

Background: ALG-2 is present both in the cytoplasm and nucleus, but little is known about its nuclear function.
Results: ALG-2 interacts with the SR superfamily protein CHERP and accumulates at nuclear speckles in a Ca2+-dependent manner.
Conclusion: ALG-2 and CHERP participate in alternative splicing.
Significance: We propose a new role of ALG-2 and CHERP in post-transcriptional processing in the nucleus.
Keywords: Alternative Splicing, Calcium, Calcium-binding Proteins, Calcium Signaling, Protein-Protein Interactions, RNA Polymerase II, Subcellular Organelles, SR Proteins, Nuclear Speckles
Abstract
The intracellular Ca2+ signaling pathway is important for the control of broad cellular processes from fertilization to cell death. ALG-2 is a Ca2+-binding protein that contains five serially repeated EF-hand motifs and interacts with various proteins in a Ca2+-dependent manner. Although ALG-2 is present both in the cytoplasm and in the nucleus, little is known about its nuclear function. Ca2+ homeostasis endoplasmic reticulum protein (CHERP) was first identified as an endoplasmic reticulum protein that regulates intracellular Ca2+ mobilization in human cells, but recent proteomics data suggest an association between CHERP and spliceosomes. Here, we report that CHERP, containing a Pro-rich region and a phosphorylated Ser/Arg-rich RS-like domain, is a novel Ca2+-dependent ALG-2-interactive target in the nucleus. Immunofluorescence microscopic analysis revealed localization of CHERP to the nucleoplasm with prominent accumulation at nuclear speckles, which are the sites of storage and modification for pre-mRNA splicing factors. Live cell time-lapse imaging showed that nuclear ALG-2 was recruited to the CHERP-localizing speckles upon Ca2+ mobilization. Results of co-immunoprecipitation assays revealed binding of CHERP to a phosphorylated form of RNA polymerase II. Knockdown of CHERP or ALG-2 in HT1080 cells resulted in generation of alternatively spliced isoforms of the inositol 1,4,5-trisphosphate receptor 1 (IP3R1) pre-mRNA that included exons 41 and 42 in addition to the major isoform lacking exons 40–42. Furthermore, binding between CHERP and IP3R1 RNA was detected by an RNA immunoprecipitation assay using a polyclonal antibody against CHERP. These results indicate that CHERP and ALG-2 participate in regulation of alternative splicing of IP3R1 pre-mRNA and provide new insights into post-transcriptional regulation of splicing variants in Ca2+ signaling pathways.
Introduction
ALG-2 (apoptosis-linked gene 2, also named programmed cell death 6; gene name, PDCD6), a 22-kDa Ca2+-binding protein, was identified by a method to select genes involved in apoptosis of murine cells (1). Although accumulating data indicate involvement of ALG-2 in apoptosis and cancer in mammals (2–6), details of its physiological functions at the molecular level have remained unclear. ALG-2 has five serially repeated EF-hand motifs (penta-EF-hand and a penta-EF-hand domain) (7, 8) and interacts with various proteins, including ALIX (9, 10), Sec31A (11, 12), and PLSCR3 (see Refs. 13 and 14 and references therein). Most ALG-2-binding proteins contain a proline-rich region (PRR),3 through which they interact with a Ca2+-bound form of ALG-2. Identification of ALG-2-binding sites in ALIX (15, 16), Sec31A (17), and PLSCR3 (13) allowed us to determine two different types of ALG-2-binding motifs as follows: type 1, PPYP(X)nYP (where X, variable; n = 4 in ALIX and PLSCR3-ABS1); type 2, PXPGF (X, variable; Sec31A and PLSCR3-ABS2). We recently developed a method to screen novel ALG-2-interacting proteins using these motifs in PRRs for an in silico search, followed by far Western blot analysis of GFP-fused PRR proteins (18). In this study, we selected one of the previously obtained positive candidates, named CHERP (Ca2+ homeostasis endoplasmic reticulum protein), for further characterization as an ALG-2-interacting protein and investigated its biological functions.
CHERP was first identified as a target of a monoclonal antibody that blocked 1,4,5-trisphosphate (IP3)-induced Ca2+ release from the isolated endoplasmic reticulum (ER) (19), and its cDNA was immunoscreened from a cDNA expression library of human erythroleukemia (HEL) cells (20). CHERP was shown to co-localize with IP3 receptors throughout the cytoplasmic and perinuclear regions in HEL cells and in Jurkat cells (20, 21). Antisense-mediated knockdown of CHERP impaired intracellular Ca2+ mobilization and cell growth and proliferation. A recent study has indicated that CHERP interacts with ryanodine receptor 1 (RyR1) and that knockdown of CHERP affects Ca2+ release from the ER (22).
However, proteomics analyses showed that CHERP was present in the fractions of 17 S U2 small nuclear ribonucleoproteins (23) and nuclear speckles (24), which are storage and assembly sites for splicing factors. Lin-Moshier et al. (25) re-investigated subcellular localization of CHERP by immunostaining with a specific antibody and by fluorescence microscopic analysis of GFP-fused CHERP, and they identified nuclear localization signals and concluded that CHERP exclusively localizes to the nucleus, including nuclear speckles. Nuclear function of CHERP, however, has not been demonstrated yet. There is a segment of Arg-Ser dipeptide repeats near the C terminus of CHERP. Ser/Arg-rich (SR) proteins containing a region of Arg-Ser dipeptide repeats (an RS domain) and RNA recognition motifs (RRMs) constitute a family of splicing regulatory factors (26–28). RS domains of SR proteins are phosphorylated at numerous serine residues, and the phosphorylation is thought to play important roles in broad phenomena of RNA processing, including alternative splicing (29). Phosphorylation of the RS-like domain of CHERP, however, has not been reported yet.
In this report, we show that ALG-2 interacts with CHERP in a Ca2+-dependent manner through at least two sites containing ALG-2-binding motif-like sequences in the PRR. ALG-2 was shown to be recruited to CHERP-positive nuclear speckles upon Ca2+ mobilization in living cells by time-lapse imaging of fluorescent protein-fused proteins. Depletion of CHERP or ALG-2 by the RNA interference method affected alternative splicing of the pre-mRNA of inositol 1,4,5-trisphosphate receptor 1 (IP3R1). Association of CHERP with IP3R1 RNA was demonstrated. These findings suggest that CHERP has a new role as an SR superfamily protein and regulates alternative splicing of IP3R1 pre-mRNA. ALG-2 may also participate in the post-transcriptional regulation of IP3R1 pre-mRNA at least in part by interacting with CHERP.
MATERIALS AND METHODS
Antibodies and Reagents
The following antibodies were purchased: mouse anti-GFP monoclonal antibody (mAb) clone B-2 (sc-9996), rabbit anti-FBP21 (WBP4) polyclonal antibody (pAb) (N-16; sc-84249), mouse anti-GAPDH mAb (clone 6C5, sc-32233), and rabbit anti-pol II pAb (N-20, sc-899) from Santa Cruz Biotechnology (Santa Cruz, CA); mouse anti-calnexin mAb (clone 37) from BD Biosciences; rabbit anti-apoptosis-inducing factor pAb (ab1998) and mouse anti-SF3A2 mAb (ab77800) from Abcam (Cambridge, UK); mouse anti-CHERP mAb (clone 2H5) from Abnova (Taipei, Taiwan); mouse anti-SC35 mAb (clone S4045) from Sigma; and mouse mAb against pan-SR proteins (clone 1H4) from Invitrogen. Affinity purification of rabbit anti-human ALG-2 pAb using the recombinant ALG-2 protein immobilized on an N-hydroxysuccinimide column (GE Healthcare) was described previously (12). Anti-human ALG-2 antiserum was also raised in a goat using glutathione S-transferase (GST)-fused ALG-2 as an antigen, and the antibody was similarly affinity-purified (18). Anti-human CHERP antisera were raised in rabbits using a CHERP C-terminal region (820–916 amino acids) protein (CHERPCt) that was fused with GST, and specific antibodies were affinity-purified using CHERPCt that was fused with maltose-binding protein. Protease inhibitors 4-(2-aminoethyl)-benzenesulfonyl fluoride hydrochloride (Pefabloc) and l-trans-epoxysuccinyl-leucylamido(4-guanidino)butane (E-64) were from Merck and the Peptide Institute (Osaka, Japan), respectively. Thapsigargin, an inhibitor of sarcoplasmic/endoplasmic reticulum Ca2+-ATPase (SERCA), was from Wako (Osaka, Japan).
Plasmid Construction
Human CHERP cDNA (clone MHS1011-76655) was purchased from Open Biosystems (Lafayette, CO), and the cDNA of full-length (FL) was inserted between the XhoI site and the SalI site of pEGFP-C1 (Clontech) or pSGFP2-C1 (kindly provided by Dr. Wada, Fukushima Medical University School of Medicine, who constructed the vector according to Ref. 30) using an In-Fusion Advantage PCR cloning Kit (Clontech). To construct plasmids for expression of GFP-fused CHERP deletion mutants designated GFP-CHERP ΔCID and GFP-CHERP ΔRS, cDNA fragments lacking CID (149–293 amino acids) or RS (725–822 amino acids) were amplified by the overlap PCR method using pEGFP-C1/CHERP FL as a template DNA and with two different pairs of specific primers, and each fragment was inserted into the XhoI/SalI site of pEGFP-C1. To construct pEGFP-C1/CHERP ΔPRR, a cDNA fragment encoding the PRR of CHERP (352–705 amino acids) was removed from pEGFP-C1/CHERP FL by cleavage at two EcoRV sites generated by site-directed mutagenesis, followed by self-ligation. Construction of pSGFP2-C1/ALG-2 and pEGFP-C3/CHERP PRR was described previously (17, 18). Other constructs were obtained by the PCR-based subcloning method with specific primers.
pmCherry-C1/ALG-2 was constructed by insertion of an EcoRI/XhoI fragment of pFLAG-ALG-2WT/RNAiR (31) into the EcoRI/SalI site of pmCherry-C1 (Clontech). The nuclear Ca2+-sensor vector CMV-NLS-R-GECO (32) was obtained from the nonprofit plasmid repository Addgene (plasmid 32462).
Cell Culture and DNA Transfection
HEK293T, HeLa SS4 (subcloned HeLa cells, see Ref. 12), and HT1080 cells were cultured in DMEM (Nissui, Tokyo, Japan) supplemented with 4 mm glutamine, 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 μg/ml streptomycin at 37 °C under humidified air containing 5% CO2. Cells were seeded and cultured for 1 day, and then they were transfected with the expression plasmid DNAs by the conventional calcium phosphate precipitation method for HEK293T cells or by using FuGENE 6 (Promega, Madison, WI) for HeLa and HT1080 cells.
Far Western Blot Analysis
Far Western (FW) blot analysis with biotin-labeled ALG-2 was performed as described previously (18, 33). Briefly, HEK293T cells that had been transfected with pEGFP-C1/CHERP FL or its deletion mutants were lysed with lysis buffer T (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1.5 mm MgCl2, and 0.2% Triton X-100 containing protease inhibitors). The cleared lysate was subjected to immunoprecipitation with GFP-TrapA (ChromoTek, Martinsried, Germany), followed by Western blot (WB) and FW analyses.
Subcellular Fractionation and Immunoprecipitation
HEK293T cells were suspended in a hypotonic buffer (10 mm HEPES-KOH, pH 7.6, 10 mm KCl, 1.5 mm MgCl2, 5 mm 2-mercaptoethanol) containing protease inhibitors (0.1 mm Pefabloc, 3 μg/ml leupeptin, 1 μm E-64, 1 μm pepstatin, and 0.1 mm phenylmethylsulfonyl fluoride) and homogenized by passing 25 times through a 26-gauge needle. After adding a solution of NaCl to 0.15 m, the homogenate was fractionated by differential successive centrifugations at 4 °C. Pellets of 600 × g (P0.6), 10,000 × g (P10), 100,000 × g (P100), and the final supernatant of 100,000 × g (S100) were subjected to WB analysis. For co-immunoprecipitation assays, P0.6 fraction was obtained without supplemental NaCl, and CHERP was extracted with lysis buffer T by brief sonication. After centrifugation at 10,000 × g for 10 min at 4 °C, the supernatant treated with RNase A (final 10 μg/ml) was subjected to immunoprecipitation with anti-CHERP or control IgG in the presence of various concentrations of CaCl2 or in the presence of 5 mm EGTA.
Size-exclusion Chromatography
HEK293T cells transiently transfected with plasmids expressing GFP-fused CHERP deletion mutants were lysed with buffer C (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1.5 mm MgCl2, 0.2% CHAPS) containing 3 μg/ml leupeptin, 1 μm pepstatin, and 0.1 mm Pefabloc. After centrifugation at 8400 × g for 10 min at 4 °C, the supernatants were treated with RNase A at a final concentration of 10 μg/ml for 60 min. About 25 μg of proteins were loaded onto a Shodex KW403-4F column (Showa Denko, Tokyo, Japan) pre-equilibrated with buffer C, and were eluted at 0.3 ml/min. The GFP signals derived from each GFP-CHERP deletion mutant were monitored with a fluorescence detector (RF-20A, Shimadzu, Kyoto, Japan). The apparent molecular weights were estimated using the calibration curve obtained from molecular mass standards and the retention times.
Immunofluorescence Microscopic Analysis and Live-cell Imaging
Immunostaining and live-cell time-lapse imaging were performed essentially as described previously (17, 18). Briefly, HeLa cells were seeded in a glass-bottom dish for time-lapse imaging (Asahi Glass, Tokyo, Japan), and the medium was replaced with Leibovitz's L15 medium (Invitrogen) containing 1% fetal bovine serum. Time-lapse images were acquired under an FV1000-D confocal laser-scanning microscope equipped with a ×100, 1.4 numerical aperture (NA) oil-immersion objective (UPLSAPO100XO, Olympus, Tokyo, Japan) before and after treatment with thapsigargin (final concentration of 2 μm) at 37 °C. Images were also processed with ImageJ to make avi movie files.
RNA Interference and RNA Sample Preparation
All siRNAs (siCHERP.1, 5′-gcuagacaugaacgaguuugacaAC-3′; siCHERP.2, 5′-gguuuaggucuagaaagaagaauAC-3′; siALG-2.1, 5′-agcaauaaagggaauguuagacgTG-3′; siALG-2.4, 5′-aaagacaggaguggagugauaucAG-3′; negative control, 5′-cguuaaucgcguauaauacgcguAT-3′; sequences of sense strand shown; deoxyribonucleotides indicated by uppercase letters) were purchased from Integrated DNA Technologies (Coralville, IA). HT1080 cells were transfected with 10 nm siRNA duplexes targeting CHERP, ALG-2, or negative control siRNA using RNAiMAX (Invitrogen) according to the reverse transfection method described in the manufacturer's instructions. Cells were harvested 72 h after siRNA transfection. Total RNA was prepared with Sepasol Super G (Nacalai Tesque, Kyoto, Japan), and genome DNA was digested with 20 units/ml DNase (Nippon Gene, Tokyo, Japan).
Quantitative RT-PCR
RT-PCR and quantitative PCR (qPCR) were performed using a PrimeScriptTM RT reagent kit (Perfect Real Time, RR037A, Takara Bio) and FastStart Essential DNA Green Master (Roche Applied Science), respectively. Reverse transcription products were analyzed by LightCycler® Nano (Roche Applied Science) using specific primers for IP3R1 isoforms (Table 1). GAPDH mRNA was used as an internal control for all qPCRs except for RNA immunoprecipitation assay. Ratios among transcript levels of individual IP3R1 isoforms were obtained by dividing each expression level by the sum of all isoforms.
TABLE 1.
Oligonucleotide primers used for detection of RNA by RT-PCR and quantification of RNA by RT-qPCR
| Protein or RNA names (gene names) | Isoforms | Primer sequences |
|
|---|---|---|---|
| Forward | Reverse | ||
| ALG-2 (PDCD6) | 5′-tcgataaagacaggagtggagtg-3′ | 5′-caaacatggatatgatcgacctg-3′ | |
| CHERP | 5′-ccaatgtgccctacttcgat-3′ | 5′-tgtactcgtgatcttccagctt-3′ | |
| GAPDH | 5′-atgttcgtcatgggtgtgaa-3′ | 5′-ggtgctaagcagttggtggt-3′ | |
| 18S rRNA | 5′-ttgactcaacacgggaaacc-3′ | 5′-tcgctccaccaactaagaacg-3′ | |
| Histone H3 (HIST2H3) | 5′-cgcaggactttaagacgga-3′ | 5′-atgtccttgggcataatggt-3′ | |
| IP3R1 (ITPR1) | ΔE40 | 5′-tggagaaaagcttcctccag-3′ | 5′-gttgaccagaacttgcctgag-3′ |
| ΔE40/41 | 5′-tggagaaaaggagcttgaacc-3′ | 5′-ccaaagctggtaaggctctctc-3′ | |
| ΔE40/41/42 | 5′-atggagaaaagggtgaggc-3′ | 5′-ccaaagctggtaaggctctctc-3′ | |
| All | 5′-agctctgcattaaggtcctac-3′ | 5′-ctgacgtttccatagtaacgg-3′ | |
RNA Immunoprecipitation Assay
RNA immunoprecipitation was performed using an RNA immunoprecipitation assay kit (MBL, Nagoya, Japan) according to the manufacturer's instructions. Briefly, HEK293T cells were lysed in the provided lysis buffer supplemented with 1.5 mm dithiothreitol, 80 units/ml RNase inhibitor (RNaseOUTTM, Invitrogen), and protease inhibitors. An aliquot (2%) of the supernatant obtained by centrifugation at 12,000 × g was used for total RNA isolation. The remainder was incubated at 4 °C for 3 h with protein G-Sepharose beads (GE Healthcare) that were preincubated with rabbit anti-CHERP pAb or control rabbit IgG. After washing with the buffer four times, RNA in the immunoprecipitates was purified and subjected to RT-PCR and qPCR analyses using primers for the cDNAs of IP3R1, GAPDH, and histone H3 (HIST2H3) (Table 1). All RNA levels were normalized to 18 S rRNA.
RESULTS
ALG-2 Interacts with CHERP in a Ca2+-dependent Manner
We previously reported that the GFP-fused PRR of CHERP showed a positive signal in FW blot analysis with biotin-labeled ALG-2 (18), but interaction between endogenous CHERP and ALG-2 remains to be established. Because there have been conflicting reports about the subcellular localization of CHERP (ER or nucleus), we first investigated the subcellular distribution of CHERP. As shown in Fig. 1A, biochemical cell fractionation of HEK293T cells by the differential centrifugation method revealed that WB signals of CHERP were mostly present in the 600 × g pellets (P0.6 fraction) in a manner similar to that of the nuclear marker WW domain-binding protein 4 (WBP4). Detection of GAPDH (cytosolic protein) only in the 100,000 × g supernatant (S100 fraction) indicated that cells were sufficiently lysed by the homogenization procedure used in this study. Because calnexin (ER transmembrane protein) and apoptosis-inducing factor (mitochondrial inter-membrane space protein) were detected in both the P0.6 and P10 fractions, separation of nuclei from the ER and mitochondria was not complete. Signal intensities of ALG-2 were similar in the two fractions of P0.6 and S100, and a faint signal was also detected in the P10 fraction. Next, CHERP was extracted from the P0.6 fraction with sonication in the buffer containing a nonionic detergent (0.2% Triton X-100), and then the supernatant was subjected to co-immunoprecipitation assay. As shown in Fig. 1B, an ALG-2-specific band was detected in the immunoprecipitation (IP) product with an antibody against CHERP (anti-CHERP) in the presence of 10 μm CaCl2 but not in the presence of 5 mm EGTA. No ALG-2 bands were observed in the case of control IgG in either condition, IgG bands of light and heavy chains (IgGL and IgGH) were detected with similar intensities in the two conditions. Faster migrating bands detected with anti-CHERP (upper panel) probably correspond to degradation products of CHERP.
FIGURE 1.

Ca2+-dependent interaction between ALG-2 and CHERP. A, subcellular fractionation of endogenous CHERP. HEK293T cells were homogenized and fractionated by differential successive centrifugations at 4 °C. Pellets of 600 × g (P0.6), 10,000 × g (P10), 100,000 × g (P100), and the final supernatant of 100,000 × g (S100) were subjected to WB with antibodies against CHERP, ALG-2, WW domain-binding protein 4 (WBP4), calnexin, apoptosis-inducing factor (AIF), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). B, co-immunoprecipitation (IP) of endogenous CHERP and ALG-2. CHERP was extracted from the P0.6 fraction by brief sonication with lysis buffer T (50 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1.5 mm MgCl2, and 0.2% Triton X-100 containing protease inhibitors). After centrifugation at 10,000 × g for 10 min at 4 °C, the supernatant (Input) was treated with RNase A (10 μg/ml) and supplemented with 5 mm EGTA (E) or 10 μm CaCl2 (C). Each sample was subjected to immunoprecipitation by incubating first with mouse IgG (negative control, ctrl) or anti-CHERP mAb for 1 h at 4 °C and then with magnetic beads carrying protein G overnight. After the beads had been collected and washed, immunoprecipitated proteins (IP products) were subjected to SDS-PAGE using 12.5% gel, transferred to a sheet of PVDF membrane, which was cut into halves (high and low molecular weight), and subjected to WB with anti-CHERP mAb (upper panel) and with anti-ALG-2 pAb (lower panel). Single and double asterisks indicate light and heavy chains of IgG, respectively. The relative amount of cleared cell lysate proteins (Input) used for analysis of IP products was 1.5%. C, analysis of Ca2+ dependence of binding between ALG-2 and CHERP. Nuclear extracts were supplemented with 5 mm EGTA (lane 2) or with different concentrations of CaCl2 (lane 3, none; lane 4, 1 μm; lane 5, 10 μm; lane 6, 100 μm) and subjected to co-immunoprecipitation with anti-CHERP mAb. The relative amount of cleared cell lysate proteins (Input) used for analysis of IP products was 1.5%. Representative data obtained from three (A), two (B), and four (C) independent experiments are shown.
As shown in Fig. 1C, the amount of ALG-2 that was co-immunoprecipitated with CHERP was increased by increasing the concentration of supplemented CaCl2 from none (lane 3) to 100 μm (lane 6). Under the conditions used, although more than 80% of input CHERP was immunoprecipitated with the antibody (data not shown), ∼1–3% of input ALG-2 was co-immunoprecipitated (lane 1, input, 1.5%). Although the increase of ALG-2 binding at higher concentrations of CaCl2 (Fig. 1C, lanes 5 and 6) is reproducible, the degree was variable from experiment to experiment, and the detected signal of ALG-2 at the lowest supplementation of CaCl2 (Fig. 1C, lane 4, 1 μm) was comparable with that without supplementation (lane 3). The apparent Ca2+ independency at the lower Ca2+ concentration range might have been partly due to a trace of Ca2+ derived from cells or from the lysis buffer and partly due to the complex that had been already formed Ca2+-dependently within the nucleus, because addition of 5 mm EGTA (Fig. 1C, lane 2) caused disappearance of the ALG-2 band.
Multiple ALG-2-binding Sites in the PRR of CHERP
In addition to the PRR, CHERP has four distinct regions with sequences similar to those designated SURP (also named SWAP), CID (RNA polymerase II C-terminal domain-interacting domain), RS (Arg-Ser dipeptide repeats), and G-patch. The PRR of CHERP has a sequence, 565PPYPHRFDYP574, that is in accordance with the type 1 ALG-2-binding motif (conserved residues underlined). To obtain evidence that this sequence is essential for CHERP to interact with ALG-2, we first constructed GFP fusion expression vectors encoding the FL CHERP protein and a deletion mutant of this region (Δ557–585) as well as various other mutants (Fig. 2A), and then we carried out FW with biotin-labeled (bio-) ALG-2 and WB with anti-GFP to check the relative amounts of proteins resolved by SDS-PAGE (Fig. 2B). Unexpectedly, FW signals were detected for Δ557–585 (Fig. 2B, left lower panel, lane 6) with intensity only slightly weaker than that of the FL protein (lane 1), indicating that other regions also contribute to the ALG-2 binding. Because mutants with deletion of CID (Fig. 2B, ΔCID, lane 2) and deletion of the RS domain (ΔRS, lane 4) retained binding abilities, but the PRR-deletion mutant (ΔPRR, lane 3) did not, we focused on the PRR for further search for ALG-2-binding sites. The N-terminal end of the PRR has a unique Pro-Ala repeat sequence (354TPPPPAPPPAPAPAPAIPP372), but deletion of this sequence had little effect on the binding (Fig. 2B, Δ354–372, lane 5). Although it is not in accordance with the previously proposed type 2 motif (PXPGF), the PRR has a similar sequence of 614PPPHGF619. To determine the importance of this type 2 motif-like sequence for ALG-2 binding, we further deleted this region (Δ557–585/Δ613–623) or replaced Phe619 with Ser (Δ557–585/F619S) in addition to the deletion of type 1 motif-like sequence (Δ557–585). These mutations caused a significant decrease in signal intensity in FW, but faint bands still remained (Fig. 2B, lanes 7 and 8). As shown in the right panels in Fig. 2B, compared with GFP-PRR, intensity of FW signals of GFP-PRR decreased by truncation of the C-terminal region containing the type 2 motif-like sequence (GFP(343–599)), or FW signals disappeared in GFP(343–462). Because signal intensity of GFP(550–636) was weaker than that of GFP PRR (Fig. 2B, lane 10 versus lane 13), there may exist at least one more important site in either side of 550–636 for ALG-2-binding.
FIGURE 2.

Identification of ALG-2-binding sites and subcellular localization of GFP-CHERP mutants. A, schematic diagrams of GFP-fused CHERP and its deletion mutants. CHERP has five distinct regions containing sequences similar to those designated SURP, CID (RNA polymerase II C-terminal domain-interacting domain), PRR (Pro-rich region), RS (Arg/Ser-rich region), and G-patch. Approximate positions of three motifs (PA repeat, PPYPHRFDYP, and PPPHGF) are indicated at the top bar and marked with small filled boxes in black and gray in each expression construct. Filled arrowhead indicates replacement of Phe619 with Ser (F619S mutation). B, FW blot analysis of GFP-fused CHERP and its deletion mutants with biotin-labeled ALG-2. HEK293T cells expressing GFP-fused CHERP FL and mutant proteins were lysed and centrifuged at 10,000 × g for 10 min. GFP fusion proteins were immunoprecipitated with GFP-TrapA from the cleared cell lysates and subjected to FW blot analysis with biotin-labeled ALG-2 (bio-ALG-2) and WB analysis using anti-GFP mAb. C, HeLa cells expressing GFP-fused CHERP FL and deletion mutants were fixed in 4% paraformaldehyde in PBS and permeabilized with 0.1% Triton X-100. Then the cells were immunostained with anti-ALG-2 pAb. Nuclear DNA was stained with Hoechst 33342. Fluorescence signals in the merged images are represented in pseudocolors as follows: Hoechst, blue; GFP, green; ALG-2, red. Bars, 10 μm. D, HeLa cells expressing GFP-CHERP ΔRS were fixed in 4% paraformaldehyde in PBS and permeabilized with 0.1% Triton X-100. Then the cells were double-immunostained with anti-ALG-2 pAb (red) and anti-Sec31A mAb (blue). Bars, 5 μm. Lower panels are magnified images of upper panels. Arrows, Puncta of GFP-CHERP ΔRS are co-localized with those of ALG-2 and Sec31A. Solid arrowheads, puncta of GFP-CHERP ΔRS are co-localized only with those of ALG-2. Unfilled arrowheads, puncta of ALG-2 are co-localized only with those of Sec31A. Representative data obtained from at least two independent experiments are shown for B–D.
CHERP Is Present in the Nucleus and Concentrated at Nuclear Speckles
We performed fluorescence microscopic analysis of GFP-CHERP mutant proteins expressed in HeLa cells. Although the full-length and the mutants deleted in CID or PRR showed exclusively nuclear localization, a deletion mutant of RS (GFP-CHERP ΔRS) showed cytoplasmic as well as nuclear localization (Fig. 2C). All cytoplasmic punctate signals of GFP-CHERP ΔRS merged with those of ALG-2 and partially overlapped with those of Sec31A, a component of COPII (Fig. 2D). To investigate the subnuclear distribution of endogenous CHERP, we carried out triple immunostaining of HeLa cells with rabbit anti-CHERP pAb, goat anti-ALG-2 pAb, and mouse mAb against SC35 (alternatively called SRSF2), which is a splicing factor that mediates specific interactions between U1 and U2 small nuclear ribonucleoprotein particles at the 3′ splice site (26), and which is a well known marker of nuclear speckles (34). Effects of pre-permeabilization with digitonin before fixation were also investigated. Immunofluorescence signals of CHERP were observed in the nucleus (Fig. 3), and the pre-permeabilization had little effect (b and f). Dense signals of CHERP in the nucleus partially overlapped with those for SC35 (Fig. 3, c and g), indicating partial localization of CHERP at nuclear speckles. Nuclear ALG-2 signals were rather uniformly diffused in the nucleoplasm compared with those of CHERP without pre-permeabilization (Fig. 3, a) but became punctate by pre-permeabilization with digitonin (e).
FIGURE 3.
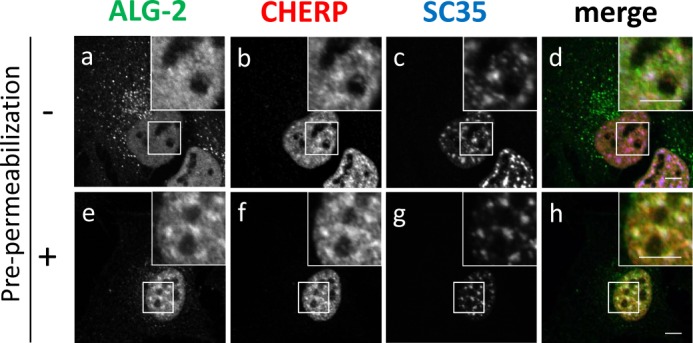
Localization of endogenous CHERP at nuclear speckles by immunofluorescence microscopic analysis. HeLa cells were cultured on coverslips, fixed, and processed for pre-permeabilization with 50 μg/ml digitonin in buffer D (20 mm HEPES-KOH, pH 7.6, 50 mm NaCl, 2 mm MgCl2, 250 mm sucrose, 1 mm DTT) (e–h) on ice for 10 min and washed with buffer D or unprocessed for pre-permeabilization (a–d), fixed in 4% paraformaldehyde in PBS, and permeabilized with 0.1% Triton X-100. Then, the cells were triple-immunostained with goat anti-ALG-2 pAb (green), rabbit anti-CHERP pAb (red), and mouse anti-SC35 mAb (blue). The fluorescence signals were analyzed with a confocal laser-scanning microscope and are represented in black and white. Merged images are shown in d and h in color. The small boxed areas are magnified in the respective large boxed areas. Bars, 5 μm. Representative data obtained from three independent experiments are shown.
ALG-2 Transiently Accumulates at Nuclear Speckles in Response to Ca2+ Mobilization
By live-cell imaging, ALG-2 has been shown to translocate to the Sec31A-positive ER exit sites transiently in response to Ca2+ mobilization (17, 35). We previously noticed the appearance of ALG-2 puncta in the nucleus but left them uncharacterized (17). Now we know that ALG-2 binds to CHERP Ca2+-dependently, we have decided to re-investigate whether fluorescent protein-fused ALG-2 in living cells responds to thapsigargin (a SERCA pump inhibitor and Ca2+-elevating agonist) and accumulates at nuclear speckles by co-localizing with CHERP. First, we expressed SGFP2-ALG-2 and NLS-R-GECO, which is a nuclear Ca2+-indicator protein (32). Thapsigargin (TG) administration resulted in redistribution of cytoplasmic and nuclear ALG-2 concomitantly with an increase in NLS-R-GECO signals (Fig. 4, A, panels a–d, and B, and supplemental Movie S1). Fluorescent signals of SGFP2-ALG-2 at nucleoplasmic puncta were increased transiently (ROI-1 and ROI-2), whereas the signals remained constant in the nucleoplasm (ROI-3) and were decreased in the nucleolus (ROI-4). Next, cells expressing SGFP2-CHERP and mCherry-ALG-2 at low to middle levels were used for time-lapse analysis. Like SGFP2-ALG-2, mCherry-ALG-2 localized throughout the cytoplasm and nucleus, uniformly in the nucleoplasm outside of nucleoli before stimulation with thapsigargin (Fig. 4C, panel f). However, SGFP2-CHERP localized within the nucleoplasm and accumulated at nuclear speckles (Fig. 4C, panels e and g) in a manner similar to that for immunostaining of fixed cells with anti-CHERP pAb (Fig. 3b). Thapsigargin treatment induced enhancement of nuclear mCherry-ALG-2 signals in a speckled pattern, and these speckled signals of mCherry-ALG-2 partially merged with those of SGFP2-CHERP (Fig. 4C, panels g and h, and supplemental Movie S2). No significant alteration of SGFP2-CHERP was observed after stimulation with thapsigargin (Fig. 4D).
FIGURE 4.

Transient translocation of GFP-ALG-2 to nuclear speckles by treatment with thapsigargin. A and C, HeLa cells co-expressing SGFP2-ALG-2 and NLS-R-GECO (A) or SGFP2-CHERP and mCherry-ALG-2 (C) were incubated for 15 min in Leibovitz L15 medium containing 1% fetal bovine serum. Time-lapse images were acquired using a confocal microscope. The corresponding movies (A, supplemental Movie S1; C, supplemental Movie S2) are shown in the supplemental material. Panels a and b and c and d correspond to images before (at 160 s) and after (at 276 s) addition of TG (final concentration of 2 μm at 196 s) in A, respectively (time after starting acquisition of images indicated in parentheses). Panels e and f and g and h correspond to images before (at 231 s) and after (at 603 s) addition of TG (at 276 s) in C, respectively. Bars, 5 μm. B and D, temporal changes in fluorescence intensities of four areas (regions of interest, ROI) indicated by white boxes shown in panels a and b in A and those in panels e and f in C were measured by Olympus FV10-ASW software (B, ROI-1, speckle; ROI-2, speckle; ROI-3, nucleoplasm; ROI-4, nucleolus; D, ROI-5, speckle; ROI-6, speckle; ROI-7, nucleoplasm; ROI-8, nucleolus). Changes in relative fluorescence intensity (ΔF/F0) derived from SGFP2-ALG-2 and NLS-R-GECO (B) and in that derived from SGFP2-CHERP and mCherry-ALG-2 (D) are represented with black and gray curves for each ROI as indicated. The time of addition of TG and the time points at which images are shown in A and C are indicated by arrowheads and arrows, respectively. Representative data obtained from three independent experiments are shown.
Arg/Ser-rich Region in CHERP Is Constitutively Phosphorylated
An RS domain is defined as a region of at least 50 amino acids with >40% Arg/Ser content characterized by consecutive Arg-Ser or Ser-Arg dipeptide repeats (28). CHERP has a similar Arg/Ser-rich region (718–817) and the dipeptides span 737–817 amino acids with 25 repeats (62% RS content). The RS domains of SR proteins, which are defined as RNA-binding proteins with RS domains and RRMs, are known to be constitutively phosphorylated (29). To investigate whether the Arg/Ser-rich region of CHERP is similarly phosphorylated, CHERP was immunoprecipitated with rabbit anti-CHERP pAb from HEK293T cells, and then the immunoprecipitates were subjected to WB analysis with rabbit anti-CHERP pAb and with commercially available mouse mAb against pan-SR proteins (clone 1H4), which recognizes a broad range of phosphorylated RS domains (36, 37). As shown in Fig. 5, upper panel, CHERP-specific bands detected with anti-CHERP shifted downward after treatment with calf intestine alkaline phosphatase (CIAP) for 4 h in the absence of phosphatase inhibitors, but the presence of inhibitors partially blocked the downward shift. Anti-SR mAb recognized endogenous CHERP proteins that were not treated with CIAP, treated with phosphatase inhibitors alone, and treated with CIAP in the presence of phosphatase inhibitors (Fig. 5, lower panel). As expected, this antibody did not detect the downward-shifted band of CHERP after CIAP treatment, strongly suggesting that the RS domain of CHERP is phosphorylated at Ser residues in a manner similar to the RS domains of SR proteins.
FIGURE 5.
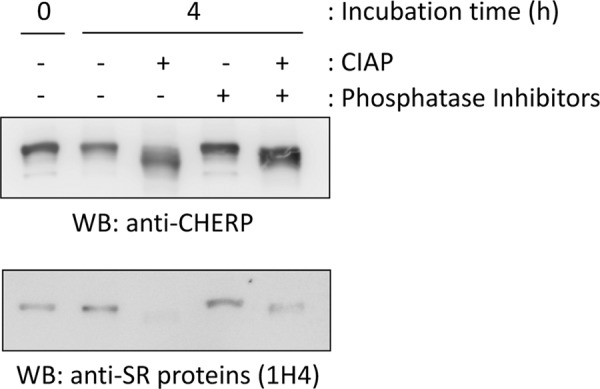
Detection of CHERP with anti-phosphorylated RS domains by WB. HEK293T cells were lysed with buffer T, and the cleared cell lysate was subjected to immunoprecipitation with rabbit anti-CHERP pAb. The IP products were treated with or without 0.5 units/μl CIAP in a reaction buffer (50 mm Tris-HCl, pH 8.0, 0.1 mm Zn(OAc)2, 1 mm MgCl2) in the presence or absence of phosphatase inhibitors (10 mm NaF, 1 mm NaVO4, 5 mm sodium pyrophosphate, and 10 mm β-glycerophosphate) at 37 °C for 4 h and then subjected to WB with rabbit anti-CHERP pAb (upper panel) and mouse mAb against pan-SR proteins (1H4) (lower panel). Representative data obtained from two independent experiments are shown.
CHERP Interacts with Phosphorylated RNA Polymerase II
According to the annotation in the protein database of Uniprot/Swiss-Prot, CHERP contains a CTD (C-terminal domain of RNA polymerase II)-interacting domain (CID) based on the primary sequence similarity. To experimentally investigate whether CHERP interacts with RNA polymerase II (pol II), we performed a co-immunoprecipitation assay with anti-pol II pAb or with anti-CHERP pAb in the presence or absence of Ca2+ using the nuclear extract from HEK293T cells. As shown in the upper panel of Fig. 6A, CHERP-specific bands were detected in the immunoprecipitates (IP) with anti-CHERP but not with control IgG. Two specific bands were detected by WB with anti-pol II in the total and nuclear extract fractions. The upper bands (pol IIO), absent in the post-nuclear supernatant, correspond to the phosphorylated transcriptionally engaged forms of pol II, and the lower bands (pol IIA) correspond to the unphosphorylated and transcriptionally unengaged forms (38). Only pol IIO bands were detected in the immunoprecipitates with anti-CHERP both in the presence of 10 μm CaCl2 and in the presence of 5 mm EGTA, indicating that CHERP and pol IIO interact Ca2+-independently. However, ALG-2 bands were observed in the immunoprecipitates with anti-CHERP in the presence of 10 μm CaCl2 but not in the presence of 5 mm EGTA. We performed a complementary co-immunoprecipitation assay with anti-pol II for immunoprecipitation and detected CHERP in the immunoprecipitates (Fig. 6B). ALG-2 was also co-immunoprecipitated with anti-pol II pAb but only in the presence of CaCl2.
FIGURE 6.
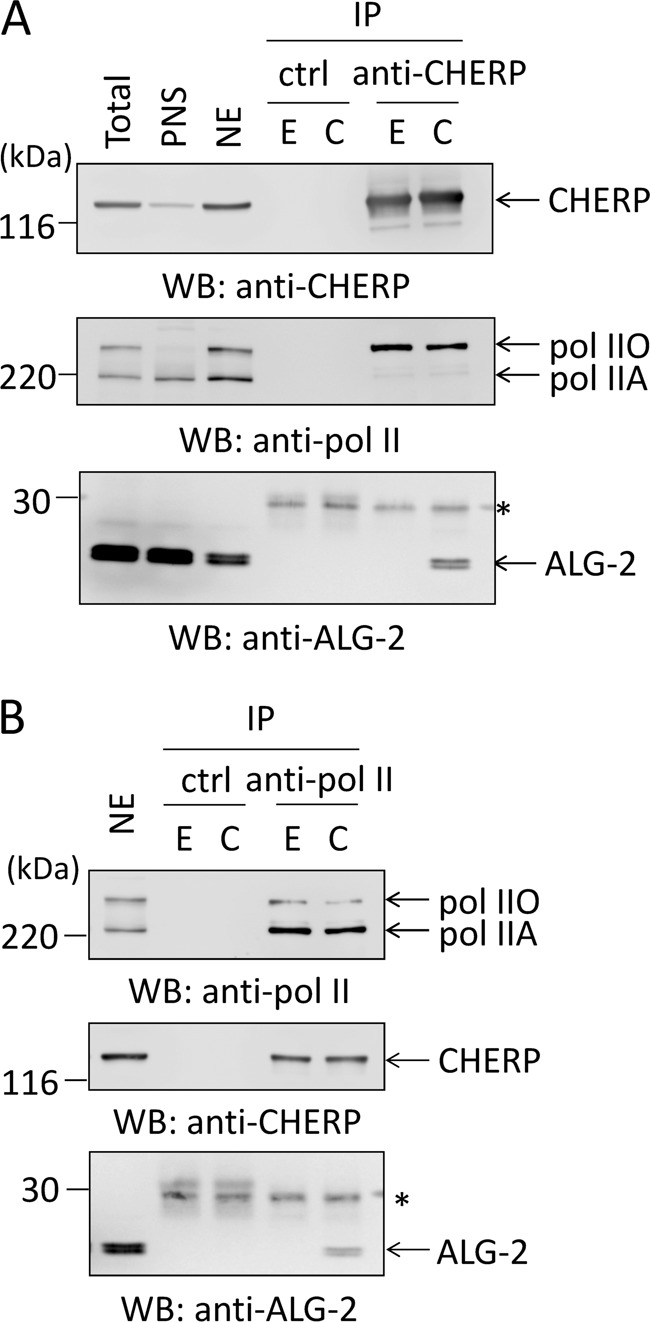
Binding between CHERP and RNA polymerase II. The crude nuclear fraction (P0.6 fraction) was prepared from HEK293T cells, and the nuclear extract (NE) was treated with 10 μg/ml RNase A. After addition of 5 mm EGTA (E) or 10 μm CaCl2 (C), the nuclear extract was subjected to immunoprecipitation with rabbit anti-CHERP pAb (A) or rabbit anti-pol II pAb as well as with rabbit IgG as a negative control (B). The immunoprecipitated proteins (IP) were subjected to WB with rabbit anti-CHERP pAb, anti-pol II pAb, or rabbit anti-ALG-2 pAb. The relative amounts of cleared cell lysate proteins (nuclear extract) used for analysis of IP products were 5% except for ALG-2 (2.5%) in B. PNS, post-nuclear supernatant; pol IIO, phosphorylated form of RNA polymerase II; pol IIA, unphosphorylated form of RNA polymerase II. Asterisk, IgG light chain. Representative data obtained from two (A) and three (B) independent experiments are shown.
PRR of CHERP Associates with RNA Polymerase II
Next, we examined whether the database-annotated CID of CHERP is essential for interaction between CHERP and RNA pol II by expressing various deletion mutants of GFP-CHERP and carrying out immunoprecipitation with anti-pol II pAb, followed by WB with anti-GFP mAb. As shown in Fig. 7, unexpectedly, GFP-CHERP ΔCID was co-immunoprecipitated equally well in comparison with GFP-CHERP FL. The remaining PRR within GFP-CHERP ΔCID is likely responsible for the binding, because the PRR of CHERP alone was sufficient for the association with RNA pol II, whereas the deletion of the PRR (GFP-CHERP ΔPRR) failed to bind to RNA pol II.
FIGURE 7.
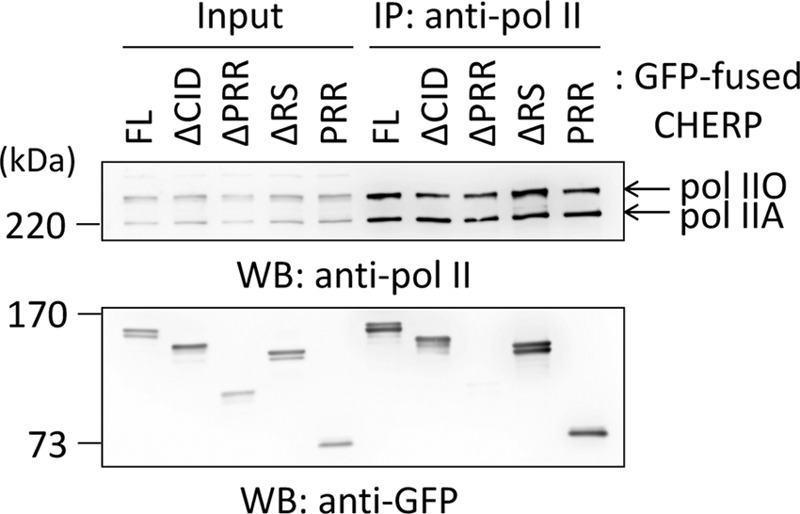
Identification of RNA polymerase II-binding region in CHERP. The nuclear extract (NE) prepared from HEK293T cells expressing GFP-fused CHERP FL or each deletion mutant was treated with 10 μg/ml RNase A and subjected to immunoprecipitation with rabbit anti-pol II pAb in the presence of phosphatase inhibitors. The immunoprecipitated proteins (IP) were subjected to WB using rabbit anti-pol II pAb and mouse anti-GFP mAb. The relative amount of nuclear extract proteins used for analysis of IP products was 5%. pol IIO, phosphorylated form; pol IIA, unphosphorylated form. Representative data obtained from four independent experiments are shown.
We further investigated the state of complex formation of GFP-CHERP deletion mutants by size-exclusion chromatography. As shown in Fig. 8A, the results revealed that a majority of the GFP fusion protein of the full-length was eluted at ∼163 kDa, slightly greater than the molecular mass of monomeric protein calculated from the amino acid sequence (132.7 kDa, Table 2). A second peak appeared faster (>669 kDa), suggesting that some portion of CHERP population exists in an aggregate oligomeric form or in a complex with other proteins, probably including RNA pol II and RNA processing factors. GFP-CHERP ΔCID also exhibited peaks corresponding to a monomeric form (116 kDa) and aggregates (>669 kDa). Because a single band of the expected size was observed by Western blotting (Fig. 8B), the appearance of additional multiple peaks of smaller molecular masses suggests a higher susceptibility of the deletion mutant to proteolysis during the analytical size-exclusion chromatography that was performed at room temperature. GFP-CHERP ΔRS was eluted at ∼172 kDa, slightly greater than a monomeric form (121.5 kDa). GFP-CHERP-PRR (68 kDa) was eluted at ∼81 kDa. In contrast, GFP-CHERP ΔPRR was exclusively eluted at higher molecular masses (>669, 395, and 204 kDa in an approximate peak height ratio of 3:11:8) than the monomeric form (93 kDa), suggesting occurrence as an aggregate oligomeric form or in a complex with other proteins. Effects of deletions in CHERP displayed a different pattern in the size-exclusion chromatography, suggesting that each region has a different structural role in protein-protein interactions. Although the PRR itself is likely to be sufficient for interaction with pol II, potential misfolding of the GFP-CHERP proteins caused by deletion should be also considered, and we cannot exclude the possibility that other regions, including CID, also have the capacity to interact with pol II independently of the PRR.
FIGURE 8.

Size-exclusion chromatography profiles of GFP-fused CHERP deletion mutants. A, HEK293T cells transiently expressing indicated GFP-fused CHERP deletion mutants were lysed, and cell extracts treated with RNase A were resolved by size-exclusion chromatography. The GFP signals derived from each GFP-CHERP deletion mutant were monitored with a fluorescence detector. A number indicated on the top of each GFP signal peak stand for the retention time. The positions of molecular mass standards (in kilodaltons, kDa) are indicated in the panel for control (No transfection). B, cell extracts used for A were analyzed by WB with anti-GFP mAb.
TABLE 2.
Size-exclusion chromatography analyses of GFP-fused CHERP deletion mutants
| Sample | Mr (×103) | Peak retention time | Peak height (>100) | Calculated Mr (×103) |
|---|---|---|---|---|
| min | ||||
| GFP | 29 | 11.039 | 131,836 | 42.9 |
| GFP-CHERP FL | 132.7 | 6.795 | 463 | >669 |
| 8.385 | 247 | 315.9 | ||
| 9.263 | 1497 | 163.3 | ||
| 10.929 | 357 | 46.6 | ||
| GFP-CHERP DCID | 116 | 6.298 | 118 | >669 |
| 9.578 | 345 | 128.8 | ||
| 10.29 | 615 | 75.4 | ||
| 10.91 | 330 | 47.3 | ||
| GFP-CHERP DPRR | 93 | 7.169 | 3376 | >669 |
| 8.087 | 11116 | 395.3 | ||
| 8.966 | 8086 | 204.1 | ||
| 11.008 | 145 | 44.0 | ||
| GFP-CHERP DRS | 121.5 | 7.174 | 1168 | >669 |
| 8.276 | 1708 | 342.9 | ||
| 9.195 | 20,076 | 171.8 | ||
| GFP-CHERP-PRR | 68 | 8.979 | 201 | 202.1 |
| 10.198 | 1832 | 80.8 | ||
| 11.003 | 1223 | 44.1 |
CHERP Knockdown Increases Inclusion of Exons 41 and 42 in IP3R1 Pre-mRNA
CHERP was first identified as a modulator of the IP3 receptor and was shown to regulate Ca2+ homeostasis (19, 20). Because CHERP localizes partly at nuclear speckles, contains the phosphorylated RS domain, and interacts with RNA pol II, we speculated that abnormality in Ca2+ homeostasis caused by CHERP depletion in the previous reports might have been due to indirect effects of nuclear events. Hence, in this study, we analyzed the roles of CHERP and ALG-2 on alternative splicing of pre-mRNAs of Ca2+-related proteins. IP3R1 has three segments (S1, S2, and S3) that are reported to be variable in the splicing variants (39–41). S2 corresponds to exons 40–42. To investigate whether knockdown of CHERP causes changes in a splicing pattern of variable segments of S2 in IP3R1 pre-mRNA, we performed RT-PCR using total RNA obtained from HT1080 cells that were transfected with siRNAs targeting CHERP (siCHERP.1 and .2) or negative control siRNA (NC) and with the use of S2-specific primers 1F and 1R (see Fig. 9A). Amplicons that skip exons 40–42 were exclusively produced from cells of mock transfection (data not shown) and NC siRNA, and amplicons that lack only exon 40 (ΔE40) or both exons 40 and 41 (ΔE40/41) were increased by transfection with siCHERP.1 and siCHERP.2 compared with NC (Fig. 9B). No significant alteration of splicing patterns was observed in the S1 and S3 segments (data not shown). To further quantitatively analyze the effects of knockdown of CHERP or ALG-2 on alternative splicing in S2, we performed real time qPCR using each isoform-specific primer, which corresponds to the splice junction sequence (Fig. 9A). Transfection of HT1080 cells with siCHERP.1 or siCHERP.2 reduced the level of CHERP mRNA to ∼25% as shown in the lower panel of Fig. 9C. We observed statistically significant increases in percentage of isoforms ΔE40 and ΔE40/41 in CHERP knockdown cells compared with the control (percentage of ΔE40: NC, 0.0410 ± 0.0063; siCHERP.1, 0.659 ± 0.149; siCHERP.2, 0.506 ± 0.0625; percentage of ΔE40/41: NC, 1.16 ± 0.0820; siCHERP.1, 4.60 ± 0.597; siCHERP.2, 4.64 ± 0.579) (Fig. 9C, upper panel). Likewise, increases in ΔE40 and ΔE40/41 were observed (percentage of ΔE40: NC, 0.0434 ± 0.00440; siALG-2.1, 0.186 ± 0.0242; siALG-2.4, 0.288 ± 0.0367; percentage of ΔE40/41: NC, 1.11 ± 0.114; siALG-2.1, 2.16 ± 0.199; siALG-2.4, 2.65 ± 0.227) (Fig. 9D, upper panel) in ALG-2 knockdown cells, in which ALG-2 mRNA was reduced to less than 20% of the negative control (Fig. 9D, lower panel).
FIGURE 9.

Effects of CHERP or ALG-2 knockdown on alternative splicing of IP3R1 pre-mRNA. A, schematic diagram of the alternative spliced region (S2 splice site, exons 40–42) of IP3R1 pre-mRNA. Arrows indicate PCR primers used in this study. B, total RNA was extracted from HT1080 cells transfected with siRNAs specific for CHERP (siCHERP.1 and siCHERP.2) or negative control siRNA (NC) and analyzed by RT-PCR with forward (1F) and reverse (1R) primers to detect all isoforms. Representative data obtained from three independent experiments are shown. C and D, total RNA was extracted from HT1080 cells that were transfected with siRNAs specific for CHERP, ALG-2 (siALG-2.1 and siALG-2.4) or negative control (NC). After reverse transcription, real time quantitative PCR (RT-qPCR) was performed with primers for the detection of each splicing isoform of IP3R1 pre-mRNA (ΔE40: 2F and 2R; ΔE40/41: 3F and 3R; ΔE40/41/42: 4F and 3R) as well as the mRNA levels of CHERP and ALG-2. *, p < 0.05; **, p < 0.01; ***, p < 0.001 versus each control (analysis of variance followed by Dunnett's test; mean ± S.E.; n = 4). All RNA levels were normalized to GAPDH mRNA that was used as a reference RNA.
CHERP Physically Associates with IP3R1 RNA
To investigate more direct contribution of CHERP to alternative splicing of IP3R1 pre-mRNA, we performed an RNA immunoprecipitation assay with anti-CHERP pAb and analyzed associated RNAs by RT-PCR. A CHERP-specific WB band was detected in the IP product of anti-CHERP pAb but not in that of rabbit control IgG (Fig. 10A). The cDNA fragment, amplicon of 118 bp, corresponding to segment S2 in IP3R1 was detected by 5% PAGE followed by ethidium bromide staining in the total fraction and in the IP product of anti-CHERP pAb after reverse transcription (+RT) but not without reverse transcription (−RT) (Fig. 10B). Furthermore, we carried out RT-qPCR to measure the relative abundance of IP3R1 mRNA contained in the IP product of anti-CHERP pAb. As shown in Fig. 10C, the degree of enrichment of IP3R1 mRNA from total RNA by immunoprecipitation with anti-CHERP pAb (2.38 ± 0.245) was ∼8-fold greater than that by immunoprecipitation with control IgG (0.311 ± 0.176), whereas mRNAs of histone H3 and GAPDH were not enriched.
FIGURE 10.
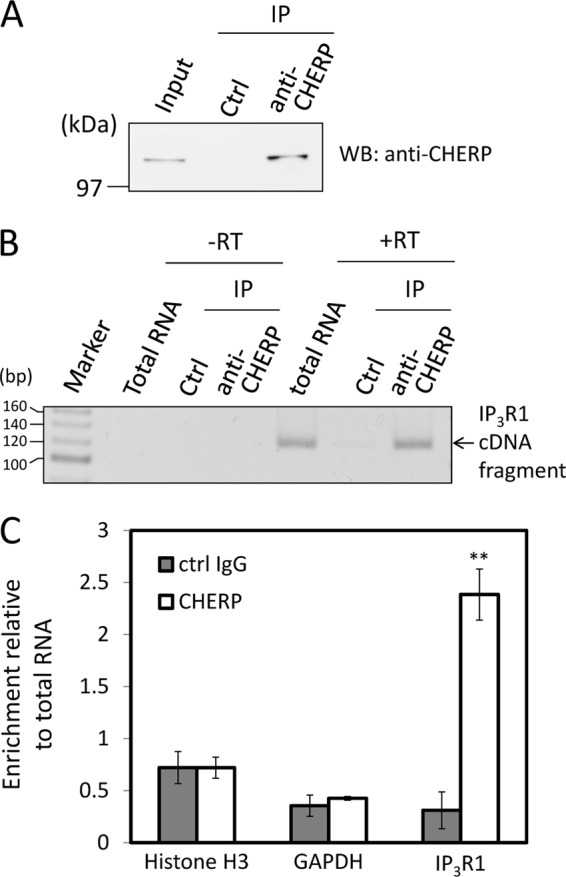
Physical association of CHERP with IP3R1 RNA. A, RNA immunoprecipitation (RIP) was performed with rabbit IgG (control, ctrl) or rabbit anti-CHERP pAb. Immunoprecipitated (IP) proteins were subjected to WB using rabbit anti-CHERP pAb. The relative amount of cleared cell lysate proteins (Input) used for analysis of IP products was 1.5%. B, RNAs were isolated from whole cells (total RNA) or immunoprecipitates (IP) and subjected to RT-PCR after reverse transcription (+RT) or without reverse transcription (−RT) for detection of the IP3R1 segment S2 with the primers of 1F and 1R as indicated in Fig. 9A. RT-PCR products were analyzed by 5% PAGE and stained with ethidium bromide. C, relative amounts of RNA in the immunoprecipitates were measured by RT-qPCR for histone H3, GAPDH, and IP3R1 RNAs using 18 S rRNA as a reference RNA. Relative degree of enrichment of analyzed RNA in the immunoprecipitates compared with total RNA is expressed in the ordinate. **, p < 0.01 versus each control IgG (Student's t test; mean ± S.E.; n = 3). Representative data obtained from three independent experiments are shown.
DISCUSSION
CHERP was first identified as an ER-localizing protein that modulates the IP3 receptor (19, 20). Lin-Moshier et al. (25) recently re-investigated the subcellular localization of CHERP and concluded that CHERP is a nuclear protein, and they identified four NLS as follows: one in the boundary of the RS domain, two within the RS domain, and one near the C terminus. We also observed predominant nuclear localization of endogenous CHERP in several types of cells, including HeLa, HEK293, HT1080, and Jurkat cells by immunostaining with a specific antibody (Fig. 3, data not shown). GFP-fused CHERP, fused at the N or C terminus, also showed nuclear localization by fluorescence microscopic analysis (Fig. 2 and data not shown). Although immunostaining with a commercially available monoclonal antibody against CHERP also showed predominant nuclear localization of CHERP, it displayed weak signals of CHERP throughout the cytoplasm (data not shown). Expression of GFP-CHERP ΔRS resulted in partial distribution of the mutant in the cytoplasm (Fig. 2C). Interestingly, a fraction of cytoplasmic GFP-CHERP ΔRS co-localized with Sec31A (Fig. 2D), a COPII component, which is found co-localized with ALG-2 at ER exit sites (11, 12). We found that CHERP is phosphorylated and is recognized by a monoclonal antibody against pan-SR proteins with phosphorylated RS domains (Fig. 5). A recent study has indicated that CHERP is likely to be a substrate of SRPK1 and SRPK2 (42), which are SR-specific protein kinases and control nuclear entry and speckle formation of SR proteins (29). CHERP may shuttle between the nucleus and the cytoplasm under regulation by serine-arginine protein kinases. Because an ALG-2 dimer has the capacity to bridge two different interacting proteins as a Ca2+-dependent adaptor (31), CHERP may have an intrinsic property of association with the ER membrane through the ALG-2·Sec31A complex under certain physiological or nonphysiological conditions, including in vitro biochemical experiments.
In the protein database of UniProt, CHERP is also designated SR-related RNA polymerase II CTD-associated factor 6 (SCAF6) as an alternative name, and it has been assigned to contain a CID (a CTD-interacting domain). The CID of RBM16/SCAF8 was previously shown to bind to the phosphorylated form of the CTD of pol II (43). The CID of CHERP exhibits a lower degree of similarity with SCAF4 (31% identity) and with SCAF8 (26% identity) than the similarity (81% identity) between SCAF8 and SCAF4 at the primary structural level, and it remains to be clarified whether CHERP binds to the CTD of pol II directly. Some SR proteins interact with pol II directly or via nascent pre-mRNAs and function in coupling transcription to splicing (44). Because CHERP does not have an RRM common to SR proteins, CHERP might associate with RNA via interacting with other proteins that possess RNA-binding motifs such as SR140, a potential interacting protein of CHERP (25). Results of our preliminary experiments showed that another RRM-containing protein, SCAF4, was also co-immunoprecipitated with an antibody against CHERP (data not shown). However, we cannot completely rule out the possibility of direct interaction between CHERP and RNA. Because the PRR of CHERP is well conserved in vertebrates (data not shown), it might have a functional role in protein-protein or protein-RNA interactions in addition to the presence of the direct binding sites for ALG-2 and direct or indirect binding sites for RNA pol II.
In contrast to the punctate distribution of CHERP and SC35 at the nuclear speckles, nuclear ALG-2 showed a diffuse pattern by immunofluorescence microscopic analysis (Fig. 3). Pre-permeabilization of cells with digitonin before fixation, however, enhanced the relative signal intensity of ALG-2 at nuclear speckles, probably due to washing out of nucleoplasmic free ALG-2 through nuclear pores but anchoring of ALG-2 to speckles via association with speckle components (Fig. 3). We demonstrated thapsigargin-induced Ca2+-dependent transient accumulation of ALG-2 at the nuclear speckles by time-lapse live-cell imaging (Fig. 4 and supplemental Movies S1 and S2). SGFP2-ALG-2 co-localized with mCherry-fused splicing factor 3A subunit 2 (SF3A2), which is a component of U2 small nuclear ribonucleoprotein (supplemental Movie S3). An increase in nuclear Ca2+ caused no obvious change in nuclear speckle accumulation of CHERP and SF3A2, indicating that Ca2+/ALG-2 is not essential for subnuclear localization of these proteins. Knockdown of CHERP by siRNA did not abolish the Ca2+-dependent accumulation of ALG-2 (data not shown). It remains unknown whether depletion of CHERP by the siRNA method was merely insufficient or whether ALG-2 associates with other nuclear speckle-localizing proteins in addition to CHERP. Studies are in progress to search for additional nuclear proteins that associate with ALG-2 in a Ca2+-dependent manner.
RBM22, a spliceosome-associated RNA-binding protein, was previously shown to induce translocation of ALG-2 from the cytoplasm to the nucleus by overexpression experiments, and interaction with ALG-2 was proposed (45). RBM22 contains the sequence 373PPPPPGF379, which matches with an ALG-2-binding motif type 2. We previously reported that GFP-RBM22 showed a strong signal by far Western blot analysis with biotin-labeled ALG-2 but that GFP-RBM22 was not pulled down with GST-ALG-2 in the presence of Ca2+, suggesting that RBM22 exists in a form inaccessible to ALG-2, for instance tightly bound with RNA, in the cell (18). In this study, we also performed live-cell imaging of GFP-RBM22 (supplemental Movie S4), but this protein showed relatively diffuse nucleoplasmic localization in accordance with the previous report (45). Co-localization at the nuclear speckles with ALG-2 after Ca2+ stimulation was not evident for RBM22. RBM22 is evolutionarily conserved, and human RBM22 as well as the yeast ortholog Cwc2 have recently been shown to function in a catalytic center of the spliceosome by making contact with catalytically important RNA elements, including the U6 internal stem-loop and regions of U6 and the pre-mRNA intron near the 5′ splice site (46). However, CHERP is not found in fungi, and PRRs are lacking in the CHERP proteins from Caenorhabditis elegans and Drosophila, suggesting that Ca2+-dependent interaction between CHERP and ALG-2 is a unique phenomenon in vertebrates.
Variants of IP3R1 contain or lack three alternatively spliced exons (S1, S2, and S3), and expression of variants is regulated in a tissue-specific and temporally specific manner (39, 40, 47). The S2-containing form is predominant in neurons and absent from peripheral tissues (48). S2 is located between the two PKA phosphorylation sites, and alternative splicing of S2 is thought to contribute to the susceptibility of IP3R1 to phosphorylation by PKA (39, 49), but the major effect of alternative splicing of IP3R1 pre-mRNA on its physiological functions is still unknown. The levels of the alternatively spliced transcripts of IP3R1 mRNA generated by the knockdown of CHERP or ALG-2 were very low compared with the major ΔE40/41/42 transcript (Fig. 9, B–D). Alternatively spliced isoforms might have differences in sensitivities against post-translational modifications or have much longer half-lives, and they contribute to the altered IP3R1 functions more than the apparent mRNA levels. It would be interesting to know whether the degree of knockdown effects of CHERP or ALG-2 on alternative splicing is different in cell lines and cells at different developmental stages. We investigated the effects of CHERP knockdown on alternative splicing of other pre-mRNAs that encode proteins involved in Ca2+ homeostasis regulation, including sarcoplasmic reticulum Ca2+-ATPase 1 (SERCA1) and CRACR2A, but no significant changes were observed in these pre-mRNAs (data not shown). Decrease in the CHERP mRNA by the antisense method induced growth arrest in HEL cells (20). In addition to IP3R1 pre-mRNA, CHERP and ALG-2 may have proper targets of pre-mRNAs for proteins that regulate cell growth, proliferation, and Ca2+ mobilization.
Ca2+-dependent alternative splicing has been shown for pre-mRNAs of several proteins, including BK channel slo, IP3R1, plasma membrane Ca2+-ATPase, neurexin IIα, and NMDA receptor 1 (50–54). Ca2+/calmodulin-dependent protein kinase IV, a calmodulin-dependent kinase, is involved in alternative splicing influenced by Ca2+ elevation that is triggered by depolarization in neuronal cells (47, 50). Identification of CHERP as an ALG-2-interacting protein and as a splicing modulator in this study has added new players in the Ca2+-dependent alternative splicing system and should contribute to an understanding of the sophisticated post-transcriptional regulation in mammalian cells.
Acknowledgments
We thank Dr. Seiji Masuda (Kyoto University) for valuable suggestions. We also thank Keiko Yamamoto for technical assistance.
This work was supported in part by Grant-in-aid for Scientific Research (B) Project No. 23380056 from the Japan Society for the Promotion of Science (to M. M.) and Grant-in-aid from the Japan Society for the Promotion of Science Fellows Project No. 25-3584 (to K. S.).

This article contains supplemental Movies S1–S4.
- PRR
- proline-rich region
- FW
- far Western
- IP3R1
- inositol 1,4,5-trisphosphate receptor type 1
- pAb
- polyclonal antibody
- qPCR
- quantitative PCR
- RRM
- RNA recognition motif
- TG
- thapsigargin
- WB
- Western blotting
- ER
- endoplasmic reticulum
- CHERP
- Ca2+ homeostasis endoplasmic reticulum protein
- CTD
- C-terminal domain
- CID
- C-terminal domain-interacting domain
- pol
- polymerase
- SERCA
- sarcoplasmic/endoplasmic reticulum Ca2+-ATPase
- FL
- full-length
- ROI
- region of interest
- NC
- negative control
- HEL
- human erythroleukemia
- IP
- immunoprecipitation
- CIAP
- calf intestine alkaline phosphatase
- NLS
- nuclear localization signal.
REFERENCES
- 1. Vito P., Lacanà E., D'Adamio L. (1996) Interfering with apoptosis: Ca2+-binding protein ALG-2 and Alzheimer's disease gene ALG-3. Science 271, 521–525 [DOI] [PubMed] [Google Scholar]
- 2. Rao R. V., Poksay K. S., Castro-Obregon S., Schilling B., Row R. H., del Rio G., Gibson B. W., Ellerby H. M., Bredesen D. E. (2004) Molecular components of a cell death pathway activated by endoplasmic reticulum stress. J. Biol. Chem. 279, 177–187 [DOI] [PubMed] [Google Scholar]
- 3. Draeby I., Woods Y. L., la Cour J. M., Mollerup J., Bourdon J. C., Berchtold M. W. (2007) The calcium binding protein ALG-2 binds and stabilizes Scotin, a p53-inducible gene product localized at the endoplasmic reticulum membrane. Arch. Biochem. Biophys. 467, 87–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mahul-Mellier A. L., Strappazzon F., Petiot A., Chatellard-Causse C., Torch S., Blot B., Freeman K., Kuhn L., Garin J., Verna J. M., Fraboulet S., Sadoul R. (2008) Alix and ALG-2 are involved in tumor necrosis factor receptor 1-induced cell death. J. Biol. Chem. 283, 34954–34965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yamada Y., Arao T., Gotoda T., Taniguchi H., Oda I., Shirao K., Shimada Y., Hamaguchi T., Kato K., Hamano T., Koizumi F., Tamura T., Saito D., Shimoda T., Saka M., Fukagawa T., Katai H., Sano T., Sasako M., Nishio K. (2008) Identification of prognostic biomarkers in gastric cancer using endoscopic biopsy samples. Cancer Sci. 99, 2193–2199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aviel-Ronen S., Coe B. P., Lau S. K., da Cunha Santos G., Zhu C. Q., Strumpf D., Jurisica I., Lam W. L., Tsao M. S. (2008) Genomic markers for malignant progression in pulmonary adenocarcinoma with bronchioloalveolar features. Proc. Natl. Acad. Sci. U.S.A. 105, 10155–10160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maki M., Narayana S. V., Hitomi K. (1997) A growing family of the Ca2+-binding proteins with five EF-hand motifs. Biochem. J. 328, 718–720 [PMC free article] [PubMed] [Google Scholar]
- 8. Maki M., Kitaura Y., Satoh H., Ohkouchi S., Shibata H. (2002) Structures, functions and molecular evolution of the penta-EF-hand Ca2+-binding proteins. Biochim. Biophys. Acta 1600, 51–60 [DOI] [PubMed] [Google Scholar]
- 9. Missotten M., Nichols A., Rieger K., Sadoul R. (1999) Alix, a novel mouse protein undergoing calcium-dependent interaction with the apoptosis-linked-gene 2 (ALG-2) protein. Cell Death Differ. 6, 124–129 [DOI] [PubMed] [Google Scholar]
- 10. Vito P., Pellegrini L., Guiet C., D'Adamio L. (1999) Cloning of AIP1, a novel protein that associates with the apoptosis-linked gene ALG-2 in a Ca2+-dependent reaction. J. Biol. Chem. 274, 1533–1540 [DOI] [PubMed] [Google Scholar]
- 11. Yamasaki A., Tani K., Yamamoto A., Kitamura N., Komada M. (2006) The Ca2+-binding protein ALG-2 is recruited to endoplasmic reticulum exit sites by Sec31A and stabilizes the localization of Sec31A. Mol. Biol. Cell 17, 4876–4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Shibata H., Suzuki H., Yoshida H., Maki M. (2007) ALG-2 directly binds Sec31A and localizes at endoplasmic reticulum exit sites in a Ca2+-dependent manner. Biochem. Biophys. Res. Commun. 353, 756–763 [DOI] [PubMed] [Google Scholar]
- 13. Shibata H., Suzuki H., Kakiuchi T., Inuzuka T., Yoshida H., Mizuno T., Maki M. (2008) Identification of Alix-type and non-Alix-type ALG-2-binding sites in human phospholipid scramblase 3: differential binding to an alternatively spliced isoform and amino acid-substituted mutants. J. Biol. Chem. 283, 9623–9632 [DOI] [PubMed] [Google Scholar]
- 14. Maki M., Suzuki H., Shibata H. (2011) Structure and function of ALG-2, a penta-EF-hand calcium-dependent adaptor protein. Sci. China Life Sci. 54, 770–779 [DOI] [PubMed] [Google Scholar]
- 15. Shibata H., Yamada K., Mizuno T., Yorikawa C., Takahashi H., Satoh H., Kitaura Y., Maki M. (2004) The penta-EF-hand protein ALG-2 interacts with a region containing PxY repeats in Alix/AIP1, which is required for the subcellular punctate distribution of the amino-terminal truncation form of Alix/AIP1. J. Biochem. 135, 117–128 [DOI] [PubMed] [Google Scholar]
- 16. Suzuki H., Kawasaki M., Inuzuka T., Okumura M., Kakiuchi T., Shibata H., Wakatsuki S., Maki M. (2008) Structural basis for Ca2+-dependent formation of ALG-2/Alix peptide complex: Ca2+/EF3-driven arginine switch mechanism. Structure 16, 1562–1573 [DOI] [PubMed] [Google Scholar]
- 17. Shibata H., Inuzuka T., Yoshida H., Sugiura H., Wada I., Maki M. (2010) The ALG-2 binding site in Sec31A influences the retention kinetics of Sec31A at the endoplasmic reticulum exit sites as revealed by live-cell time-lapse imaging. Biosci. Biotechnol. Biochem. 74, 1819–1826 [DOI] [PubMed] [Google Scholar]
- 18. Osugi K., Suzuki H., Nomura T., Ariumi Y., Shibata H., Maki M. (2012) Identification of the P-body component PATL1 as a novel ALG-2-interacting protein by in silico and far-Western screening of proline-rich proteins. J. Biochem. 151, 657–666 [DOI] [PubMed] [Google Scholar]
- 19. O'Rourke F., Soons K., Flaumenhauft R., Watras J., Baio-Larue C., Matthews E., Feinstein M. B. (1994) Ca2+ release by inositol 1,4,5-trisphosphate is blocked by the K+-channel blockers apamin and tetrapentylammonium ion, and a monoclonal antibody to a 63 kDa membrane protein: reversal of blockade by K+ ionophores nigericin and valinomycin and purification of the 63 kDa antibody-binding protein. Biochem. J. 300, 673–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Laplante J. M., O'Rourke F., Lu X., Fein A., Olsen A., Feinstein M. B. (2000) Cloning of human Ca2+ homeostasis endoplasmic reticulum protein (CHERP): regulated expression of antisense cDNA depletes CHERP, inhibits intracellular Ca2+ mobilization and decreases cell proliferation. Biochem. J. 348, 189–199 [PMC free article] [PubMed] [Google Scholar]
- 21. O'Rourke F. A., LaPlante J. M., Feinstein M. B. (2003) Antisense-mediated loss of calcium homeostasis endoplasmic reticulum protein (CHERP; ERPROT213-21) impairs Ca2+ mobilization, nuclear factor of activated T-cells (NFAT) activation and cell proliferation in Jurkat T-lymphocytes. Biochem. J. 373, 133–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ryan T., Sharma P., Ignatchenko A., MacLennan D. H., Kislinger T., Gramolini A. O. (2011) Identification of novel ryanodine receptor 1 (RyR1) protein interaction with calcium homeostasis endoplasmic reticulum protein (CHERP). J. Biol. Chem. 286, 17060–17068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Will C. L., Urlaub H., Achsel T., Gentzel M., Wilm M., Lührmann R. (2002) Characterization of novel SF3b and 17S U2 snRNP proteins, including a human Prp5p homologue and an SF3b DEAD-box protein. EMBO J. 21, 4978–4988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Saitoh N., Spahr C. S., Patterson S. D., Bubulya P., Neuwald A. F., Spector D. L. (2004) Proteomic analysis of interchromatin granule clusters. Mol. Biol. Cell 15, 3876–3890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lin-Moshier Y., Sebastian P. J., Higgins L., Sampson N. D., Hewitt J. E., Marchant J. S. (2013) Re-evaluation of the role of calcium homeostasis endoplasmic reticulum protein (CHERP) in cellular calcium signaling. J. Biol. Chem. 288, 355–367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fu X. D., Maniatis T. (1992) The 35-kDa mammalian splicing factor SC35 mediates specific interactions between U1 and U2 small nuclear ribonucleoprotein particles at the 3′ splice site. Proc. Natl. Acad. Sci. U.S.A. 89, 1725–1729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zahler A. M., Lane W. S., Stolk J. A., Roth M. B. (1992) SR proteins: a conserved family of pre-mRNA splicing factors. Genes Dev. 6, 837–847 [DOI] [PubMed] [Google Scholar]
- 28. Manley J. L., Krainer A. R. (2010) A rational nomenclature for serine/arginine-rich protein splicing factors (SR proteins). Genes Dev. 24, 1073–1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ghosh G., Adams J. A. (2011) Phosphorylation mechanism and structure of serine-arginine protein kinases. FEBS J. 278, 587–597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kremers G. J., Goedhart J., van den Heuvel D. J., Gerritsen H. C., Gadella T. W., Jr. (2007) Improved green and blue fluorescent proteins for expression in bacteria and mammalian cells. Biochemistry 46, 3775–3783 [DOI] [PubMed] [Google Scholar]
- 31. Okumura M., Ichioka F., Kobayashi R., Suzuki H., Yoshida H., Shibata H., Maki M. (2009) Penta-EF-hand protein ALG-2 functions as a Ca2+-dependent adaptor that bridges Alix and TSG101. Biochem. Biophys. Res. Commun. 386, 237–241 [DOI] [PubMed] [Google Scholar]
- 32. Zhao Y., Araki S., Wu J., Teramoto T., Chang Y. F., Nakano M., Abdelfattah A. S., Fujiwara M., Ishihara T., Nagai T., Campbell R. E. (2011) An expanded palette of genetically encoded Ca2+ indicators. Science 333, 1888–1891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Osugi K., Shibata H., Maki M. (2013) Biochemical and immunological detection of physical interactions between penta-EF-hand protein ALG-2 and its binding partners. Methods Mol. Biol. 963, 187–200 [DOI] [PubMed] [Google Scholar]
- 34. Spector D. L., Lamond A. I. (2011) Nuclear speckles. Cold Spring Harb. Perspect. Biol. 3, pii: a000646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. la Cour J. M., Mollerup J., Berchtold M. W. (2007) ALG-2 oscillates in subcellular localization, unitemporally with calcium oscillations. Biochem. Biophys. Res. Commun. 353, 1063–1067 [DOI] [PubMed] [Google Scholar]
- 36. Neugebauer K. M., Roth M. B. (1997) Distribution of pre-mRNA splicing factors at sites of RNA polymerase II transcription. Genes Dev. 11, 1148–1159 [DOI] [PubMed] [Google Scholar]
- 37. Mathew R., Hartmuth K., Möhlmann S., Urlaub H., Ficner R., Lührmann R. (2008) Phosphorylation of human PRP28 by SRPK2 is required for integration of the U4/U6-U5 tri-snRNP into the spliceosome. Nat. Struct. Mol. Biol. 15, 435–443 [DOI] [PubMed] [Google Scholar]
- 38. Muñoz M. J., de la Mata M., Kornblihtt A. R. (2010) The carboxy terminal domain of RNA polymerase II and alternative splicing. Trends Biochem. Sci. 35, 497–504 [DOI] [PubMed] [Google Scholar]
- 39. Danoff S. K., Ferris C. D., Donath C., Fischer G. A., Munemitsu S., Ullrich A., Snyder S. H., Ross C. A. (1991) Inositol 1,4,5-trisphosphate receptors: distinct neuronal and nonneuronal forms derived by alternative splicing differ in phosphorylation. Proc. Natl. Acad. Sci. U.S.A. 88, 2951–2955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Nakagawa T., Shiota C., Okano H., Mikoshiba K. (1991) Differential localization of alternative spliced transcripts encoding inositol 1,4,5-trisphosphate receptors in mouse cerebellum and hippocampus: in situ hybridization study. J. Neurochem. 57, 1807–1810 [DOI] [PubMed] [Google Scholar]
- 41. Nucifora F. C., Jr., Li S. H., Danoff S., Ullrich A., Ross C. A. (1995) Molecular cloning of a cDNA for the human inositol 1,4,5-trisphosphate receptor type 1, and the identification of a third alternatively spliced variant. Brain. Res. Mol. Brain Res. 32, 291–296 [DOI] [PubMed] [Google Scholar]
- 42. Varjosalo M., Keskitalo S., Van Drogen A., Nurkkala H., Vichalkovski A., Aebersold R., Gstaiger M. (2013) The protein interaction landscape of the human CMGC kinase group. Cell Rep. 3, 1306–1320 [DOI] [PubMed] [Google Scholar]
- 43. Becker R., Loll B., Meinhart A. (2008) Snapshots of the RNA processing factor SCAF8 bound to different phosphorylated forms of the carboxyl-terminal domain of RNA polymerase II. J. Biol. Chem. 283, 22659–22669 [DOI] [PubMed] [Google Scholar]
- 44. Das R., Yu J., Zhang Z., Gygi M. P., Krainer A. R., Gygi S. P., Reed R. (2007) SR proteins function in coupling RNAP II transcription to pre-mRNA splicing. Mol. Cell 26, 867–881 [DOI] [PubMed] [Google Scholar]
- 45. Montaville P., Dai Y., Cheung C. Y., Giller K., Becker S., Michalak M., Webb S. E., Miller A. L., Krebs J. (2006) Nuclear translocation of the calcium-binding protein ALG-2 induced by the RNA-binding protein RBM22. Biochim. Biophys. Acta 1763, 1335–1343 [DOI] [PubMed] [Google Scholar]
- 46. Rasche N., Dybkov O., Schmitzová J., Akyildiz B., Fabrizio P., Lührmann R. (2012) Cwc2 and its human homologue RBM22 promote an active conformation of the spliceosome catalytic centre. EMBO J. 31, 1591–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Choi J. Y., Beaman-Hall C. M., Vallano M. L. (2004) Granule neurons in cerebellum express distinct splice variants of the inositol trisphosphate receptor that are modulated by calcium. Am. J. Physiol. Cell Physiol. 287, C971–C980 [DOI] [PubMed] [Google Scholar]
- 48. Schell M. J., Danoff S. K., Ross C. A. (1993) Inositol (1,4,5)-trisphosphate receptor: characterization of neuron-specific alternative splicing in rat brain and peripheral tissues. Brain Res. Mol. Brain Res. 17, 212–216 [DOI] [PubMed] [Google Scholar]
- 49. Wagner L. E., 2nd, Li W. H., Yule D. I. (2003) Phosphorylation of type-1 inositol 1,4,5-trisphosphate receptors by cyclic nucleotide-dependent protein kinases: a mutational analysis of the functionally important sites in the S2+ and S2− splice variants. J. Biol. Chem. 278, 45811–45817 [DOI] [PubMed] [Google Scholar]
- 50. Xie J., Black D. L. (2001) A CaMK IV responsive RNA element mediates depolarization-induced alternative splicing of ion channels. Nature 410, 936–939 [DOI] [PubMed] [Google Scholar]
- 51. Rozic-Kotliroff G., Zisapel N. (2007) Ca2+-dependent splicing of neurexin IIα. Biochem. Biophys. Res. Commun. 352, 226–230 [DOI] [PubMed] [Google Scholar]
- 52. Li Q., Lee J. A., Black D. L. (2007) Neuronal regulation of alternative pre-mRNA splicing. Nat. Rev. Neurosci. 8, 819–831 [DOI] [PubMed] [Google Scholar]
- 53. Xie J. (2008) Control of alternative pre-mRNA splicing by Ca2+ signals. Biochim. Biophys. Acta 1779, 438–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Krebs J., Groenendyk J., Michalak M. (2011) Ca2+-signaling, alternative splicing and endoplasmic reticulum stress responses. Neurochem. Res. 36, 1198–1211 [DOI] [PubMed] [Google Scholar]