a 5′-32P-labeled telomeric sequence probe (TTAGGG)4 was added to the prehybridized membrane and incubated at 50 °C for 2 h, followed by 24–120 h of exposure to a PhosphorImager screen. Telomere length was analyzed using ImageQuant TL7.0 software (GE Healthcare). After removing the background signal from the total lane signal, the half-maximal peak heights were determined. Telomere length was taken to be the midpoint between the two half-maximal heights, calculated by comparison to the molecular weight markers (λ DNA/HindIII digest ladder) present in the gel.
Annexin V Assay
Cells were cultured in 6-well plates. At 70% confluency, the growth medium from each well was collected. Next, cells were washed with PBS, and the PBS from each well after washing was collected. The cells were dislodged using trypsin, and the cells from each well were pooled together with the growth medium collected previously and PBS for that well. The pooled cells were pelleted by centrifugation and resuspended in 100 μl of annexin-binding buffer (10 mm HEPES (pH 7.4), 140 mm NaCl, and 2.5 mm CaCl2) containing 5 μl of annexin V conjugate (Alexa Fluor 680, Invitrogen) and 21 μl of 5 μg/ml propidium iodide, followed by incubation for 15 min in the dark. The cells were analyzed using a BD AccuriTM C6 flow cytometer (BD Biosciences).
Immunoblotting
For sample preparation, cells were harvested and lysed with radioimmune precipitation assay buffer (25 mm Tris-HCl (pH 7.6), 150 mm NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, and 0.1% SDS) containing protease inhibitor mixture (Thermo Scientific). Cell lysates were treated with 0.25 units/μl Benzonase® (Merck4Biosciences) for 5 min at room temperature, and 10–20 μg of total protein was loaded on a 4–12% BisTris gel (Invitrogen), followed by standard SDS-PAGE and immunoblotting analysis. The antibodies used were as follows: anti-FLAG (Sigma, catalog no. A8592, 1:1000), anti-β-actin (Sigma, catalog no. A5441, 1:10,000), and anti-cleaved caspase 3 (Cell Signaling Technology, Inc., catalog no. 9661, 1:1000).
Flow Cytometry Analysis
Cells were cultured in 6-well plates, trypsinized, resuspended in DMEM, and analyzed using MoFlo® (Beckman Coulter, Inc.).
Quantitative RT-PCR
Quantitative RT-PCR was performed as described previously (18). The mRNA level of endogenous TPP1 was normalized to the 18 S rRNA level in the same total RNA preparation.
Immunofluorescence
Immunofluorescence was performed as described previously (18). In short, cells were cultured on coverslips in 12-well plates. At ∼60% confluency, cells were fixed in PBS containing 4% formaldehyde for 10 min, washed with PBS, permeabilized in PBS containing 0.5% Triton X-100 for 5 min, and incubated with primary antibodies (mouse monoclonal anti-TRF2 (Imgenex, catalog no. IMG-124A, 1:500) and rabbit polyclonal anti-53BP1 (Novus Biologicals, catalog no. NB100-304, 1:1000)) for 1 h. After removal of the primary antibodies and three wash steps, cells were incubated with secondary antibodies (Alexa Fluor 568-conjugated anti-mouse IgG (Invitrogen, 1:500) and Alexa Fluor 647-conjugated anti-rabbit IgG (Invitrogen, 1:500)) for 30 min in the dark. After removal of the secondary antibodies and three subsequent wash steps, the coverslips were mounted on microscope slides using Vectashield mounting medium with DAPI (Vector Laboratories), sealed with transparent nail polish, and stored in the dark at −20 °C until imaging was performed.
Cell Imaging
For immunofluorescence, imaging was performed using the Nikon TE2000-U inverted fluorescence microscope equipped with Photometrics Cascade II EM-CCD camera and Yokogawa spinning disc confocal (CSU-Xm2) (Nikon Instruments, Inc.) with a ×60 oil objective. Images were acquired using Metamorph software. For brightfield imaging, the Axiovert 200 M inverted fluorescence microscope (Zeiss) equipped with a Cascade 512B CCD camera (Roper Scientific) and a Xenon arc lamp (XBO75) was used with a ×20 objective. Images were acquired by Metafluor software (Universal Imaging). All microscope-derived images were processed and analyzed using the ImageJ (Rasband, W.S., National Institutes of Health, http://imagej.nih.gov/ij/, 1997–2012) and Photoshop (Adobe) programs.
RESULTS
Inhibition of Telomerase Decreases HeLa Cell Proliferation, Especially in TEL Patch Mutant TPP1 Cell Lines
Given that the TEL patch of TPP1 is essential for the action of telomerase in vivo (18, 19), mutations in the TEL patch are expected to shorten telomeres, disrupt replicative immortality, and impede cancer cell growth. In our previous work, however, these effects appeared to be compensated by the overexpression of TPP1 in the engineered HeLa cell lines, presumably because the TPP1 recruited more telomerase and led to unnatural telomere extension (18). We therefore envisioned that inhibition of telomerase activity in the TEL patch mutant background might accentuate the repression of cell proliferation. We utilized the previously documented HeLa-EM2–11ht-derived stable cell lines that carry gene cassettes expressing both shTPP1 (for knockdown of endogenous TPP1 protein) and shRNA-resistant FLAG-tagged TPP1 in a doxycycline-dependent manner (18) (Fig. 1A). The E169A/E171A (TEL mut1) and L212A (TEL mut2) TEL patch mutants were chosen for this analysis because they have been shown previously to substantially inhibit telomerase recruitment, telomerase enzyme processivity, and telomere lengthening (18) (Fig. 1B). As reported previously (18), TPP1-FLAG versions of WT and TEL patch mutant TPP1 proteins are expressed at comparable levels in these cell lines.
FIGURE 1.

Cell proliferation decreases upon mutation of the TEL patch of TPP1 and upon inhibition of telomerase activity. A, schematic of the gene cassette stably integrated in the HeLa-EM2–11ht cell line for coexpressing a shRNA targeting endogenous TPP1 (shTPP1), a shRNA-resistant FLAG-tagged version of the TPP1 protein (TPP1-FLAG), and GFP with internal ribsome entry site (IRES) in a doxycycline-dependent manner (using a bidirectional tetracycline-responsive promoter (Ptet-bi)). B, description of the HeLa-EM2–11ht-derived stable cell lines used in this study. C, growth curves for the indicated cell lines in the absence (−BIBR) or presence (+BIBR) of 20 μm BIBR1532. The dashed lines below TEL mut1 and 2 indicate the time window during which TPP1-FLAG expression is lost, and arrows indicate that expression remains off.
For telomerase inhibition, we used BIBR1532, a selective, non-competitive inhibitor of both native and recombinant human telomerase (20, 21). Using a direct enzyme assay in extracts of cells overexpressing telomerase reverse transcriptase and telomerase RNA (22), we confirmed that the drug inhibits telomerase activity in a dose-dependent manner in vitro, both in the presence (IC50 = 70 nm) and absence (IC50 = 30 nm) of POT1-TPP1.6 Akin to reports in the literature (21), we observed that increasing doses of BIBR1532 led to a progressive decline in cell proliferation and determined 20 μm BIBR1532 to be the drug concentration at which substantial inhibition of growth of the parental HeLa cells was observed. All stable cell lines were passaged twice every 7 days, and at every passage, 20 μm BIBR1532 as well as 200 ng/ml doxycycline were freshly added.
As is evident from the growth curves (Fig. 1C), cell proliferation decreased in all cell lines in the presence of BIBR1532. Intriguingly, suppression of cell proliferation by BIBR1532 was more prominent in the TEL patch mutant cell lines than in the parental or WT cell lines. (See the first 35 days. It will be shown below that cells that have lost TPP1-FLAG expression take over the culture at later time points.) Note that at each transfer, the same number of live cells was plated, so when the curves maintain what looks like a modest difference in “population doublings” over many days, this means that with every passage the +BIBR cells grew or survived much less than the −BIBR cells. For example, in the TEL mut2 data at day 10, the cell counts were 62 × 104 and 47 × 104 cell/ml for −BIBR and +BIBR, respectively, and at day 14, 153 × 104 and 113 × 104 cell/ml, respectively. Thus, the growth curves for +BIBR and −BIBR are very different at early time points. Of note, even in the absence of BIBR, cell growth was retarded, especially in TEL mut1 (10–30 days).
Similar effects of BIBR and TEL patch mutations were observed in four additional sets of biological replicates, one of which is shown in Fig. 2A. The effect of BIBR was less drastic in the two experiments in which the cells were treated less frequently (twice every 8 days) with fresh BIBR. In all biological replicates, the decrease in proliferation of TEL patch mutant cell lines was accompanied by gross changes in cell morphology that were possibly indicative of cell death (Fig. 2B). These results indicate that mutation of the TEL patch and inhibition of telomerase work together to block cell proliferation at early time points. The two processes appear to be synergistic because the combined effect exceeds the sum of the two individual effects.
FIGURE 2.

Cell proliferation recovers at later time points in TEL patch mutant cell lines. A, growth curves from long-term experiments (∼100 days in culture) of the indicated cell lines in the absence (−BIBR) or presence (+BIBR) of 20 μm BIBR1532. Note that the effect of BIBR1532 on cell growth is less drastic here than in Fig. 1, presumably because of less frequent drug treatment (twice every 8 days here and twice every 7 days in Fig. 1). B, brightfield images of parental cells (left) and TEL mut1 cells captured at the indicated time points. Arrowheads indicate damaged/dead cells.
At later time points (∼60 days), TEL patch mutant cell lines exhibited recovery of cell proliferation that coincided with the reappearance of a more normal cell morphology (Fig. 2B). As detailed below, this correlates with cells that have lost expression of TPP1-FLAG taking over the population.
Inhibition of HeLa Cell Proliferation Correlates with Suppression of Telomere Elongation
To determine whether the decrease in cell proliferation observed with the mutation of the TEL patch and with inhibition of telomerase correlated with a decrease in telomere length, we measured the telomere length as a function of the days in culture. As shown previously (18), in the absence of BIBR1532, telomere length was maintained in parental cells (Fig. 3A) and increased in cells expressing WT TPP1 (B), whereas TEL patch mutant cell lines failed to stimulate telomere elongation in the first 24–40 days (C and D). However, in the presence of BIBR1532, maintenance and elongation of telomeres were suppressed in all cell lines (Fig. 3, A–D), confirming that elongation of their telomeres is telomerase-dependent.
FIGURE 3.

Both TEL patch mutation and telomerase inhibition suppress telomere elongation at early time points. TRF Southern blot analysis (top panel) and mean TRF length versus days in culture (bottom panel) of parental (A), WT (B), TEL mut1 (C), and TEL mut2 (D) cell lines. All samples were derived from the experiment shown in Fig. 2A.
Because the telomeres of the TEL mut1 clone were shorter (∼2 kb) than those of any of the other tested cell lines, we analyzed another clone of TEL mut1 (TEL mut1 clone #5), which has longer telomeres (∼4 kb). This clone also exhibited BIBR-dependent telomere shortening, with recovery seen starting around day 50 (Fig. 4).
FIGURE 4.
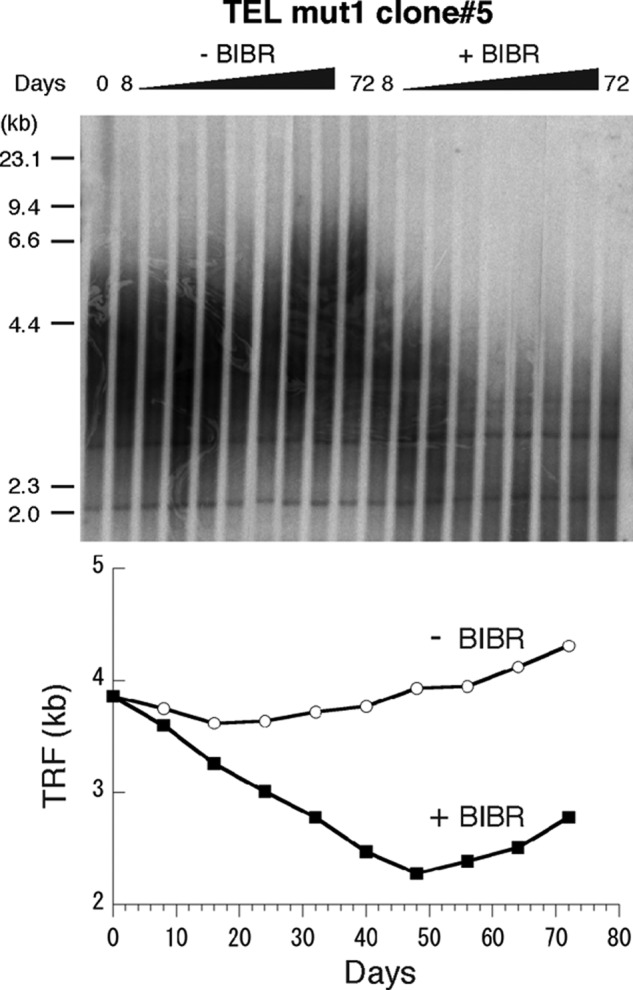
A second TEL mut1 clone that possesses longer telomeres also showed suppression of telomere elongation at early time points. TRF Southern blot analysis (top panel) and mean TRF length plot (bottom panel) of TEL mut1 clone #5.
These results show that telomere elongation in cells overexpressing WT TPP1 is telomerase-dependent (a previously untested assumption) and that the suppression of telomere lengthening in TEL patch mutant cell lines correlates strongly with the inhibition of their cell growth. Detailed comparison of the kinetics of telomere length changes and cell proliferation is not straightforward because only live cells were used for DNA extraction and TRF analysis. Thus, the higher number of dead cells (potentially with shorter telomeres) in the +BIBR population made no contribution to telomere length measurements, whereas they contributed to the cell proliferation measurements. Interestingly, at later time points (> 28–40 days) in the absence of BIBR1532, TEL patch mutant cell lines showed a recovery of telomere elongation (Fig. 3, C and D), further highlighting the strong correlation between suppression of telomere lengthening and inhibition of cell proliferation (Fig. 2A).
TEL Patch Mutations Induce Apoptosis in HeLa Cells
To determine the pathway responsible for the decrease in cell proliferation at early time points, we initially examined cell morphology in this growth period. In addition to cell death, we observed cells exhibiting shrinkage and rounding in TEL patch mutant cultures (Fig. 2B), suggesting that at least a subset of these cells may be undergoing apoptosis. Indeed, cleavage of the executioner caspase-3, an indicator of apoptosis, was detected at day 28 only in TEL patch mutant cell lines (Fig. 5A). The apoptotic phenotype was further corroborated by the increased number of annexin V-positive cells, another marker for apoptosis, only in TEL patch mutant cell lines grown for a similar duration (Fig. 5B). As evident from both these assays, apoptosis was exacerbated in the presence of BIBR1532, correlating with the cell growth kinetics at early time points (∼30 days, Figs. 1C and 2A). We did not observe apoptosis of parental HeLa in the presence of BIBR1532, probably because the drug is less effective, compared with TEL patch mutations, in inhibiting telomerase function and causing cell death. From these observations we conclude that apoptosis accounts for at least one mechanism for the decrease in cell proliferation observed in cells containing TEL patch mutations and BIBR1532.
FIGURE 5.
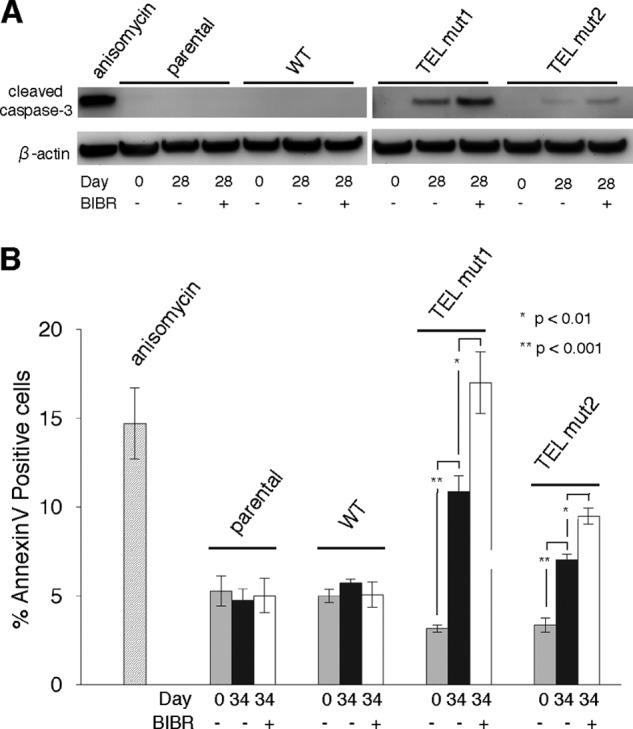
TEL patch mutations induce apoptosis of HeLa cells. A, Western blot analysis probing cleaved caspase-3 (indicative of apoptosis) in TEL mut1 and TEL mut2 cells after 28 days in culture in the absence (−) or presence (+) of BIBR. B, the percentage of annexin V-positive cells (indicative of apoptosis) for the indicated cell lines measured after 34 days in culture. Error bars represent mean ± S.D (n = 3; Student's t test; *, p < 0.01; **, p < 0.001, respectively). The α level for all tests was defined as 0.05. The anisomycin-treated cells served as a positive control for apoptosis.
To determine whether the observed apoptosis was associated with increased DNA damage response at telomeres as a consequence of acute telomere shortening, we measured the frequency of telomere dysfunction-induced foci (TIF) (23). Both WT and TEL patch mutant cell lines exhibited similarly elevated levels of TIF compared with the parental HeLa cell line, in agreement with previous observations (18) (Fig. 6, A and B). This is presumably due to some sequestration of essential shelterin factors away from telomeres by the excess TPP1-FLAG protein as a consequence of its overexpression (18). In contrast to the absence of a significant effect of TEL patch mutations on TIF formation, BIBR1532 clearly increased the DNA damage response at telomeres (Fig. 6B).
FIGURE 6.

TIF analysis. A, DAPI staining (nuclear staining, blue) and indirect immunofluorescence against TRF2 (telomere marker, red) and 53BP1 (DNA damage marker, green) for the indicated cell lines. Colocalization of TRF2 and 53BP1 indicates TIF (arrowheads). B, the percentage of TIF-positive cells in the indicated cell lines. Quantification was performed independently by two individuals using 15 fields of view for each sample, and the average of the two analyses was plotted. Error bars represent mean ± S.D.; n = 2.
Mechanism of Recovery of TEL Patch Mutant Cell Proliferation and Telomere Elongation
How do survivors arise in TEL patch mutant cell lines at later time points? To address this question, we first sequenced the TPP1-FLAG genes after PCR amplification using genomic DNA from the cell lines as templates. The TPP1-FLAG genes were retained in WT and TEL patch mutant cell lines at later time points, and so were the mutations themselves,6 ruling out the possibility that the population of TEL patch mutant survivors were devoid of the DNA coding for TEL patch mutant proteins. Next, we examined the protein expression from the integrated gene cassettes. Surprisingly, surviving TEL patch mutant cell lines displayed drastically reduced levels of TPP1-FLAG expression starting around day 40. For all three cell lines, TPP1-FLAG expression was more reduced for cells growing in the presence of BIBR1532 (Fig. 7A). Likewise, GFP expression from the same gene cassette (Fig. 1A) was greatly diminished, especially in TEL patch mutant cell lines at day 60 (Fig. 7C).
FIGURE 7.

Survivor TEL patch mutant cells have reduced expression of TPP1-FLAG, shTPP1, and GFP at later time points. A, Western blot analysis showing protein levels of TPP1-FLAG (anti-FLAG antibody) and loading control (β-actin) as a function of time in culture for the experiment shown in Fig. 1C. Samples from day 85 are derived from the experiment shown in Fig. 2A. B, quantitative RT-PCR analysis showing endogenous TPP1 mRNA levels in the indicated cell lines at the indicated time points. Endogenous TPP1 mRNA levels completely recovered after 96 days of culture. 18 S rRNA was used as the internal control during quantification. Error bars represent mean ± S.D.; n = 3. Reproducibility was high, so the error bars are difficult to see. Samples of days 7 and 38 are from the experiment shown in Fig. 1C, and samples of days 70 and 96 are from the experiment shown in Fig. 2A. C, flow cytometry analysis of GFP expressed from the stably integrated gene cassette (see Fig. 1A for a description of the gene cassette) in the indicated cell lines. GFP expression is no longer detectible in TEL mut1 and TEL mut2 cells that have been cultured for 60 days. All samples are derived from the experiment shown in Fig. 2A.
The above results could all be explained by silencing of the expression cassette at later time points. This raised an important question. Was the endogenous TPP1 no longer knocked down by shTPP1 at these later time points? Indeed, endogenous TPP1 mRNA levels recovered to almost parental HeLa levels for both TEL patch mutant cell lines at day 96 (Fig. 7B). These results suggest that there is a strong selective growth advantage for cells not expressing TEL patch mutant TPP1, further highlighting the critical importance of the TEL patch in maintaining telomeres and facilitating cancer cell survival.
DISCUSSION
Several recent studies have revealed the importance of the TEL patch of the shelterin protein TPP1 in telomerase recruitment, telomerase processivity, and telomere elongation (17–19). However, none of these studies directly evaluated the requirement of the TEL patch for cell survival. Here, we demonstrate that the TEL patch is critical for cancer cell viability. Mutation of the TEL patch decreases HeLa cell proliferation, and this phenotype is exacerbated upon concomitant inhibition of telomerase activity by the potent telomerase inhibitor BIBR1532 (Figs. 1C and 2A). The decrease in cell proliferation in the presence of TEL patch mutations coincides with telomere shortening (Fig. 3, C and D), consistent with a causative role for telomere maintenance by the TEL patch in cancer cell survival. Thus, the TEL patch qualifies as a potential target for cancer treatment, especially in concert with therapeutics that inhibit telomerase activity.
We investigated the cause of decreased proliferation of TEL patch mutant cell lines and identified apoptosis as one major pathway. Apoptosis, like telomere shortening and repression of cell proliferation, was significantly enhanced upon BIBR1532 treatment of TEL patch mutants (Fig. 5). The simplest explanation for these correlations is that short telomeres induce apoptosis. Because telomere dysfunction-induced apoptosis has been reported in some mammalian cells (24, 25), we tested whether the incidence of TIF correlated with the appearance of apoptosis. However, TIF levels were similarly elevated in WT and TEL patch mutant cell lines (Fig. 6B). It is possible that we underestimated the percentage of TIF-positive TEL patch mutant cells because of our inability to detect the telomeric signal of severely short telomeres in TEL patch mutant cells and because floating dead cells will evade detection by immunofluorescence (which was done by fixing adherent cells). In the absence of a clear correlation between TIF appearance and apoptosis, the pathway by which telomere shortening in TEL patch mutant cell lines is transduced to the repression of cell division remains undetermined. Although apoptosis is one cause for a decrease in cell proliferation of TEL patch mutant cell lines, it is possible that a subpopulation of cells undergo senescence by virtue of possessing one or more critically short telomeres.
Although the TEL patch mutant cell lines provide a well characterized system for evaluating the importance of telomerase recruitment for cancer cell survival, there are caveats that must be recognized. First, the basis for the strong telomere-elongation phenotype observed upon ectopic expression of WT TPP1, but not TEL patch mutant TPP1, has not been established. We proposed previously that it results from the excess TPP1 recruiting more telomerase (18). Our finding here that this elongation is so dramatically inhibited by BIBR1532 indicates that the extension is due to telomerase, consistent with our proposal. Another possible explanation for the telomere elongation phenotype is that excess TPP1 might titrate POT1 and/or TIN2 away from the very ends of chromosomes, allowing more access to telomerase. Although we cannot disregard this possibility, the fact that the observed telomere elongation depends on the intact TEL patch would then be unexpected because the TEL patch has not been shown to interact with POT1 or TIN2.
Another caveat regarding our study is the possibility of off-target effects of BIBR1532. High-dose BIBR (30–120 μm) has been reported to inhibit cell proliferation of telomerase-negative human fibroblasts but not hematopoietic progenitor cells (26). We do not know what BIBR concentration would affect proliferation of our HeLa cell lines through telomerase-independent pathways, so we cannot be certain that our 20 μm dose is safe in this regard. However, we consider it unlikely that an off-target effect of BIBR would differentially inhibit the growth of cells expressing TEL patch mutant TPP1 relative to WT TPP1.
Although we clearly observed a decrease in cell proliferation and an elevation of cell death of HeLa cells expressing TEL patch mutant TPP1 at early time points (up to 28–40 days in culture), cell growth (Fig. 2A) and average telomere length (Fig. 3) both recovered at later time points. One initial thought was that cells employing a telomerase-independent alternative lengthening of telomeres (27) mechanism might have taken over the population. However, the mechanism of survival turned out to be much simpler. The stably integrated gene cassette expressing TEL patch mutant TPP1 was intact but transcriptionally silenced, possibly via the formation of repressive chromatin. We conclude that this shutdown of TPP1-FLAG expression enabled cell survival and proliferation in the TEL patch mutant cell line cultures, followed by positive selection for such cells over cells that were expressing substantial amounts of the deleterious TEL patch mutant TPP1 proteins.
Given that the chromosome end protection function of TPP1 is essential for cell survival and that the TEL patch mutant cell lines are capable of accomplishing this function, we wondered how cells that have shutdown TEL patch mutant TPP1 but still contain the shTPP1 gene continue to divide. Quantitative RT-PCR analysis clearly demonstrated that the expression of shTPP1 (as inferred from the decrease in endogenous TPP1 mRNA expression) correlated directly with that of TPP1-FLAG in all cell lines (Fig. 7, A and B). Hence, survival of TEL patch mutant cells at later time points is facilitated by simultaneous TEL patch mutant TPP1 suppression (that relieves telomerase inhibition) and rescue of endogenous TPP1 expression (that allows for telomerase action and chromosome end protection).
The rescue mechanism employed by the TEL patch mutant cell lines here is specific to our recombinant expression system and highlights the essentiality of the TEL patch in telomere maintenance and cell survival. However, it is important to note that such a rescue mechanism would not be applicable to cancer cells that were treated with a potential small molecule targeting the TEL patch. Therefore, our data indicating that TEL patch mutations of TPP1 reduce HeLa cell proliferation and induce cell death encourage the development of small molecule-based strategies to target this surface for cancer treatment. Furthermore, because the combined inhibition of the TEL patch and telomerase activity accelerated the onset of cell death, the addition of TEL patch inhibition to existing anti-telomerase therapies may increase their efficacy.
Acknowledgments
We thank A. Zaug and A. Dalby for technical assistance regarding telomerase activity assays and S. C. Lin and Y. Nakashima for useful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grants K99CA167644 (to J. N.), R01GM099705 (to T. R. C.), and R01CA117907 (to J. M. E.).
M. Nakashima, J. Nandakumar, and T. R. Cech, unpublished observations.
- BIBR
- BIBR1532
- TRF
- telomeric restriction fragment
- TIF
- telomere dysfunction-induced focus/foci.
REFERENCES
- 1. Palm W., de Lange T. (2008) How shelterin protects mammalian telomeres. Annu. Rev. Genet. 42, 301–334 [DOI] [PubMed] [Google Scholar]
- 2. Levy M. Z., Allsopp R. C., Futcher A. B., Greider C. W., Harley C. B. (1992) Telomere end-replication problem and cell aging. J. Mol. Biol. 225, 951–960 [DOI] [PubMed] [Google Scholar]
- 3. Kim N. W., Piatyszek M. A., Prowse K. R., Harley C. B., West M. D., Ho P. L., Coviello G. M., Wright W. E., Weinrich S. L., Shay J. W. (1994) Specific association of human telomerase activity with immortal cells and cancer. Science 266, 2011–2015 [DOI] [PubMed] [Google Scholar]
- 4. Killela P. J., Reitman Z. J., Jiao Y., Bettegowda C., Agrawal N., Diaz L. A., Jr., Friedman A. H., Friedman H., Gallia G. L., Giovanella B. C., Grollman A. P., He T. C., He Y., Hruban R. H., Jallo G. I., Mandahl N., Meeker A. K., Mertens F., Netto G. J., Rasheed B. A., Riggins G. J., Rosenquist T. A., Schiffman M., Shih I. M., Theodorescu D., Torbenson M. S., Velculescu V. E., Wang T. L., Wentzensen N., Wood L. D., Zhang M., McLendon R. E., Bigner D. D., Kinzler K. W., Vogelstein B., Papadopoulos N., Yan H. (2013) TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. U.S.A. 110, 6021–6026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huang F. W., Hodis E., Xu M. J., Kryukov G. V., Chin L., Garraway L. A. (2013) Highly recurrent TERT promoter mutations in human melanoma. Science 339, 957–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Horn S., Figl A., Rachakonda P. S., Fischer C., Sucker A., Gast A., Kadel S., Moll I., Nagore E., Hemminki K., Schadendorf D., Kumar R. (2013) TERT promoter mutations in familial and sporadic melanoma. Science 339, 959–961 [DOI] [PubMed] [Google Scholar]
- 7. Liu X., Bishop J., Shan Y., Pai S., Liu D., Murugan A. K., Sun H., El-Naggar A. K., Xing M. (2013) Highly prevalent TERT promoter mutations in aggressive thyroid cancers. Endocr.-Relat. Cancer 20, 603–610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu X., Wu G., Shan Y., Hartmann C., von Deimling A., Xing M. (2013) Highly prevalent TERT promoter mutations in bladder cancer and glioblastoma. Cell Cycle 12, 1637–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Arita H., Narita Y., Fukushima S., Tateishi K., Matsushita Y., Yoshida A., Miyakita Y., Ohno M., Collins V. P., Kawahara N., Shibui S., Ichimura K. (2013) Upregulating mutations in the TERT promoter commonly occur in adult malignant gliomas and are strongly associated with total 1p19q loss. Acta Neuropathol. 126, 267–276 [DOI] [PubMed] [Google Scholar]
- 10. Lingner J., Hughes T. R., Shevchenko A., Mann M., Lundblad V., Cech T. R. (1997) Reverse transcriptase motifs in the catalytic subunit of telomerase. Science 276, 561–567 [DOI] [PubMed] [Google Scholar]
- 11. Meyerson M., Counter C. M., Eaton E. N., Ellisen L. W., Steiner P., Caddle S. D., Ziaugra L., Beijersbergen R. L., Davidoff M. J., Liu Q., Bacchetti S., Haber D. A., Weinberg R. A. (1997) hEST2, the putative human telomerase catalytic subunit gene, is up-regulated in tumor cells and during immortalization. Cell 90, 785–795 [DOI] [PubMed] [Google Scholar]
- 12. Venteicher A. S., Abreu E. B., Meng Z., McCann K. E., Terns R. M., Veenstra T. D., Terns M. P., Artandi S. E. (2009) A human telomerase holoenzyme protein required for Cajal body localization and telomere synthesis. Science 323, 644–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cristofari G., Adolf E., Reichenbach P., Sikora K., Terns R. M., Terns M. P., Lingner J. (2007) Human telomerase RNA accumulation in Cajal bodies facilitates telomerase recruitment to telomeres and telomere elongation. Mol. Cell 27, 882–889 [DOI] [PubMed] [Google Scholar]
- 14. Xin H., Liu D., Wan M., Safari A., Kim H., Sun W., O'Connor M. S., Songyang Z. (2007) TPP1 is a homologue of ciliate TEBP-beta and interacts with POT1 to recruit telomerase. Nature 445, 559–562 [DOI] [PubMed] [Google Scholar]
- 15. Abreu E., Aritonovska E., Reichenbach P., Cristofari G., Culp B., Terns R. M., Lingner J., Terns M. P. (2010) TIN2-tethered TPP1 recruits human telomerase to telomeres in vivo. Mol. Cell. Biol. 30, 2971–2982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nandakumar J., Cech T. R. (2013) Finding the end. Recruitment of telomerase to telomeres. Nat. Rev. Mol. Cell Biol. 14, 69–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sexton A. N., Youmans D. T., Collins K. (2012) Specificity requirements for human telomere protein interaction with telomerase holoenzyme. J. Biol. Chem. 287, 34455–34464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nandakumar J., Bell C. F., Weidenfeld I., Zaug A. J., Leinwand L. A., Cech T. R. (2012) The TEL patch of telomere protein TPP1 mediates telomerase recruitment and processivity. Nature 492, 285–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhong F. L., Batista L. F., Freund A., Pech M. F., Venteicher A. S., Artandi S. E. (2012) TPP1 OB-fold domain controls telomere maintenance by recruiting telomerase to chromosome ends. Cell 150, 481–494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pascolo E., Wenz C., Lingner J., Hauel N., Priepke H., Kauffmann I., Garin-Chesa P., Rettig W. J., Damm K., Schnapp A. (2002) Mechanism of human telomerase inhibition by BIBR1532, a synthetic, non-nucleosidic drug candidate. J. Biol. Chem. 277, 15566–15572 [DOI] [PubMed] [Google Scholar]
- 21. Damm K., Hemmann U., Garin-Chesa P., Hauel N., Kauffmann I., Priepke H., Niestroj C., Daiber C., Enenkel B., Guilliard B., Lauritsch I., Müller E., Pascolo E., Sauter G., Pantic M., Martens U. M., Wenz C., Lingner J., Kraut N., Rettig W. J., Schnapp A. (2001) A highly selective telomerase inhibitor limiting human cancer cell proliferation. EMBO J. 20, 6958–6968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Latrick C. M., Cech T. R. (2010) POT1-TPP1 enhances telomerase processivity by slowing primer dissociation and aiding translocation. EMBO J. 29, 924–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Takai H., Smogorzewska A., de Lange T. (2003) DNA damage foci at dysfunctional telomeres. Curr. Biol. 13, 1549–1556 [DOI] [PubMed] [Google Scholar]
- 24. Karlseder J., Broccoli D., Dai Y., Hardy S., de Lange T. (1999) p53- and ATM-dependent apoptosis induced by telomeres lacking TRF2. Science 283, 1321–1325 [DOI] [PubMed] [Google Scholar]
- 25. Eller M. S., Puri N., Hadshiew I. M., Venna S. S., Gilchrest B. A. (2002) Induction of apoptosis by telomere 3′ overhang-specific DNA. Exp. Cell Res. 276, 185–193 [DOI] [PubMed] [Google Scholar]
- 26. El-Daly H., Kull M., Zimmermann S., Pantic M., Waller C. F., Martens U. M. (2005) Selective cytotoxicity and telomere damage in leukemia cells using the telomerase inhibitor BIBR1532. Blood 105, 1742–1749 [DOI] [PubMed] [Google Scholar]
- 27. Bryan T. M., Englezou A., Dalla-Pozza L., Dunham M. A., Reddel R. R. (1997) Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nat. Med. 3, 1271–1274 [DOI] [PubMed] [Google Scholar]