

Background: The Warburg effect in cancer is manifested by increased glycolysis and decreased respiration. Our goal was to determine how mitochondria are suppressed in osteosarcoma (OS).
Results: OS cells showing the Warburg effect have markers of the mitochondrial permeability transition (MPT).
Conclusion: MPT plays a possible role in suppression of mitochondrial function in OS.
Significance: Our data implicate the MPT in metabolic reprogramming in cancer.
Keywords: Cancer, Glycolysis, Mitochondria, Mitochondrial Permeability Transition, Osteosarcoma, Warburg Effect, Cyclophilin D
Abstract
Metabolic reprogramming in cancer is manifested by persistent aerobic glycolysis and suppression of mitochondrial function and is known as the Warburg effect. The Warburg effect contributes to cancer progression and is considered to be a promising therapeutic target. Understanding the mechanisms used by cancer cells to suppress their mitochondria may lead to development of new approaches to reverse metabolic reprogramming. We have evaluated mitochondrial function and morphology in poorly respiring LM7 and 143B osteosarcoma (OS) cell lines showing the Warburg effect in comparison with actively respiring Saos2 and HOS OS cells and noncancerous osteoblastic hFOB cells. In LM7 and 143B cells, we detected markers of the mitochondrial permeability transition (MPT), such as mitochondrial swelling, depolarization, and membrane permeabilization. In addition, we detected mitochondrial swelling in human OS xenografts in mice and archival human OS specimens using electron microscopy. The MPT inhibitor sanglifehrin A reversed MPT markers and increased respiration in LM7 and 143B cells. Our data suggest that the MPT may play a role in suppression of mitochondrial function, contributing to the Warburg effect in cancer.
Introduction
Under adequate oxygenation, normal cells rely on mitochondrial oxidative phosphorylation to generate ATP and switch to the less favorable anaerobic pathway of glycolysis when exposed to hypoxia. However, many types of cancer cells survive and proliferate by generating ATP via glycolysis rather than oxidative phosphorylation even when oxygenated, as was postulated by Otto Warburg (1, 2). It has been suggested that cancer cells must be able to overcome oxygen inhibition of glycolysis (3). Corresponding with this increase in glycolysis is a suppression of mitochondrial activity in cancer. Potential mechanisms leading to the suppression of mitochondrial activity in cancer are being actively investigated. Because of the heterogeneous nature of cancer cells, different mechanisms may be involved, as was reviewed previously (4, 5). For instance, changes in the number of mitochondria in cancer cells have been observed by various groups, including ours (6–8). Some tumors show increased activity of pyruvate dehydrogenase kinase, which phosphorylates and inhibits pyruvate dehydrogenase and thus prevents pyruvate oxidation (9). Increased expression and mitochondrial binding of hexokinase II in cancer leading to reprogramming toward glycolysis have been observed (10). The transcription factor p53, a major tumor suppressor that is inactivated in nearly 50% of cancers (11), has been shown to play an important role in the regulation of cell metabolism (12, 13) by inducing SCO2, a positive regulator of mitochondrial cytochrome oxidase subunit 2 (14), and TIGAR, a suppressor of glycolysis (15). Therefore, inactivation of p53 may lead to down-regulation of both SCO2 and TIGAR, inhibition of mitochondria, and activation of glycolysis. Finally, voltage-dependent anion channel blockade by free tubulin leads to a decrease in oxidative phosphorylation and is enhanced by voltage-dependent anion channel phosphorylation by cytosolic kinases (16). In cancer cells, free tubulin is high and cytosolic kinases are hyperactive, potentially leading to closure of voltage-dependent anion channels, suppression of mitochondrial function, and the Warburg effect (17).
In this work, we focused on cancerous and normal cells of similar origin, osteosarcoma (OS)2 and osteoblasts, to avoid variability due to heterogeneity between different lineages. We present our data indicating the involvement of the mitochondrial permeability transition (MPT) (18, 19) in mitochondrial dysfunction in OS cells showing the Warburg effect. The MPT is a nonselective mitochondrial pore that is sensitive to cyclosporin A and its derivatives; its molecular composition is not yet fully elucidated. Various mitochondrial proteins were suggested to play a role in the MPT; however, only cyclophilin D (CypD) was shown to be a major regulator of the pore (20, 21). The MPT can be activated by various stimuli, including calcium overload and reactive oxygen species, and plays a major role in mitochondrial dysfunction in various pathologies (18, 22); however, the role of the MPT in mitochondrial dysfunction in cancer has not been shown before and is a subject of this study.
EXPERIMENTAL PROCEDURES
Materials
Cell culture growth media, ingredients, antibiotics, and fluorescent probes were from Invitrogen. DMEM powder was from Sigma. Human fetal osteoblasts (hFOB) and OS cells (Saos2, HOS, and 143B) were obtained from American Type Culture Collection. LM7 cells (23) were a gift of Dr. Eugenie Kleinerman (MD Anderson Cancer Center, Austin, TX). The cytochrome c (CytC) ELISA kit was from MitoSciences. Anti-Bcl-2 antibody was from Epitomics.
Cell Culture and Treatments
hFOB cells were cultured in DMEM/F-12 medium, and Saos2, HOS, LM7, and 143B cells were cultured in DMEM at 37 °C. All media were supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin mixture. Where indicated, cells were continuously treated with sanglifehrin A (SfA) at 0.5 μm for 1, 2, 4, or 7 days with media changes every other day.
Oxygen Consumption and Glycolysis Rate Assay
Cellular bioenergetics was measured in intact cells using the Seahorse XF24 apparatus (Seahorse Bioscience). The device measures the oxygen consumption rate (OCR) and glycolysis (extracellular acidification rate (ECAR)) by intact cells on a 24-well plate. Cells were plated 1 day before at a density of 2.5 × 104/well. Before the experiment, the regular cell medium was replaced with an unbuffered DMEM-/XF24 medium suitable for ECAR measurements. This DMEM/XF24 medium was freshly prepared from DMEM powder and contained 5 mm glucose, 1 mm glutamine, and no serum or pyruvate. At the end, cells were trypsinized and counted using a Cellometer automated cell counter (Nexcelom Bioscience). OCR and ECAR were then normalized to cell numbers.
Mitochondrial Morphology Assessment
To assess mitochondrial morphology in live cells, cells were grown on 25-mm round glass coverslips to 70% confluency, and mitochondria were stained with the MitoTracker Green (MTG) fluorescent dye, which accumulates in mitochondria due to membrane potential and once accumulated is retained in mitochondria due to formation of S–H bonds (24). MTG therefore has serious limitations as a dynamic potentiometric probe but is appropriate for visualization of mitochondrial morphology in live cells. MTG at 100 nm was loaded for 30 min at 37 °C, cells were washed with fresh medium, and coverslips were mounted in chambers for microscopy. To visualize MTG-stained mitochondria, we used an Axiovert 40 CFL inverted fluorescence microscope equipped with an AxioCam MRc digital camera, AxioVision software, and a Zeiss 100× objective. On average, 30 cells from each experimental group were photographed.
Electron Microscopy
Cultured cells or tissue pieces of xenografted OS tumors produced as described in our previous study (25) were fixed in 2.5% glutaraldehyde in 0.1 m sodium cacodylate buffer, post-fixed in 1.0% OsO4, dehydrated with ethanol, and transitioned into propylene oxide for infiltration and embedding into EPON/Araldite epoxy resin. Thin sections were cut onto grids, stained with uranyl acetate and lead citrate, examined with a Hitachi H-7650 transmission electron microscope, and digitally photographed. Archival human OS epoxy-embedded tissue blocks were thin-sectioned and photographed. Mitochondrial morphology was assessed, and the number of mitochondria in normal orthodox conformation and in swollen conformation was counted. An orthodox conformation is characterized by an electron-dense and crista-rich matrix, and a swollen conformation is characterized by an increased surface area and a less electron-dense matrix with fewer cristae. A total of 30 cells/sample were analyzed. Three samples of xenografted OS or archival human OS were used.
Mitochondrial Membrane Potential Assay
To measure mitochondrial membrane potential (ΔΨm), we used flow cytometry and a potentiometric fluorescent probe, 3,3′-dihexyloxacarbocyanine iodide (DiOC6(3)), which was previously found to be suitable for ΔΨm assay by flow cytometry (26). Cells were incubated with DiOC6(3) at 20 nm for 30 min at 37 °C, washed, lifted from plates using a cell scraper, resuspended in Hanks' balanced salt solution, and assayed using the FACSCanto II flow cytometer (BD Biosciences). In negative controls, ΔΨm was dissipated by incubating cells with carbonyl cyanide p-trifluoromethoxyphenylhydrazone at 1 μm. Data were verified using tetramethylrhodamine ethyl ester (TMRE) and fluorescence microscopy, where cells were incubated with TMRE at 10 nm for 30 min at 37 °C and visualized using the abovementioned Axiovert imaging system.
MPT Assay
To assess the MPT, mitochondrial membrane integrity was measured using a method of calcein quenching by cobalt as described by Petronilli et al. (27). The assay is based on the fact that calcein accumulated in the cytosol is quenched after co-loading with cobalt, whereas calcein accumulated in mitochondria is not accessible to cobalt and therefore not quenched unless mitochondrial membranes are permeabilized. Cells were loaded with calcein acetoxymethyl ester at 1 μm in the presence of CoCl2 at 1 mm for 30 min at 37 °C, washed, lifted from plates using a cell scraper, resuspended in Hanks' balanced salt solution, and assayed using the FACSCanto II flow cytometer. In negative controls, calcein- and cobalt-loaded cells were exposed to ionomycin at 1 μm, leading to permeabilization of plasma membranes to external calcium, calcium overload, loss of mitochondrial membrane integrity, and dissipation of the calcein signal.
Cytochrome c Release and Active Caspase-3 Assays
To measure CytC released from mitochondria to the cytosol, cells were washed with PBS, plasma membranes were permeabilized with digitonin at 0.01% in KCl-based buffer (125 mm KCl, 2 mm K2HPO4, 10 mm K-HEPES (pH 7.4), 1 mm MgCl2, 5 mm potassium succinate, and 1 μm rotenone), and cytosolic fractions were collected and spun down. These cytosolic fractions were then subjected to CytC ELISA. To measure active caspase-3, cell lysates were mixed with the caspase-3-specific fluorogenic substrate acetyl-DEVD-7-amino-4-methylcoumarin (Calbiochem) at 20 μm in a total volume of 0.2 ml in a 96-well plate and incubated for 30 min at 37 °C. The fluorescence at 440 nm (excitation at 380 nm) from the 7-amino-4-methylcoumarin tag cleaved by caspase-3 was measured in a BioTek plate reader.
Immunoblotting
Cell lysates at 25 μg/sample were electrophoresed and electroblotted onto PVDF membranes (Bio-Rad). Blots were blocked in 5% nonfat dry milk in PBS and 0.05% Tween 20, probed with a primary antibody at 1 μg/ml and then with the corresponding HRP-conjugated secondary antibody at 0.2 μg/ml, developed using SuperSignal West Pico chemiluminescent substrate (Pierce), and photographed. Blots were stripped in ReBlot Plus stripping buffer (Chemicon) and reprobed with anti-β-actin antibody to verify equal loading.
Statistical Analysis
Experiments were repeated three to five times. Data were analyzed using Prism 5.01 (GraphPad Software). Means ± S.E. were calculated, and the statistical significance was established using either Student's t test or one-way analysis of variance, as appropriate. Data with p < 0.05 were considered statistically significant.
RESULTS
Presence of the Warburg Effect in LM7 and 143B Osteogenic Sarcoma Cells
To minimize variability due to different genetic, signaling, and microenvironmental backgrounds, we studied normal and cancerous cells of similar (mesenchymal) origin, such as noncancerous osteoblasts (hFOB) and OS cell lines (Saos2, LM7, HOS, and 143B). These OS cell lines have been extensively characterized by our group, as evident from our previous studies (8, 25, 28). Importantly, the LM7 cell line is an invasive and metastatic subtype of the parental Saos2 cell line (29), and the 143B cell line is an invasive and metastatic subtype of the parental HOS cell line (30). To detect the presence of the Warburg effect in these cells, we measured the antimycin A-sensitive (i.e. mitochondrial) OCR and glycolytic ECAR due to lactate production using the Seahorse XF24 apparatus. Fig. 1 shows that while Saos2 and HOS cells had OCRs similar to that found in hFOB cells, LM7 and 143B cells have significantly lower OCRs. Meanwhile, the glycolysis rates (ECARs) were significantly increased in LM7 and 143B cells (Fig. 1). Therefore, we observed suppression of mitochondrial function and up-regulation of glycolysis in LM7 and 143B cells compared with Saos2 and HOS cells and noncancerous hFOB cells under aerobic conditions. This indicates the presence of the Warburg effect in LM7 and 143B osteogenic sarcoma cells.
FIGURE 1.
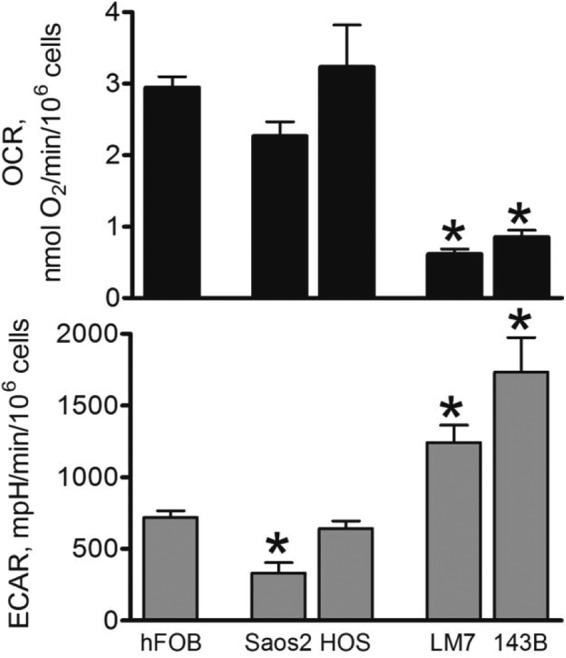
Assessment of cellular bioenergetics in osteoblasts and OS cells. Cells were seeded in Seahorse XF24-compatible 24-well plates. After 24 h, the cell medium was replaced with DMEM/XF24 medium, and the basal OCR (upper panel) and ECAR (lower panel) were assayed as measures of mitochondrial function and glycolysis, respectively, using the Seahorse XF24 apparatus. Afterward, cells were trypsinized and counted. Rates were normalized to cell numbers. Data are means ± S.E. (n = 5). *, p < 0.05 versus hFOB values. mpH, milli-pH (pH × 10−3).
Mitochondrial Swelling in LM7 and 143B Cells in Vitro, OS Xenografts in Vivo, and OS Clinical Specimens
The focus of our study was the mechanism of mitochondrial dysfunction in cancer cells showing the Warburg effect. To elucidate such a mechanism, we first assessed mitochondrial morphology in the above cell lines in vitro. To avoid possible artifacts during cell fixation and embedding, we performed live cell imaging using high-resolution fluorescence microscopy after staining with MTG, a mitochondrion-specific fluorescent probe. Fig. 2A demonstrates that actively respiring noncancerous hFOB cells and Saos2 and HOS OS cells had elongated mitochondria, whereas LM7 and 143B OS cells with the Warburg effect had many rounded organelles. This indicates mitochondrial swelling in LM7 and 143B cells. It should be noted that the MTG signal in LM7 and 143B cells was significantly weaker, although this is not shown in Fig. 2A because the image brightness was adjusted to better visualize mitochondrial morphology. Because MTG is to some extent a potential-sensitive probe (24), this decrease in signal could be due to mitochondrial depolarization, which was later verified using more appropriate potential-sensitive probes. The swelling of mitochondria in LM7 and 143B OS cells showing the Warburg effect was verified using EM. Fig. 2B shows that compared with hFOB cells, LM7 and 143B cells had enlarged and rounded mitochondria with a less electron-dense matrix, an indication of organelle swelling. Thus, both high-resolution fluorescence microscopy and EM showed the presence of mitochondrial swelling in poorly respiring LM7 and 143B OS cells.
FIGURE 2.
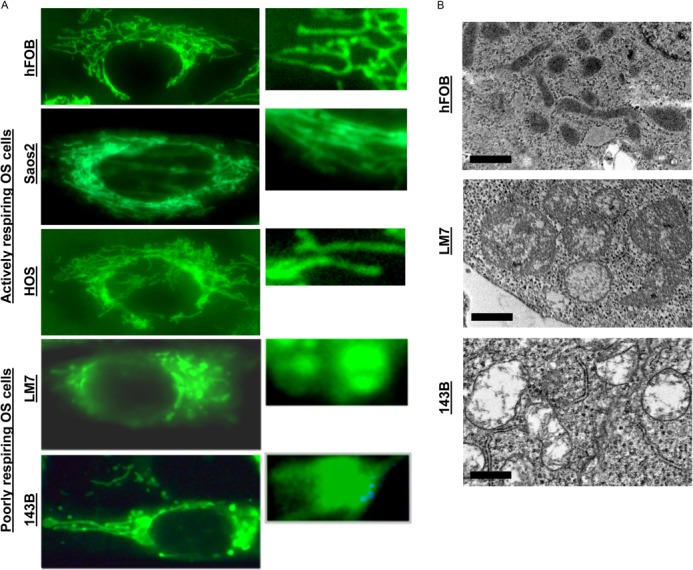
Mitochondrial morphology in osteoblasts and OS cells. A, cells plated on glass coverslips 1 day before were stained with MTG as described under “Experimental Procedures” and mounted on microscopy chambers, and live cell fluorescent images were taken. The right panels are magnified regions of the cells shown in the left panels. Images are representatives of 20–30 cells/cell line. B, electron micrographs of hFOB, LM7, and 143B cells. Images are representatives of 20 cells/cell line.
To confirm that mitochondrial swelling is not just an in vitro phenomenon, we performed EM with tissue samples of 143B xenografts in mice prepared in our previous study (8) and archival OS clinical specimens and evaluated the mitochondrial morphology. Fig. 3A shows whole cell images of a 143B xenograft and an OS clinical specimen in which both orthodox (arrows) and swollen (arrowheads) conformations of mitochondria were present. Higher magnification images show examples of orthodox and swollen mitochondria characterized by an increased size and a less electron-dense and crista-free matrix in these samples (Fig. 3B, upper panels). In addition, we examined mitochondrial morphology in archival human OS diagnostic biopsy specimens using EM. The analysis revealed a significant number of mitochondria in a swollen conformation (Fig. 3, A and B, lower panels). It should be noted that to avoid morphological changes associated with tumor necrosis, we made sure to include only necrosis-free tumor areas in our analysis. Fig. 3C presents the quantitation of our EM analysis. In OS xenografts or human OS tumor specimens, 76 and 35% of mitochondria, respectively, had a swollen conformation. Importantly, these were untreated OS xenografts in mice or diagnostic biopsies of naive human OS tumors before any treatments that could affect mitochondrial morphology were done. Thus, our data indicate that mitochondrial swelling is not solely a phenomenon observed in OS cells in vitro but is a feature present in mouse OS tumor models in vivo and in actual human OS tumors.
FIGURE 3.

Mitochondrial morphology in OS xenografts and clinical biopsy specimens. A, representative electron micrographs of 143B xenografts in mice (upper panel) or human OS clinical biopsy specimens (lower panel) showing mitochondria in either orthodox (arrows) or swollen (arrowheads) conformation. B, higher magnification electron micrographs of orthodox or swollen mitochondria in tumor samples. Arrowheads indicate mitochondria. C, quantitation of mitochondrial morphology in electron micrographs. Values are a percentage of the total number of mitochondria from 30 cells.
Mitochondrial Membrane Depolarization and Permeabilization in LM7 and 143B Cells
To further assess mitochondrial function in the cells studied, we measured mitochondrial membrane potential and integrity. We assessed mitochondrial membrane potential using the DiOC6(3) dye and flow cytometry. Although not without flaws, this fluorescent probe is optimal for flow cytometry, as was determined earlier by Kroemer and co-workers (26). Fig. 4A shows a significant decrease in DiOC6(3) signal intensity in LM7 and 143B cells, indicating mitochondrial depolarization. We verified our data using TMRE and fluorescence microscopy in intact attached cells. Fig. 4B shows that similar to the DiOC6(3) signal, the TMRE signal was significantly weaker in LM7 and 143B cells, indicating decreased membrane potential.
FIGURE 4.
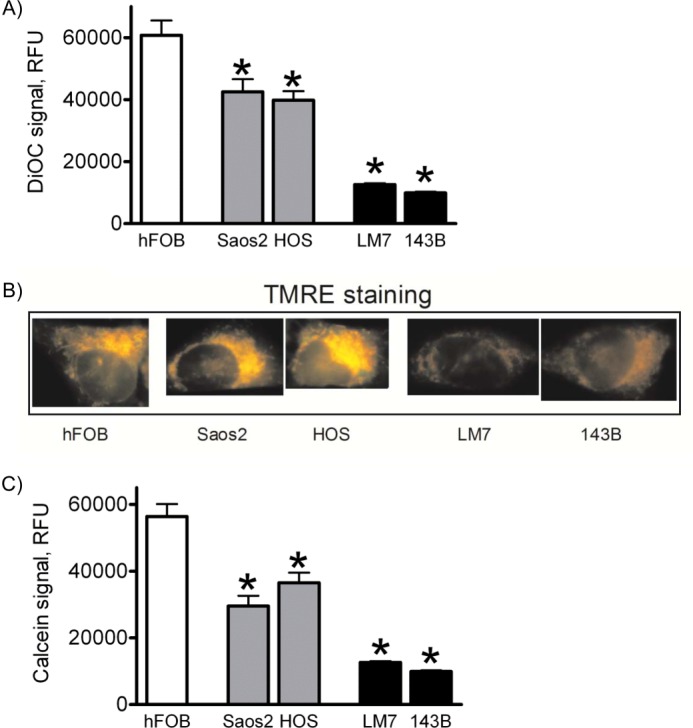
Mitochondrial membrane potential and membrane integrity in osteoblasts and OS cells. A, mitochondrial membrane potential was measured using DiOC6(3) and flow cytometry. RFU, relative fluorescence units. B, DiOC6(3) data were verified via TMRE staining of live cells grown on coverslips and fluorescence microscopy. C, mitochondrial membrane integrity was measured by the calcein signal in the presence of cobalt. Values in A and C are means ± S.E. (n = 4). *, p < 0.05 versus hFOB values.
We then examined the integrity of the mitochondrial membrane using a previously established method of calcein quenching by cobalt (27). Briefly, when cells are co-loaded with calcein and CoCl2, the calcein signal is quenched in the cytosol by cobalt and remains only in intact mitochondria that are not readily permeable to cobalt unless the mitochondrial membrane integrity is compromised. Flow cytometry analysis (Fig. 4C) revealed a significant decrease in calcein signal in LM7 and 143B cells, indicative of permeabilization of the mitochondrial membrane. In summary, all of the above changes (mitochondrial swelling, depolarization, and loss of membrane integrity) are clear indicators of the increased activity of the MPT in LM7 and 143B OS cells showing the Warburg effect (18, 31).
Release of Cytochrome c in LM7 and 143B Cells Is Not Sufficient to Induce Apoptosis
Mitochondrial swelling and permeabilization of the outer mitochondrial membrane may lead to the release of CytC to the cytosol, subsequent activation of caspases, and initiation of apoptosis. We therefore measured the presence of CytC in the cytosol and activation of caspase-3. Fig. 5A (upper panel) shows that the cytosolic CytC content was lower in Saos2 and HOS cells with active mitochondria compared with noncancerous hFOB cells as measured by ELISA of cytosolic fractions. The cytosolic CytC content was higher in poorly respiring LM7 and 143B cells showing signs of the MPT compared with Saos2 and HOS cells but was not significantly higher than in noncancerous hFOB cells. It should be noted that the total CytC expression in all of these cell lines was not different (data not shown). Caspase-3 activity was not significantly increased in LM7 and 143B cells as measured using a fluorogenic substrate cleavage assay (Fig. 5A, lower panel). These data indicate that in OS cells not showing the Warburg effect, CytC was retained better in mitochondria likely due to overexpression of Bcl-2 (Fig. 5B), whereas in LM7 and 143B OS cells showing the Warburg effect and the MPT, cytosolic CytC was increased but did not exceed the level found in the noncancerous osteoblasts. As a consequence, caspase-3 activity was not higher than in hFOB cells, and thus, the basal rates of apoptosis were not increased.
FIGURE 5.
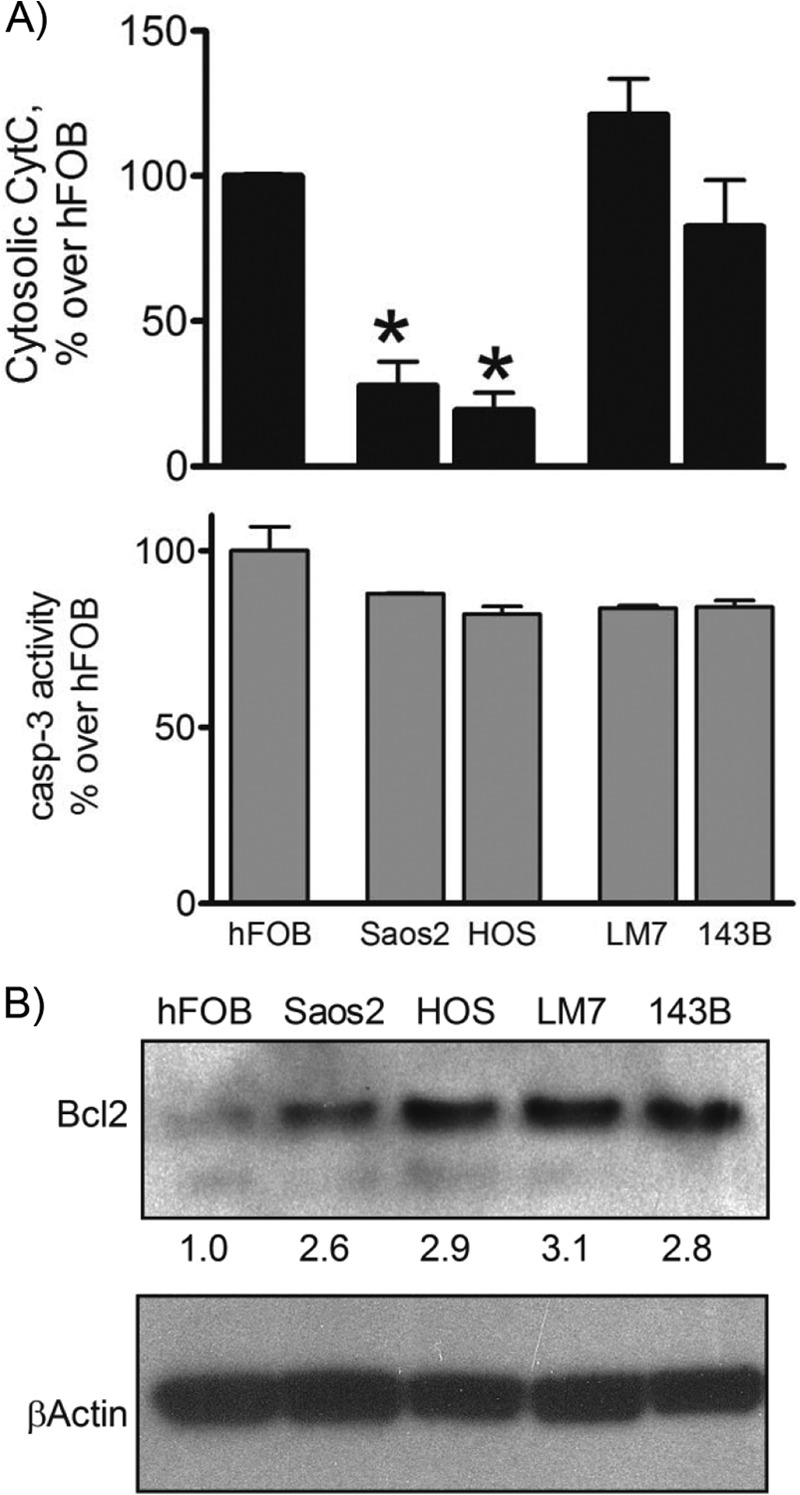
Cytochrome c release and caspase-3 activity in osteoblasts and OS cells. A, CytC in cytosolic fractions (upper panel) and caspase-3 (casp-3) activity in cell lysates (lower panel) were measured by ELISA and fluorogenic substrate cleavage assay, respectively. Values are means ± S.E. (n = 3). *, p < 0.05 versus hFOB values. B, Bcl-2 levels in cell lysates were assayed by immunoblotting. Blots were reprobed for β-actin to verify equal loading. The numbers under the upper panel indicate the relative intensities of each band as measured by densitometry. Blots are representatives of three.
Mitochondrial Dysfunction and the Warburg Effect Are Reversed in LM7 and 143B Cells after Treatment with the MPT Inhibitor SfA
As stated above, mitochondrial dysfunction in LM7 and 143B cells is associated with markers of the MPT. To further confirm the involvement of the MPT, we studied the effect of the MPT inhibitor SfA on mitochondrial morphology and function in these cells. We preferred SfA to the more commonly used cyclosporin A because SfA was shown to be more specific and less immunosuppressive (32). We treated LM7 and 143B cells with SfA for various times (data not shown) and found that a 7-day incubation produced the most stable effects. Fig. 6A shows that after a 7-day treatment of LM7 and 143B cells with SfA, we could no longer observe mitochondrial swelling. Both mitochondrial membrane potential and integrity were significantly increased as indicated by an increase in DiOC6(3) signal and calcein signal in the presence of cobalt, respectively (Fig. 6, B and C). These data indicate that SfA effectively inhibited MPT in the cells studied.
FIGURE 6.

Effect of SfA on mitochondria in LM7 and 143B OS cells. Cells were treated with SfA at 0.5 μm for 7 days, and mitochondrial morphology and function were assessed. A, cells plated on glass coverslips were stained with MTG, and live cell fluorescent images were taken. The right panels are magnified regions of the cells shown in the left panels. Images are representatives of 30 cells/cell line. B, mitochondrial membrane potential was measured by DiOC6(3) signal and flow cytometry as described in the legend for Fig. 4A. C, mitochondrial membrane integrity was measured by the calcein signal in the presence of cobalt and flow cytometry as described in the legend for Fig. 4B. The cellular OCR (D) and ECAR (E) were measured using a Seahorse XF24 machine. Data in B–E are expressed as a percentage of the control in the absence of SfA. Values are means ± S.E. (n = 4). *, p < 0.05 versus control values.
Next, we examined whether inhibition of the MPT can improve mitochondrial function and reverse metabolic reprogramming in LM7 and 143B OS cells. Fig. 6 shows a significant increase in cell respiration (panel D) and a decrease in cell glycolysis rate (panel E) as measured by the OCR and ECAR, respectively. These data indicate that inhibition of the MPT with SfA improved mitochondrial function and reversed metabolic reprogramming and the Warburg effect in the OS cells studied. Altogether, our work indicates that in the studied OS cells showing the Warburg effect and metabolic reprogramming, mitochondrial dysfunction is associated with the increased MPT activity and that inhibition of the MPT leads to the reversal of the Warburg effect in these cells.
DISCUSSION
In this work, we studied an array of cell lines of similar origin, such as normal osteoblasts and four cancerous OS cells. Two of the four OS cell lines (Saos2 and HOS) are actively respiring, whereas the other two (LM7 and 143B) are poorly respiring and highly glycolytic, indicating the presence of the Warburg effect in these cells. We performed various assays of mitochondrial structure and function and found that LM7 and 143B OS cells showing the Warburg effect had a large number of swollen mitochondria. Importantly, the mitochondrial swelling found in LM7 and 143B OS cells in vitro was also seen in mouse OS tumor models in vivo and in human OS tumors. This suggests that the described phenomenon is not restricted to OS cell lines in vitro but is also found in OS in vivo. Consistent with our finding, mitochondrial swelling was previously detected in other tumor types by EM (33, 34). Further analysis of mitochondrial function indicated depolarization and membrane permeabilization in LM7 and 143B cells. Together, these markers (mitochondrial swelling, depolarization, and membrane permeabilization) indicate the presence of the MPT. Furthermore, the use of SfA, a known MPT inhibitor, reversed mitochondrial swelling, depolarization and membrane permeabilization in LM7 and 143B cells. This is further proof that the MPT may play a role in the suppression of mitochondrial function in the OS cells studied. Importantly, reversal of the abovementioned MPT markers with SfA was concomitant with increased oxygen consumption and decreased glycolysis in LM7 and 143B cells, indicating reversal of the Warburg effect. We therefore concluded that the MPT plays a possible role in the suppression of mitochondrial function in these OS cell lines showing the Warburg phenotype.
An important question is how LM7 and 143B cells tolerate the MPT, which has been attributed to both apoptotic and necrotic death signaling (22). The role of the MPT in apoptosis is still debated, and CypD knock-out mouse studies indicate that the MPT is not involved in the regulation of apoptosis. Nevertheless, the MPT flickering or prolonged opening in only a portion of cell mitochondria may lead to mitochondrial swelling, rupture of the outer mitochondrial membrane, and release of CytC, leading to programmed cell death without significant ATP depletion, which would preclude the progression of apoptosis (35). Indeed, in our poorly respiring LM7 and 143B OS cells showing the MPT, we detected increased CytC in cytosolic fractions compared with the actively respiring Saos2 and HOS cells not showing the MPT. It should be noted that cytosolic CytC in Saos2 and HOS cells was significantly lower than in noncancerous hFOB cells, which can likely be attributed to the overexpression of Bcl-2 and which needs further investigation. Anti-apoptotic Bcl-2 proteins, such as Bcl-2 itself, limit CytC release from mitochondria via a mechanism that is not yet fully understood (36, 37). Although the release of CytC from mitochondria was increased in LM7 and 143B OS cells, it did not exceed the levels found in noncancerous hFOB cells and was likely insufficient to induce any significant activation of caspase-3. Further extensive studies are needed to validate these hypotheses and are the subject of our future research efforts. As for the necrotic death, massive prolonged opening of the MPT pore in noncancerous cells causes cessation of oxidative phosphorylation, ATP depletion, and cell demise due to bioenergetic collapse (35). In our model system, LM7 and 143B OS cells use primarily the glycolytic pathway and are less dependent on mitochondrial ATP production, therefore avoiding the MPT-mediated ATP depletion and necrotic death. Thus, the death-inducing effect of the MPT may be negated in the OS cells studied due to the reasons described above. The results from this study are also in agreement with previous data from our group (38) and from other laboratories (39, 40) describing a pro-oncogenic role of a major positive regulator of the MPT, CypD, which contradicts the pro-death effect of the MPT. CypD overexpression found in different tumors should sensitize mitochondria to the MPT, but despite that, no signs of increased apoptosis or necrosis were found in these tumors (39). If the MPT is needed to shut down mitochondrial function for bioenergetic reprogramming and establishing the Warburg effect in cancer, the pro-oncogenic role of CypD can be easily explained.
Another important question raised by our study is what can trigger the MPT in cancer cells. One possible trigger is the increased oxidative stress and reactive oxygen species production found in cancer cells (41). This oxidative stress may be associated with the hypoxic tumor microenvironment found frequently within solid tumor masses and also within bone marrow during hematologic malignancies (42). Moreover, repetitive hypoxia interposed with reoxygenation, a mode of hypoxia shown to cause the most pronounced mitochondrial dysfunction and the MPT in cardiovascular pathologies (43, 44), is most relevant to cancer because it was shown that a significantly higher percentage of tumor cells are exposed to hypoxia-reoxygenation than to hypoxia alone (45, 46). Hypoxia-reoxygenation is caused by irregular blood supply within tumors, an immature and leaky vasculature, and an abnormal and constantly changing vessel network architecture (47, 48). The exposure of originally actively respiring cancer cells to hypoxia-reoxygenation may eventually lead to MPT induction, mitochondrial dysfunction, and metabolic reprogramming. The connection between hypoxia-reoxygenation and tumor cell metabolism is also the subject of our ongoing studies.
Overall, our study suggests a novel mechanism of mitochondrial dysfunction in cancer cells showing the Warburg effect: the MPT. These findings provide a rationale for elucidating the role of the MPT in other types of cancer and for further attempts to use MPT inhibitors to reverse the Warburg effect.
Acknowledgments
We gratefully acknowledge Dr. Paolo Bernardi (University of Padova) and Drs. Thomas Gunter and George Porter (University of Rochester) for expertise and fruitful discussions. We also thank Dr. Eugenie Kleinerman for providing LM7 cells.
This work was supported, in whole or in part, by National Institutes of Health Grant KL2 RR024136 from the National Center for Research Resources and Grants R03 AR061515 and P30 AR061307 from NIAMS. This work was also supported by the Center for Musculoskeletal Research, BeatSarcoma, and the Karen D'Amico Foundation.
- OS
- osteosarcoma
- MPT
- mitochondrial permeability transition
- CypD
- cyclophilin D
- CytC
- cytochrome c
- SfA
- sanglifehrin A
- OCR
- oxygen consumption rate
- ECAR
- extracellular acidification rate
- MTG
- MitoTracker Green
- DiOC6(3)
- 3,3′-dihexyloxacarbocyanine iodide
- TMRE
- tetramethylrhodamine ethyl ester.
REFERENCES
- 1. Warburg O. (1956) On the origin of cancer cells. Science 123, 309–314 [DOI] [PubMed] [Google Scholar]
- 2. Warburg O. (1930) The Metabolism of Tumours, Constable Press, London [Google Scholar]
- 3. Gatenby R. A., Gillies R. J. (2004) Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 4, 891–899 [DOI] [PubMed] [Google Scholar]
- 4. Wenner C. E. (2012) Targeting mitochondria as a therapeutic target in cancer. J. Cell. Physiol. 227, 450–456 [DOI] [PubMed] [Google Scholar]
- 5. Ward P. S., Thompson C. B. (2012) Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell 21, 297–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brandon M., Baldi P., Wallace D. C. (2006) Mitochondrial mutations in cancer. Oncogene 25, 4647–4662 [DOI] [PubMed] [Google Scholar]
- 7. Czarnecka A. M., Czarnecki J. S., Kukwa W., Cappello F., Scińska A., Kukwa A. (2010) Molecular oncology focus–is carcinogenesis a ‘mitochondriopathy’? J. Biomed. Sci. 17, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Shapovalov Y., Hoffman D., Zuch D., de Mesy Bentley K., Eliseev R. A. (2011) Mitochondrial dysfunction in cancer cells due to aberrant mitochondrial replication. J. Biol. Chem. 286, 22331–22338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cairns R. A., Harris I. S., Mak T. W. (2011) Regulation of cancer cell metabolism. Nat. Rev. Cancer 11, 85–95 [DOI] [PubMed] [Google Scholar]
- 10. Pedersen P. L. (2007) Warburg, me and hexokinase 2: multiple discoveries of key molecular events underlying one of cancers' most common phenotypes, the “Warburg Effect”, i.e., elevated glycolysis in the presence of oxygen. J. Bioenerg. Biomembr. 39, 211–222 [DOI] [PubMed] [Google Scholar]
- 11. Vousden K. H., Lane D. P. (2007) p53 in health and disease. Nat. Rev. Mol. Cell Biol. 8, 275–283 [DOI] [PubMed] [Google Scholar]
- 12. Vousden K. H., Ryan K. M. (2009) p53 and metabolism. Nat. Rev. Cancer 9, 691–700 [DOI] [PubMed] [Google Scholar]
- 13. Vaseva A. V., Moll U. M. (2009) The mitochondrial p53 pathway. Biochim. Biophys. Acta 1787, 414–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Matoba S., Kang J. G., Patino W. D., Wragg A., Boehm M., Gavrilova O., Hurley P. J., Bunz F., Hwang P. M. (2006) p53 regulates mitochondrial respiration. Science 312, 1650–1653 [DOI] [PubMed] [Google Scholar]
- 15. Bensaad K., Tsuruta A., Selak M. A., Vidal M. N., Nakano K., Bartrons R., Gottlieb E., Vousden K. H. (2006) TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 126, 107–120 [DOI] [PubMed] [Google Scholar]
- 16. Rostovtseva T. K., Bezrukov S. M. (2012) VDAC inhibition by tubulin and its physiological implications. Biochim. Biophys. Acta 1818, 1526–1535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maldonado E. N., Sheldon K. L., DeHart D. N., Patnaik J., Manevich Y., Townsend D. M., Bezrukov S. M., Rostovtseva T. K., Lemasters J. J. (2013) Voltage-dependent anion channels modulate mitochondrial metabolism in cancer cells. Regulation by free tubulin and erastin. J. Biol. Chem. 288, 11920–11929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rasola A., Sciacovelli M., Pantic B., Bernardi P. (2010) Signal transduction to the permeability transition pore. FEBS Lett. 584, 1989–1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hunter D. R., Haworth R. A. (1979) The Ca2+-induced membrane transition in mitochondria *1, *2I. The protective mechanisms. Arch. Biochem. Biophys. 195, 453–459 [DOI] [PubMed] [Google Scholar]
- 20. Baines C. P., Kaiser R. A., Purcell N. H., Blair N. S., Osinska H., Hambleton M. A., Brunskill E. W., Sayen M. R., Gottlieb R. A., Dorn G. W. (2005) Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 434, 658–662 [DOI] [PubMed] [Google Scholar]
- 21. Basso E., Fante L., Fowlkes J., Petronilli V., Forte M. A., Bernardi P. (2005) Properties of the permeability transition pore in mitochondria devoid of cyclophilin D. J. Biol. Chem. 280, 18558–18561 [DOI] [PubMed] [Google Scholar]
- 22. Zamzami N., Larochette N., Kroemer G. (2005) Mitochondrial permeability transition in apoptosis and necrosis. Cell Death Differ. 12, 1478–1480 [DOI] [PubMed] [Google Scholar]
- 23. Lafleur E. A., Koshkina N. V., Stewart J., Jia S.-F., Worth L. L., Duan X., Kleinerman E. S. (2004) Increased Fas expression reduces the metastatic potential of human osteosarcoma cells. Clin. Cancer Res. 10, 8114–8119 [DOI] [PubMed] [Google Scholar]
- 24. Macho A., Decaudin D., Castedo M., Hirsch T., Susin S. A., Zamzami N., Kroemer G. (1996) Chloromethyl X rosamine is an aldehyde fixable potential sensitive fluorochrome for the detection of early apoptosis. Cytometry 25, 333–340 [DOI] [PubMed] [Google Scholar]
- 25. Shapovalov Y., Benavidez D., Zuch D., Eliseev R. A. (2010) Proteasome inhibition with bortezomib suppresses growth and induces apoptosis in osteosarcoma. Int. J. Cancer 127, 67–76 [DOI] [PubMed] [Google Scholar]
- 26. Métivier D., Dallaporta B., Zamzami N., Larochette N., Susin S. A., Marzo I., Kroemer G. (1998) Cytofluorometric detection of mitochondrial alterations in early CD95/Fas/APO-1-triggered apoptosis of Jurkat T lymphoma cells. Comparison of seven mitochondrion-specific fluorochromes. Immunol. Lett. 61, 157–163 [DOI] [PubMed] [Google Scholar]
- 27. Petronilli V., Miotto G., Canton M., Brini M., Colonna R., Bernardi P., Di Lisa F. (1999) Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys. J. 76, 725–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Eliseev R. A., Zuscik M. J., Schwarz E. M., O'Keefe R. J., Drissi H., Rosier R. N. (2005) Increased radiation-induced apoptosis of Saos2 cells via inhibition of NFκB: a role for c-Jun N-terminal kinase. J. Cell Biochem. 96, 1262–1273 [DOI] [PubMed] [Google Scholar]
- 29. Jia S.-F., Worth L. L., Kleinerman E. S. (1999) A nude mouse model of human osteosarcoma lung metastases for evaluating new therapeutic strategies. Clin. Exp. Metastasis 17, 501–506 [DOI] [PubMed] [Google Scholar]
- 30. Luu H. H., Kang Q., Park J. K., Si W., Luo Q., Jiang W., Yin H., Montag A. G., Simon M. A., Peabody T. D., Haydon R. C., Rinker-Schaeffer C. W., He T. C. (2005) An orthotopic model of human osteosarcoma growth and spontaneous pulmonary metastasis. Clin. Exp. Metastasis 22, 319–329 [DOI] [PubMed] [Google Scholar]
- 31. Petit P. X., Goubern M., Diolez P., Susin S. A., Zamzami N., Kroemer G. (1998) Disruption of the outer mitochondrial membrane as a result of large amplitude swelling: the impact of irreversible permeability transition. FEBS Lett. 426, 111–116 [DOI] [PubMed] [Google Scholar]
- 32. Gregory M. A., Bobardt M., Obeid S., Chatterji U., Coates N. J., Foster T., Gallay P., Leyssen P., Moss S. J., Neyts J. (2011) Preclinical characterization of naturally occurring polyketide cyclophilin inhibitors from the sanglifehrin family. Antimicrob. Agents Chemother. 55, 1975–1981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Arismendi-Morillo G. (2009) Electron microscopy morphology of the mitochondrial network in human cancer. Int. J. Biochem. Cell Biol. 41, 2062–2068 [DOI] [PubMed] [Google Scholar]
- 34. Gasparre G., Romeo G., Rugolo M., Porcelli A. M. (2011) Learning from oncocytic tumors: why choose inefficient mitochondria? Biochim. Biophys. Acta 1807, 633–642 [DOI] [PubMed] [Google Scholar]
- 35. Rasola A., Bernardi P. (2011) Mitochondrial permeability transition in Ca2+-dependent apoptosis and necrosis. Cell Calcium 50, 222–233 [DOI] [PubMed] [Google Scholar]
- 36. Adams J. M., Cory S. (2007) The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene 26, 1324–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Deveraux Q. L., Reed J. C. (1999) IAP family proteins—suppressors of apoptosis. Genes Dev. 13, 239–252 [DOI] [PubMed] [Google Scholar]
- 38. Eliseev R. A., Malecki J., Lester T., Zhang Y., Humphrey J., Gunter T. E. (2009) Cyclophilin D interacts with Bcl-2 and exerts an anti-apoptotic effect. J. Biol. Chem. 284, 9692–9699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schubert A., Grimm S. (2004) Cyclophilin D, a component of the permeability transition-pore, is an apoptosis repressor. Cancer Res. 64, 85–93 [DOI] [PubMed] [Google Scholar]
- 40. Machida K., Ohta Y., Osada H. (2006) Suppression of apoptosis by cyclophilin D via stabilization of hexokinase II mitochondrial binding in cancer cells. J. Biol. Chem. 281, 14314–14320 [DOI] [PubMed] [Google Scholar]
- 41. Nieminen A. L., Byrne A. M., Herman B., Lemasters J. J. (1997) Mitochondrial permeability transition in hepatocytes induced by t-BuOOH: NAD(P)H and reactive oxygen species. Am. J. Physiol. Cell Physiol. 272, C1286–C1294 [DOI] [PubMed] [Google Scholar]
- 42. Semenza G. L. (2003) Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 3, 721–732 [DOI] [PubMed] [Google Scholar]
- 43. Lesnefsky E. J., Hoppel C. L. (2003) Ischemia-reperfusion injury in the aged heart: role of mitochondria. Arch. Biochem. Biophys. 420, 287–297 [DOI] [PubMed] [Google Scholar]
- 44. Halestrap A. P. (2006) Calcium, mitochondria and reperfusion injury: a pore way to die. Biochem. Soc. Trans. 34, 232–237 [DOI] [PubMed] [Google Scholar]
- 45. Kimura H., Braun R. D., Ong E. T., Hsu R., Secomb T. W., Papahadjopoulos D., Hong K., Dewhirst M. W. (1996) Fluctuations in red cell flux in tumor microvessels can lead to transient hypoxia and reoxygenation in tumor parenchyma. Cancer Res. 56, 5522–5528 [PubMed] [Google Scholar]
- 46. Yasui H., Matsumoto S., Devasahayam N., Munasinghe J. P., Choudhuri R., Saito K., Subramanian S., Mitchell J. B., Krishna M. C. (2010) Low-field magnetic resonance imaging to visualize chronic and cycling hypoxia in tumor-bearing mice. Cancer Res. 70, 6427–6436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dewhirst M. W., Cao Y., Moeller B. (2008) Cycling hypoxia and free radicals regulate angiogenesis and radiotherapy response. Nat. Rev. Cancer 8, 425–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Matsumoto S., Yasui H., Mitchell J. B., Krishna M. C. (2010) Imaging cycling tumor hypoxia. Cancer Res. 70, 10019–10023 [DOI] [PMC free article] [PubMed] [Google Scholar]