

Background: Carboxypeptidase M (CPM) generates agonist for the kinin B1 receptor (B1R).
Results: CPM binds to B1R on the cell membrane and allosterically increases B1R agonist affinity.
Conclusion: CPM is a positive allosteric modulator of the B1R, thereby increasing receptor signaling.
Significance: Interfering with CPM binding to B1R could be a novel approach to inhibit deleterious effects of B1R signaling in inflammation.
Keywords: Allosteric Regulation, Carboxypeptidase, G Protein-coupled Receptors (GPCR), Glycosyl Phosphatidylinositol Anchors, Peptidases, Peptide Hormones, Bradykinin, Kallidin
Abstract
Ligand binding to extracellular domains of G protein-coupled receptors can result in novel and nuanced allosteric effects on receptor signaling. We previously showed that the protein-protein interaction of carboxypeptidase M (CPM) and kinin B1 receptor (B1R) enhances B1R signaling in two ways; 1) kinin binding to CPM causes a conformational activation of the B1R, and 2) CPM-generated des-Arg-kinin agonist is efficiently delivered to the B1R. Here, we show CPM is also a positive allosteric modulator of B1R signaling to its agonist, des-Arg10-kallidin (DAKD). In HEK cells stably transfected with B1R, co-expression of CPM enhanced DAKD-stimulated increases in intracellular Ca2+ or phosphoinositide turnover by a leftward shift of the dose-response curve without changing the maximum. CPM increased B1R affinity for DAKD by ∼5-fold but had no effect on basal B1R-dependent phosphoinositide turnover. Soluble, recombinant CPM bound to HEK cells expressing B1Rs without stimulating receptor signaling. CPM positive allosteric action was independent of enzyme activity but depended on interaction of its C-terminal domain with the B1R extracellular loop 2. Disruption of the CPM/B1R interaction or knockdown of CPM in cytokine-treated primary human endothelial cells inhibited the allosteric enhancement of CPM on B1R DAKD binding or ERK1/2 activation. CPM also enhanced the DAKD-induced B1R conformational change as detected by increased intramolecular fluorescence or bioluminescence resonance energy transfer. Thus, CPM binding to extracellular loop 2 of the B1R results in positive allosteric modulation of B1R signaling, and disruption of this interaction could provide a novel therapeutic approach to reduce pathological B1R signaling.
Introduction
Carboxypeptidase M (CPM)3 is a member of the metallocarboxypeptidase family of peptidases (1), also known as funnelins based on the shape of the entrance to their active site (2). According to its genomic structure (3), sequence (4), and x-ray crystal structure (5), CPM belongs to the “regulatory” or CPN/E subfamily, also designated as Clan MC, family M14B according to the MEROPS classification (1), consisting of eight members, five of which are active (2, 6, 7). They are distinguished from the CPA/B (M14A) subfamily by the lack of a propeptide activation sequence and the presence of an additional C-terminal domain consisting of a seven-stranded β barrel with topological similarity to the plasma protein transthyretin (2, 6, 7). CPM is an ectoenzyme anchored to the plasma membrane of cells by a glycosylphosphatidylinositol (GPI) anchor (8, 9) and is widely distributed in the body, for example in lung and placental microvilli, kidney, blood vessels, intestine, brain, and peripheral nerves and can be found in soluble form in various body fluids (1, 10, 11). CPM has a strict specificity, cleaving only C-terminal Arg or Lys residues (12), and although >60 endogenous human peptides or proteins have been identified as potential CPM substrates, only 22 have been tested and shown to have altered activity after C-terminal cleavage (10). These include growth factors, chemokines, plasminogen binding proteins, complement proteins, kinins, and opioid peptides (1, 6, 10).
The seven-transmembrane domain kinin B1 receptor (B1R) couples to both Gαq and Gαi, and although it is constitutively expressed in a few cell types, its expression is inducible in many cells under pathological and inflammatory conditions (13, 14). The B1R plays key roles in nociception, inflammation, and renal and cardiovascular diseases (14, 15). For example, in the cardiovascular system, B1R signaling can affect multiple physiological and pathological processes including blood pressure control, arteriogenesis, ischemic pre- and post-conditioning, myocardial infarction, heart failure, diabetic cardiomyopathy, and septic shock (16–23). B1R activation is also involved in the therapeutic effects of angiotensin converting enzyme (ACE) inhibitors and angiotensin type 1 receptor blockers, which are extensively used to treat not only hypertension but various cardiovascular and renal diseases (24–26).
Endogenous peptide agonists of the B1R, des-Arg9-bradykinin and des-Arg10-kallidin (DAKD), are derived from precursor kininogens after initial cleavage by plasma or tissue kallikreins to release bradykinin (BK) or kallidin (KD) followed by removal of their C-terminal Arg by a B-type carboxypeptidase (1, 11, 14). Although several mammalian carboxypeptidases have the proper specificity to carry out this last step in vitro, fulfilling this role effectively in vivo would also depend on properties such as pH optimum, localization, access to kinin substrate, and proximity to the B1R (11, 27–29). CPM has ideal properties in this regard, being a GPI-anchored plasma membrane protein with a neutral pH optimum exhibiting a specificity for C-terminal Arg and the lowest Km for BK of the physiological substrates tested (1, 8, 12, 30). In addition to its wide distribution, endotoxin or cytokines that induce B1R expression (14) also increase CPM expression (31–33). Thus, it is likely that cells expressing B1Rs also express CPM. The crystal structure of CPM and molecular modeling suggest that CPM adopts a favorable orientation for generating peptide products near the membrane by interaction of its positively charged residues in the C-terminal domain with phospholipid head groups (5). We found that CPM does indeed play a critical role in B1R signaling. CPM and B1Rs are co-localized in lipid raft domains and interact on the cell surface as evidenced by FRET analysis, cross-linking, and coimmunoprecipitation (34). Disruption of lipid rafts or the CPM/B1R interaction by CPM monoclonal antibody greatly reduced B1R signaling in response to administration of BK or KD (29, 34). We initially attributed this to more efficient delivery of the CPM-cleaved products (i.e. B1R agonists) to the receptor. Although this is clearly an important component of the CPM effect, we recently discovered that the CPM/B1R interaction mediates a second, novel mechanism of B1R activation that results from substrate binding to CPM to cause a conformational change and allosteric activation of the B1R without generation of B1R agonist (29, 35).
Allosteric modulation of GPCR signaling is currently an area of great interest because of the large repertoire of potential unique allosteric binding sites on GPCR extracellular domains and the possibility of affecting receptor signaling in a more specific and biased fashion (36, 37). In the present study we investigated whether the basal interaction between CPM and B1R allosterically modulates B1R responses to its own agonists. We found that CPM enhances B1R signaling by increasing the affinity of the receptor for its endogenous agonist, DAKD. The allosteric effect depended on the interaction between extracellular loop 2 (EL2) of the B1R and the C-terminal domain of CPM. The CPM/B1R interaction could be disrupted by a CPM monoclonal antibody or a peptide containing the antibody epitope and reduced B1R agonist affinity and signaling in primary and transfected cells. Thus, CPM is an endogenous positive allosteric modulator of B1R signaling to its orthosteric agonist.
EXPERIMENTAL PROCEDURES
Materials
Low glucose Dulbecco's modified Eagle's medium (DMEM) was obtained from Invitrogen. Fetal bovine serum (FBS) was from Atlanta Biologicals. DAKD and polylysine were from Sigma. CT peptide (Ac-KGQVFDQNGNPLPN-NH2) was synthesized by Chi Scientific. Fura-2/AM was from Molecular Probes. The TC-FlAsHTM II in-Cell Tetracysteine Tag Detection kit was from Invitrogen. ViviRenTM live cell substrate was from Promega. [3H]DAKD was from PerkinElmer Life Sciences. Myo-[3H]inositol was from American Radiolabeled Chemicals, Inc. Anti-CPM monoclonal antibody was from Novocastra. Anti-B1R and B2R polyclonal antibodies were from Santa Cruz Biotechnology. ERK1/2 and anti-phosphorylated ERK1/2 antibodies were from Cell Signaling. Goat anti-mouse and anti-rabbit IgG conjugated-HRP were from Pierce. 5-Dimethylaminonaphthalene-1-sulfonyl-l-alanyl-l-arginine (dansyl-Ala-Arg) was synthesized and purified as described previously (38).
Cells
Primary human lung microvascular endothelial cells (HLMVEC) from Lonza were cultured in T-25 or T-75 flasks coated with 0.1% gelatin in endothelial cell basal medium (EBM®-2, Lonza) supplemented with EGM®-2 SingleQuots® kit (Lonza) and 10% fetal bovine serum (Atlanta Biologicals). Cells between passage 3 and 6 were used for assay. Human embryonic kidney (HEK) 293 cells from the American Type Culture Collection were maintained in DMEM containing 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% FBS at 37 °C in a humidified atmosphere of 5% CO2.
Generation of Receptor and Carboxypeptidase Constructs
The cDNAs for human B1R (a kind gift from Dr. Fredrik Leeb-Lundberg, University of Lund, Sweden) and human CPM (4, 39) were cloned into pcDNA3 or pcDNA6 vectors (Invitrogen) for expression in mammalian cells. The B1R-TC-CFP and CPD-DIII constructs were generated as described previously (35). B1R-TC cDNA was digested with NheI and BamHI and cloned into pRluc-N1 to generate B1R-TC-Rluc. The CPM mutants CPM-E264Q, CPM-S180N, and CPM-S180N/E264Q were produced by site-directed mutagenesis as described (35, 39). GPI-anchored CPN catalytic subunit (50 kDa) (CPN-GPI) was generated by cloning the human CPN 50 kDa catalytic subunit cDNA upstream of the CPM GPI anchor signal sequence (encoding the C-terminal 24 residues of CPM) into pcDNA3 with techniques similar to those used to generate ACE-GPI (40). All PCR fragments were amplified using high fidelity TaqDNA polymerase, and constructs were verified by DNA sequencing performed by the DNA Services Facility of the Research Resources Center, University of Illinois at Chicago.
Transfection and Establishment of Stable Cell Lines
HEK293 cells stably expressing constructs used in this study were generated as described previously (35). For transient expression, HEK cells in 12-well plates were transfected with 1 μg of DNA using SuperFect (Qiagen) or 0.30 μg of DNA using Lipofectamine 2000 (Invitrogen) unless otherwise indicated. Cells were used for experiments 24 h after transfection.
CPM Knockdown by siRNA
HLMVEC at ∼80% confluence in 12-well plates were transfected with specific CPM siRNA (SMARTpool® plus, Dharmacon) or nonspecific control siRNA (Ambion) using the Amaxa Nucleofector (Lonza, Walkersville, MD) and the manufacturer's kit and protocol optimized for HLMVEC. After 48 h cells were treated with 5 ng/ml IL-1β and 200 units/ml IFN-γ for 16 h and then used for experiments.
Cell Binding of Purified CPM
A soluble form of recombinant human CPM (rCPM) lacking the C-terminal GPI anchor was expressed in baculovirus-infected insect cells and purified to homogeneity as we previously described (5, 39). The purified rCPM (500 ng/ml in PBS) was added to control cells or cells stably expressing B1R or CPM and then incubated at 37 °C for 30 min. After incubation, cells were washed twice with ice-cold PBS. The lysates from the washed cells were analyzed by Western blotting to assess the binding of rCPM, which has a lower molecular mass (∼50 kDa) than CPM expressed in human cells and tissues (∼62 kDa) (5, 39).
Measurement of Intracellular Ca2+
Increases in intracellular calcium concentration ([Ca2+]i) were determined with fura-2/AM using a PTI Deltascan microspectrofluorometer as described (34, 35). Responses were quantified by integrating the area under the curve using Origin 8.0 software (OriginLab Corp.). The EC50 was calculated by plotting the dose-response curve based on the area under the curve of the calcium response using GraphPad Prism 5.0 software (GraphPad Software, Inc.).
Phosphoinositide Turnover Assay
Phosphoinositide (PI) turnover was determined as previously described (41) with slight modifications. Cells at about 80% confluence in 12-well plates were labeled for 18–24 h with 1 μCi/ml of myo-[3H]inositol in DMEM with 2% dialyzed FBS. After loading, the cells were incubated with 15 mm LiCl for 60 min at 37 °C, then stimulated with agonists for 30 min at 37 °C followed by termination with 0.5 ml of ice-cold 20 mm formic acid. After 30 min on ice, the supernatant was combined with another 0.5 ml of 20 mm formic acid, alkalinized with 0.2 ml of 3% ammonium hydroxide, and then applied to an AG 1-X8 anion exchange column. The column was washed with 2 ml of 20 mm formic acid, 4 ml of 50 mm ammonium hydroxide, and then 4 ml of 40 mm ammonium formate containing 0.1 m formic acid. After washing, inositol triphosphate (IP3) was eluted using 5 ml of buffer containing 2 m ammonium formate and 0.1 m formic acid. IP3 radioactivity was determined in Beckman liquid scintillation counter after adding 10 ml of scintillation fluid.
Immunoprecipitation
Control HEK cells or HEK cells stably expressing B1R, B2R, B1R(B2R-EL1), or B1R(B2R-EL2) were transfected with 0.30 μg of CPM cDNA for 24 h, and then the B1R was immunoprecipitated, and precipitates were analyzed for CPM by Western blotting as previously described (34, 35).
Determination of ERK1/2 Phosphorylation
Cells (80–90% confluent) stably expressing B1R and CPM in 24-well plates were preincubated without or with 50 μm CT peptide for 30 min at 37 °C. Cells were then stimulated with various concentrations of DAKD for 5 min and then lysed in 200 μl of radioimmune precipitation assay buffer (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, 1% sodium deoxycholate) containing 1% protease inhibitor mixture (Sigma) and 1 mm Na3VO4 for 5 min on ice followed by centrifugation at 14,000 × g for 10 min at 4 °C. Supernatants were collected, and phosphorylated ERK1/2 and total ERK1/2 were identified by Western blotting and quantified by densitometry using software Quantity One (4.4.0, Bio-Rad). Phosphorylated ERK1/2 was normalized to the corresponding total ERK1/2 band. Data are expressed as -fold increase over the value for the non-treated control. EC50 was calculated by plotting dose-response curves with GraphPad Prism 5.0.
Western Blotting
Cells were lysed in radioimmune precipitation assay buffer containing a 1% protease inhibitor mixture followed by centrifugation at 14,000 × g for 10 min. Supernatants were collected and separated on an 8% SDS-polyacrylamide gel followed by Western blotting as described (34, 35).
Measurement of Changes in B1R Intramolecular FRET or Bioluminescence Resonance Energy Transfer (BRET)
Intramolecular FRET or BRET between the fluorescein arsenical hairpin binder (FlAsH)-labeled CCPGCC in the B1R intracellular loop 3 and C-terminal CFP or Rluc was determined as described (18, 35, 42). Briefly, HEK cells stably expressing B1R-TC-CFP or B1R-TC-Rluc with or without CPM were incubated with 2 μm FlAsH-EDT2 for 30 min at 37 °C. After incubation, the cells were washed with 250 μm 2,3-dimercaptopropanol in buffer three times (5 min each) to remove nonspecific FlAsH binding, and then the cells were resuspended in Hanks' balanced salt solution. For detection of the change in FRET, the emission at 530 nm (FlAsH emission) was recorded continuously after stimulation with B1R agonists while exciting at 430 nm (Aminco Bowman Series 2 spectrofluorometer). The FRET change was expressed as Δ emission at 530 nm. For detection of BRET, luciferase substrate (6 μm ViviRen) was added, and the emissions from both FlAsH (500–600 nm) and bioluminescence (400–480 nm) were recorded on an Infinite M200 Pro (Tecan). The BRET change was calculated using net BRET after stimulation with B1R agonist minus the BRET before stimulation.
Binding Assay
Monolayers (80% confluent) of HEK cells stably expressing B1R with or without CPM or HLMVEC in 24-well plates were rinsed 3 times with PBS and incubated with 4 nm of [3H]DAKD or 100 nm DAKD (4 nm [3H]DAKD + 96 nm cold DAKD) for 90 min on ice. The incubation was stopped by rinsing the cells 3 times with ice-cold PBS which were then lysed in 0.4 ml of 0.2 m NaOH and transferred to a liquid scintillation vial. The bound [3H]DAKD was measured in a Beckman liquid scintillation counter (41).
Determination of CPM Activity
CPM activity was measured using dansyl-Ala-Arg substrate as described (38, 43) with modifications for measurement in live cells (34, 35). The activity of CPM is expressed as fluorescent units/min/106 cells.
Statistical Analysis
Data are expressed as the means ± S.E. Statistical analysis was performed using Student's t test (GraphPad Prism 5.0). Values of p < 0.05 were considered significant.
RESULTS
CPM Enhances B1R Signaling at Submaximal Agonist Concentration
Based on our previous work, CPM heterodimerizes with the B1R on the membrane, and this interaction enhances B1R signaling in two ways (29, 34, 35). First, the B2R agonists BK or KD can activate B1R signaling by binding to CPM as substrates, resulting in a conformational change in the B1R mediated by CPM/B1R protein-protein interactions and subsequent downstream activation of calcium or nitric oxide (NO) signaling (29, 35). Second, removal of the C-terminal Arg of BK or KD by CPM generates B1R agonists (e.g. des-Arg9-BK or DAKD) that can further activate the associated receptor or additional B1Rs (29, 34).
In the present study we investigated whether basal CPM/B1R interactions could affect B1R signaling stimulated by B1R agonists (which are not CPM substrates). Indeed, we found CPM co-expression enhanced the B1R-dependent increase in intracellular Ca2+ ([Ca2+]i) in response to agonist stimulation with DAKD (Fig. 1A). This resulted from enhanced sensitivity of the B1R to DAKD as evidenced by a leftward shift in the dose-response curve without an increase in the maximal response (Fig. 1B). The EC50 of DAKD for inducing B1R-dependent increase in [Ca2+]i was reduced from 3.6 nm in the HEK cells stably expressing B1R alone to 1.3 nm in stably expressing B1R and CPM. The affinity of the B1R for DAKD was about 5-fold higher in HEK cells stably expressing B1R and CPM than that in cells stably expressing the B1R alone (Fig. 1C). To exclude the possibility that the effect was due to increased B1R expression in the stably transfected cells, CPM was transiently transfected into cells stably expressing B1R alone. After transfection, CPM was well expressed and had comparable activity (Fig. 1D) with cells stably expressing B1R and CPM (34). In the transiently transfected cells, CPM increased B1R-dependent PI turnover, reducing the EC50 of DAKD from 13.1 to 1.9 nm after CPM transfection (Fig. 1E). We also tested the effect of increasing amounts of CPM cDNA transiently transfected into cells stably expressing B1R, which resulted in a dose-dependent increase in CPM expression (Fig. 2A) and activity (Fig. 2B). Transfection of the control pcDNA3 vector had no effect on basal expression or activity of CPM (Fig. 2, A and B). Moreover, CPM transfection did not affect B1R expression in the stably transfected cells because the maximal binding of B1R agonist [3H]DAKD was the same in cells transfected with increasing doses of CPM cDNA or pcDNA3 vector control (Fig. 2C). Increasing CPM expression resulted in a corresponding enhancement of B1R-dependent PI turnover with submaximal (10 nm DAKD) B1R agonist stimulation (Fig. 2D). In contrast, CPM expression had no effect on PI turnover stimulated with saturating levels of DAKD (500 nm) (Fig. 2E). This is consistent with the lack of effect of CPM on maximal binding of B1R agonist (Fig. 2C), maximal B1R-dependent calcium signaling (Fig. 1B), or PI turnover (Fig. 1E). In contrast, when increasing doses of B1R cDNA were transfected into cells stably expressing CPM, saturation binding of B1R with its agonist was correspondingly elevated as expected (Fig. 3A). The expression of CPM was not affected at any level of B1R transfection (Fig. 3B). However, stable expression of CPM enhanced the increase in PI turnover in response to 10 nm DAKD when increasing amounts of B1R cDNA were transiently transfected (Fig. 3C). Taken together, these data indicate that CPM allosterically enhances agonist affinity and B1R signaling at submaximal agonist doses.
FIGURE 1.

CPM enhances B1R signaling and agonist binding affinity. A and B, HEK cells stably co-expressing B1R and CPM or B1R alone were stimulated with various doses of B1R agonist DAKD, and the increase in [Ca2+]i was recorded. Representative tracings are shown in A, and the increase in [Ca2+]i at different doses was quantified (B) as described under “Experimental Procedures.” The data are representative results from three experiments. C, binding of various concentrations of [3H]DAKD to HEK cells stably expressing B1R with or without CPM was determined as described under “Experimental Procedures,” and binding constants were determined by a non-linear fit of one site binding using Prism 5.0. The Kd is expressed as the mean ± S.E. (n = 3). *, p < 0.05 versus B1R (Student's t test). D, HEK cells stably expressing B1Rs were transfected with plasmid encoding the cDNA of CPM or empty vector (pcDNA3), and after 24 h the expression and activity of CPM was determined. The inset is representative of CPM expression from three experiments, and the activity is expressed as the mean ± S.E. (n = 3). *, p < 0.05 versus pcDNA3 (Student's t test). E, the dose-dependent increase in PI turnover in response to DAKD was measured in HEK cells transfected as in D. Data shown are mean ± S.E. (n = 3).
FIGURE 2.

CPM expression increases B1R-dependent PI turnover at low agonist concentration. A and B, HEK cells stably expressing B1Rs were transfected for 24 h with increasing concentrations of CPM cDNA or empty vector (pcDNA3), and the expression (A) and activity (B) of CPM were measured. Cells transfected as in A and B were used to determine the effect of CPM on B1R binding of 100 nm DAKD (C) or B1R-dependent PI turnover in response to 10 nm DAKD (D) or 500 nm DAKD (E). The solid bar denotes basal PI turnover without DAKD stimulation. In C, DALKD = binding in the presence of 1 μm des-Arg10-Leu9-kallidin (B1R antagonist). The data in A, D, and E are representative examples from three experiments, and in B and C are expressed as the mean ± S.E. (n = 3).
FIGURE 3.

CPM enhances low dose DAKD-stimulated PI turnover at different levels of B1R expression. Control HEK cells or HEK cells stably expressing CPM were transfected with increasing concentrations of B1R cDNA for 24 h, and B1R binding to 100 nm DAKD (4 nm [3H]DAKD + 96 nm cold DAKD) (A) and CPM expression (B) were determined. C, B1R-dependent PI turnover was measured in response to 10 nm DAKD in cells transfected as in A. The data in A and C are expressed as mean ± S.E. (n = 3) and in B are representative from three experiments.
CPM Does Not Alter B1R Constitutive Activity and Is Not a Direct B1R Agonist
The B1R was previously reported to exhibit constitutive activity (44). One way CPM could enhance B1R signaling is by altering this basal level of activity. To investigate this, various doses of B1R cDNA were transfected into control HEK cells or HEK cells stably expressing CPM. In B1R-transfected cells, basal PI turnover increased with transfection of increasing concentrations of B1R cDNA but not in control vector pcDNA3-transfected cells (Fig. 4A), consistent with previous reports (44). However, expression of CPM did not alter basal PI turnover at increasing doses of B1R cDNA (Fig. 4A). Furthermore, the basal PI turnover was not affected in cells stably expressing B1R after transfection with increasing doses of CPM cDNA or vector control, but PI turnover was similarly elevated by stimulation with saturating (500 nm) B1R agonist DAKD in both cases (Fig. 4B). We further explored whether CPM could directly activate the B1R as a protein agonist. To determine whether soluble CPM could bind to the B1R, cells were incubated with purified, soluble (lacking the GPI anchor) rCPM (500 ng/ml) for 30 min, washed, and then lysed and blotted for CPM. The baculovirus-expressed rCPM can be distinguished from membrane-bound CPM expressed in HEK cells because of its lower molecular mass due to reduced glycosylation in insect cells (5, 39). rCPM bound to cells stably expressing B1Rs but not to control HEK cells or cells stably expressing CPM (Fig. 4C), indicating it can bind to the B1R. However, rCPM (500 ng/ml) did not induce a detectable increase in [Ca2+]i in HEK cells stably expressing the B1R (Fig. 4D). Thus, soluble rCPM does not act as a direct protein agonist, and CPM expression does not alter the constitutive activity of the B1R.
FIGURE 4.

CPM does not alter constitutive B1R-dependent PI turnover and is not a direct B1R agonist. A, control HEK cells or HEK cells stably expressing CPM were transfected with various concentrations of B1R cDNA or pcDNA3 vector for 24 h and basal PI turnover was measured. B, HEK cells stably expressing B1R were transfected with increasing concentrations of CPM cDNA or empty vector pcDNA3 for 24 h transfection, and basal PI turnover was measured as well as 500 nm DAKD-stimulated PI turnover at the highest CPM dose. C, control HEK cells or cells stably expressing B1R or CPM were incubated with 500 ng/ml rCPM and washed, and the rCPM attached to cells was detected by Western blotting. D, HEK cells stably expressing B1R were stimulated with 500 ng/ml rCPM and then 1 μm DAKD (positive control), and the increase in [Ca2+]i was recorded. The data in A and B are expressed as the mean ± S.E. (n = 3) and in C and D are representative examples from three experiments.
The Effect of CPM on B1R Signaling Is Independent of Its Enzyme Activity and Substrate Specificity
To determine whether CPM catalytic activity was required for its basal enhancement of B1R signaling, we made use of a CPM mutant (E264Q) that is catalytically inactive but still binds substrates, which we have characterized before (34, 39). In HEK cells stably expressing B1Rs and CPM-E264Q, the dose-response curve for the increase in [Ca2+]i in response to DAKD was shifted to the left compared with cells expressing only B1R (Fig. 5A) as we found with wtCPM (Fig. 1B). The EC50 for DAKD decreased from 21.9 nm in cells stably expressing B1Rs alone to 5.5 nm in cells stably expressing B1R and CPM-E264Q (Fig. 5A).
FIGURE 5.

CPM enhances B1R signaling independent of its enzyme activity and substrate specificity. The dose-dependent increase in [Ca2+]i stimulated by DAKD was measured in HEK cells stably expressing B1R alone or B1R with CPM-E264Q, a mutant that lacks enzyme activity (A), CPM S180N, an active mutant that reverses the preference of CPM for cleaving C-terminal Arg to Lys (B), and CPM S180N E264Q, a mutant with Lys specificity for substrate binding, but without enzyme activity (C). The data shown are representative examples from three experiments.
CPM specifically cleaves only C-terminal Arg or Lys from peptides but has a clear preference for Arg (12, 38). We explored whether CPM substrate preference is associated with its enhancement of B1R signaling by mutating Ser-180 to Asn, which we previously determined to decrease the kcat/Km for C-terminal Arg substrate by ∼100-fold and increased that for C-terminal Lys by ∼2-fold4 (35). HEK cells stably co-expressing B1R and CPM-S180N also showed an increase in the B1R-dependent calcium response induced by stimulation with various doses of DAKD compared with cells expressing B1Rs alone (Fig. 5B). The corresponding EC50 for DAKD decreased from 18.6 nm in cells stably expressing B1R alone to 3.2 nm in cells stably expressing B1R and CPM-S180N (Fig. 5B). We further examined the effect of a CPM-S180N/E264Q double mutant on B1R signaling and found that it also enhanced the B1R-dependent increase in [Ca2+]i induced by DAKD (Fig. 5C), reducing the EC50 for DAKD from 17.4 nm in cells stably expressing B1R alone to 4.7 nm in cells stably expressing B1R and CPM-S180N/E264Q. These results indicate that the CPM-mediated enhancement of B1R signaling does not require binding or cleavage of a peptide substrate by CPM.
CPD-DIII or GPI-anchored CPN Catalytic Subunit Do Not Regulate B1R Signaling
We investigated whether CPD, another membrane-anchored carboxypeptidase, could enhance B1R signaling. Human CPD consists of 3 carboxypeptidase domains with sequence identity to CPM ranging from 27 to 45% (45). Domains I and II have enzymatic activity, but domain III (DIII) does not because the residue equivalent to the catalytic Glu-264 in CPM is Tyr (46, 47). Because CPD-DIII contains the C-terminal transmembrane anchor and CPM enhancement of B1R signaling did not require enzyme activity, we investigated whether the CPD-DIII could enhance B1R signaling. As shown in Fig. 6A, the dose-response curve to DAKD in stimulating a B1R-dependent increase in [Ca2+]i was identical in cells stably expressing the B1R or stably co-expressing the B1R and CPD-DIII. This lack of effect is consistent with our previous finding that the B1R and CPD-DIII do not interact or co-immunoprecipitate when co-expressed in HEK cells (35). This could be due to membrane microdomain localization of transmembrane CPD-DIII that differs from CPM, which is localized in lipid raft membrane microdomains by virtue of its GPI anchor (8, 34). We previously showed this membrane microdomain localization was important because CPM/B1R interaction was reduced when lipid rafts were disrupted with methyl-β-cyclodextrin (34). To determine whether a related carboxypeptidase localized in lipid rafts might also enhance B1R signaling, we generated a GPI-anchored version of the CPN catalytic domain (CPN-GPI) containing the C-terminal GPI anchor signal sequence of CPM. We showed that CPM and the catalytic subunit of CPN share 41% sequence identity (4) and have similar overall three-dimensional structures (5, 48). Although wtCPN is secreted into the medium when expressed in HEK cells, CPN-GPI was expressed in the cell membrane as expected (data not shown). However, CPN-GPI did not enhance the B1R-dependent increase in [Ca2+]i induced by DAKD stimulation when stably co-expressed with B1R (Fig. 6B). Thus lipid raft localization of a carboxypeptidase is not sufficient to enhance B1R signaling, which involves a specific interaction site(s) in CPM not shared by CPN.
FIGURE 6.

CPD-DIII or GPI-anchored CPN catalytic subunit do not enhance B1R signaling. The dose-dependent increase in [Ca2+]i stimulated by DAKD was measured in HEK cells stably expressing B1R alone or B1R with CPD-DIII, the third carboxypeptidase domain of human CPD containing the transmembrane anchor (A), or CPN-GPI, a GPI-anchored form of the 50-kDa active catalytic subunit of CPN (B). The data shown are representative of three experiments.
Extracellular Loop 2 of the B1R and a CPM C-terminal Domain Epitope Are Involved in the Allosteric Enhancement of B1R Signaling
Our previous studies showed that CPM or CPM-E264Q and B1R form a complex on the cell membrane (29, 34, 35). Thus we further investigated which domains of CPM and B1R contributed to the allosteric interaction. Because CPM is a GPI-anchored ectoenzyme, it must interact with the B1R extracellular domain. There is accumulating evidence that a major site for allosteric regulation of GPCRs is in their second extracellular loop (EL2) (49–51). To investigate this we replaced the B1R EL2 with the B2R EL2 to generate chimeric receptor B1R(B2R-EL2) (Fig. 7A) as we found that CPM does not interact with the B2R as determined by co-immunoprecipitation (Fig. 7I). As a control, another chimeric receptor B1R(B2R-EL1) was generated by replacing EL1 of the B1R with the corresponding B2R sequence (Fig. 7B). Stimulation of HEK cells stably expressing either B1R(B2R-EL2) or B1R(B2R-EL1) with DAKD resulted in a similar dose-dependent increase in PI turnover (Fig. 7, C and D). However, whereas co-expression of CPM shifted the does-response curve to the left in cells expressing B1R(B2R-EL1) (Fig. 7D), it had no effect in cells expressing B1R(B2R-EL2) (Fig. 7C). Transfection of increasing amounts of CPM cDNA into cells stably expressing the chimeric constructs (which increased CPM expression and activity; not shown) increased submaximal binding of DAKD and DAKD-dependent PI turnover in HEK cells stably expressing B1R(B2R-EL1) (Fig. 7, F and H) similar to wild type B1R but not in cells expressing B1R(B2R-EL2) (Fig. 7, E and G). Consistent with these functional results, CPM could not form a complex with the B1R(B2R-EL2) as shown by a lack of co-immunoprecipitation, whereas the B1R(B2R-EL1) chimera co-immunoprecipitated with CPM as well as the wtB1R (Fig. 7I). Thus, EL2 of B1R plays a key role in the allosteric interaction of B1R with CPM.
FIGURE 7.

Extracellular loop 2 of the B1R is important for CPM allosteric enhancement of B1R signaling. B1R chimeras were generated by replacing EL2 or EL1 of B1R with the corresponding B2R sequence as shown schematically in B1R(B2R-EL2) (A) and B1R(B2R-EL1) (B) where the dashed line represents the replaced sequence. HEK cells stably expressing B1R(B2R-EL2) (C) or B1R(B2R-EL1) (D) were transfected with 0.30 μg of CPM cDNA or empty vector pcDNA3 for 24 h, and the dose-dependent increase in PI turnover stimulated by DAKD was measured. E and F, HEK cells stably expressing B1R(B2R-EL2) (E) or B1R(B2R-EL1) (F) were transfected with increasing concentrations of CPM cDNA or empty vector pcDNA3 for 24 h, and the binding of 4 nm [3H]DAKD was measured. G and H, HEK cells stably expressing B1R(B2R-EL2) (G) or B1R(B2R-EL1) (H) were transfected with increasing concentrations of CPM cDNA or empty vector pcDNA3 for 24 h transfection, and then PI turnover was assessed. I, B1R interaction with CPM depends on EL2. Control HEK cells or HEK cells stably expressing B1R, B2R, B1R(B2R-EL1), or B1R(B2R-EL2) were transfected with 0.30 μg of CPM cDNA for 24 h, and then the B1R was immunoprecipitated (IP), and precipitates were analyzed for CPM by Western blotting (IB). The data shown are representative of three experiments.
We previously found that a CPM monoclonal antibody specific for an 11-amino acid epitope (residues 302–312) on the CPM C-terminal domain and a peptide (“CT peptide”) containing this epitope (Ac-KGQVFDQNGNPLPN-NH2) both disrupted the B1R-CPM interaction and reduced CPM-dependent B1R signaling to BK or KD (34, 35). We thus investigated the effect of CT peptide on CPM allosteric enhancement of B1R-dependent ERK activation by DAKD. As shown in Fig. 8, A and B, in HEK cells stably expressing B1R and CPM the CT peptide decreased ERK1/2 phosphorylation, shifting the dose-response curve for DAKD to the right. The EC50 for ERK1/2 phosphorylation induced by DAKD increased from 0.9 to 6.6 nm in the presence of the CT peptide. The CT peptide also shifted the dose-response curve for an DAKD-induced increase in [Ca2+]i to the right in cells co-expressing B1R and CPM (Fig. 8C), increasing the EC50 from 3.1 nm to 10.3 nm, but had no effect on the response in cells stably expressing B1R alone (Fig. 8D). Thus, the CPM C-terminal domain containing residues 299–312 is involved in the allosteric interaction between B1R and CPM.
FIGURE 8.

Disruption of CPM/B1R interaction with CT peptide decreases B1R-dependent signaling. HEK cells stably expressing B1R and CPM were incubated without or with 50 μm CT peptide for 10 min. Cells were stimulated with various concentrations of B1R agonist DAKD for 5 min, and total and phospho-ERK1/2 were determined by Western blotting (A). Bands were quantified by densitometry, and phospho-ERK1/2 values were normalized to the density of total ERK1/2 (B). HEK cells stably co-expressing B1R and CPM (C) or B1R alone (D) were incubated without or with 50 μm CT peptide for 10 min after Fura-2/AM loading, and B1R-dependent increase in [Ca2+]i was recorded in response to various concentrations of DAKD. The data are representative of three experiments.
Evidence for an Allosteric Interaction between CPM and B1R in Primary Cells
CPM is present in human endothelial cells, and inflammatory cytokines can increase its expression 2–3-fold, conditions that also up-regulate B1R expression (6, 14, 31–33, 52). We thus used cytokine-treated HLMVEC to examine the allosteric effect of CPM on B1R function. We confirmed that CPM was expressed and active in cytokine-treated HLMVEC (Fig. 9, A and B) as was the B1R as indicated by [3H]DAKD binding (Fig. 9C). The CT peptide and CPM monoclonal antibody that disrupt CPM/B1R interaction significantly decreased B1R binding of DAKD at submaximal concentrations (4 nm) but had minimal effect on DAKD binding at a saturating concentration (100 nm) (Fig. 9C). The CT peptide and CPM monoclonal antibody did not reduce CPM activity or expression (Fig. 9, A and B). As an alternate approach, we knocked down CPM expression using siRNA. CPM activity and expression were significantly decreased in cytokine-treated HLMVEC 24 h after transfection with CPM siRNA compared with negative control siRNA (Fig. 9, D and E). The non-saturated binding of [3H]DAKD (4 nm) with B1R was decreased by ∼50% in CPM siRNA-treated cells, whereas binding of saturating concentrations of DAKD (100 nm) was not affected (Fig. 9D). These data indicate that CPM is a positive allosteric modulator of B1R function in primary cells at native expression levels.
FIGURE 9.

CPM enhances B1R binding of its agonist at low concentration in primary human endothelial cells. HLMVEC were pretreated with 5 ng/ml IL-1β and 200 units/ml IFN-γ for 16 h (“cytokine-treated”) and then incubated with 50 μm CT peptide or 500 ng/ml CPM monoclonal antibody for 90 min. CPM expression (A) and activity (B) were determined. Inhibition by MGTA (dl-2-mercaptomethyl-3-guanidinoethylthiopropanoic acid) was used as a positive control. C, cytokine-treated HLMVEC were then incubated with 50 μm CT peptide or 500 ng/ml CPM monoclonal antibody and 4 nm [3H]DAKD or 100 nm DAKD (4 nm [3H]DAKD+ 96 nm cold DAKD) for 90 min. Cells were washed, and specific binding was determined as described under “Experimental Procedures.” The data are expressed as % of control binding without CT-peptide or antibody. D and E, HLMVEC were transfected with specific CPM siRNA or nonspecific control (NC) siRNA using an Amaxa nucleofector. After 48 h cells were cytokine-treated as above, and CPM expression (D) and activity (E) were determined as well as the binding of B1R with its agonist, 4 nm [3H]DAKD or 100 nm DAKD (4 nm [3H]DAKD + 96 nm cold DAKD), for 90 min (F). The data in B, C, E, and F are expressed as the mean ± S.E. (n = 3). * = p < 0.05 versus control (Student's t test). The data in A and D are representative of three experiments.
CPM Enhances B1R Conformational Change in Response to Agonist
GPCR-positive allosteric modulators that cause a leftward shift in the dose-response curve can do so by increasing agonist affinity and/or by lowering the energy barrier for transition from the inactive to active state by stabilizing an intermediate receptor conformation (49, 53, 54). To investigate the possible effect of CPM on B1R receptor conformation, we utilized an intramolecular FRET approach (55, 56) that was previously used to investigate the activation-dependent conformational change in the B1R by us (35). A tetracysteine motif was inserted into intracellular loop 3 that binds the FRET acceptor, a small molecule called FlAsH, and CFP (B1R-TC-CFP) was attached to the C terminus as the FRET donor. As we found previously, the addition of 10 nm DAKD increased the intramolecular FRET in HEK cells stably expressing B1R-TC-CFP alone (Fig. 10A), consistent with a decrease in the distance between intracellular loop 3 and the C terminus. Co-expression of CPM did not alter basal FRET but significantly increased the FRET induced by 10 nm DAKD (Fig. 10A). We further confirmed the results using intramolecular BRET by using B1R-TC-Rluc, which was generated by replacing GFP with Renilla luciferase. DAKD increased the intramolecular BRET in the HEK cells stably expressing B1R-TC-Rluc alone (Fig. 10B). CPM co-expression had no effect on basal BRET of B1R-TC-Rluc but further increased the BRET induced by DAKD in the same cells (Fig. 10B). These data indicate that basal binding of CPM does not induce a conformational change in the B1R toward a more activated state, consistent with the lack of effect of CPM expression on constitutive activity of the B1R (Fig. 4, A and B). However, CPM did enhance the conformational change induced with sub-saturating concentrations of DAKD, consistent with our finding of enhanced agonist affinity (Fig. 1).
FIGURE 10.

CPM increased agonist-mediated conformational change of the B1R. A, HEK cells stably expressing B1R-TC-CFP alone or stably co-expressing CPM were stimulated with 10 nm DAKD, and the change in intramolecular FRET was determined as described under “Experimental Procedures.” FU, fluorescent units. B, HEK cells stably expressing B1R-TC-Rluc alone or stably co-expressing CPM were stimulated with 10 nm DAKD, and the change in net BRET was determined. The data are expressed as the mean ± S.E. (n = 3). * = p < 0.05 versus Vehicle; # = p < 0.05 versus control without CPM (Student's t test).
DISCUSSION
Cell surface peptidases regulate peptide signaling by activating, modulating, or inactivating peptide hormones via the cleavage of specific peptide bonds. For example, BK and KD released from kininogen are B2R agonists, and carboxypeptidase-mediated removal of their C-terminal Arg residue inactivates them as B2R agonists while simultaneously generating B1R agonists (6, 11, 14). Although B1R agonists can potentially be produced by several carboxypeptidases in various cellular and subcellular locations (6, 11, 14), if the site of generation is not close to the receptor, the agonist concentration will be reduced by diffusion and short peptide half-life due to exposure to cellular and blood proteases and filtration by the kidney. Thus, peptide agonist generation in close proximity to its corresponding receptor is most likely to lead to a meaningful cellular response. Indeed, we found that co-expression of CPM and B1R in the same cell results in their assembly into a functional protein complex critical for efficient generation of a B1R signal when low concentrations of B2R agonists BK or KD are applied to the cell (34). However, co-expression in the same cell alone was not sufficient for the most robust signaling response as disruption of the CPM/B1R interaction (without affecting the expression level or activity of the two proteins in the same cell) reduced the response (34). Although generation of B1R agonist in close proximity to the receptor is undoubtedly important for this effect (Fig. 11), we found a second, novel mechanism by which CPM enhances B1R signaling. Substrate binding to CPM to causes a conformational change and activation of the B1R via protein-protein interaction without generation of B1R agonist (29, 35) (Fig. 11). Thus, cells co-expressing B1R and a catalytically inactive CPM mutant (E2264Q) that still binds substrate gave a B1R response to B2R agonists BK and KD which was affected by CPM mutations that alter substrate binding affinity (35). In the present study we found a third mechanism by which CPM enhances B1R signaling (Fig. 11), acting as a positive allosteric modulator of B1R signaling stimulated by B1R agonists, which are not CPM substrates.
FIGURE 11.
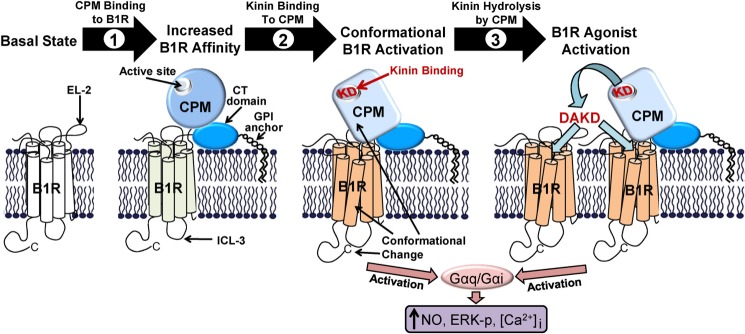
Schematic diagram showing the ways in which CPM interacts with the B1R to facilitate kinin signaling. A model of GPI-anchored CPM and its potential membrane orientation based on its crystal structure and interactions with the B1R are shown. Based on our studies, CPM enhances B1R signaling in three ways; 1) basal binding of CPM allosterically enhances B1R affinity for its des-Arg-kinin agonist, 2) kinin substrate (i.e. BK or KD) binding to the CPM active site causes a conformational change in CPM that is transmitted via protein-protein interaction to the B1R, resulting in G protein coupling and activation of calcium, nitric oxide (NO), or ERK signaling, and 3) cleavage of the C-terminal Arg of KD (or BK) by CPM generates B1R agonist (e.g. DAKD) that can further activate the associated receptor or additional B1Rs.
GPCRs already represent targets for ∼40% of all Food and Drug Administration-approved drugs, but these agents typically bind to the orthosteric binding site, which due to similarities between different receptor subtypes, limits their specificity (36, 37, 57, 58). Allosteric modulators of GPCR function are becoming popular targets for drug development because they represent novel agents with potential advantages over current drugs targeting orthosteric binding sites. For example, allosteric modulators that lack agonistic activity only have effects in the presence of endogenous orthosteric agonists, thus maintaining the appropriate timing and localization of physiological signaling and limiting toxicity that might occur with agents interacting directly with the orthosteric site (53, 59, 60). In addition, allosteric agents that interact with the extracellular surface, where there is little sequence identity between receptors, can exhibit enhanced specificity for receptor subtypes and can alter coupling between the extracellular, transmembrane, and intracellular domains to affect a subset of a GPCR downstream pathways leading to more specific biased signaling (53, 60). Allosteric modulators fall into four main categories (53); positive allosteric modulators that enhance receptor signaling, negative allosteric modulators that inhibit receptor function, allosteric agonists that can stimulate receptor signaling in the absence of another agonist, and silent allosteric modifiers that have no effect on their own but can compete for the same allosteric binding site and inhibit the effect of other allosteric agents. Most positive allosteric modulators enhance GPCR function by causing a leftward shift in the dose-response curve to agonist through enhanced agonist affinity and/or by lowering the energy barrier for transition to the active state by stabilizing an intermediate receptor conformation, whereas some increase the maximal response (53). We found that the positive allosteric effect of CPM was caused by a leftward shift in the B1R dose-response curve to DAKD mediated by a ∼5-fold increase in agonist affinity for the receptor without affecting the maximal response. This is consistent with our finding of increased intramolecular FRET and BRET in our B1R-TC-CFP or B1R-TC-Rluc constructs co-expressed with CPM in response to a sub-saturating dose of agonist DAKD. The lack of effect of CPM expression on basal FRET or BRET in B1R-TC-CFP or B1R-TC-Rluc without agonist would argue against CPM, causing a change in basal receptor conformation to a more activated state. However, we can not rule out the possibility that CPM does cause a change in conformation in the extracellular domain sufficient to lower the transition energy barrier that does not change the distance between intracellular loop 3 and the C terminus that would be required to see a change in basal FRET or BRET.
Investigations of allosteric modulators of GPCR signaling have primarily focused on the effects of synthetic small molecules (36, 53, 60). The only well studied examples of endogenous allosteric regulation by membrane proteins are GPCRs that form homo- or heterodimers and receptor activity-modifying proteins 1, 2, and 3, whose coexpression and binding interaction determine the agonist specificity of the calcitonin and calcitonin receptor-like GPCRs (59, 61). CPM is unique in this regard as it can allosterically enhance and stimulate B1R signaling as well as generate the endogenous orthosteric agonist as described above. In addition, as it is membrane-bound by a GPI anchor, the protein portion of CPM exists in the extracellular space and would thus have to interact with an extracellular domain(s) of the B1R to enhance receptor function (Fig. 11). It is now clear that conformational coupling between GPCR extracellular domains, transmembrane orthosteric binding sites, and intracellular domains is essential for activation; thus, agents that interact with a GPCR extracellular domains can alter receptor signaling (51, 57, 62, 63). Of the four extracellular domains of GPCRs, EL-2 is a prime target for allosteric modulators (50) as supported by the following considerations. Typically, EL-2 is the longest and most structured extracellular loop in family A GPCRs (63). In addition, a highly conserved disulfide bond found in most GPCRs links EL-2 to the top of transmembrane domain (TM)3 (Cys-110–Cys-189 in the B1R), stabilizing the conformation and allowing binding-induced conformational changes of EL-2 to be transmitted to TM3, an important structural and functional hub due to its interactions with other TM domains, ligands, G proteins, and ICL2 (51, 57, 62, 63). EL-2 is also the site of autoantibody binding to GPCRs, resulting in persistent activation in a variety of autoimmune diseases (64), and mutations in EL-2 can cause GPCRs to become constitutively active, indicating it can stabilize the inactive conformation of the receptor in the basal state (51, 65). EL-2 can also affect orthosteric ligand binding by directly interacting with the ligand and/or regulating access or egress of orthosteric ligands to the transmembrane binding pocket by forming a “lid” structure (51, 63). Consistent with these findings, we found that the B1R EL-2 was important for the allosteric effect of CPM to be manifested. Replacement of the B1R EL-2 with that of the B2R, with which CPM does not interact, eliminated the ability of CPM to enhance B1R function. Furthermore, disruption of the CPM/B1R interaction by the CPM CT peptide or mAb indicates that interaction of the B1R EL-2 with the C-terminal domain of CPM is important for this effect.
Activation of the B1R is typically associated with the harmful effects of chronic inflammation; however, it can also be beneficial in various disease states. For example, B1R stimulation enhances inflammation and fibrosis in diabetic cardiomyopathy (66) and B1R knock-out protects mice from lipopolysaccharide-induced hypotension and reduces pain in response to thermal or chemical stimuli (67). Conversely, B1R activation is protective in renal ischemia/reperfusion injury (68), reduces renal fibrosis and cardiac remodeling (69, 70), and promotes neovascularization and angiogenesis during wound healing (67, 71). B1R signaling also contributes to the beneficial cardiovascular effects of ACE inhibitors and angiotensin type 1 receptor blockers (72, 73). Many times pathological effects are seen with high, uncontrolled activation of GPCRs, whereas lower levels of physiological signaling are beneficial. Thus, the typical drug development approach to inhibit all receptor signaling with an antagonist or inverse agonist targeted to the orthosteric binding site may not be the best approach for maximum therapeutic effectiveness. A potential major advantage of allosteric antagonists is the ability to dampen receptor signaling underlying the pathological response while maintaining physiological signaling (53, 59, 60). One novel approach to develop allosteric antagonists of the B1R would be to design agents to inhibit the positive allosteric effects of CPM on B1R signaling by targeting the interaction site as we did with the CT peptide. This would eliminate the basal positive allosteric effect of CPM and its conformational activation of the B1R via substrate binding and reduce the local concentration of B1R agonist generated by CPM while maintaining a lower level of B1R activation, potentially preserving beneficial effects. A further level of reduction of B1R activity could be achieved by adding a specific CPM inhibitor to further reduce agonist generation without completely eliminating it from other sources. Because ACE inhibitors can also act as direct allosteric agonists of B1Rs (24, 74), disruption of the CPM/B1R interaction could potentially affect the therapeutic and/or side effects of these drugs as well.
In conclusion, we found that GPI-anchored CPM is an endogenous positive allosteric modulator of the kinin B1R in addition to its previously described ability to conformationally activate B1R signaling in response to substrate binding and efficiently generate the des-Arg-kinin B1R agonists from BK or KD in close proximity to the receptor (29, 34, 35). These functions depend on formation of a CPM/B1R complex that involves EL-2 of the B1R and the non-catalytic C-terminal domain of CPM. Thus, CPM plays a key role in facilitating B1R signaling in response to low endogenous levels of kinin peptides, and disruption of the CPM/B1R interaction could potentially provide a novel therapeutic approach to reducing pathological B1R signaling in chronic inflammatory conditions without completely abrogating basal signaling that could be beneficial.
Acknowledgment
We thank Svitlana Brovkovych for help with the CPM knockdown studies.
This work was supported, in whole or in part, by National Institutes of Health Grants RO1 DK41431 and PO1 HL60678.
F. Tan, P. A. Deddish, and R. A. Skidgel, unpublished information.
- CPM
- carboxypeptidase M
- rCPM
- recombinant CPM
- B1R
- kinin B1 receptor
- CFP
- cyan fluorescent protein
- BRET
- bioluminescence resonance energy transfer
- [Ca2+]i
- intracellular calcium concentration
- GPI
- glycosylphosphatidylinositol
- ACE
- angiotensin I converting enzyme
- GPCR
- G protein-coupled receptor
- DAKD
- des-Arg10-kallidin
- CT peptide
- Ac-KGQVFDQNGNPLPN-NH2
- dansyl-Ala-Arg
- 5-dimethylaminonaphthalene-1-sulfonyl-l-alanyl-l-arginine
- TC
- tetracysteine
- FlAsH
- fluorescein arsenical hairpin binder
- HLMVEC
- human lung microvascular endothelial cell
- CPD
- carboxypeptidase D
- DIII
- domain III
- CPN
- carboxypeptidase N
- PI
- phosphoinositide
- KD
- kallidin
- BK
- bradykinin
- IP3
- inositol triphosphate
- EL2
- extracellular loop 2.
REFERENCES
- 1. Zhang X., Skidgel R. (2013) in Handbook of Proteolytic Enzyme (Rawlings N. D., Salvesen G. S., eds) pp. 1357–1366, 3rd Ed., Academic Press, Oxford [Google Scholar]
- 2. Gomis-Rüth F. X. (2008) Structure and mechanism of metallocarboxypeptidases. Crit. Rev. Biochem. Mol. Biol. 43, 319–345 [DOI] [PubMed] [Google Scholar]
- 3. Li J., Rehli M., Timblin B., Tan F., Krause S. W., Skidgel R. A. (2002) Structure of the human carboxypeptidase M gene. Identification of a proximal GC-rich promoter and a unique distal promoter that consists of repetitive elements. Gene 284, 189–202 [DOI] [PubMed] [Google Scholar]
- 4. Tan F., Chan S. J., Steiner D. F., Schilling J. W., Skidgel R. A. (1989) Molecular cloning and sequencing of the cDNA for human membrane-bound carboxypeptidase M. Comparison with carboxypeptidases A, B, H, and N. J. Biol. Chem. 264, 13165–13170 [PubMed] [Google Scholar]
- 5. Reverter D., Maskos K., Tan F., Skidgel R. A., Bode W. (2004) Crystal structure of human carboxypeptidase M, a membrane-bound enzyme that regulates peptide hormone activity. J. Mol. Biol. 338, 257–269 [DOI] [PubMed] [Google Scholar]
- 6. Skidgel R. A., Erdös E. G. (1998) Cellular carboxypeptidases. Immunol. Rev. 161, 129–141 [DOI] [PubMed] [Google Scholar]
- 7. Reznik S. E., Fricker L. D. (2001) Carboxypeptidases from A to Z. Implications in embryonic development and Wnt binding. Cell. Mol. Life Sci. 58, 1790–1804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Deddish P. A., Skidgel R. A., Kriho V. B., Li X. Y., Becker R. P., Erdös E. G. (1990) Carboxypeptidase M in Madin-Darby canine kidney cells. Evidence that carboxypeptidase M has a phosphatidylinositol glycan anchor. J. Biol. Chem. 265, 15083–15089 [PubMed] [Google Scholar]
- 9. Li X. Y., Skidgel R. A. (1999) Release of glycosylphosphatidylinositol-anchored carboxypeptidase M by phosphatidylinositol-specific phospholipase C up-regulates enzyme synthesis. Biochem. Biophys. Res. Commun. 258, 204–210 [DOI] [PubMed] [Google Scholar]
- 10. Deiteren K., Hendriks D., Scharpé S., Lambeir A. M. (2009) Carboxypeptidase M. Multiple alliances and unknown partners. Clin. Chim. Acta 399, 24–39 [DOI] [PubMed] [Google Scholar]
- 11. Skidgel R. A. (1988) Basic carboxypeptidases. Regulators of peptide hormone activity. Trends Pharmacol. Sci. 9, 299–304 [DOI] [PubMed] [Google Scholar]
- 12. Skidgel R. A., Davis R. M., Tan F. (1989) Human carboxypeptidase M. Purification and characterization of a membrane-bound carboxypeptidase that cleaves peptide hormones. J. Biol. Chem. 264, 2236–2241 [PubMed] [Google Scholar]
- 13. Kuhr F., Lowry J., Zhang Y., Brovkovych V., Skidgel R. A. (2010) Differential regulation of inducible and endothelial nitric oxide synthase by kinin B1 and B2 receptors. Neuropeptides 44, 145–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Leeb-Lundberg L. M., Marceau F., Müller-Esterl W., Pettibone D. J., Zuraw B. L. (2005) International union of pharmacology. XLV. Classification of the kinin receptor family. From molecular mechanisms to pathophysiological consequences. Pharmacol. Rev. 57, 27–77 [DOI] [PubMed] [Google Scholar]
- 15. Talbot S., Chahmi E., Dias J. P., Couture R. (2010) Key role for spinal dorsal horn microglial kinin B1 receptor in early diabetic pain neuropathy. J. Neuroinflammation 7, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Su J. B. (2006) Kinins and cardiovascular diseases. Curr. Pharm. Des. 12, 3423–3435 [DOI] [PubMed] [Google Scholar]
- 17. Duka I., Duka A., Kintsurashvili E., Johns C., Gavras I., Gavras H. (2003) Mechanisms mediating the vasoactive effects of the B1 receptors of bradykinin. Hypertension 42, 1021–1025 [DOI] [PubMed] [Google Scholar]
- 18. Hillmeister P., Gatzke N., Dülsner A., Bader M., Schadock I., Hoefer I., Hamann I., Infante-Duarte C., Jung G., Troidl K., Urban D., Stawowy P., Frentsch M., Li M., Nagorka S., Wang H., Shi Y., le Noble F., Buschmann I. (2011) Arteriogenesis is modulated by bradykinin receptor signaling. Circ. Res. 109, 524–533 [DOI] [PubMed] [Google Scholar]
- 19. Merino V. F., Todiras M., Campos L. A., Saul V., Popova E., Baltatu O. C., Pesquero J. B., Bader M. (2008) Increased susceptibility to endotoxic shock in transgenic rats with endothelial overexpression of kinin B(1) receptors. J. Mol. Med. 86, 791–798 [DOI] [PubMed] [Google Scholar]
- 20. Pesquero J. B., Araujo R. C., Heppenstall P. A., Stucky C. L., Silva J. A., Jr., Walther T., Oliveira S. M., Pesquero J. L., Paiva A. C., Calixto J. B., Lewin G. R., Bader M. (2000) Hypoalgesia and altered inflammatory responses in mice lacking kinin B1 receptors. Proc. Natl. Acad. Sci. U.S.A. 97, 8140–8145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cayla C., Todiras M., Iliescu R., Saul V. V., Gross V., Pilz B., Chai G., Merino V. F., Pesquero J. B., Baltatu O. C., Bader M. (2007) Mice deficient for both kinin receptors are normotensive and protected from endotoxin-induced hypotension. FASEB J. 21, 1689–1698 [DOI] [PubMed] [Google Scholar]
- 22. Alhenc-Gelas F., Bouby N., Richer C., Potier L., Roussel R., Marre M. (2011) Kinins as therapeutic agents in cardiovascular and renal diseases. Curr. Pharm. Des. 17, 2654–2662 [DOI] [PubMed] [Google Scholar]
- 23. Potier L., Waeckel L., Vincent M. P., Chollet C., Gobeil F., Jr., Marre M., Bruneval P., Richer C., Roussel R., Alhenc-Gelas F., Bouby N. (2013) Selective kinin receptor agonists as cardioprotective agents in myocardial ischemia and diabetes. J. Pharmacol. Exp. Ther. 346, 23–30 [DOI] [PubMed] [Google Scholar]
- 24. Erdös E. G., Tan F., Skidgel R. A. (2010) Angiotensin I-converting enzyme inhibitors are allosteric enhancers of kinin B1 and B2 receptor function. Hypertension 55, 214–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tschöpe C., Spillmann F., Altmann C., Koch M., Westermann D., Dhayat N., Dhayat S., Bascands J. L., Gera L., Hoffmann S., Schultheiss H. P., Walther T. (2004) The bradykinin B1 receptor contributes to the cardioprotective effects of AT1 blockade after experimental myocardial infarction. Cardiovasc. Res. 61, 559–569 [DOI] [PubMed] [Google Scholar]
- 26. Vijayaraghavan K., Deedwania P. (2011) Renin-angiotensin-aldosterone blockade for cardiovascular disease prevention. Cardiol. Clin. 29, 137–156 [DOI] [PubMed] [Google Scholar]
- 27. Skidgel R. A. (1996) in Zinc Metalloproteases in Health and Disease (Hooper N. M., ed) pp. 241–283, Taylor & Francis Ltd., London [Google Scholar]
- 28. Skidgel R. A., Erdös E. G. (1998) in Pro-inflammatory and Anti-inflammatory Peptides.(Said S. I., ed)pp 459–476, Marcel Dekker, Inc., New York [Google Scholar]
- 29. Zhang X., Tan F., Brovkovych V., Zhang Y., Lowry J. L., Skidgel R. A. (2013) Carboxypeptidase M augments kinin B1 receptor signaling by conformational crosstalk and enhances endothelial nitric oxide output. Biol. Chem. 394, 335–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McGwire G. B., Skidgel R. A. (1995) Extracellular conversion of epidermal growth factor (EGF) to des-Arg53-EGF by carboxypeptidase M. J. Biol. Chem. 270, 17154–17158 [DOI] [PubMed] [Google Scholar]
- 31. Hadkar V., Sangsree S., Vogel S. M., Brovkovych V., Skidgel R. A. (2004) Carboxypeptidase-mediated enhancement of nitric oxide production in rat lungs and microvascular endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 287, L35–L45 [DOI] [PubMed] [Google Scholar]
- 32. Sangsree S., Brovkovych V., Minshall R. D., Skidgel R. A. (2003) Kininase I-type carboxypeptidases enhance nitric oxide production in endothelial cells by generating bradykinin B1 receptor agonists. Am. J. Physiol. Heart Circ. Physiol. 284, H1959–H1968 [DOI] [PubMed] [Google Scholar]
- 33. Schremmer-Danninger E., Offner A., Siebeck M., Roscher A. A. (1998) B1 bradykinin receptors and carboxypeptidase M are both up-regulated in the aorta of pigs after LPS infusion. Biochem. Biophys. Res. Commun. 243, 246–252 [DOI] [PubMed] [Google Scholar]
- 34. Zhang X., Tan F., Zhang Y., Skidgel R. A. (2008) Carboxypeptidase M and kinin B1 receptors interact to facilitate efficient B1 signaling from B2 agonists. J. Biol. Chem. 283, 7994–8004 [DOI] [PubMed] [Google Scholar]
- 35. Zhang X., Tan F., Brovkovych V., Zhang Y., Skidgel R. A. (2011) Cross-talk between carboxypeptidase M and the kinin B1 receptor mediates a new mode of G protein-coupled receptor signaling. J. Biol. Chem. 286, 18547–18561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wang C. I., Lewis R. J. (2013) Emerging opportunities for allosteric modulation of G-protein coupled receptors. Biochem. Pharmacol. 85, 153–162 [DOI] [PubMed] [Google Scholar]
- 37. Kenakin T. P. (2009) 7TM receptor allostery. Putting numbers to shapeshifting proteins. Trends Pharmacol. Sci. 30, 460–469 [DOI] [PubMed] [Google Scholar]
- 38. Tan F., Deddish P. A., Skidgel R. A. (1995) Human carboxypeptidase M. Methods Enzymol. 248, 663–675 [DOI] [PubMed] [Google Scholar]
- 39. Tan F., Balsitis S., Black J. K., Blöchl A., Mao J. F., Becker R. P., Schacht D., Skidgel R. A. (2003) Effect of mutation of two critical glutamic acid residues on the activity and stability of human carboxypeptidase M and characterization of its signal for glycosylphosphatidylinositol anchoring. Biochem. J. 370, 567–578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Marcic B., Deddish P. A., Skidgel R. A., Erdös E. G., Minshall R. D., Tan F. (2000) Replacement of the transmembrane anchor in angiotensin I-converting enzyme (ACE) with a glycosylphosphatidylinositol tail affects activation of the B2 bradykinin receptor by ACE inhibitors. J. Biol. Chem. 275, 16110–16118 [DOI] [PubMed] [Google Scholar]
- 41. Kalatskaya I., Schüssler S., Blaukat A., Müller-Esterl W., Jochum M., Proud D., Faussner A. (2004) Mutation of tyrosine in the conserved NPXXY sequence leads to constitutive phosphorylation and internalization, but not signaling, of the human B2 bradykinin receptor. J. Biol. Chem. 279, 31268–31276 [DOI] [PubMed] [Google Scholar]
- 42. Pfleger K. D., Seeber R. M., Eidne K. A. (2006) Bioluminescence resonance energy transfer (BRET) for the real-time detection of protein-protein interactions. Nat. Protoc. 1, 337–345 [DOI] [PubMed] [Google Scholar]
- 43. Deddish P. A., Skidgel R. A., Erdös E. G. (1989) Enhanced Co2+ activation and inhibitor binding of carboxypeptidase M at low pH. Similarity to carboxypeptidase H (enkephalin convertase). Biochem. J. 261, 289–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Leeb-Lundberg L. M., Kang D. S., Lamb M. E., Fathy D. B. (2001) The human B1 bradykinin receptor exhibits high ligand-independent, constitutive activity. Roles of residues in the fourth intracellular and third transmembrane domains. J. Biol. Chem. 276, 8785–8792 [DOI] [PubMed] [Google Scholar]
- 45. Tan F., Rehli M., Krause S. W., Skidgel R. A. (1997) Sequence of human carboxypeptidase D reveals it to be a member of the regulatory carboxypeptidase family with three tandem active site domains. Biochem. J. 327, 81–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Aloy P., Companys V., Vendrell J., Aviles F. X., Fricker L. D., Coll M., Gomis-Rüth F. X. (2001) The crystal structure of the inhibitor-complexed carboxypeptidase D domain II and the modeling of regulatory carboxypeptidases. J. Biol. Chem. 276, 16177–16184 [DOI] [PubMed] [Google Scholar]
- 47. Eng F. J., Novikova E. G., Kuroki K., Ganem D., Fricker L. D. (1998) gp180, a protein that binds duck hepatitis B virus particles, has metallocarboxypeptidase D-like enzymatic activity. J. Biol. Chem. 273, 8382–8388 [DOI] [PubMed] [Google Scholar]
- 48. Keil C., Maskos K., Than M., Hoopes J. T., Huber R., Tan F., Deddish P. A., Erdös E. G., Skidgel R. A., Bode W. (2007) Crystal structure of the human carboxypeptidase N (kininase I) catalytic domain. J. Mol. Biol. 366, 504–516 [DOI] [PubMed] [Google Scholar]
- 49. May L. T., Leach K., Sexton P. M., Christopoulos A. (2007) Allosteric modulation of G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 47, 1–51 [DOI] [PubMed] [Google Scholar]
- 50. Avlani V. A., Gregory K. J., Morton C. J., Parker M. W., Sexton P. M., Christopoulos A. (2007) Critical role for the second extracellular loop in the binding of both orthosteric and allosteric G protein-coupled receptor ligands. J. Biol. Chem. 282, 25677–25686 [DOI] [PubMed] [Google Scholar]
- 51. Unal H., Karnik S. S. (2012) Domain coupling in GPCRs. The engine for induced conformational changes. Trends Pharmacol. Sci. 33, 79–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Skidgel R. A., Johnson A. R., Erdös E. G. (1984) Hydrolysis of opioid hexapeptides by carboxypeptidase N. Presence of carboxypeptidase in cell membranes. Biochem. Pharmacol. 33, 3471–3478 [DOI] [PubMed] [Google Scholar]
- 53. Burford N. T., Watson J., Bertekap R., Alt A. (2011) Strategies for the identification of allosteric modulators of G-protein-coupled receptors. Biochem. Pharmacol. 81, 691–702 [DOI] [PubMed] [Google Scholar]
- 54. Canals M., Sexton P. M., Christopoulos A. (2011) Allostery in GPCRs. “MWC” revisited. Trends Biochem. Sci. 36, 663–672 [DOI] [PubMed] [Google Scholar]
- 55. Hoffmann C., Gaietta G., Bünemann M., Adams S. R., Oberdorff-Maass S., Behr B., Vilardaga J. P., Tsien R. Y., Ellisman M. H., Lohse M. J. (2005) A FlAsH-based FRET approach to determine G protein-coupled receptor activation in living cells. Nat. Methods 2, 171–176 [DOI] [PubMed] [Google Scholar]
- 56. Nakanishi J., Takarada T., Yunoki S., Kikuchi Y., Maeda M. (2006) FRET-based monitoring of conformational change of the β2 adrenergic receptor in living cells. Biochem. Biophys. Res. Commun. 343, 1191–1196 [DOI] [PubMed] [Google Scholar]
- 57. Venkatakrishnan A. J., Deupi X., Lebon G., Tate C. G., Schertler G. F., Babu M. M. (2013) Molecular signatures of G-protein-coupled receptors. Nature 494, 185–194 [DOI] [PubMed] [Google Scholar]
- 58. Jacoby E., Bouhelal R., Gerspacher M., Seuwen K. (2006) The 7 TM G-protein-coupled receptor target family. Chem. Med. Chem. 1, 761–782 [DOI] [PubMed] [Google Scholar]
- 59. Smith N. J., Milligan G. (2010) Allostery at G protein-coupled receptor homo- and heteromers. Uncharted pharmacological landscapes. Pharmacol. Rev. 62, 701–725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Conn P. J., Christopoulos A., Lindsley C. W. (2009) Allosteric modulators of GPCRs. A novel approach for the treatment of CNS disorders. Nat. Rev. Drug Discov. 8, 41–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ritter S. L., Hall R. A. (2009) Fine-tuning of GPCR activity by receptor-interacting proteins. Nat. Rev. Mol. Cell Biol. 10, 819–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bokoch M. P., Zou Y., Rasmussen S. G., Liu C. W., Nygaard R., Rosenbaum D. M., Fung J. J., Choi H. J., Thian F. S., Kobilka T. S., Puglisi J. D., Weis W. I., Pardo L., Prosser R. S., Mueller L., Kobilka B. K. (2010) Ligand-specific regulation of the extracellular surface of a G-protein-coupled receptor. Nature 463, 108–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wheatley M., Wootten D., Conner M. T., Simms J., Kendrick R., Logan R. T., Poyner D. R., Barwell J. (2012) Lifting the lid on GPCRs. The role of extracellular loops. Br. J. Pharmacol. 165, 1688–1703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Unal H., Jagannathan R., Karnik S. S. (2012) Mechanism of GPCR-directed autoantibodies in diseases. Adv. Exp. Med. Biol. 749, 187–199 [DOI] [PubMed] [Google Scholar]
- 65. Klco J. M., Wiegand C. B., Narzinski K., Baranski T. J. (2005) Essential role for the second extracellular loop in C5a receptor activation. Nat. Struct. Mol. Biol. 12, 320–326 [DOI] [PubMed] [Google Scholar]
- 66. Westermann D., Walther T., Savvatis K., Escher F., Sobirey M., Riad A., Bader M., Schultheiss H. P., Tschöpe C. (2009) Gene deletion of the kinin receptor B1 attenuates cardiac inflammation and fibrosis during the development of experimental diabetic cardiomyopathy. Diabetes 58, 1373–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pesquero J. B., Bader M. (2006) Genetically altered animal models in the kallikrein-kinin system. Biol. Chem. 387, 119–126 [DOI] [PubMed] [Google Scholar]
- 68. Kakoki M., McGarrah R. W., Kim H. S., Smithies O. (2007) Bradykinin B1 and B2 receptors both have protective roles in renal ischemia/reperfusion injury. Proc. Natl. Acad. Sci. U.S.A. 104, 7576–7581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Moniwa N., Agata J., Hagiwara M., Ura N., Shimamoto K. (2006) The role of bradykinin B1 receptor on cardiac remodeling in stroke-prone spontaneously hypertensive rats (SHR-SP). Biol. Chem. 387, 203–209 [DOI] [PubMed] [Google Scholar]
- 70. Hagiwara M., Murakami H., Ura N., Agata J., Yoshida H., Higashiura K., Shimamoto K. (2004) Renal protective role of bradykinin B1 receptor in stroke-prone spontaneously hypertensive rats. Hypertens. Res. 27, 399–408 [DOI] [PubMed] [Google Scholar]
- 71. Emanueli C., Bonaria Salis M., Stacca T., Pintus G., Kirchmair R., Isner J. M., Pinna A., Gaspa L., Regoli D., Cayla C., Pesquero J. B., Bader M., Madeddu P. (2002) Targeting kinin B(1) receptor for therapeutic neovascularization. Circulation 105, 360–366 [DOI] [PubMed] [Google Scholar]
- 72. Duka A., Kintsurashvili E., Duka I., Ona D., Hopkins T. A., Bader M., Gavras I., Gavras H. (2008) Angiotensin-converting enzyme inhibition after experimental myocardial infarct. Role of the kinin B1 and B2 receptors. Hypertension 51, 1352–1357 [DOI] [PubMed] [Google Scholar]
- 73. Xu J., Carretero O. A., Shesely E. G., Rhaleb N. E., Yang J. J., Bader M., Yang X. P. (2009) The kinin B1 receptor contributes to the cardioprotective effect of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers in mice. Exp. Physiol. 94, 322–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Ignjatovic T., Tan F., Brovkovych V., Skidgel R. A., Erdös E. G. (2002) Novel mode of action of angiotensin I converting enzyme inhibitors. Direct activation of bradykinin B1 receptor. J. Biol. Chem. 277, 16847–16852 [DOI] [PubMed] [Google Scholar]