

Background: Transforming growth factor (TGF)-β1 treatment decreases human trophoblast invasion.
Results: Smad-dependent up-regulation of Snail mediates TGF-β1-induced down-regulation of VE-cadherin.
Conclusion: TGF-β1 decreases human trophoblast invasion by down-regulating VE-cadherin.
Significance: Our results provide important insights into the molecular mechanisms mediating TGF-β1-induced down-regulation of VE-cadherin and decreased cell invasion in human trophoblast cells.
Keywords: Cadherins, Invasion, SMAD Transcription Factor, Transforming Growth Factor-β (TGFβ), Trophoblast, Snail, VE-cadherin
Abstract
Human trophoblast cells express transforming growth factor-β (TGF-β) and TGF-β receptors. It has been shown that TGF-β1 treatment decreases the invasiveness of trophoblast cells. However, the molecular mechanisms underlying TGF-β1-decreased trophoblast invasion are still not fully understood. In the current study, we demonstrated that treatment of HTR-8/SVneo human trophoblast cells with TGF-β1 decreased cell invasion and down-regulated the expression of vascular endothelial cadherin (VE-cadherin). In addition, the inhibitory effect of TGF-β1 on VE-cadherin was confirmed in primary cultures of human trophoblast cells. Moreover, knockdown of VE-cadherin using siRNA decreased the invasiveness of HTR-8/SVneo cells and primary cultures of trophoblast cells. Treatment with TGF-β1 induced the activation of Smad-dependent signaling pathways and the expression of Snail and Slug. Knockdown of Smads attenuated TGF-β1-induced up-regulation of Snail and Slug and down-regulation of VE-cadherin. Interestingly, depletion of Snail, but not Slug, attenuated TGF-β1-induced down-regulation of VE-cadherin. Furthermore, overexpression of Snail suppressed VE-cadherin expression. Chromatin immunoprecipitation analyses showed the direct binding of Snail to the VE-cadherin promoter. These results provide evidence that Snail mediates TGF-β1-induced down-regulation of VE-cadherin, which subsequently contributed to TGF-β1-decreased trophoblast cell invasion.
Introduction
Trophoblast invasion is a tightly regulated event that occurs during placental development. It has been shown that insufficient invasion of trophoblast cells into the uterine decidua and inadequate remodeling of the uterine vasculature is associated with preeclampsia, which is a dangerous medical condition characterized by high blood pressure and high levels of protein in the urine of pregnant women (1–3). Thus, a better understanding of the molecular mechanisms that regulate trophoblast invasion will help to delineate the processes involved in the establishment of pregnancy under normal and pathological conditions.
In mammals, transforming growth factor-β (TGF-β) consists of three isoforms, TGF-β1, -β2, and -β3, each of which is encoded by a different gene and is expressed in a tissue-specific manner (4). TGF-βs regulate diverse cellular functions including cell proliferation, apoptosis, differentiation, migration, and invasion (5). However, the effects of TGF-βs are largely dependent on the type and the differentiation state of its target cells (6). TGF-βs initiate downstream signaling pathways and exert their cellular processes by binding to transmembrane type I (TβRI)2 and type II (TβRII) receptors (7). In the canonical pathway, upon TGF-β binding to TβRII, TβRI is recruited and phosphorylated, which results in TβRI activation. Activation of TβRI leads to phosphorylation of the receptor-regulated Smad proteins, Smad2 and Smad3. Phosphorylated Smad2 and Smad3 then bind to the common-mediator Smad, Smad4, and translocate into the nucleus. Nuclear Smad complexes then bind to Smad-specific binding elements in the promoter region to mediate TGF-β-regulated gene expression (8).
Vascular endothelial-cadherin (VE-cadherin), also known as cadherin-5, is a transmembrane protein that plays very important roles in maintaining and controlling endothelial adherens junctions (9). Similar to most cadherins, the cytoplasmic domain of VE-cadherin interacts with p120-catenin, β-catenin, and plakoglobin (10). An interaction between β-catenin and α-catenin anchors the cadherins to the actin cytoskeleton, which strengthen the cadherin-mediated cell-cell adhesion (11). VE-cadherin has been identified in human placenta and is expressed in endothelial cells of placental vessels, cytotrophoblasts in cell columns, extravillous trophoblast cells, and decidua (12, 13). Importantly, cultured human cytotrophoblast cells isolated from chorionic villi treated with an antibody against VE-cadherin significantly decreases cell invasion (12). Moreover, in preeclampsia patients, VE-cadherin is not detected in any of the cytotrophoblast cells of the placenta (14). These results demonstrate that VE-cadherin is required to maintain the invasive capacity of human trophoblast cells.
Changes in the cadherin expression profiles are important for normal trophoblast cell invasion, and aberrant expression of cadherins has been shown to be associated with placental disorders such as preeclampsia (13). TGF-β superfamily members are expressed in the human endometrium and placenta where they modulate many biological functions to establish a successful pregnancy (15). All three TGF-β isoforms and their receptors are detected in trophoblast cells, although the expression of TGF-β3 remains controversial due to differing antibody affinities or specificities (15). Treatments with TGF-β1, -β2, and -β3 decrease the invasiveness of human placenta explant-derived extravillous trophoblast cells via down-regulating the activity of proteases (16). However, it is not known whether VE-cadherin is involved in TGF-β-decreased trophoblast cell invasion.
In the current study, we tested the hypothesis that VE-cadherin mediated the TGF-β1-induced suppression of trophoblast cell invasion. Our results indicated that TGF-β1 treatment decreased invasiveness in the human trophoblast cell line HTR-8/SVneo. In addition, treatment with TGF-β1 down-regulated VE-cadherin expression in HTR-8/SVneo cells and in primary cultures of human trophoblast cells. Furthermore, the effects of TGF-β1 on the down-regulation of VE-cadherin were mediated by the transcription factor, Snail, via activation of the Smad-dependent signaling pathway.
EXPERIMENTAL PROCEDURES
Cell Culture
The human trophoblast cell line, HTR-8/SVneo was kindly provided by Dr. Charles Graham (Queen's University, Kingston, Ontario, Canada). HTR-8/Svneo is an SV40 large T antigen immortalized first trimester short-lived extravillous trophoblast cell line (17). Cells were grown in a 1:1 (v/v) mixture of DMEM/F-12 medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS; Hyclone Laboratories Inc., Logan, UT). Cultures were maintained at 37 °C in a humidified atmosphere of 5% CO2 in air.
Primary Trophoblast Cell Culture
Human trophoblast cells were isolated from first trimester placental tissue explants as previously described (18). Briefly, chorionic villi were washed with cold medium and finely minced. Fragments of the chorionic villi were allowed to adhere for 2–3 days, after which any non-adherent material was removed. These tissue explants were further cultured for 10–14 days during which the culture medium was changed every 2 days. Trophoblast cells were separated from the villous explants by a brief trypsin digestion.
Antibodies and Reagents
Polyclonal anti-TGF-β receptor I (number 3712), anti-phospho-Smad2 (number 3103) and anti-Smad4 (number 9515), monoclonal anti-Smad2 (number 3103), anti-phospho-Smad3 (number 9520) anti-Smad3 (number 9523), anti-Snail (number 3895), and snit-Slug (number 9585) antibodies were obtained from Cell Signaling (Danvers, MA). A monoclonal anti-VE-cadherin antibody (number 610251) was obtained from BD Biosciences. A monoclonal anti-α-tubulin antibody was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Horseradish peroxidase-conjugated goat anti-mouse and goat anti-rabbit IgG were obtained from Bio-Rad. Recombinant human TGF-β1 was obtained from R&D Systems (Minneapolis, MN). SB431542 was obtained from Sigma.
Small Interfering RNA (siRNA) Transfection
To knockdown endogenous TGF-β receptor I, Smad2, Smad3, Smad4, Snail, and Slug cells were transfected with 50 nm ON-TARGETplus SMARTpool siRNA that targets a specific gene (Dharmacon, Lafayette, CO) using Lipofectamine RNAiMAX (Invitrogen). The siCONTROL NON-TARGETING pool siRNA (Dharmacon) was used as the transfection control. The knockdown efficiency was examined using RT-qPCR or Western blot analysis.
Snail Overexpression
Cells were seeded in 6-well plates and transfected with Lipofectamine 2000 (Invitrogen) and 1 μg of empty pCMV vector or vector encoding a full-length human Snail (GeneCopoeia, Rockville, MD).
Western Blot Analysis
Cells were lysed in cell lysis buffer (Cell Signaling). Equal amounts of protein were separated by SDS-polyacrylamide gel electrophoresis and transferred onto PVDF membranes. After 1 h of blocking with 5% nonfat dry milk in Tris-buffered saline (TBS), the membranes were incubated overnight at 4 °C with primary antibodies, which were diluted in 3% bovine serum albumin (BSA)/TBS. Following primary antibody incubation, the membranes were incubated with the appropriate HRP-conjugated secondary antibody. Immunoreactive bands were detected using an enhanced chemiluminescent substrate. Membranes were stripped with stripping buffer at 50 °C for 30 min and reprobed with anti-α-tubulin as a loading control.
Reverse Transcription-Quantitative Real-time PCR (RT-qPCR)
Total RNA was extracted using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. Reverse transcription was performed with 3 μg of RNA, random primers, and Moloney murine leukemia virus reverse transcriptase (Promega, Madison, WI). The primers used for SYBR Green reverse transcription-qPCR (RT-qPCR) were the following: TGF-β receptor I, 5′-GTT AAG GCC AAA TAT CCC AAA CA-3′ (sense) and 5′-ATA ATT TTA GCC ATT ACT CTC AAG G-3′ (antisense); VE-cadherin, 5′-CAG CCC AAA GTG TGT GAG AA-3′ (sense) and 5′-CGG TCA AAC TGC CCA TAC TT-3′ (antisense); Smad2, 5′-GCC TTT ACA GCT TCT CTG AAC AA-3′ (sense) and 5′-ATG TGG CAA TCC TTT TCG AT-3′ (antisense); Smad3, 5′-CCC CAG CAC ATA ATA ACT TGG-3′ (sense) and 5′-AGG AGA TGG AGC ACC AGA AG-3′ (antisense); Smad4, 5′-TGG CCC AGG ATC AGT AGG T-3′ (sense) and 5′-CAT CAA CAC CAA TTC CAG CA-3′ (antisense); Snail, 5′-CCC CAA TCG GAA GCC TAA CT-3′ (sense) and 5′-GCT GGA AGG TAA ACT CTG GAT TAG A-3′ (antisense); Slug, 5′-TTC GGA CCC ACA CAT TAC CT-3′ (sense) and 5′-GCA GTG AGG GCA AGA AAA AG-3′ (antisense); and GAPDH, 5′-GAG TCA ACG GAT TTG GTC GT-3′ (sense) and 5′-GAC AAG CTT CCC GTT CTC AG-3′ (antisense). RT-qPCR was performed using the Applied Biosystems 7300 Real-time PCR System (PerkinElmer Life Sciences) equipped with a 96-well optical reaction plate. All of the RT-qPCR experiments were run in triplicate, and a mean value was used to determine the mRNA levels. Relative quantification of the mRNA levels was performed using the comparative Ct method with GAPDH as the reference gene and the formula 2−ΔΔCt.
Invasion Assay
Invasion assays were performed in Boyden chambers according to a previously published method, with minor modifications (19). Transwell filters (8-μm pore size, 24 wells, BD Biosciences) were coated with 1 mg/ml of growth factor-reduced Matrigel (BD Biosciences). Cells in DMEM/F-12 medium that was supplemented with 0.1% FBS were incubated for 48 h against a gradient of 10% FBS. Cells that penetrated the membrane were fixed with cold methanol, and the cell nuclei were stained with Hoechst 33258 and quantified using an epifluorescence microscopy and Northern Eclipse 6.0 software (Empix Imaging, Mississauga, ON). Triplicate inserts were used for each individual experiment, and five microscopic fields were quantified per insert.
Chromatin Immunoprecipitation (ChIP)
A ChIP assay was performed using the ChIP-IT kit (Active Motif, Carlsbad, CA) according to the manufacturer's protocol. Briefly, cells were fixed in 1% formaldehyde at room temperature for 10 min; the fixation reaction was stopped by adding the glycine stop-fix solution to the dish at room temperature for 5 min. After washing, the cells were resuspended in lysis buffer and incubated for 30 min on ice. The cells were homogenized, and the nuclei were resuspended in shearing buffer and subjected to pre-optimized ultrasonic disruption conditions to yield 200–1500-bp DNA fragments. Sheared chromatin was incubated in ChIP buffer with protein G magnetic beads and Snail antibody or mouse IgG (Cell Signaling) as a negative control on a rolling shaker overnight at 4 °C. The immunoprecipitated chromatin was purified from the chromatin/antibody mixture with several washing steps, and the chromatin-immunoprecipitated DNA was eluted in elution buffer. Cross-linking was reversed by adding reverse cross-link buffer. The purified DNA was subjected to PCR amplification (one cycle of 94 °C for 3 min and 40 cycles of 94 °C for 20 s, 64 °C for 30 s, and 72 °C for 30 s) for the E-box within the VE-cadherin promoter using specific forward (5′-GGG TGG ACA AGC ACC TTA AA-3′) and reverse (5′-CAG CTC TGG GAC TCT GAA CC-3′) primers. The selected primers were confirmed using an in silico PCR program (GENOME) to ensure the generation of a single amplicon from the human genomic DNA. The PCR products (166 bp) were resolved by electrophoresis in a 1% agarose gel and visualized by ethidium bromide staining.
Statistical Analysis
The results were presented as the mean ± S.E. of at least three independent experiments. Statistical evaluation was performed using a t test for paired data. Multiple comparisons were first analyzed using one-way analysis of variance, followed by Tukey's multiple comparison tests. A significant difference was defined as p < 0.05.
RESULTS
TGF-β1 Decreases Human Trophoblast Cell Invasion
To examine the effect of TGF-β1 on human trophoblast invasion, HTR-8/SVneo cells were treated with different concentrations (1, 5 and 10 ng/ml) of recombinant human TGF-β1. A Matrigel invasion assay showed that treatment with 5 and 10 ng/ml of TGF-β1 for 48 h significantly decreased HTR-8/SVneo cell invasion (Fig. 1A), which was consistent with previous studies that showed anti-migratory and anti-invasive effects of TGF-β1 on HTR-8/SVneo cells (20, 21). SB431542 is a potent and specific TβRI inhibitor (22). Treatment with SB431542 abolished the TGF-β1-decreased HTR-8/SVneo cell invasion (Fig. 1B). To further confirm the requirement of the TGF-β receptor in the TGF-β1-induced anti-invasive effect, TβRI siRNA was used to knockdown endogenous TβRI expression. As shown in Fig. 1C, TβRI siRNA significantly down-regulated TβRI protein levels in HTR-8/SVneo cells. Moreover, the TGF-β1 effects on cell invasion were abolished using siRNA-mediated depletion of TβRI (Fig. 1D). It has been shown that TGF-β1 treatment decreases [3H]thymidine incorporation in HTR-8/SVneo cells (17), but does not affect proliferation and apoptosis in extravillous trophoblast cells derived from first trimester placental explants (16). To confirm that the inhibitory effect of TGF-β1 on cell invasion was not due to differences in cell growth, cell proliferation after TGF-β1 treatment was examined using a trypan blue exclusion assay. As shown in Fig. 1E, TGF-β1 treatment only decreased cell proliferation after 72 h in culture. Moreover, the effect of TGF-β1 on cell morphology was microscopically examined. As shown in Fig. 1F, treatment with 5 ng/ml of TGF-β1 for 24 and 48 h did not significantly change the morphology of HTR-8/SVneo cells.
FIGURE 1.

TGF-β1 decreases HTR-8/SVneo cell invasion. A, cells were treated with increasing concentrations of TGF-β1 (1, 5, and 10 ng/ml) and seeded onto Matrigel-coated transwell inserts. After 48 h of incubation, non-invading cells were wiped from the upper side of the filter, and the nuclei of the invading cells were stained with Hoechst 33258. The top panels show representative images of the invasion assays. The scale bar represents 200 μm. The bottom panels show the summarized quantitative results. B, cells were treated with 5 ng/ml of TGF-β1 in combination with SB431542 (10 μm). Cell invasion was examined using the Matrigel invasion assay. C, cells were transfected with 50 nm control siRNA (si-Ctrl) or TβRI siRNA (si-TβRI) for 48 h. The protein levels of TβRI were examined using Western blot analyses. D, after TβRI knockdown, cells were treated with 5 ng/ml of TGF-β1 and cell invasion was examined using the Matrigel invasion assay. E, cells were treated with 5 ng/ml of TGF-β1 every 24 h, and the number of cells was quantified using the trypan blue exclusion assay. F, cells were treated with 5 ng/ml of TGF-β1 for 24 and 48 h. The cell morphology was microscopically examined. The results of the invasion assay were expressed as the mean ± S.E. of at least three independent experiments. Values without a common letter were significantly different (p < 0.05).
TGF-β1 Down-regulates VE-cadherin Expression in Human Trophoblast Cells
To examine whether TGF-β1 treatment affects the expression of VE-cadherin, HTR-8/SVneo cells were treated with different concentrations of TGF-β1 for 24 h. RT-qPCR analysis showed that treatment with TGF-β1 down-regulated VE-cadherin mRNA levels in HTR-8/SVneo cells (Fig. 2A). In addition, treatment with 5 ng/ml of TGF-β1 down-regulated VE-cadherin mRNA levels in a time-dependent manner (Fig. 2B). Similarly, Western blot analysis showed the inhibitory effects of TGF-β1 on VE-cadherin protein levels in HTR-8/SVneo cells (Fig. 2C). Importantly, treatment with TGF-β1 also down-regulated VE-cadherin protein levels in primary trophoblast cells that were isolated from three different first trimester placental tissue explants (Fig. 2D). Treatment with SB431542 abolished TGF-β1-induced down-regulation of VE-cadherin mRNA and protein levels (Fig. 3, A and B). Moreover, siRNA-mediated depletion of TβRI attenuated TGF-β1-induced down-regulation of VE-cadherin mRNA and protein levels (Fig. 3, C and D). TGF-β1 has been shown to induce EMT by down-regulating E-cadherin and up-regulating N-cadherin (23). To examine whether TGF-β1 regulates E-cadherin and N-cadherin expression, we examined the expression levels of E-cadherin in HTR-8/SVneo cells and primary cultures of trophoblast cells. Our results showed that both HTR-8/SVneo cells and primary cultures of trophoblast cells expressed N-cadherin, whereas E-cadherin was not detected in both types of cells (Fig. 3E). Treatment of HTR-8/SVneo cells with TGF-β1 up-regulated N-cadherin protein levels but did not affect E-cadherin protein levels (Fig. 3F).
FIGURE 2.

TGF-β1 down-regulates VE-cadherin in human trophoblast cells. A, HTR-8/SVneo cells were treated with increasing concentrations of TGF-β1 (1, 5, and 10 ng/ml), and the mRNA levels of VE-cadherin were examined using RT-qPCR. B, HTR-8/SVneo cells were treated with 5 ng/ml of TGF-β1, and the mRNA levels of VE-cadherin were analyzed at different time points using RT-qPCR. C, HTR-8/SVneo cells were treated with 1, 5, and 10 ng/ml of TGF-β1 for 24 and 48 h. The protein levels of VE-cadherin were examined by Western blot. D, three different primary cultures of human trophoblast cells (primary trophoblast cell (PTC); # 1, 2, and 3) were treated with 5 ng/ml of TGF-β1 for 24 h, and the protein levels of VE-cadherin were examined by Western blot. The RT-qPCR results were expressed as the mean ± S.E. of at least three independent experiments. Values without a common letter were significantly different (p < 0.05).
FIGURE 3.

TGF-β type I receptor is required for TGF-β1-induced down-regulation of VE-cadherin. A and B, HTR-8/SVneo cells were treated with 5 ng/ml of TGF-β1 in combination with SB431542 (10 μm). The mRNA (A) and protein levels (B) of VE-cadherin were examined using RT-qPCR and Western blot, respectively. C and D, HTR-8/SVneo cells were transfected with 50 nm control siRNA (si-Ctrl) or TβRI siRNA (si-TβRI) for 48 h and then treated with 5 ng/ml of TGF-β1 for 24 h. The mRNA (C) and protein (D) levels of VE-cadherin and TβRI were examined using RT-qPCR and Western blot, respectively. E, the endogenous protein levels of E-cadherin and N-cadherin in HTR-8/SVneo and two different primary human trophoblast cultures (PTC)(PTC1 and PTC2) were examined using Western blot. The human choriocarcinoma cell line JEG3 was used as a positive control to detect E-cadherin. F, HTR-8/SVneo cells were treated with 5 ng/ml of TGF-β1 for 24 h. The protein levels of E-cadherin and N-cadherin were examined using Western blot. The RT-qPCR results were expressed as the mean ± S.E. of at least three independent experiments. Values without a common letter were significantly different (p < 0.05). C indicates control, and T indicates TGF-β1.
VE-cadherin Is Required to Maintain the Invasive Capacity of Human Trophoblast Cells
To directly examine the role of VE-cadherin in human trophoblast cell invasion; we knocked down endogenous VE-cadherin in HTR-8/SVneo and primary trophoblast cells. Cells treated with VE-cadherin siRNA for 24 and 48 h showed significantly down-regulated VE-cadherin protein levels (Fig. 4A). In addition, a Matrigel invasion assay revealed that down-regulation of VE-cadherin decreased cell invasion in both HTR-8/SVneo and primary trophoblast cells (Fig. 4B). To test whether TGF-β1 further decreases cell invasion in VE-cadherin-deficient trophoblasts, VE-cadherin was knocked down in HTR-8/SVneo cells and the effect of TGF-β1 on cell invasion was examined. As shown in Fig. 4C, treatment with TGF-β1 and VE-cadherin siRNA showed the comparable effects on the down-regulation of VE-cadherin protein levels. Treatment with TGF-β1 only slightly down-regulated the VE-cadherin protein levels in VE-cadherin siRNA-treated HTR-8/SVneo cells (Fig. 4C). However, TGF-β1 significantly decreased cell invasion in VE-cadherin siRNA-treated HTR-8/SVneo cells (Fig. 4D). Taken together, these results indicated that VE-cadherin played an important role in maintaining the invasive capacity of human trophoblast cells. Moreover, our results suggested the presence of additional factors that mediated TGF-β1-decreased trophoblast cell invasion, which indicated that regulation may be multifactorial.
FIGURE 4.

VE-cadherin is required to maintain the invasive capacity of human trophoblast cells. A, HTR-8/SVneo and primary trophoblast culture cells were transfected with 50 nm control siRNA (si-Ctrl) or VE-cadherin siRNA (si-VE) for 24 and 48 h. The TβRI protein levels were examined by Western blot. B, after VE-cadherin knockdown, cell invasion was examined using the Matrigel invasion assay. C, HTR-8/SVneo cells were transfected with 50 nm control siRNA (si-Ctrl) or VE-cadherin siRNA (si-VE) for 24 h and then treated with 5 ng/ml of TGF-β1 for 24 h. The VE-cadherin protein levels were examined using Western blot. D, after VE-cadherin knockdown, HTR-8/SVneo cells were treated with 5 ng/ml of TGF-β1 and cell invasion was examined using the Matrigel invasion assay. The results of the invasion assay were expressed as the mean ± S.E. of at least three independent experiments. Values without a common letter were significantly different (p < 0.05). C indicates control, and T indicates TGF-β1.
Smad-dependent Pathways Are Involved in TGF-β1-induced Down-regulation of VE-cadherin
TGF-β-regulated cellular functions are mediated by activation of the Smad signaling pathway. Western blot analysis showed that TGF-β1 treatment induced Smad2 and Smad3 phosphorylation in a time-dependent manner in HTR-8/SVneo cells (Fig. 5A). To examine the involvement of Smad signaling in TGF-β1-induced down-regulation of VE-cadherin, siRNA was used to knockdown the common mediator Smad4. As shown in Fig. 5, B and C, down-regulation of Smad4 abolished the TGF-β1-induced down-regulation of VE-cadherin mRNA and protein levels. Although Smad2 and Smad3 are highly homologous, they can mediate TGF-β-regulated cellular functions redundantly and differentially in a context-dependent manner (24). To further examine the role of Smad2 and Smad3 in the TGF-β1-induced down-regulation of VE-cadherin, Smad2 and Smad3 were knocked down using specific siRNA. As shown in Fig. 5, D and E, down-regulation of Smad2 or Smad3 alone attenuated TGF-β1-induced down-regulation of VE-cadherin mRNA and protein levels. Interestingly, knockdown of Smad2 exhibited a greater inhibitory effect on TGF-β1-down-regulated VE-cadherin compared with Smad3.
FIGURE 5.

Smad-dependent signaling pathways are required for TGF-β1-induced down-regulation of VE-cadherin. A, HTR-8/SVneo cells were treated with 5 ng/ml of TGF-β1 for the indicated durations. The Smad2 and Smad3 phosphorylation levels were examined using Western blot with antibodies specific for the phosphorylated, activated forms of Smad2 (p-Smad2) and Smad3 (p-Smad3). B and C, HTR-8/SVneo cells were transfected with 50 nm control siRNA (si-Ctrl) or Smad4 siRNA (si-Smad4) for 48 h and then treated with 5 ng/ml of TGF-β1 for 24 h. The mRNA (B) and protein (C) levels of VE-cadherin and Smad4 were examined using RT-qPCR and Western blot, respectively. D and E, HTR-8/SVneo cells were transfected with 50 nm control siRNA (si-Ctrl), Smad2 siRNA (si-Smad2), or Smad3 siRNA (si-Smad3) for 48 h and then treated with 5 ng/ml of TGF-β1 for 24 h. The mRNA (D) and protein (E) levels of VE-cadherin, Smad2, and Smad3 were examined using RT-qPCR and Western blot, respectively. The RT-qPCR results were expressed as the mean ± S.E. of at least three independent experiments. Values without a common letter were significantly different (p < 0.05). C indicates control, and T indicates TGF-β1.
TGF-β1 Up-regulates Snail and Slug
TGF-β1 has been shown to up-regulate the expression of many transcriptional factors including Snail, Slug, Twist, and ZEB1 (25). Previous study has shown that Snail, Slug, and Twist can bind to the human VE-cadherin promoter and suppress its promoter activity (26). In HTR-8/SVneo cells, treatment with TGF-β1 up-regulated Snail and Slug mRNA levels in time- and dose-dependent manners (Fig. 6, A and B). However, the expression levels of Twist and ZEB1 were not affected by TGF-β1 (Fig. 6A). Similarly, Western blot analysis showed the stimulatory effects of TGF-β1 on Snail and Slug protein levels in time- and dose-dependent manners (Fig. 6, C and D). Moreover, TGF-β1-induced up-regulation of Snail and Slug was both abolished by treatment with SB431542 (Fig. 6, E and F) and knockdown of TβRI (Fig. 6, G and H).
FIGURE 6.

TGF-β1 induces Snail and Slug expression. A, HTR-8/SVneo cells were treated with 5 ng/ml of TGF-β1 for the indicated durations. The Snail, Slug, Twist, and ZEB1 mRNA levels were examined by RT-qPCR. B, HTR-8/SVneo cells were treated with increasing concentrations of TGF-β1 (1, 5, and 10 ng/ml) for 24 h. The mRNA levels of Snail and Slug were examined by RT-qPCR. C, HTR-8/SVneo cells were treated with 5 ng/ml of TGF-β1 for the indicated durations. The protein levels of Snail and Slug were examined using Western blot. D, HTR-8/SVneo cells were treated with increasing concentrations of TGF-β1 (1, 5, and 10 ng/ml) for 24 h. The protein levels of Snail and Slug were examined by Western blot. E and F, HTR-8/SVneo cells were treated with 5 ng/ml of TGF-β1 in combination with SB431542 (10 μm). The mRNA (E) and protein (F) levels of Snail and Slug were examined using RT-qPCR and Western blot, respectively. G and H, HTR-8/SVneo cells were transfected with 50 nm control siRNA (si-Ctrl) or TβRI siRNA (si-TβRI) for 48 h and then treated with 5 ng/ml of TGF-β1 for 24 h. The mRNA (G) and protein (H) levels of Snail and Slug were examined using RT-qPCR and Western blot, respectively. The RT-qPCR results were expressed as the mean ± S.E. of at least three independent experiments. Values without a common letter were significantly different (p < 0.05). C indicates control, and T indicates TGF-β1.
Smad-dependent Signaling Pathways Are Involved in TGF-β1-induced Up-regulation of Snail and Slug
To investigate whether the activation of Smads is required for TGF-β1-induced up-regulation of Snail and Slug, Smad4 was knocked down in HTR-8/SVneo cells. Knockdown of Smad4 abolished TGF-β1-induced up-regulation of Snail and Slug mRNA and protein levels (Fig. 7, A and B). Similarly, Smad2 and Smad3 were also knocked down to examine the involvement of specific Smads in TGF-β1-induced up-regulation of Snail and Slug. Knockdown of Smad2 and Smad3 both attenuated TGF-β1-induced up-regulation of Snail and Slug mRNA and protein levels (Fig. 7, C and D). Interestingly, our results showed that TGF-β1-up-regulated Snail was primarily mediated by Smad2, whereas Smad3 was largely involved in the TGF-β1-induced up-regulation of Slug (Fig. 7, C and D).
FIGURE 7.

Smad-dependent signaling pathways are required for the TGF-β1-induced up-regulation of Snail and Slug. A and B, HTR-8/SVneo cells were transfected with 50 nm control siRNA (si-Ctrl) or Smad4 siRNA (si-Smad4) for 48 h and then treated with 5 ng/ml of TGF-β1 for 24 h. The mRNA (A) and protein (B) levels of Snail and Slug were examined using RT-qPCR and Western blot, respectively. C and D, HTR-8/SVneo cells were transfected with 50 nm control siRNA (si-Ctrl), Smad2 siRNA (si-Smad2), or Smad3 siRNA (si-Smad3) for 48 h and then treated with 5 ng/ml of TGF-β1 for 24 h. The mRNA (C) and protein (D) levels of Snail and Slug were examined using RT-qPCR and Western blot, respectively. The RT-qPCR results were expressed as the mean ± S.E. of at least three independent experiments. Values without a common letter were significantly different (p < 0.05). C indicates control, and T indicates TGF-β1.
Snail Is Required for TGF-β1-induced Down-regulation of VE-cadherin
Next, we used loss- and gain-of-function approaches to evaluate the contributions of Snail and Slug in TGF-β1-induced down-regulation of VE-cadherin. HTR-8/SVneo cells treated with Snail and Slug siRNAs down-regulated basal levels and attenuated TGF-β1-induced up-regulation of Snail and Slug, respectively (Fig. 8, A and B). Interestingly, only the inhibition of Snail, but not Slug, attenuated the TGF-β1-induced down-regulation of VE-cadherin (Fig. 8, A and B). In contrast, overexpression of Snail significantly down-regulated VE-cadherin mRNA and protein levels in HTR-8/SVneo cells (Fig. 8, C and D). Interestingly, neither knockdown nor overexpression of Snail significantly affected the basal levels of cell invasion (Fig. 8E). Previous study using the electrophoretic mobility shift assay (EMSA) showed that Snail could bind to the human VE-cadherin promoter (26). However, it was not known whether Snail could bind to the VE-cadherin promoter within the natural chromatin context of the cell. Thus, we sought to determine whether Snail could directly bind to the VE-cadherin promoter using a chromatin immunoprecipitation (ChIP) assay with an anti-Snail antibody followed by PCR and primers specific for the VE-cadherin promoter (Fig. 8F). ChIP analyses showed that in HTR-8/SVneo cells, Snail specifically bound to the VE-cadherin promoter after 24 h of TGF-β1 treatment (Fig. 8G). These results demonstrated that Snail was required for TGF-β1-induced down-regulation of VE-cadherin in human trophoblast cells.
FIGURE 8.

Snail mediates the TGF-β1-induced down-regulation of VE-cadherin. A and B, HTR-8/SVneo cells were transfected with 50 nm control siRNA (si-Ctrl), Snail siRNA (si-Snail), or Slug siRNA (si-Slug) for 48 h and then treated with 5 ng/ml of TGF-β1 for 24 h. The mRNA (A) and protein (B) levels of VE-cadherin, Snail, and Slug were examined using RT-qPCR and Western blot, respectively. C and D, HTR-8/SVneo cells were transfected with control vector (pCMV) or vector containing full-length Snail cDNA (Snail) for 24 and 48 h. The mRNA (C) and protein (D) levels of VE-cadherin and Snail were examined using RT-qPCR and Western blot, respectively. E, after knockdown and overexpression of Snail in the HTR-8/SVneo cells, cell invasion was examined using the Matrigel invasion assay. F, the Snail binding site in the human VE-cadherin promoter is highlighted by a box. The primers for the ChIP assay are underlined. G, HTR-8/SVneo cells were treated with 5 ng/ml of TGF-β1 for 24 h before being subjected to ChIP analysis. Anti-Snail or IgG antibodies were used to immunoprecipitate DNA-containing complexes. Subsequent PCR was performed with primers complementary to the VE-cadherin promoter region containing the Snail binding site. The PCR products were resolved by electrophoresis in a 1% agarose gel and visualized by ethidium bromide staining. M indicates the 100-bp DNA ladder. C indicates control, and T indicates TGF-β1.
DISCUSSION
TGF-β1 has been shown to decrease human trophoblast cell invasion (15, 16). However, whether VE-cadherin is involved in TGF-β1-decreased human trophoblast cell invasion still remains unknown. In the present study, we reported that TGF-β1 treatment down-regulated VE-cadherin in a human trophoblast cell line, HTR-8/SVneo, and in primary cultures of human trophoblast cells. TGF-β1 induced Snail and Slug expression via Smad-dependent signaling pathways. In addition, our results indicated that Snail, but not Slug, bound directly to the VE-cadherin promoter and suppressed VE-cadherin expression, which subsequently contributed to TGF-β1-decreased human trophoblast cell invasion (Fig. 9).
FIGURE 9.
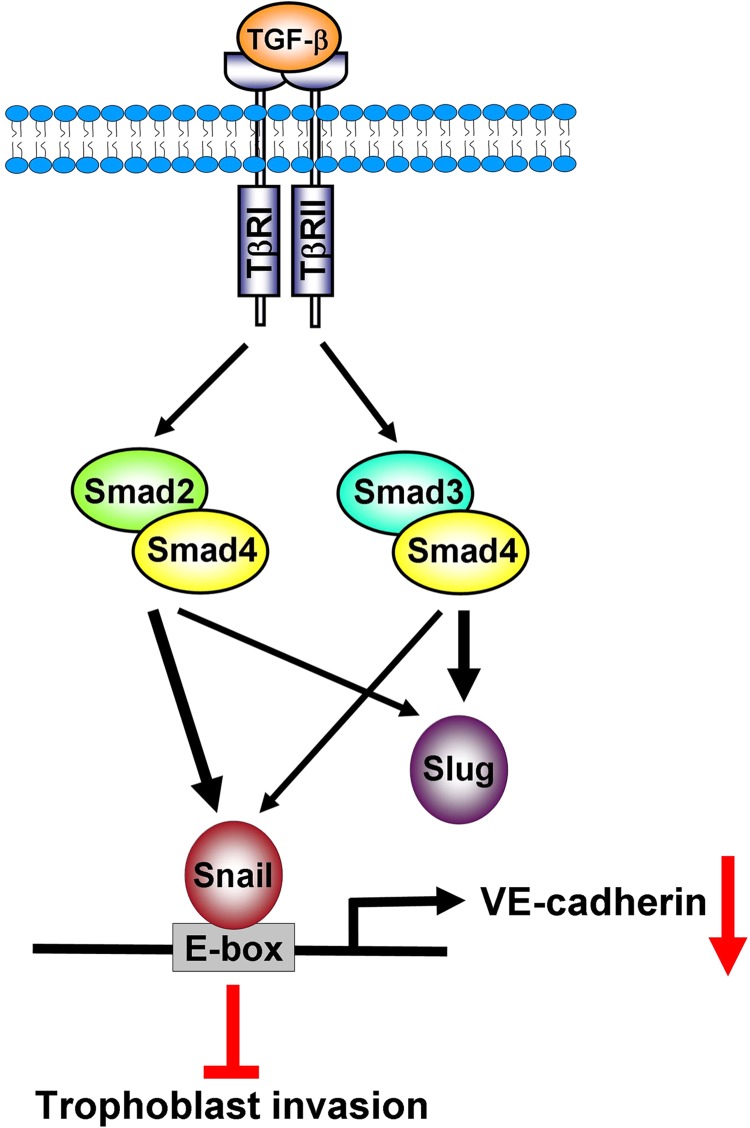
A schematic illustrating the signaling pathways for TGF-β1-induced down-regulation of VE-cadherin and the inhibition of cell invasion in human trophoblast cells. TGF-β1 treatment resulted in the activation of Smad2 and Smad3, which up-regulated the expression of Snail and Slug. The Smad2-dependent pathway is more involved in TGF-β1-induced up-regulation of Snail that Slug (thick versus thin arrow). In contrast, TGF-β1-induced up-regulation of Slug is primarily mediated by the Smad3-dependent pathway. Up-regulated Snail but not Slug subsequently binds to the E-box on the human VE-cadherin promoter and contributes to the TGF-β1-induced down-regulation of VE-cadherin and the inhibition of trophoblast cell invasion.
Human trophoblast invasion is a tightly regulated process that plays an important role in placental development. However, the regulatory mechanisms of trophoblast cell invasion have not been fully characterized. TGF-β1 and TGF-β receptors are expressed in human trophoblast cells and have been demonstrated to regulate human trophoblast cell proliferation, differentiation, and invasion (15, 27). In preeclampsia, a placental disease that is caused by inadequate or incomplete trophoblast cell invasion, increased levels of TGF-β1 are detected in the serum and placental tissue of patients and are also associated with the risk of the disease (28, 29). In addition, a systematic study on the expression of TGF-β signaling proteins shows that TGF-β1, TβRI, and TβRII levels in human trophoblasts are higher in the first trimester than the second and third trimesters (30). Importantly, the levels of TGF-β1 and TβRI are significantly decreased in choriocarcinoma, which is consistent with a previous study suggesting that disruption of the TGF-β signaling pathway may contribute to the malignancy of trophoblast cells (27, 30). TGF-β1 has also been shown to inhibit trophoblast cell invasion. However, the mechanisms of TGF-β1-decreased trophoblast cell invasion have not been fully defined. Many previous studies have demonstrated that TGF-β1-decreased human trophoblast cell invasion is mainly acting via the regulation of protease activity. TGF-β1 has been shown to inhibit the activity and expression of matrix metalloproteinase 9 and the urokinase-type plasminogen activator (16, 31, 32). In addition, tissue inhibitors of metalloproteinase and plasminogen activator inhibitor-1 are up-regulated by TGF-β1 (32, 33). To the best of our knowledge, the current study is the first that demonstrated that TGF-β1 decreases human trophoblast cell invasion by down-regulating the cell-cell adhesion molecule, VE-cadherin, thus providing a novel mechanism for the effects of TGF-β1 on trophoblast invasion.
The epithelial-mesenchymal transition (EMT) plays a very important role during embryonic development and has also been implicated in cancer where it contributes to tumor progression and metastasis (23, 34). Loss of E-cadherin is a hallmark of EMT (35). E-cadherin has been shown to reduce trophoblast cell invasion, and increased levels of E-cadherin have been detected in the trophoblast cells of preeclampsia patients (14, 36). Thus, it has been proposed that the process by which trophoblasts are localized at the tip of chorionic villi involves the conversion from a coherently attached phenotype to a migratory and invasive phenotype, which invades the endometrium, resembling EMT (13). Interestingly, in human placenta, positive expression of VE-cadherin is associated with the down-regulation of E-cadherin expression (14). Inhibition of VE-cadherin with antibody treatment results in a decrease in trophoblast cell invasion (12). In the present study, siRNA-mediated knockdown of VE-cadherin resulted in the down-regulation of VE-cadherin, which decreased the basal invasiveness of human trophoblast cells. Our approach provided specific evidence of the role of VE-cadherin in trophoblast cell invasion, and our results were consistent with a previous finding that indicated that VE-cadherin expression was required to maintain the invasive capacity of human trophoblast cells.
TGF-β1 stimulates cancer cell invasion by inducing EMT (37). However, our results demonstrated the inhibitory effect of TGF-β1 on trophoblast cell invasion. Actually, TGF-β1 has been shown to suppress cell migration and invasion in other cell types (38–42). These results emphasize that the effects on cell migration and invasion mediated by TGF-β1 vary depending on the target cell type. Furthermore, TGF-β1 can down-regulate E-cadherin and up-regulate N-cadherin (23). In the current study, we showed that the E-cadherin protein was not detected nor was it affected by TGF-β1 in both HTR-8/SVneo and primary human trophoblast cultures; however, TGF-β1 treatment up-regulated N-cadherin in HTR-8/SVneo cells. The results demonstrating the absence of E-cadherin expression are consistent with a recent study showing that the E-cadherin promoter is hypermethylated in HTR-8/SVneo cells (43). Our recent study showed that TGF-β1 does not affect the expression of N-cadherin in primary trophoblast cell cultures, although the direct knockdown of N-cadherin decreases cell invasion (44). To the best of our knowledge, to date, the direct involvement of N-cadherin in TGF-β1-regulated trophoblast cell invasion remains unknown. Thus, whereas previous studies suggest roles for N-cadherin in TGF-β1-induced cancer cell invasion, future studies will be required to determine whether these effects extend to human trophoblast cells.
Currently, only a few growth factors have been shown to regulate VE-cadherin. Treatment with vascular endothelial growth factor (VEGF) up-regulates VE-cadherin expression by activating the ERK and p38 MAPK signaling pathways in human umbilical vein endothelial cells and HTR-8/SVneo cells (45). TGF-β1 down-regulates VE-cadherin in human multilineage progenitor cells and mouse pulmonary endothelial cells (46, 47). However, in microvascular endothelial cells from the bovine corpus luteum, TGF-β1 treatment alters the localization of VE-cadherin in cellular junctions without affecting its total expression levels (48). Although TGF-β1 has been shown to regulate VE-cadherin expression, the regulatory effect of TGF-β1 in human trophoblast cells and its underlying molecular mechanisms still remain unknown. Our study demonstrated the inhibitory effect of TGF-β1 on VE-cadherin expression in human trophoblast cells. In addition, using siRNA-mediated knockdown approaches, our results indicated that Smad-dependent signaling pathways were involved in TGF-β1-induced down-regulation of VE-cadherin. Moreover, our results showed that both Smad2 and Smad3 siRNAs attenuated TGF-β1-induced down-regulation of VE-cadherin, and interestingly, inhibition of Smad2 exhibited greater effects compared with those caused by Smad3 inhibition. Examination of the Smad signaling pathway showed that both Smad2 and Smad3 were required for TGF-β1-induced Snail and Slug expression, whereas Smad2 was more responsible for up-regulation of Snail, and Smad3 was more involved in TGF-β1-induced Slug expression. Therefore, these results may explain why the knockdown of Smad2 demonstrated stronger inhibitory effects on TGF-β1-induced down-regulation of VE-cadherin.
Several transcription factors have been found to bind to the VE-cadherin promoter and regulate its gene expression. Sp1, TAL-1, and the ETS family of transcription factors bind to and enhance VE-cadherin promoter activity (49–51). In contrast, Snail, Slug, and Twist transcription factors are responsible for the down-regulation of VE-cadherin promoter activity by binding to the E-box of the promoter region (26). The binding between these transcription factors and the human VE-cadherin promoter were shown in an in vitro system using EMSA (26). Thus, whether Snail, Slug, or Twist can bind to human VE-cadherin promoter within a natural chromatin cellular context and whether the binding of these transcription factors can directly regulate VE-cadherin mRNA and protein levels still remain unknown. In the current study, our results indicated that TGF-β1 treatment induced Snail and Slug, but not Twist, expression. Interestingly, only Snail siRNA attenuated TGF-β1-induced down-regulation of VE-cadherin mRNA and protein levels. In addition, overexpression of Snail confirmed the inhibitory effect of Snail on VE-cadherin expression. Moreover, ChIP analysis showed that Snail could bind to the VE-cadherin promoter and suppress its expression in HTR-8/SVneo cells. Taken together, these results indicated that Snail was required for the TGF-β1-induced down-regulation of VE-cadherin in human trophoblast cells. Importantly, the partial attenuation of TGF-β1-induced down-regulation of VE-cadherin by Snail siRNA indicated the presence of additional mediators and suggested that this regulation might be multifactorial.
Overexpression of Snail has been shown to be associated with cell invasion in many types of human cancers (52). Therefore, we also examined the effects of Snail on trophoblast cell invasion. Our results showed that the knockdown and overexpression of Snail did not significantly affect the basal levels of HTR-8/SVneo cell invasion. Interestingly, a recent study showed that ethanol induces Snail expression but decreases cell invasion without affecting E-cadherin expression in immortalized human pancreatic ductal epithelial cells (53). These results indicated that up-regulation of Snail may not always be associated with increased cell invasion. Moreover, although overexpression of Snail down-regulated VE-cadherin expression in HTR-8/SVneo cells, basal cell invasiveness was not affected. These results suggested that the overexpression of Snail modulates other unknown molecules to counteract the down-regulation of VE-cadherin-decreased cell invasion. Furthermore, our results also suggested that upon TGF-β1 treatment, activation of other factors and signaling pathways cooperates with Snail, which may modulate its regulatory roles that ultimately contribute to TGF-β1-decreased trophoblast cell invasion.
In summary, our study demonstrated that Snail is a transcription factor that directly binds to the VE-cadherin promoter and mediated TGF-β1-induced down-regulation of VE-cadherin expression. The down-regulation of VE-cadherin was involved in TGF-β1-decreased human trophoblast cell invasion. Moreover, our data indicated that the activation of Smad-dependent signaling pathways contributed to TGF-β1-induced Snail expression and the down-regulation of VE-cadherin. These results provided important insights into the molecular mechanisms mediating TGF-β1-induced down-regulation of VE-cadherin and decreased cell invasion in human trophoblast cells.
This work was supported by an operating grant from the Canadian Institutes of Health Research (to P. C. K. L.).
- TβRI and TβRII
- transmembrane type I and II receptors
- VE-cadherin
- vascular endothelial-cadherin
- qPCR
- quantitative PCR
- EMT
- epithelial-mesenchymal transition.
REFERENCES
- 1. Chaddha V., Viero S., Huppertz B., Kingdom J. (2004) Developmental biology of the placenta and the origins of placental insufficiency. Semin. Fetal Neonatal Med. 9, 357–369 [DOI] [PubMed] [Google Scholar]
- 2. Lim K. H., Zhou Y., Janatpour M., McMaster M., Bass K., Chun S. H., Fisher S. J. (1997) Human cytotrophoblast differentiation/invasion is abnormal in pre-eclampsia. Am. J. Pathol. 151, 1809–1818 [PMC free article] [PubMed] [Google Scholar]
- 3. Brosens I., Pijnenborg R., Vercruysse L., Romero R. (2011) The “Great Obstetrical Syndromes” are associated with disorders of deep placentation. Am. J. Obstet. Gynecol. 204, 193–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Böttner M., Krieglstein K., Unsicker K. (2000) The transforming growth factor-βs. Structure, signaling, and roles in nervous system development and functions. J. Neurochem, 75, 2227–2240 [DOI] [PubMed] [Google Scholar]
- 5. Blobe G. C., Schiemann W. P., Lodish H. F. (2000) Role of transforming growth factor β in human disease. N. Engl. J. Med. 342, 1350–1358 [DOI] [PubMed] [Google Scholar]
- 6. Krieglstein K., Rufer M., Suter-Crazzolara C., Unsicker K. (1995) Neural functions of the transforming growth factors β. Int. J. Dev. Neurosci. 13, 301–315 [DOI] [PubMed] [Google Scholar]
- 7. Shi Y., Massagué J. (2003) Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell 113, 685–700 [DOI] [PubMed] [Google Scholar]
- 8. Heldin C. H., Miyazono K., ten Dijke P. (1997) TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature 390, 465–471 [DOI] [PubMed] [Google Scholar]
- 9. Vestweber D. (2008) VE-cadherin. The major endothelial adhesion molecule controlling cellular junctions and blood vessel formation. Arterioscler. Thromb. Vasc. Biol. 28, 223–232 [DOI] [PubMed] [Google Scholar]
- 10. Le Guelte A., Dwyer J., Gavard J. (2011) Jumping the barrier. VE-cadherin, VEGF and other angiogenic modifiers in cancer. Biol. Cell 103, 593–605 [DOI] [PubMed] [Google Scholar]
- 11. Kobielak A., Fuchs E. (2004) α-Catenin. At the junction of intercellular adhesion and actin dynamics. Nat. Rev. Mol. Cell Biol. 5, 614–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhou Y., Fisher S. J., Janatpour M., Genbacev O., Dejana E., Wheelock M., Damsky C. H. (1997) Human cytotrophoblasts adopt a vascular phenotype as they differentiate. A strategy for successful endovascular invasion? J. Clin. Invest. 99, 2139–2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kokkinos M. I., Murthi P., Wafai R., Thompson E. W., Newgreen D. F. (2010) Cadherins in the human placenta–epithelial-mesenchymal transition (EMT) and placental development. Placenta 31, 747–755 [DOI] [PubMed] [Google Scholar]
- 14. Zhou Y., Damsky C. H., Fisher S. J. (1997) Preeclampsia is associated with failure of human cytotrophoblasts to mimic a vascular adhesion phenotype. One cause of defective endovascular invasion in this syndrome? J. Clin. Invest. 99, 2152–2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jones R. L., Stoikos C., Findlay J. K., Salamonsen L. A. (2006) TGF-β superfamily expression and actions in the endometrium and placenta. Reproduction 132, 217–232 [DOI] [PubMed] [Google Scholar]
- 16. Lash G. E., Otun H. A., Innes B. A., Bulmer J. N., Searle R. F., Robson S. C. (2005) Inhibition of trophoblast cell invasion by TGFB1, -2, and -3 is associated with a decrease in active proteases. Biol. Reprod. 73, 374–381 [DOI] [PubMed] [Google Scholar]
- 17. Graham C. H., Hawley T. S., Hawley R. G., MacDougall J. R., Kerbel R. S., Khoo N., Lala P. K. (1993) Establishment and characterization of first trimester human trophoblast cells with extended lifespan. Exp. Cell Res. 206, 204–211 [DOI] [PubMed] [Google Scholar]
- 18. Graham C. H., Lysiak J. J., McCrae K. R., Lala P. K. (1992) Localization of transforming growth factor-β at the human fetal-maternal interface. Role in trophoblast growth and differentiation. Biol. Reprod. 46, 561–572 [DOI] [PubMed] [Google Scholar]
- 19. Woo M. M., Salamanca C. M., Minor A., Auersperg N. (2007) An improved assay to quantitate the invasiveness of cells in modified Boyden chambers. In Vitro Cell Dev. Biol. Anim 43, 7–9 [DOI] [PubMed] [Google Scholar]
- 20. Gleeson L. M., Chakraborty C., McKinnon T., Lala P. K. (2001) Insulin-like growth factor-binding protein 1 stimulates human trophoblast migration by signaling through α5β1 integrin via mitogen-activated protein kinase pathway. J. Clin. Endocrinol. Metab. 86, 2484–2493 [DOI] [PubMed] [Google Scholar]
- 21. Lala P. K., Graham C. G. (2001) TGFB-responsive human trophoblast-derived cell lines. Placenta 22, 889–890 [DOI] [PubMed] [Google Scholar]
- 22. Inman G. J., Nicolás F. J., Callahan J. F., Harling J. D., Gaster L. M., Reith A. D., Laping N. J., Hill C. S. (2002) SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 62, 65–74 [DOI] [PubMed] [Google Scholar]
- 23. Thiery J. P., Acloque H., Huang R. Y., Nieto M. A. (2009) Epithelial-mesenchymal transitions in development and disease. Cell 139, 871–890 [DOI] [PubMed] [Google Scholar]
- 24. Brown K. A., Pietenpol J. A., Moses H. L. (2007) A tale of two proteins. Differential roles and regulation of Smad2 and Smad3 in TGF-β signaling. J. Cell. Biochem. 101, 9–33 [DOI] [PubMed] [Google Scholar]
- 25. Saitoh M., Miyazawa K. (2012) Transcriptional and post-transcriptional regulation in TGF-β-mediated epithelial-mesenchymal transition. J. Biochem. 151, 563–571 [DOI] [PubMed] [Google Scholar]
- 26. Lopez D., Niu G., Huber P., Carter W. B. (2009) Tumor-induced upregulation of Twist, Snail, and Slug represses the activity of the human VE-cadherin promoter. Arch. Biochem. Biophys. 482, 77–82 [DOI] [PubMed] [Google Scholar]
- 27. Xu G., Chakraborty C., Lala P. K. (2001) Expression of TGF-β signaling genes in the normal, premalignant, and malignant human trophoblast. Loss of smad3 in choriocarcinoma cells. Biochem. Biophys. Res. Commun. 287, 47–55 [DOI] [PubMed] [Google Scholar]
- 28. Benian A., Madazli R., Aksu F., Uzun H., Aydin S. (2002) Plasma and placental levels of interleukin-10, transforming growth factor-β1, and epithelial-cadherin in preeclampsia. Obstet. Gynecol. 100, 327–331 [DOI] [PubMed] [Google Scholar]
- 29. Muy-Rivera M., Sanchez S. E., Vadachkoria S., Qiu C., Bazul V., Williams M. A. (2004) Transforming growth factor-β1 (TGF-β1) in plasma is associated with preeclampsia risk in Peruvian women with systemic inflammation. Am. J. Hypertens. 17, 334–338 [DOI] [PubMed] [Google Scholar]
- 30. Xuan Y. H., Choi Y. L., Shin Y. K., Ahn G. H., Kim K. H., Kim W. J., Lee H. C., Kim S. H. (2007) Expression of TGF-β signaling proteins in normal placenta and gestational trophoblastic disease. Histol. Histopathol. 22, 227–234 [DOI] [PubMed] [Google Scholar]
- 31. Meisser A., Chardonnens D., Campana A., Bischof P. (1999) Effects of tumour necrosis factor-α, interleukin-1α, macrophage colony stimulating factor and transforming growth factor β on trophoblastic matrix metalloproteinases. Mol. Hum. Reprod. 5, 252–260 [DOI] [PubMed] [Google Scholar]
- 32. Graham C. H. (1997) Effect of transforming growth factor-beta on the plasminogen activator system in cultured first trimester human cytotrophoblasts. Placenta 18, 137–143 [DOI] [PubMed] [Google Scholar]
- 33. Karmakar S., Das C. (2002) Regulation of trophoblast invasion by IL-1β and TGF-β1. Am. J. Reprod. Immunol. 48, 210–219 [DOI] [PubMed] [Google Scholar]
- 34. Lim J., Thiery J. P. (2012) Epithelial-mesenchymal transitions. Insights from development. Development 139, 3471–3486 [DOI] [PubMed] [Google Scholar]
- 35. Voulgari A., Pintzas A. (2009) Epithelial-mesenchymal transition in cancer metastasis. Mechanisms, markers, and strategies to overcome drug resistance in the clinic. Biochim. Biophys. Acta 1796, 75–90 [DOI] [PubMed] [Google Scholar]
- 36. Li H. W., Cheung A. N., Tsao S. W., Cheung A. L., O W. S. (2003) Expression of E-cadherin and β-catenin in trophoblastic tissue in normal and pathological pregnancies. Int. J. Gynecol. Pathol. 22, 63–70 [DOI] [PubMed] [Google Scholar]
- 37. Katsuno Y., Lamouille S., Derynck R. (2013) TGF-β signaling and epithelial-mesenchymal transition in cancer progression. Curr. Opin. Oncol. 25, 76–84 [DOI] [PubMed] [Google Scholar]
- 38. Fritzmann J., Morkel M., Besser D., Budczies J., Kosel F., Brembeck F. H., Stein U., Fichtner I., Schlag P. M., Birchmeier W. (2009) A colorectal cancer expression profile that includes transforming growth factor β inhibitor BAMBI predicts metastatic potential. Gastroenterology 137, 165–175 [DOI] [PubMed] [Google Scholar]
- 39. Humbert L., Lebrun J. J. (2013) TGF-β inhibits human cutaneous melanoma cell migration and invasion through regulation of the plasminogen activator system. Cell Signal. 25, 490–500 [DOI] [PubMed] [Google Scholar]
- 40. Hölting T., Zielke A., Siperstein A. E., Clark O. H., Duh Q. Y. (1994) Transforming growth factor-β1 is a negative regulator for differentiated thyroid cancer. Studies of growth, migration, invasion, and adhesion of cultured follicular and papillary thyroid cancer cell lines. J. Clin. Endocrinol. Metab. 79, 806–813 [DOI] [PubMed] [Google Scholar]
- 41. Wang X., Sun W., Zhang C., Ji G., Ge Y., Xu Y., Zhao Y. (2011) TGF-β1 inhibits the growth and metastasis of tongue squamous carcinoma cells through Smad4. Gene 485, 160–166 [DOI] [PubMed] [Google Scholar]
- 42. Zhao M. R., Qiu W., Li Y. X., Zhang Z. B., Li D., Wang Y. L. (2006) Dual effect of transforming growth factor β1 on cell adhesion and invasion in human placenta trophoblast cells. Reproduction 132, 333–341 [DOI] [PubMed] [Google Scholar]
- 43. Chen Y., Wang K., Leach R. (2013) 5-Aza-dC treatment induces mesenchymal-to-epithelial transition in 1st trimester trophoblast cell line HTR8/SVneo. Biochem. Biophys. Res. Commun. 432, 116–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ng Y. H., Zhu H., Leung P. C. (2012) Twist modulates human trophoblastic cell invasion via regulation of N-cadherin. Endocrinology 153, 925–936 [DOI] [PubMed] [Google Scholar]
- 45. Lala N., Girish G. V., Cloutier-Bosworth A., Lala P. K. (2012) Mechanisms in decorin regulation of vascular endothelial growth factor-induced human trophoblast migration and acquisition of endothelial phenotype. Biol. Reprod. 87, 59. [DOI] [PubMed] [Google Scholar]
- 46. Li Z., Jimenez S. A. (2011) Protein kinase Cδ and c-Abl kinase are required for transforming growth factor-β induction of endothelial-mesenchymal transition in vitro. Arthritis Rheum. 63, 2473–2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yang H., Zhang L., Weakley S. M., Lin P. H., Yao Q., Chen C. (2011) Transforming growth factor-β increases the expression of vascular smooth muscle cell markers in human multi-lineage progenitor cells. Med. Sci. Monit. 17, BR55–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Maroni D., Davis J. S. (2011) TGFB1 disrupts the angiogenic potential of microvascular endothelial cells of the corpus luteum. J. Cell Sci. 124, 2501–2510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Birdsey G. M., Dryden N. H., Amsellem V., Gebhardt F., Sahnan K., Haskard D. O., Dejana E., Mason J. C., Randi A. M. (2008) Transcription factor Erg regulates angiogenesis and endothelial apoptosis through VE-cadherin. Blood 111, 3498–3506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Deleuze V., Chalhoub E., El-Hajj R., Dohet C., Le Clech M., Couraud P. O., Huber P., Mathieu D. (2007) TAL-1/SCL and its partners E47 and LMO2 up-regulate VE-cadherin expression in endothelial cells. Mol. Cell. Biol. 27, 2687–2697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Gory S., Dalmon J., Prandini M. H., Kortulewski T., de Launoit Y., Huber P. (1998) Requirement of a GT box (Sp1 site) and two Ets binding sites for vascular endothelial cadherin gene transcription. J. Biol. Chem. 273, 6750–6755 [DOI] [PubMed] [Google Scholar]
- 52. Becker K. F., Rosivatz E., Blechschmidt K., Kremmer E., Sarbia M., Höfler H. (2007) Analysis of the E-cadherin repressor Snail in primary human cancers. Cells Tissues Organs 185, 204–212 [DOI] [PubMed] [Google Scholar]
- 53. Ward S. T., Dangi-Garimella S., Shields M. A., Collander B. A., Siddiqui M. A., Krantz S. B., Munshi H. G. (2011) Ethanol differentially regulates snail family of transcription factors and invasion of premalignant and malignant pancreatic ductal cells. J. Cell. Biochem. 112, 2966–2973 [DOI] [PMC free article] [PubMed] [Google Scholar]