

Background: The cellular DNA repair machinery completes the 5′-end gap repair of HIV-1 integration.
Results: The 5′-end DNA gap repair of HIV-1 integration depends on dNTP availability in macrophages, monocytes, and CD4+ T cells.
Conclusion: 5′-End DNA gap repair is a rate-limiting step in HIV-1 integration in macrophages and monocytes due to limited cellular dNTPs.
Significance: Availability of cellular dNTP and polymerase controls HIV-1 integration kinetics.
Keywords: DNA Polymerase, DNA Repair, HIV-1, Macrophages, Monocytes, DNA Gap Filling, Integration, dNTP
Abstract
HIV-1 proviral DNA integration into host chromosomal DNA is only partially completed by the viral integrase, leaving two single-stranded DNA gaps with 5′-end mismatched viral DNA flaps. It has been inferred that these gaps are repaired by the cellular DNA repair machinery. Here, we investigated the efficiency of gap repair at integration sites in different HIV-1 target cell types. First, we found that the general gap repair machinery in macrophages was attenuated compared with that in dividing CD4+ T cells. In fact, the repair in macrophages was heavily reliant upon host DNA polymerase β (Pol β). Second, we tested whether the poor dNTP availability found in macrophages is responsible for the delayed HIV-1 proviral DNA integration in this cell type because the Km value of Pol β is much higher than the dNTP concentrations found in macrophages. Indeed, with the use of a modified quantitative AluI PCR assay, we demonstrated that the elevation of cellular dNTP concentrations accelerated DNA gap repair in macrophages at HIV-1 proviral DNA integration sites. Finally, we found that human monocytes, which are resistant to HIV-1 infection, exhibited severely restricted gap repair capacity due not only to the very low levels of dNTPs detected but also to the significantly reduced expression of Pol β. Taken together, these results suggest that the low dNTP concentrations found in macrophages and monocytes can restrict the repair steps necessary for HIV-1 integration.
Introduction
HIV-1 belongs to the Lentivirus genus in the family Retroviridae. Lentiviruses are unique in their ability to infect both dividing and non-dividing cells (1), whereas many other retroviruses can exclusively infect dividing cells. After HIV-1 reverse transcriptase (RT)2 completes reverse transcription, which converts the viral ssRNA genome into proviral dsDNA in the cytosol (2), the proviral dsDNA is transported into the nucleus and integrated into host chromosomal DNA. The integration process is catalyzed by a virally encoded integrase that carries out two enzymatic reactions: 3′-end processing and strand transfer (3). The 3′-end processing reaction of integrase results in the cleavage of two nucleotides from both 3′-ends of the proviral DNA to expose 3′-OH groups. The free 3′-OH groups are then oriented through the mechanism of strand transfer to allow nucleophilic attack on chromosomal DNA. This covalently links only one of the two viral DNA strands to the host DNA and creates a single-stranded gap between the two DNA species, as well as 5′-end mismatched flaps of viral DNA (4).
It is thought that 5′-end joining is completed by cellular enzymes specifically found in the base excision repair (BER) and single-strand break repair pathways (5–7). BER involves three sequential enzymatic reactions. First, host cell DNA polymerases, such as DNA polymerase β (Pol β) or δ, use cellular dNTPs to fill in the single-stranded gap. Next, the 5′-end mismatched flap is cleaved by host nucleases, such as FEN1 (flap endonuclease 1). The resulting DNA fragments are then joined together by one of the two host cell ligases, I or III (6). Previously, it was shown that recombinant BER proteins can repair a synthetic viral/host DNA gapped substrate in vitro (6). Additionally, it has been demonstrated that cells with knocked down Pol β have lower viral DNA integration efficiency (7).
Both reverse transcription and 5′-end DNA gap repair steps require cellular dNTPs. However, this metabolite can vary greatly between cell types (8). We reported previously that the dNTP levels in terminally differentiated human primary macrophages are extremely low, with concentrations ranging from 20 to 60 nm (9). This concentration is ∼200 times lower than the dNTP concentrations found in activated/dividing CD4+ T cells (5 μm) (8). In addition, our laboratory and others have reported that cellular SAMHD1 (sterile alpha motif and HD domain-containing protein 1), a dNTP triphosphohydrolase, contributes to the low dNTP levels found in macrophages and other myeloid lineage cells (10, 11). Importantly, the dNTP discrepancy between macrophages and active CD4+ T cells has been extensively studied with regard to the effect that the cellular concentration has on the efficiency of HIV-1 reverse transcription (9, 12–15).
HIV-1 and other lentiviruses uniquely complete reverse transcription at low dNTP concentrations, which are found in non-dividing cell types. A number of factors promote this unique ability of lentiviruses. First, HIV-1 RT has a very tight binding affinity for dNTPs compared with other oncoretrovirus RTs (16), allowing HIV-1 RT to complete reverse transcription in environments with low substrate availability (8). In addition, recent reports have shown that HIV-2 and several simian immunodeficiency viruses can enhance reverse transcription by elevating the cellular dNTP concentrations in macrophages. This is accomplished through the action of Vpx (viral protein X) which targets SAMHD1, a host protein, for proteasomal degradation. This in turn leads to an increase in cellular dNTP levels. We and several groups have reported that treatment of macrophages, dendritic cells, and resting CD4+ T cells with virus-like particles expressing Vpx enhances HIV-1 infection (10, 11, 17). However, although many of the recent SAMHD1 studies focused exclusively on how this increase in dNTPs affects viral reverse transcription (8, 9, 12–14, 18, 19), we hypothesized that cellular dNTP concentrations also influence DNA gap repair during viral DNA integration into the host genome.
Here, we demonstrate that HIV-1 DNA gap repair in monocyte-derived macrophages (MDMs) was greatly reduced compared with CD4+ T cells and that this difference was due to the reduced repair capability of enzymes expressed in macrophages and the low cellular dNTP concentration in this cell type. Indeed, HIV-1 5′-end gap repair remained incomplete at integration sites in macrophages, but the amount of fully repaired integration sites was increased when cellular dNTP levels were elevated. This supports our hypothesis that cellular dNTP availability influences the process of HIV-1 integration in addition to the process of reverse transcription.
EXPERIMENTAL PROCEDURES
Cells and HIV-1 Vector
Human monocytes were isolated from buffy coats from multiple donors (New York Blood Center) through density gradient centrifugation. Cells were further column-purified using anti-CD14 microbeads (MACS, Miltenyi Biotec). Monocytes were differentiated to macrophages for 7 days and supplemented with 100 units/ml GM-CSF (R&D Systems, Minneapolis, MN) at 37 °C in RPMI 1640 medium (HyClone, Logan, UT) containing heat-inactivated 10% FBS (Atlanta Biologicals, Lawrenceville, GA) and 1% penicillin/streptomycin (Invitrogen). CD4+ T cells were isolated from buffy coats using anti-CD4 microbeads (MACS, Miltenyi Biotec). CD4+ T cells were activated with 6 μg/ml phytohemagglutinin (J-Oils Mills, Inc., Tokyo, Japan) (20) and 20 units/ml IL-2 (Miltenyi Biotec).
The pD3HIV-GFP vector expresses enhanced GFP and all of the HIV-1 NL4-3 proteins except for Nef and Env. The vector was pseudotyped with vesicular stomatitis virus glycoprotein as described previously (14). A viral vector was made by transfection of 293FT cells (Invitrogen) with pD3HIV-GFP and pVSV-G as described previously (19). Briefly, cells were washed after 24 h, and the medium was collected at 72 h. The medium containing the vector was treated with TURBO DNase (Ambion, Austin, TX) for 30 min at 37 °C.
Preparations of Nuclear Extracts
Nuclear extracts from monocytes, macrophages, naïve, and activated CD4+ T cells were prepared from multiple donors as described previously (21). Briefly, cells were washed with Dulbecco's PBS (Invitrogen) and then resuspended in hypotonic buffer before Dounce homogenization. Lysed cells were centrifuged at 3300 × g for 15 min. The pellet containing the cell nuclei was then mixed with high salt buffer (1.6 m KCl) and incubated on ice. Extracts were centrifuged at 22,065 × g for 30 min. The supernatant was collected, dialyzed overnight, and saved as nuclear extracts at −80 °C. Nuclear extracts were normalized to total protein concentration as measured by the Bradford assay (Bio-Rad).
In Vitro Gap Repair Assay
Oligonucleotides previously published (6), primer Key35 (5′-ATTCGAGCTATCCTTGCGCG-3′), primer Key31 (5′-ACTGCTAGAGATTTTCCACACTGACTA-3′), template Key36 (5′-TAGTCAGTGTGGAAAATCTCTAGCAGGCCCCGCGCAAGGATAGCTCGAAT-3′), and a new primer for 3′-end labeling (Key36+dC, 5′-CTAGTCAGTGTGGAAAATCTCTAGCAGGCCCCGCGCAAGGATAGCTCGAAT-3′) were ordered from Integrated DNA Technologies (22). Key35 was 5′-end-labeled with γ-32P using T4 polynucleotide kinase for 30 min at 37 °C. Gapped substrate i (see Fig. 1A) was constructed with primers at the following concentrations: Key35, 800 nm; Key31, 2.4 μm; and Key36, 1.6 μm (6). To 3′-end label Key31, the primer was first annealed to Key36+dC, incubated with recombinant RT and α-32P-labeled dC for 30 min at 37 °C, and then run on a Tris-free Micro Bio-Spin P-30 column (Bio-Rad). The purified primer-template was then annealed to Key35. The substrate was constructed with 3′-end-labeled Key31 (2.4 μm), Key36+dC (1.6 μm), and Key35 (800 nm).
FIGURE 1.

In vitro DNA gap repair activity of nuclear extracts from human primary activated CD4+ T cells and MDMs. A, 32P-5′-end-labeled (substrate i) or 32P-3′-end-labeled (substrate ii) gapped DNA substrate was used in this study as described previously (6). Substrate i consists of a 20-mer upstream primer (blue) and a 27-mer downstream primer (red), which are annealed to a 50-mer template, creating a four-nucleotide gap (gray) with a 5′-end flap of the downstream primer. B, nuclear extracts normalized to 2 μg/μl total protein from either macrophages (lane 2) or CD4+ T cells (lane 3) from three donors (data from one donor are shown) were incubated with the 5′-end gapped substrate for 30 min at 37 °C. Reactions were quenched with 40 mm EDTA and subjected to 14% urea-PAGE. Gap filling generated a 24-mer product, followed by a fully repaired 50-mer product as indicated. A 10-mer ladder was run in lane 1. C, repair capacity comparison of macrophages and activated CD4+ T cells from three donors. The percent of the fully repaired 50-mer product in the total loading in each lane was quantified by densitometry, and the -fold difference between CD4+ T cells and macrophages was calculated. D, substrate ii containing the 3′-end-labeled 28-mer downstream primer was incubated with nuclear extracts normalized to 2 μg/μl total protein from macrophages in the presence (lane 2) or absence (lane 3) of 250 μm dNTPs for 30 min at 37 °C. Lane 1 is the control reaction without nuclear extract. Reactions were quenched with 40 mm EDTA and subjected to 14% urea-PAGE. The unrepaired 28-mer downstream primer and the fully repaired 51-mer product are indicated. A 10-mer ladder (lane 4) and the 28-mer control downstream primer (lane 5) are also shown.
Gap repair reactions (20 μl) contained 20 nm substrate i or ii (see Fig. 1A) and 75 μm (dT)20, 2 mm ATP, dNTPs (60 nm, 2.5 μm, or 250 μm) and reaction buffer. Reactions were initiated by the addition of 4 μl of diluted nuclear extract from multiple donors at an initial concentration of 0.4 or 2 μg/μl and incubated at 37 °C for 30, 60, 90, or 120 min. Reactions were terminated using 40 mm EDTA (Invitrogen) and 99% formamide and immediately denatured by incubation at 95 °C for 5 min. To monitor for higher molecular weight repair products, 4 μl of the 30-μl final reaction was separated by 14% urea-PAGE (SequaGel, National Diagnostics). Data were captured by PhosphorImager analysis (PerkinElmer Life Sciences) and quantitated using Quantity One software. Importantly, the repair activities analyzed by the quantitation of the final repair products throughout this study were reproducibly linear to both the amounts of the extracts (supplemental Fig. S1) used and the incubation time (supplemental Fig. 2) as illustrated in the legends of the supplemental figures.
Pol β Depletion Assay
Nuclear extracts from MDMs and CD4+ T cells from two donors were depleted as described previously (21). Briefly, 30 μl of nuclear extract (normalized to 4 μg/μl) was added to 10 μl of Protein A/G Plus-agarose beads (Santa Cruz Biotechnology) along with 3 μl of anti-Pol β (Abcam) or anti-α-tubulin (Santa Cruz Biotechnology) antibody. Reactions were agitated overnight at 4 °C and then centrifuged for 1 min at 10,000 × g. The supernatant was collected and analyzed by Western blotting to monitor the completeness of depletion prior to testing these extracts in the repair reactions.
Western Blotting
Anti-α-actin (Santa Cruz Biotechnology), anti-α-tubulin, and anti-Pol β antibodies were used for Western blotting. Nuclear extracts were prepared from two donors as described above and normalized by the Bradford assay for total protein. Samples were mixed with SDS and incubated at 95 °C for 5 min to denature the proteins. The lysate was then subjected to 12% SDS-PAGE and transferred to a Hybond ECL membrane (Amersham Biosciences). Primary and secondary antibodies were used at a 1:1000 dilution. Gels were visualized by chemiluminescence (SuperSignal West Femto maximum sensitivity substrate, Thermo Scientific) and imaged using a Bio-Rad molecular imager.
In Vivo 5′-End Joining Assay
As shown in Fig. 6, macrophages from three donors were transduced with pD3HIV-GFP for 24 h. The cells were washed, incubated for a consecutive 24 h, and then treated with 5 μm nevirapine and 1 μm raltegravir for 6 h. After the 6-h treatments (“day 0”), 1 million macrophages were washed with Dulbecco's PBS and scraped. Cells were then centrifuged at low speed for 5 min and resuspended in 200 μl of Dulbecco's PBS. Cells were monitored for GFP expression using a C6 flow cytometer (Accuri). The remaining cells were pelleted and resuspended in 100 μl of lysis buffer (Wizards SV genomic purification system, Promega). Following this day 0 analysis, 1 mm deoxynucleosides (dNs) or medium was added to the cells, which were analyzed by flow cytometry 24, 48, 72 or 96 h after dN treatment. The medium was changed at 48 h, and both inhibitors and the dNs were replaced. Percent transduction was determined using FlowJo software (version 8.8.5, Tree Star Inc.). The remaining cells were pelleted and resuspended in 100 μl of lysis buffer. The genomic extracts were also monitored for integrated proviral DNA by AluI PCR as described previously (23). Briefly, pHR-CMV-GFP was transduced and stably integrated into 293FT cells, and genomic DNA was extracted as described previously (23). 293FT genomic preparations containing the integrated pHR-CMV-GFP genome were diluted 1:2, 1:4, 1:8, 1:16, and 1:24 with genomic extract from non-transduced 293FT cells. These extracts were then assayed for the amount of late RT DNA products using the following primers: late forward primer for the pHR vector, 5′-TGTGTGCCCGTCTGTTGTGT-3′; late RT reverse primer, 5′-GAGTCCTGCGTCGAGAGAGC-3′; and late RT probe, 5′-6-carboxyfluorescein-CAGTGGCGCCCGAACAGGGA-carboxytetramethylrhodamine-3′. The amount of signal from each dilution was compared with the signal from a known amount of diluted pD3HIV-GFP with the only variation in primers used for D3 being the late RT reverse primer (5′-GAGTCCTGCGTCGAGAGATC-3′). The diluted and quantified 293FT extract was then used as a standard to measure the amount of integrated proviral DNA using the following primers: AluI forward primer, 5′-AACTAGGGAACCCACTGCTTAAG-3′; AluI reverse primer, 5′-TGCTGGGATTACAGGCGTGAG-3′; and AluI probe, 5′-6-carboxyfluorescein-ACACTACTTGAAGCACTCAAGGCAAGCTTT-carboxytetramethylrhodamine-3′.
FIGURE 6.

Effect of cellular dNTP on in vivo HIV-1 LTR 5′-end DNA gap repair in human primary macrophages. A, diagram for the experimental procedure. Human primary macrophages from four donors were transduced with the pD3HIV-GFP vector for 2 days. The transduced macrophages were then washed and treated with 1 μm raltegravir (Ralt.) and 5 μm nevirapine (Nev.) for 6 h. The treated macrophages were cultured for 96 h in the presence and absence of 1 mm dNs, and the cells were collected at 0, 24, 48, 72, and 96 h. B, quantitative AluI PCR. Genomic DNAs were prepared from the collected macrophages and analyzed by quantitative AluI PCR for monitoring the HIV gap DNA repair at the integration sites.
RESULTS
HIV-1 LTR 5′-End Gap Repair in Nuclear Extracts Prepared from Macrophages and Activated CD4+ T Cells
We first compared the DNA gap repair capability of human primary MDMs and activated CD4+ T cells by employing an in vitro assay that can directly monitor the three enzymatic steps to complete 5′-end gap repair: DNA synthesis to fill the ssDNA gap, 5′-end mismatch flap removal, and DNA ligation. This assay utilizes nuclear extracts prepared from cells (24) and synthetic HIV-1 DNA gap repair substrates. As shown in Fig. 1A, the gap substrate consists of two primers: a 20-mer upstream primer (blue) and a 27-mer primer that encodes the 5′-end sequence of the viral 5′-LTR (red) for substrate i. These two primers were annealed to a 50-mer DNA template, creating a four-nucleotide single-stranded gap region with a 5′-end single nucleotide mismatch flap (6). The 20-mer and 28-mer primers in substrate ii were labeled with γ-32P at the 5′-end of the upstream primer (Fig. 1A, *) (25). Gap filling by cellular DNA polymerases with substrate i generated a four-nucleotide extension of the upstream primer (Fig. 1A, gray). This was followed by mismatch flap removal and ligation between the 24-mer extended upstream primer and the processed 26-mer downstream primer, which in turn generated a 50-mer repair product. To verify the absence of the strand displacement activity in the assay, which can also generate the same 50-mer product simply by extending the labeled upstream primer, we constructed a similar gap substrate with a γ-32P-3′-end-labeled downstream primer by incorporating a single radioactive dGTP to form the 51-mer template (substrate ii in Fig. 1, A and D).
Next, we prepared nuclear extracts from human primary macrophages and activated CD4+ T cells and normalized each extract to 2 μg/μl total protein. To compare the DNA gap repair capability, we incubated each extract with substrate i for 30 min at 37 °C at saturating dNTP concentrations (250 μm). The reactions were then quenched with 20 mm EDTA and analyzed by 14% urea-PAGE to monitor the amount of repair. As shown in Fig. 1B, nuclear extracts from both macrophages (lane 2) and activated CD4+ T cells (lane 3) extended the 20-mer upstream primer (blue), generating products with sizes between 21 and 24 nucleotides (gray lines). The gap-filled product was then ligated to the downstream primer (Fig. 1B, red), generating the 50-mer repair product. Next, to compare the gap repair capacity of each cell type, we quantified the amount of 50-mer repair product in reactions with extracts from multiple donors and normalized for the amount of total protein in each reaction. As shown in Fig. 1C, nuclear extracts from macrophages were 3.4-fold lower in their ability to generate the 50-mer products compared with nuclear extracts from activated CD4+ T cells, indicating that macrophages have reduced repair capability.
As discussed previously, it is possible that the 50-mer product observed in Fig. 1B could also be generated by the strand displacement activity of host cellular polymerases and not by ligation between the 24-mer extended upstream primer and processed 26-mer downstream primer (26, 27). To rule out this alternative mechanism, we incubated substrate ii with dNTPs alone, nuclear extract from macrophages, or recombinant RT, which is known to have strand displacement activity (28). With this substrate, only the ligation between the two primers can generate the radioactively labeled 51-mer product, whereas upstream primer extension and strand displacement cannot generate the radioactively labeled 51-mer product. As shown in Fig. 1D, only nuclear extract from macrophages displayed the 51-mer repair product. These data validate that the 50-mer products observed in Fig. 1B were in fact products of gap filling and ligation between the extended upstream and processed downstream primers and not strand displacement.
HIV-1 LTR DNA Gap Repair Is Dependent on Pol β
Previously, it was shown that proteins within the BER pathway are important for HIV-1 infection (5, 6). More specifically, DNA Pol β helps in HIV-1 DNA integration (7) and has also been shown to be preferentially used in the BER pathway in terminally differentiated non-dividing cells (29, 30). To determine whether human primary CD4+ T cells and macrophages depend on this polymerase to complete HIV-1 LTR 5′-end gap repair, we initially immunodepleted DNA Pol β from nuclear extracts of macrophages and CD4+ T cells using anti-DNA Pol β antibody. We also treated the extracts with α-tubulin antibody as a control. Western blot analysis for DNA Pol β showed the successful depletion of Pol β from the extracts (Fig. 2A, lower panel, lanes 3, 5, 7, and 9), whereas anti-α-tubulin antibody did not affect the Pol β level (lanes 2, 4, 6, and 8). Next, we tested the repair capabilities of each nuclear extract using the assay described in Fig. 1B with 250 μm dNTPs to saturate the assay. Note that the level of cellular actin protein in each sample (Fig. 2A, lower panel) was used to normalize the total extract amount. As shown in the α-tubulin control reactions in Fig. 2A, nuclear extracts from two CD4+ T cell donors (lanes 6 and 8) generated a greater amount of 50-mer repair products compared with extracts from two macrophage donors (lanes 2 and 4). However, upon depletion of Pol β from the nuclear extracts from both donors of macrophages (Fig. 2A, lanes 3 and 5) and CD4+ T cells (lanes 7 and 9), the generation of the 50-mer repair product was greatly reduced for both cell types. The amount of 24-mer gap-filled product (Fig. 2B) and fully repaired 50-mer product (Fig. 2C) in each sample were quantified and normalized to the level of actin in each extract. As shown in Fig. 2 (B and C, see asterisks), there was a significant reduction in both the 24-mer gap-filled product and 50-mer ligated product. These data suggest that DNA Pol β is utilized predominantly by both CD4+ T cells and macrophages for HIV-1 LTR 5′-end DNA gap repair. Additionally, due to the variation in amount of repair observed between cell types, other factors could contribute to the differences seen between cell types, such as expression of repair enzymes.
FIGURE 2.

Roles of DNA Pol β in in vitro LTR DNA gap repair activity of nuclear extracts from human primary macrophages and activated CD4+ T cells. A, nuclear extracts from human macrophages and activated CD4+ T cells from two donors (d1 and d2) were normalized to 4 μg/μl total protein and immunodepleted (Im. Depl.) for Pol β or control anti-α-tubulin antibody. The depleted extracts were incubated with substrate i for 30 min with 250 μm dNTPs (upper panel). The depleted extract was Western-blotted (WB) for the presence of either DNA Pol β or actin (lower panels). The lanes in the lower panels correspond to the immunodepletion indicated in the upper panel. The 24-mer gap-filled product (B) and the fully repaired 50-mer product (C) in the reactions with macrophages and T cells immunodepleted with DNA Pol β or anti-α-tubulin (Tub) antibody were quantitated against the total product. As shown in Fig. 2 (B and C, see single and double asterisks), there was a significant reduction in both the 24-mer gap-filled product and the 50-mer ligated product.
HIV-1 LTR 5′-End DNA Gap Repair Is Dependent on dNTP Concentrations
After confirming the role of Pol β in the 5′-end gap repair pathway in both CD4+ T cells and macrophages, we next tested whether the efficiency of this repair pathway is also influenced by the physiological dNTP concentrations found in both cell types. In fact, the Km and Kd values of Pol β for dNTPs are ∼5 and 2 μm, respectively. Both of these values are much higher than the concentration of dNTPs found in macrophages (20–60 nm), but they are close to the concentration found in activated CD4+ T cells (2–5 μm) and other dividing cells (8, 31, 32). Therefore, it is possible that, in macrophages, the dNTP incorporation/gap filling step by Pol β could be a rate-limiting step during HIV-1 LTR 5′-end DNA gap repair. We predicted that if the concentration of dNTPs correlated with gap filling efficiency, then gap repair would be efficient at saturating dNTP concentrations (250 μm compared with concentrations below the Kd of Pol β (5 μm)). To explore this possibility, we prepared nuclear extracts from CD4+ T cells and macrophages from multiple donors and assayed for the efficiency of gap repair with substrate i at dNTP concentrations found in macrophages (60 nm) and CD4+ T cells (2.5 μm) and also at saturating dNTP concentrations (250 μm) (Fig. 3A, M, T, and S, respectively). As shown in Fig. 3A, nuclear extracts from both macrophages and CD4+ T cells were able to efficiently complete 5′-end gap repair at saturating dNTP concentrations (50-mer band). The repair product was reduced in CD4+ T cell extracts at the dNTP concentration found in the T cells (2.5 μm). Furthermore, nuclear extracts from both macrophages and CD4+ T cells failed to execute the 5′-end gap repair at 60 nm dNTP concentrations. The repair by nuclear extracts from macrophages was found to be 3.5-fold lower than that of nuclear extracts from CD4+ T cells when 250 μm dNTPs was used, which was also observed in Fig. 1. However, the macrophage extracts displayed a 11.5-fold lower repair capability than the CD4+ T cell extracts when 2.5 μm dNTPs was used (Fig. 3B). Similar results were observed in reactions utilizing substrate ii, which was labeled with RT, which unfortunately produced a doublet due to terminal transferase activity (Fig. 3C, control (C)). This produced a doublet 51-mer/52-mer band in the repair reactions; however, these results still confirmed that the repair observed was due to the joining of both primers and not strand displacement (Fig. 3C). Collectively, these data support a model in which HIV-1 LTR 5′-end DNA gap repair is dependent on the cellular dNTP availability and cellular enzymes, including Pol β. They also support that DNA gap filling may be a rate-limiting step for HIV-1 proviral DNA integration into the genome of macrophages.
FIGURE 3.

DNA gap repair activity of nuclear extracts from human primary macrophages and activated CD4+ T is dependent on dNTP concentrations. A, nuclear extracts from macrophages and CD4+ T cells were normalized to 2 μg/μl total protein. The normalized extracts were incubated with substrate i for 30 min at 37 °C with set dNTP concentrations of 60 nm (M), 2.5 μm (T), and 250 μm (S). The unrepaired gapped substrate produced a labeled 20-mer upstream primer in the control (C), and the fully repaired substrate produced a 50-mer band. B, -fold differences between the repair capabilities of macrophage and T cell extracts at different dNTP concentrations were calculated using the percent repair determined by densitometry of the repaired 50-mer product. C, substrate ii containing the 3′-end-labeled 28-mer downstream primer was incubated with nuclear extracts from macrophages or activated CD4+ T cells from two donors (d1 and d2; 2 μg/μl total protein) for 30 min at 37 °C with three different dNTP concentrations (60 nm (M), 2.5 μm (T), and 250 μm (S)). The unrepaired 28-mer downstream primer (C), the fully repaired 51-mer product, and the 10-mer ladder (L) are marked.
Nuclear Extracts from Macrophages Fail to Repair the Gapped Substrate during Extended Incubation Times
Next, we tested whether DNA gap repair can be detected at longer incubation times using substrate i in the presence of nuclear extracts from macrophages from two donors. Although there was variability displayed between donors, which is common, gap repair with 60 nm dNTPs (Fig. 4A, M) was not restored when the incubation time was extended to 60, 90, or 120 min. Additionally, the 50-mer repair product level increased at T cell and saturating dNTP concentrations. This was observed with both donors and further confirms that the limited dNTP levels contribute to the attenuated 5′-end gap repair observed in macrophages.
FIGURE 4.
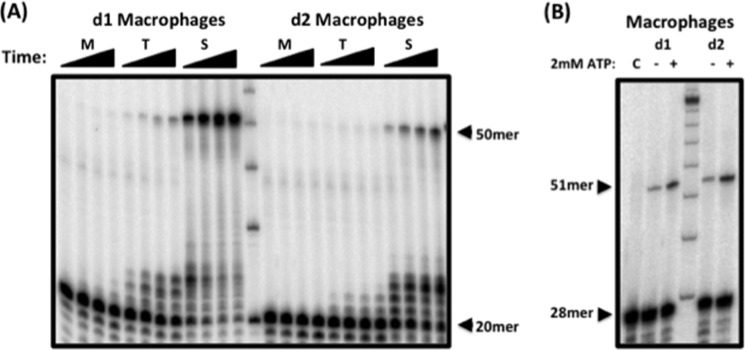
Nuclear extracts from human primary macrophages fail to repair substrate at physiological dNTP concentrations. Nuclear extracts from macrophages from two donors (d1 and d2), which were normalized to 2 μg/μl total protein, were incubated with substrate i for 30, 60, 90, or 120 min at 60 nm (M), 2.5 μm (T), or 250 μm (S) dNTP. The unrepaired 20-mer upstream primer and the fully repaired 50-mer product are indicated. B, nuclear extracts from macrophages from two donors were incubated with substrate ii containing the 3′-end-labeled unligated downstream primer at 250 μm dNTP and with (+) or without (−) 2 mm ATP. The 51-mer ligated product is indicated. C, control.
Abundant ATP in Macrophages Supports Efficient Ligation of the 5′-End Gap
Unlike cellular dNTP levels, ATP concentrations range between ∼6 and ∼2 mm for peripheral blood mononuclear cells and macrophages (9, 33, 34). ATP is a necessary cofactor for DNA ligases. Therefore, we tested whether 5′-end DNA gap repair is also dependent on the concentration of ATP. We incubated nuclear extract from macrophages with substrate ii at the saturating dNTP concentration (250 μm) with or without 2 mm ATP. As shown in Fig. 4B, 5′-end gap repair was minimally detected upon ATP deprivation. The amount of repair detected under this condition could be due to the presence of ATP prebound to DNA ligases in the extracts. However, upon addition of 2 mm ATP, an increase in ligated repair product was observed. These data indicate that, unlike dNTPs, the concentration of ATP in macrophages is sufficient to support the ligation step in DNA gap repair.
Gap Repair in Human Primary Monocytes
Unlike activated CD4+ T cells and macrophages, monocytes are restrictive to HIV-1 infection (35, 36). Additionally, it has been suggested that primary restriction of HIV-1 infection in monocyte occurs during a step after reverse transcription (37). Monocytes have also been shown to display reduced DNA repair compared with other cell types (38). Therefore, we examined the DNA gap repair capacity and cellular dNTP concentrations of human primary monocytes. First, we prepared nuclear extracts from monocytes and macrophages of the same donors. Nuclear extract from each cell type (2 μg/μl protein) was incubated with substrate i in the presence of 60 nm, 2.5 μm, or 250 μm dNTPs (Fig. 5B, M, T, and S, respectively). As shown in Fig. 5B, macrophages displayed significant repair only at saturating dNTP concentrations (250 μm). Notably, nuclear extract from monocytes displayed reduced repair even at saturating dNTP concentrations compared with macrophage extract. When the repair products from each extract were quantified (Fig. 5B), monocytes had ∼16-fold less DNA gap repair product at 250 μm dNTP compared with macrophages. The same result was observed in the reactions with substrate ii at 250 μm (data not shown). These data demonstrate that monocytes have even more attenuated DNA gap repair capacity than macrophages. Next, we performed Western blotting for Pol β using all nuclear extracts. As shown in Fig. 5 (C and D), monocytes harbored 4.7-fold less DNA Pol β compared with macrophages. Finally, we measured the dNTP concentrations of monocytes using our primer extension-based dNTP assay using a 32P-5′-labeled 18-mer primer annealed to a 19-mer DNA template, leaving only a one-nucleotide overhang in the template strand. This allows for a single dNTP to be incorporated by DNA polymerases, such as HIV-1 RT. We methanol-extracted cellular metabolites containing all four dNTPs from macrophages and monocytes from two donors. These cellular metabolites were then incubated with the described primer-template such that RT would extend the primer with the complementary dNTP to the template strand overhang. In this assay, the higher the dNTP concentration is within the cell, the greater the extension of the 18-mer primer to the 19-mer product (Fig. 5E, P and E, respectively). As shown in Fig. 5E, no primer extension was detected when the dNTP extract from monocytes (Mo) was applied, whereas the macrophage dNTP extract (M) and the T cell extract (T) generated detectable primer extension. This confirms that monocytes have lower dNTP concentrations compared with macrophages, which, in addition to the reduced levels of Pol β, may contribute to the block of 5′-end gap repair in monocytes. Therefore, it can be speculated that incomplete HIV-1 DNA integration in monocytes may be the primary cause of the restrictive infection in this cell type.
FIGURE 5.

Comparison of in vitro HIV LTR gap repair activity of human primary monocytes and macrophages from the same donors. Nuclear extracts were prepared from monocytes and macrophages from two donors (d1 and d2) and were normalized to 2 μg/μl total protein. A, the nuclear extracts were incubated with substrate i for 30 min with 60 nm (M), 2.5 μm (T), or 250 μm (S) dNTPs. The unrepaired (Unrep.) 20-mer primer and the fully repaired 50-mer product are indicated. B, the repair capabilities of macrophages and monocytes at 250 μm dNTP were compared. C, extracts from macrophages (lanes 1 and 2) and monocytes (lanes 3 and 4) used in A were analyzed by Western blotting (WB) for both Pol β and actin. D, the Pol β levels normalized to the corresponding actin levels in C were compared. E, dNTP assay with dNTP extracts from macrophages (M), monocytes (Mo), and activated CD4+ T cells (T) from two donors. dNTP extracts were applied to the HIV-1 RT-mediated single-nucleotide incorporation reaction using a 5′-end-labeled 18-mer (P) annealed to the 19-mer template (template used for dATP incorporation). C, no dNTP reaction; PC, reaction with saturating 1 mm dNTPs, generating a 19-mer extended product (E).
AluI PCR-based Quantification of 5′-End Gap Repair at Sites of Integration in Macrophages
Finally, we investigated whether the cellular dNTP concentration directly affects HIV-1 LTR 5′-end DNA gap repair in macrophages in cell culture during HIV-1 infection. To test this, we isolated and monitored the kinetics of the ssDNA gap repair step independently of reverse transcription and integration. For this, we designed an experimental protocol (Fig. 6A) that allows reverse transcription and partial DNA integration into host chromosomes to occur and then completely blocks both reverse transcription and integration at later time points. This allowed us to monitor only the gap repair kinetics of the partially integrated HIV-1 DNA. We transduced human primary MDMs with a vesicular stomatitis virus glycoprotein-pseudotyped pD3HIV-GFP vector, which encodes the entire HIV-1 genome except env and nef, which were replaced with enhanced GFP. Because reverse transcription in macrophages reaches completion between 24 and 72 h after initial infection (15, 39, 40), macrophages were initially transduced by HIV-1 vector for 48 h, and further reverse transcription and integration were blocked by treating the cells with 5 μm nevirapine (41) and 1 μm raltegravir, respectively (Fig. 6A). Note that these drugs do not interfere with the 5′-end gap repair machinery (42). Next, the drug-treated macrophages were cultured in the presence or absence of 1 mm dNs, which elevate cellular dNTP concentrations by >15-fold (11). Macrophages were collected, and cellular genomic DNA was isolated at 0, 24, 48, 72, and 96 h after dN treatment. We then employed quantitative AluI PCR using two primers that flank the 5′-end LTR DNA gap to measure the completion of HIV-1 LTR gap repair. We hypothesized that the low dNTP concentration in macrophages would restrict DNA gap repair and lead to a lower AluI PCR copy number compared with macrophages treated with dNs. As shown in Fig. 6B, the macrophages treated with dNs showed elevated AluI DNA copy numbers compared with the untreated macrophages, the values of which would increase if previous steps were not completely inhibited (43). This result supports our hypothesis that the limited cellular dNTP concentrations found in macrophages delay HIV-1 LTR 5′-end DNA gap repair during viral integration.
DISCUSSION
In this study, we have reported that there are two steps in the HIV-1 life cycle that are affected by the availability of cellular dNTPs. The first step, reverse transcription, has been studied extensively with regard to the low dNTP concentration found in terminally differentiated/non-dividing macrophages (9, 12–15). Furthermore, the very low dNTP levels in macrophages during viral reverse transcription have been reported to lead to 1) frequent incorporation of non-canonical nucleotides by HIV-1 RT (9, 21), 2) an increase in strand transfer of HIV-1 RT (18), and 3) an increased reliance on the central polypurine tract for completion of proviral DNA synthesis (44, 45). These three effects collectively lead to the attenuated production of virus in macrophages compared with activated CD4+ T cells (8).
Unlike reverse transcription, the effect of cellular dNTP concentrations on the 5′-end LTR DNA gap repair step of the viral life cycle has yet to be studied in detail. 5′-End gap repair has been shown to occur as quickly as 1 h after transduction with oncoretroviruses, such as Moloney murine leukemia virus (7). However, unlike HIV-1, oncoretroviruses exclusively infect dividing cells, which have high cellular dNTP levels. Therefore, the role of dNTPs in 5′-end LTR DNA gap repair would not be limiting under these cellular conditions (1, 9, 19). It has previously been shown that many of the proteins involved in the single-strand break repair and BER pathways have the ability to repair this integration intermediate (24–26). Additionally, it has been demonstrated that knocking down these proteins with siRNA can reduce integration and expression of viral genes (5, 7). Here, we have reported that 5′-end LTR DNA gap repair is heavily reliant upon both the availability of cellular repair machinery, such as DNA Pol β, and cellular dNTP abundance. Additionally, we found that in vivo viral integration in macrophages can be enhanced through the addition of dNs, which elevate the cellular dNTP concentration by ∼22-fold (11). Furthermore, we also revealed that monocytes harbor reduced 5′-end LTR DNA gap repair enzymes and cellular dNTP levels compared with macrophages. Our data support the hypothesis that these two intrinsic cellular parameters may contribute to the restrictive HIV-1 infection of monocytes.
A series of recent studies have revealed that SAMHD1, a dNTP triphosphohydrolase, is expressed in myeloid cells, including monocytes, macrophages, and dendritic cells, and that SAMHD1 contributes to regulating the low dNTP pool (10, 11, 17). Moreover, Vpx, which is known to direct SAMHD1 degradation, is found in HIV-2 and several simian immunodeficiency virus strains (46). It is plausible that, unlike HIV-1, the 5′-end LTR DNA gap repair for cells infected with HIV-2 and simian immunodeficiency virus strains may not be kinetically delayed in macrophages because Vpx-mediated degradation of SAMHD1 leads to significant increases in the cellular dNTP concentration (11, 47, 48). Moreover, the efficiency of gap repair may differ depending on the site of integration in macrophages versus activated CD4+ T cells. These possibilities will be further investigated in our future studies.
Acknowledgments
We thank Sarah Amie, Laura Nguyen, and Dr. Joseph A. Hollenbaugh for manuscript editing.
This work was supported, in whole or in part, by National Institutes of Health Grants AI049781 (to B. K.) and F31 GM095190 (to W. D.).

This article contains supplemental Figs. S1 and S2.
- RT
- reverse transcriptase
- BER
- base excision repair
- Pol β
- polymerase β
- MDM
- monocyte-derived macrophage
- dN
- deoxynucleoside.
REFERENCES
- 1. Lewis P. F., Emerman M. (1994) Passage through mitosis is required for oncoretroviruses but not for the human immunodeficiency virus. J. Virol. 68, 510–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mastro A. M., Hurley D. J., Winning R. K., Filipowski R., Ogilvie M. L., Gartner T. K. (1986) Mitogenic activity of snake venom lectins. Cell Tissue Kinet. 19, 557–566 [DOI] [PubMed] [Google Scholar]
- 3. Bushman F. D., Craigie R. (1991) Activities of human immunodeficiency virus (HIV) integration protein in vitro: specific cleavage and integration of HIV DNA. Proc. Natl. Acad. Sci. U.S.A. 88, 1339–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fujiwara T., Mizuuchi K. (1988) Retroviral DNA integration: structure of an integration intermediate. Cell 54, 497–504 [DOI] [PubMed] [Google Scholar]
- 5. Espeseth A. S., Fishel R., Hazuda D., Huang Q., Xu M., Yoder K., Zhou H. (2011) siRNA screening of a targeted library of DNA repair factors in HIV infection reveals a role for base excision repair in HIV integration. PLoS ONE 6, e17612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yoder K. E., Bushman F. D. (2000) Repair of gaps in retroviral DNA integration intermediates. J. Virol. 74, 11191–11200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yoder K. E., Espeseth A., Wang X. H., Fang Q., Russo M. T., Lloyd R. S., Hazuda D., Sobol R. W., Fishel R. (2011) The base excision repair pathway is required for efficient lentivirus integration. PLoS ONE 6, e17862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Diamond T. L., Roshal M., Jamburuthugoda V. K., Reynolds H. M., Merriam A. R., Lee K. Y., Balakrishnan M., Bambara R. A., Planelles V., Dewhurst S., Kim B. (2004) Macrophage tropism of HIV-1 depends on efficient cellular dNTP utilization by reverse transcriptase. J. Biol. Chem. 279, 51545–51553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kennedy E. M., Gavegnano C., Nguyen L., Slater R., Lucas A., Fromentin E., Schinazi R. F., Kim B. (2010) Ribonucleoside triphosphates as substrate of human immunodeficiency virus type 1 reverse transcriptase in human macrophages. J. Biol. Chem. 285, 39380–39391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goldstone D. C., Ennis-Adeniran V., Hedden J. J., Groom H. C., Rice G. I., Christodoulou E., Walker P. A., Kelly G., Haire L. F., Yap M. W., de Carvalho L. P., Stoye J. P., Crow Y. J., Taylor I. A., Webb M. (2011) HIV-1 restriction factor SAMHD1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 480, 379–382 [DOI] [PubMed] [Google Scholar]
- 11. Lahouassa H., Daddacha W., Hofmann H., Ayinde D., Logue E. C., Dragin L., Bloch N., Maudet C., Bertrand M., Gramberg T., Pancino G., Priet S., Canard B., Laguette N., Benkirane M., Transy C., Landau N. R., Kim B., Margottin-Goguet F. (2012) SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 13, 223–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Weiss K. K., Bambara R. A., Kim B. (2002) Mechanistic role of residue Gln151 in error prone DNA synthesis by human immunodeficiency virus type 1 (HIV-1) reverse transcriptase (RT). Pre-steady state kinetic study of the Q151N HIV-1 RT mutant with increased fidelity. J. Biol. Chem. 277, 22662–22669 [DOI] [PubMed] [Google Scholar]
- 13. Jamburuthugoda V. K., Chugh P., Kim B. (2006) Modification of human immunodeficiency virus type 1 reverse transcriptase to target cells with elevated cellular dNTP concentrations. J. Biol. Chem. 281, 13388–13395 [DOI] [PubMed] [Google Scholar]
- 14. Jamburuthugoda V. K., Santos-Velazquez J. M., Skasko M., Operario D. J., Purohit V., Chugh P., Szymanski E. A., Wedekind J. E., Bambara R. A., Kim B. (2008) Reduced dNTP binding affinity of 3TC-resistant M184I HIV-1 reverse transcriptase variants responsible for viral infection failure in macrophage. J. Biol. Chem. 283, 9206–9216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Collin M., Gordon S. (1994) The kinetics of human immunodeficiency virus reverse transcription are slower in primary human macrophages than in a lymphoid cell line. Virology 200, 114–120 [DOI] [PubMed] [Google Scholar]
- 16. Operario D. J., Reynolds H. M., Kim B. (2005) Comparison of DNA polymerase activities between recombinant feline immunodeficiency and leukemia virus reverse transcriptases. Virology 335, 106–121 [DOI] [PubMed] [Google Scholar]
- 17. Laguette N., Sobhian B., Casartelli N., Ringeard M., Chable-Bessia C., Ségéral E., Yatim A., Emiliani S., Schwartz O., Benkirane M. (2011) SAMHD1 is the dendritic- and myeloid-cell-specific HIV-1 restriction factor counteracted by Vpx. Nature 474, 654–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Operario D. J., Balakrishnan M., Bambara R. A., Kim B. (2006) Reduced dNTP interaction of human immunodeficiency virus type 1 reverse transcriptase promotes strand transfer. J. Biol. Chem. 281, 32113–32121 [DOI] [PubMed] [Google Scholar]
- 19. Van Cor-Hosmer S. K., Daddacha W., Kelly Z., Tsurumi A., Kennedy E. M., Kim B. (2012) The impact of molecular manipulation in residue 114 of human immunodeficiency virus type-1 reverse transcriptase on dNTP substrate binding and viral replication. Virology 422, 393–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Clapham P. R., McKnight A. (2002) Cell surface receptors, virus entry and tropism of primate lentiviruses. J. Gen. Virol. 83, 1809–1829 [DOI] [PubMed] [Google Scholar]
- 21. Kennedy E. M., Amie S. M., Bambara R. A., Kim B. (2012) Frequent incorporation of ribonucleotides during HIV-1 reverse transcription and their attenuated repair in macrophages. J. Biol. Chem. 287, 14280–14288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goldschmidt V., Marquet R. (2004) Primer unblocking by HIV-1 reverse transcriptase and resistance to nucleoside RT inhibitors (NRTIs). Int. J. Biochem. Cell Biol. 36, 1687–1705 [DOI] [PubMed] [Google Scholar]
- 23. Butler S. L., Hansen M. S., Bushman F. D. (2001) A quantitative assay for HIV DNA integration in vivo. Nat. Med. 7, 631–634 [DOI] [PubMed] [Google Scholar]
- 24. Manley J. L., Fire A., Cano A., Sharp P. A., Gefter M. L. (1980) DNA-dependent transcription of adenovirus genes in a soluble whole-cell extract. Proc. Natl. Acad. Sci. U.S.A. 77, 3855–3859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Albright A. V., Erickson-Viitanen S., O'Connor M., Frank I., Rayner M. M., González-Scarano F. (2000) Efavirenz is a potent nonnucleoside reverse transcriptase inhibitor of HIV type 1 replication in microglia in vitro. AIDS Res. Hum. Retroviruses 16, 1527–1537 [DOI] [PubMed] [Google Scholar]
- 26. Maga G., Villani G., Tillement V., Stucki M., Locatelli G. A., Frouin I., Spadari S., Hübscher U. (2001) Okazaki fragment processing: modulation of the strand displacement activity of DNA polymerase δ by the concerted action of replication protein A, proliferating cell nuclear antigen, and flap endonuclease-1. Proc. Natl. Acad. Sci. U.S.A. 98, 14298–14303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Imoto S., Bransfield L. A., Croteau D. L., Van Houten B., Greenberg M. M. (2008) DNA tandem lesion repair by strand displacement synthesis and nucleotide excision repair. Biochemistry 47, 4306–4316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Charneau P., Alizon M., Clavel F. (1992) A second origin of DNA plus-strand synthesis is required for optimal human immunodeficiency virus replication. J. Virol. 66, 2814–2820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nouspikel T., Hanawalt P. C. (2002) DNA repair in terminally differentiated cells. DNA Repair 1, 59–75 [DOI] [PubMed] [Google Scholar]
- 30. Akbari M., Peña-Diaz J., Andersen S., Liabakk N. B., Otterlei M., Krokan H. E. (2009) Extracts of proliferating and non-proliferating human cells display different base excision pathways and repair fidelity. DNA Repair 8, 834–843 [DOI] [PubMed] [Google Scholar]
- 31. Kraynov V. S., Showalter A. K., Liu J., Zhong X., Tsai M. D. (2000) DNA polymerase β: contributions of template-positioning and dNTP triphosphate-binding residues to catalysis and fidelity. Biochemistry 39, 16008–16015 [DOI] [PubMed] [Google Scholar]
- 32. Singh J., Su L., Snow E. T. (1996) Replication across O6-methylguanine by human DNA polymerase β in vitro. Insights into the futile cytotoxic repair and mutagenesis of O6-methylguanine. J. Biol. Chem. 271, 28391–28398 [DOI] [PubMed] [Google Scholar]
- 33. Johnson A., O'Donnell M. (2005) DNA ligase: getting a grip to seal the deal. Curr. Biol. 15, R90–R92 [DOI] [PubMed] [Google Scholar]
- 34. Simsek D., Jasin M. (2011) DNA ligase III: a spotty presence in eukaryotes, but an essential function where tested. Cell Cycle 10, 3636–3644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Neil S., Martin F., Ikeda Y., Collins M. (2001) Postentry restriction to human immunodeficiency virus-based vector transduction in human monocytes. J. Virol. 75, 5448–5456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vatakis D. N., Bristol G., Wilkinson T. A., Chow S. A., Zack J. A. (2007) Immediate activation fails to rescue efficient human immunodeficiency virus replication in quiescent CD4+ T cells. J. Virol. 81, 3574–3582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Triques K., Stevenson M. (2004) Characterization of restrictions to human immunodeficiency virus type 1 infection of monocytes. J. Virol. 78, 5523–5527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bauer M., Goldstein M., Christmann M., Becker H., Heylmann D., Kaina B. (2011) Human monocytes are severely impaired in base and DNA double-strand break repair that renders them vulnerable to oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 108, 21105–21110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. O'Brien W. A. (1994) HIV-1 entry and reverse transcription in macrophages. J. Leukoc. Biol. 56, 273–277 [DOI] [PubMed] [Google Scholar]
- 40. Gavegnano C., Kennedy E. M., Kim B., Schinazi R. F. (2012) The impact of macrophage nucleotide pools on HIV-1 reverse transcription, viral replication, and the development of novel antiviral agents. Mol. Biol. Int. 2012, 625983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yamashita M., Perez O., Hope T. J., Emerman M. (2007) Evidence for direct involvement of the capsid protein in HIV infection of nondividing cells. PLoS; pathog. 3, 1502–1510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Michelini Z., Galluzzo C. M., Negri D. R., Leone P., Amici R., Bona R., Summa V., Di Santo R., Costi R., Pommier Y., Marchand C., Palmisano L., Vella S., Cara A. (2010) Evaluation of HIV-1 integrase inhibitors on human primary macrophages using a luciferase-based single-cycle phenotypic assay. J. Virol. Methods 168, 272–276 [DOI] [PubMed] [Google Scholar]
- 43. Kelly J., Beddall M. H., Yu D., Iyer S. R., Marsh J. W., Wu Y. (2008) Human macrophages support persistent transcription from unintegrated HIV-1 DNA. Virology 372, 300–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Skasko M., Kim B. (2008) Compensatory role of human immunodeficiency virus central polypurine tract sequence in kinetically disrupted reverse transcription. J. Virol. 82, 7716–7720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Van Cor-Hosmer S. K., Daddacha W., Kim B. (2010) Mechanistic interplay among the M184I HIV-1 reverse transcriptase mutant, the central polypurine tract, cellular dNTP concentrations and drug sensitivity. Virology 406, 253–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Romani B., Cohen E. A. (2012) Lentivirus Vpr and Vpx accessory proteins usurp the cullin4-DDB1 (DCAF1) E3 ubiquitin ligase. Curr. Opin. Virol. 2, 755–763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Amie S. M., Noble E., Kim B. (2013) Intracellular nucleotide levels and the control of retroviral infections. Virology 436, 247–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kim B., Nguyen L. A., Daddacha W., Hollenbaugh J. A. (2012) Tight interplay among SAMHD1 protein level, cellular dNTP levels, and HIV-1 proviral DNA synthesis kinetics in human primary monocyte-derived macrophages. J. Biol. Chem. 287, 21570–21574 [DOI] [PMC free article] [PubMed] [Google Scholar]